

Coronaviruses may invade the CNS, disseminate, and participate in the induction of neurological diseases. Their neuropathogenicity is being increasingly recognized in humans, and the presence and persistence of human coronaviruses (HCoV) in human brains have been proposed to cause long-term sequelae. Using our mouse model relying on natural susceptibility to HCoV OC43 and neuronal cell cultures, we have defined the most relevant path taken by HCoV OC43 to access and spread to and within the CNS toward the brain stem and spinal cord and studied in cell culture the underlying modes of intercellular propagation to better understand its neuropathogenesis. Our data suggest that axonal transport governs HCoV OC43 egress in the CNS, leading to the exacerbation of neuropathogenesis. Exploiting knowledge on neuroinvasion and dissemination will enhance our ability to control viral infection within the CNS, as it will shed light on underlying mechanisms of neuropathogenesis and uncover potential druggable molecular virus-host interfaces.
KEYWORDS: central nervous system, coronavirus, encephalitis, neuroinvasion, neuropathogenesis, neuropropagation
ABSTRACT
Human coronaviruses (HCoVs) are recognized respiratory pathogens for which accumulating evidence indicates that in vulnerable patients the infection can cause more severe pathologies. HCoVs are not always confined to the upper respiratory tract and can invade the central nervous system (CNS) under still unclear circumstances. HCoV-induced neuropathologies in humans are difficult to diagnose early enough to allow therapeutic interventions. Making use of our already described animal model of HCoV neuropathogenesis, we describe the route of neuropropagation from the nasal cavity to the olfactory bulb and piriform cortex and then the brain stem. We identified neuron-to-neuron propagation as one underlying mode of virus spreading in cell culture. Our data demonstrate that both passive diffusion of released viral particles and axonal transport are valid propagation strategies used by the virus. We describe for the first time the presence along axons of viral platforms whose static dynamism is reminiscent of viral assembly sites. We further reveal that HCoV OC43 modes of propagation can be modulated by selected HCoV OC43 proteins and axonal transport. Our work, therefore, identifies processes that may govern the severity and nature of HCoV OC43 neuropathogenesis and will make possible the development of therapeutic strategies to prevent occurrences.
IMPORTANCE Coronaviruses may invade the CNS, disseminate, and participate in the induction of neurological diseases. Their neuropathogenicity is being increasingly recognized in humans, and the presence and persistence of human coronaviruses (HCoV) in human brains have been proposed to cause long-term sequelae. Using our mouse model relying on natural susceptibility to HCoV OC43 and neuronal cell cultures, we have defined the most relevant path taken by HCoV OC43 to access and spread to and within the CNS toward the brain stem and spinal cord and studied in cell culture the underlying modes of intercellular propagation to better understand its neuropathogenesis. Our data suggest that axonal transport governs HCoV OC43 egress in the CNS, leading to the exacerbation of neuropathogenesis. Exploiting knowledge on neuroinvasion and dissemination will enhance our ability to control viral infection within the CNS, as it will shed light on underlying mechanisms of neuropathogenesis and uncover potential druggable molecular virus-host interfaces.
INTRODUCTION
Human coronaviruses (HCoVs) are positive-stranded RNA viruses from the Coronaviridae family in the order Nidovirales that cause respiratory tract infections (1). In vulnerable patients, the infection can cause more serious pathologies, such as pneumonia, bronchiolitis, and meningitis (2–4). The medical importance of these endemic respiratory viruses circulating worldwide was long neglected until the emergence of severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome epidemics (5–8). It is now becoming clear that these viruses are not always confined to the upper respiratory tract and can indeed invade the central nervous system (CNS) under still unclear circumstances (5–10). The neuroinvasive potential of coronaviruses was further documented when RNA from endemic prototype HCoV strains OC43 and 229E was detected in human brains (11, 12). SARS-CoV particles were even found in the brains of infected patients (9). Along with their neuroinvasive properties, the neuropathogenicity of HCoV is being increasingly recognized in humans, as several recent reports associated cases of encephalitis (10), acute flaccid paralysis (13), and other neurological symptoms (14–21) with complications of acute HCoV infection. Recovery from acute infection seems not to guaranty complete clearance of the virus, as HCoV can be detected in the brains of asymptomatic healthy patients, suggesting persistence after the onset of infection (11, 12, 22). This notion is indeed supported by the findings that HCoV can chronically infect mouse brain (23, 24) and neural cell cultures (25, 26). The constant presence of the virus in the CNS and, perhaps, the concomitant inflammation were proposed to cause long-term or chronic sequelae related to the development or aggravation of chronic neurological diseases (11, 12, 22, 27–29). Given their high prevalence (4), long-term persistence, and probable neuropathogenesis, the burden of HCoV-related diseases is likely currently underestimated.
HCoV-induced neuropathologies in humans are difficult to diagnose early enough to allow therapeutic interventions. To circumvent these limitations, we developed a model of HCoV neuropathogenesis by taking advantage of the natural susceptibility of mice to neuroinvasion by the widely circulating HCoV OC43 human strain. Upon infection, mice indeed developed neurological symptoms reminiscent of the afflictions reported in several human patients (10, 13, 19–21), such as encephalitis, transient flaccid paralysis, and long-term persistence in surviving mice (23, 24, 30–33). Knowledge of the paths and underlying mechanisms governing the propagation of the virus from the upper respiratory tract to and within the CNS is currently incomplete, which hinders the elaboration of antiviral countermeasures adapted to this particular host compartment.
In our study, we defined the path taken by HCoV OC43 (34, 35) to access and spread to and within the CNS and studied the underlying modes of intercellular propagation to better understand its neuropathogenesis in both humans and mice. We present herein data showing the initial colonization of the brain at the olfactory bulbs before the spreading proceeds to the highly susceptible regions, such as the piriform cortex and other regions associated with olfaction, and then globally through the CNS. We obtained evidence supporting a critical role of the olfactory neuroepithelium during the neuroinvasion process, as chemically induced degeneration of the olfactory sensory neurons by zinc sulfate (ZnSO4) (36, 37) almost completely denied access of HCoV OC43 to the CNS. Confocal microscopy revealed virus material along axons both in brain sections and in neuronal cell cultures. We demonstrate that axonal transport is a valid propagation strategy that the virus may use to facilitate locally the infection of neuronal cells. This mode of neuron-to-neuron propagation shared mechanistic properties with the passive diffusion of released particles, although several dissimilarities were also noted. We describe for the first time the presence along axons of viral platforms whose static dynamism suggests a specialized structure polarizing locally viral release toward neighboring neurons, thus potentially fueling neuropropagation within the CNS. Our work, therefore, identifies processes that may govern the severity and nature of HCoV OC43 neuropathogenesis.
RESULTS
HCoV OC43 invades the CNS through the neuroepithelium and initiates neuropropagation at olfactory bulbs.
Intranasal inoculation of HCoV OC43 has successfully been used in mice to mimic infection of humans by aerosol droplets (33, 38, 39). To assess neuroinvasion, we took advantage of the partially immature CNS of 15-day-old mice, which results in an increased efficiency of intranasal inoculation (Table 1). We examined sagitally sectioned whole heads of intranasally infected mice. Viral antigens were detected as soon as 3 days postinfection (dpi) (Fig. 1A) in dendrite-associated cilia and the cell body of olfactory sensory neurons (OSN) (Fig. 1B and C), which reside in the olfactory epithelium in the nasal cavity. The number of infected cells further increased at 4 dpi (Fig. 1D to F). This amplification coincided with the dissemination of the virus from the initial infection from mostly the olfactory bulb and piriform cortex at day 3 postinfection (p.i.) to other regions of the CNS at day 4, including the hippocampus and brain stem, and, finally, toward the spinal cord.
TABLE 1.
Efficiency of neuroinvasion in C57BL/6 mice at different ages
| Day postinoculation | No. of positive CNS samples/total no. tested | % neuroinvasion |
|---|---|---|
| 13 | 8/12 | 66 |
| 15 | 8/12 | 66 |
| 17 | 2/12 | 17 |
| 19 | 1/12 | 8 |
FIG 1.

HCoV OC43 neuroinvasion of the CNS initiates at the olfactory bulb. Representative histological examination (confocal imaging) of viral spreading after intranasal infection of PBS-PFA-perfused 15-day-old C57BL/6 mice with 104.25 TCID50 (10 μl) of the rOC/ATCC reference virus (6 mice were tested) was performed. Detection of viral N protein (green) at 3 (A to C) and 4 (D to F) days postinfection in the nasal cavity (region 1), the olfactory bulb (region 2), (the piriform cortex region 3), the hippocampus (region 4), and the brain stem (region 5) is shown. Blue represents cell nuclei detected with DAPI (4′,6-diamidino-2-phenylindole). Successive magnifications of the insets are shown to the right of the wide views in panels A (A → B → C) and D (D → E → F).
Given the initial infection in the olfactory bulb and by analogy to other neuroinvasive viruses (40–44), we hypothesized that HCoV OC43 could access the CNS through the olfactory neuroepithelium, the subepithelial nerves surrounding the upper respiratory tract, or the bloodstream. Fifteen-day-old C57BL/6 mice (which are the best compromise to minimize swelling of the tongue while retaining high susceptibility to neuroinvasion [Table 1] [38]) were subjected to different types of inoculation, all using the same neuroinvasion-compatible dose of 104.25 50% tissue culture infective doses (TCID50)/10 μl HCoV OC43 (as defined by the control intranasal inoculation; see below). In the first group, intracerebral injection served as a positive neuroinvasion control. In the second, intranasal inoculation was used to achieve more realistic levels expected upon more natural delivery. To facilitate the contact of the virus with subepithelial nerves, the epithelial barrier was bypassed in the third group by injecting the inoculum directly into the tongue of the mice. At 5 days postinoculation, the brains were harvested, total RNA was extracted, and the virus RNA copy number per gram of tissue was determined by quantitative real-time reverse transcription-PCR (RT-PCR) using the gene encoding the membrane (M) protein as a target. Upon intranasal inoculation, 68% of the mouse brains tested were found to be positive for HCoV OC43 (Fig. 2A). As expected, the range of the absolute virus RNA copy number varied considerably from levels barely higher than the background (here determined by real-time RT-PCR from sham-infected naive brains) to very high levels reminiscent of those observed upon intracerebral injection. Subepithelial injection in the tongue led to a surprising 38% positive brains. However, the absolute viral RNA copy number remained strikingly lower than what was seen for intranasal inoculation; thus, the subepithelial nerves cannot be considered major contributors of the neuroinvasion observed upon intranasal inoculation.
FIG 2.

The olfactory bulb is the primary site of HCoV OC43 neuroinvasion. (A) Fifteen-day-old mice were inoculated with HCoV OC43 by the intracerebral (I.C.), intranasal (I.N.), or intralingual (I.L.) route. At 5 days postinoculation, the brains were harvested and the M RNA copy number was assessed by quantitative RT-PCR. Each circle represents a probed brain for a single mouse. The percentages of brains containing M RNA are shown underneath. (B) In addition to the brain, blood and livers were also collected from the same mice inoculated intranasally and probed for M RNA. (C) Mice inoculated intranasally were sacrificed after 1 to 5 days and probed for M RNA. (D) Zinc sulfate (ZnSO4) instillation 3 days before intranasal virus inoculation significantly abrogated neuroinvasion. Results are from two independent experiments. *, P < 0.05; **, P < 0.01.
We were not able to detect viral RNA from any blood samples from neuroinvaded animals at 5 days postinoculation (Fig. 2B). Viral RNA was detected in the livers of only 3 out of 9 animals whose brains were HCoV OC43 positive. Detection in the liver appeared not to be directly associated with high titers in the brain, as many mice with high titers in the brain were negative in the liver, and one liver-positive mouse had a low titer in the brain. In another experiment, 15-day-old mice were intranasally exposed to HCoV OC43 for up to 5 days. Olfactory bulbs, cortex, brain stem, spinal cord, and blood harvested at 1 to 5 days postinfection were analyzed for the presence of viral RNA (Fig. 2C). Some virus RNA was detected in blood at day 1, but the amount quickly waned to reach undetectable levels by day 2. Virus RNA was detected in the CNS of two mice at day 2 but only in the olfactory bulb, suggesting rapid neuroinvasion by the olfactory route. Surprisingly, the olfactory bulb, cortex, and brain stem of all mice were found to be positive for virus RNA by day 3, although the titers were systematically higher in the olfactory bulb. The infection globally resolved by day 5 in two mice (with a notable more durable presence in the brain stem for one of these mice) but endured in the other three mice.
We next chemically induced the destruction of the neuroepithelium through ZnSO4 exposure (36, 37). This procedure was previously shown to damage the nerve endings of the olfactory neuroepithelium (45). ZnSO4-treated mice were found to be resistant to neuroinvasion by HCoV OC43 at 5 days p.i. (Fig. 2D). Overall, these data support a direct route of neuroinvasion through the olfactory neuroepithelium and propagation through the CNS along a predictable path toward the spinal cord.
HCoV OC43 is found to be associated with axons both in vivo and in vitro.
The sequential nature of HCoV OC43 neuropropagation suggests a nonstochastic pathway of propagation within the CNS. Interestingly, immunostaining on brain sections of intracranially infected mice revealed the presence of the HCoV OC43 nucleocapsid (N) along axons, as defined by the marker βIII-tubulin (46), in several areas of the brains, such as the hippocampus, diencephalon, and cortex (Fig. 3A). Although it is more difficult to detect, accumulation of spike glycoprotein, the structural glycoprotein forming the virus crown, could also be noticed along nucleocapsid-positive axonal structures (Fig. 3B).
FIG 3.

HCoV OC43 spike and nucleocapsid proteins are associated with axons in vivo. Brains were collected from PFA-perfused mice 5 days after intracerebral inoculation, sagitally sectioned, and stained for either βIII-tubulin and nucleocapsid protein N (A) or spike glycoprotein and nucleocapsid (B) and then analyzed by confocal microscopy. Note that in panel B the image is a representative image taken in the hippocampal region of the brain. The insets on the right represent zoomed images of the areas delimited by the blue dotted boxes.
To get a deeper insight into this axonal association, the localization of HCoV was investigated in infected neuronal cell cultures. A very similar accumulation of virus material could be detected along βIII-tubulin-positive axonal structures both in infected cells of the LA-N-5 cell line (Fig. 4A), a neuroblastoma cell line previously used as a convenient model of human neurons (33, 47, 48), and in primary mouse neuronal cell (PMNC) cultures (Fig. 4B). Sequential staining for the surface spike (with polyclonal serum, before permeabilization) and total spike (with monoclonal antibody, after permeabilization) revealed strong colocalization on LA-N-5 axons (Fig. 4C, top), indicating that most spike proteins thus detected are indeed extracellularly associated with the plasma membrane of these structures. Coimmunostainings of spike proteins along with other structural viral proteins were performed to further characterize these spike-positive structures (Fig. 4C). Colocalization of N proteins with surface spike was found to be inconclusive because of the massive amount of N observed along the axons. Nevertheless, the strong colocalization of surface spike with total envelope (E) and surface hemagglutinin-esterase (HE), two other structural proteins, suggested that the aggregated signal of spike detected on axons (henceforth termed “spike platforms”) represents viral proteins coalescing in assembled particles.
FIG 4.
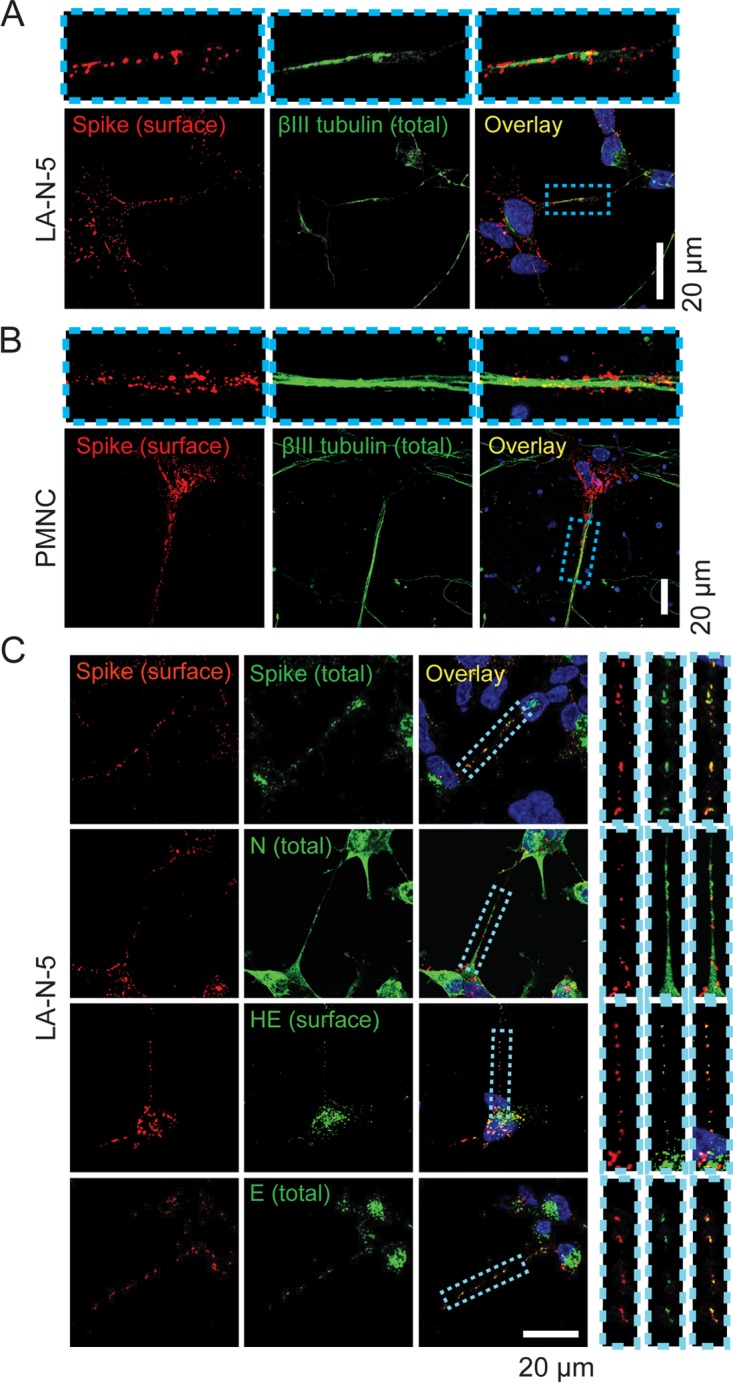
HCoV OC43 forms a viral platform at the surface of axons in neuronal cell culture. LA-N-5 cell (A) and murine primary mixed neuronal cell (B) cultures were infected at an MOI of 0.2 and 0.1, respectively. Fixed cells (on ice) were surface stained for spike glycoprotein using a specific polyclonal rabbit antibody, fixed again, permeabilized, stained for the axonal marker βIII-tubulin, and analyzed by confocal microscopy. (C) Surface spike was labeled on live LA-N-5 cells before fixation and then permeabilized (or not for E staining) and stained back for either total spike (using a monoclonal mouse antibody), total E, surface HE, or total N protein. The insets above or beside panels A to C represent zoomed images of the areas delimited by the blue dotted boxes.
HCoV OC43 spike platforms are static but temporally dynamic.
A kinetic analysis performed on synchronously infected LA-N-5 axons revealed that detection of spike at the cell body precedes detection along the axons (Fig. 5A and B), suggesting trafficking toward the axon. We next sought to determine by live cell imaging whether the surface spike platforms are mobile or not along axons. Given the difficulty with obtaining a stable infectious recombinant virus bearing a fluorescent tag on a structural protein (49, 50), we instead relied on immunostaining in LA-N-5 axons of progeny particles. Surface spike was stained by indirect immunofluorescence at 24 h postinfection (568 nm, red). At the concentration used (1/200), the polyclonal antibody was not found to be neutralizing (Fig. 5C). Serial images were taken every 5 s to generate nearly continuous 15- to 20-min videos, a compromise for the observation of both fast (∼1=μm/s) and slow (<0.1-μm/s) axonal transport events. Under these settings, spike platforms were easily perceptible. Unidirectional movement was rarely observed and never resulted in constant progression to either end of the axonal structure (Fig. 5D; see also Videos S1 and S2 in the supplemental material). Globally, the platforms exposed at the surface of axons were rather static structures that remained roughly in the same area during the span of image acquisition. Similar results were obtained in PMNCs (Video S3) and using the nonneutralizing 3-2B.2 monoclonal antibody on LA-N-5 cells (Videos S4 and S5). Restaining of the infected cells 1.5 h after the initial acquisition with a different monoclonal antispike antibody (488 nm, green) revealed dim green platforms that probably recently formed, given the lack of an older red signal (Fig. 5E, bottom, arrowheads). Several dually colored and brighter platforms (at least 1.5 h old since the time that they emitted a red signal) were observed, but many dimmer platforms emitted only a green signal. Our interpretation is that these platforms arose during the hour between the two stainings, hence representing a pool of newly formed structures. The fate of the viral platforms could be characterized using a ratiometric approach in which infected LA-N-5 cells were stained first with the polyclonal antispike antibody (red, 568 nm), chased for the indicated period of time at 37°C, and then subjected to a monoclonal antispike antibody (green, 488 nm) (Fig. 5F). To control for possible passive shedding of the bound antibodies, the same procedures were performed on infected cells fixed before the initial staining. Single-platform 568/488-nm ratios were quantified and plotted by normalization to the value at time zero. While the ratios from fixed cells remained quite stable throughout the chase period, they steadily decreased in nonfixed cells (Fig. 5G). The same downward trend was observed on both cell bodies and axons. To verify if this decline was caused by a loss of the old spike platform red signal and/or the emergence of newer green platforms, the nonratiometric absolute signal per infected cell in both channels was also plotted (normalized relative to the signal at time zero; Fig. 5H). Again, no variation in signal was observed on fixed infected cells. In contrast, the signal from the 568-nm channel decreased over time, indicating a loss and/or release of viral material, while that from the 488-nm channel remained stable. The stability of the 488-nm signal is indicative of a steady-state balance between the efflux/influx of old and new spike glycoproteins. Overall, these data suggest that spike platforms are spatially static structures that dynamically self-renew.
FIG 5.

Spike platforms on LA-N-5 axons are static along axons yet temporally dynamic. (A, B) Kinetics of spike association with axons. LA-N-5 axons were synchronously infected (MOI = 3) and then chased for the indicated time period before fixation, permeabilization, and staining for total spike using a specific monoclonal antibody. Samples were analyzed by confocal microscopy (A), and the percentage of infected cells harboring spike on their axons was then determined (B). The error bars represent the range from the means from two independent experiments. (C) Effect of the rabbit polyclonal antibody on virus propagation. LA-N-5 cells were infected at an MOI of 0.02 and cultured for 72 h in the presence of different dilutions of either a preimmune serum or the polyclonal antispike antibody. (D, E) Live cell imaging of viral platforms on axons. Infected (MOI = 0.25) LA-N-5 cells were incubated for 20 h, and then surface spike platforms (red) were immunolabeled using a polyclonal rabbit antibody followed by a red-emitting secondary antibody (Alexa Fluor 568 [AF568]). (D) Movement along the axon was assessed by live cell confocal microscopy. (E) After an hour of live cell imaging, surface spike platforms were reimmunolabeled using a specific mouse monoclonal antibody followed by a green-emitting secondary antibody (Alexa Fluor 488 [AF488]). (F) Schema representing the quantification of the fate of the platforms. (G) Ratiometric quantification of the phenomenon observed in panel E. As a control, cells were fixed immediately after the initial staining with the rabbit polyclonal antibody (red) and reimmunolabeled after the indicated time period. Red/green signal ratios were plotted from whole fields or only cell bodies or axons, as indicated. (H) Overall fluorescence decay, as observed in panel E. The absolute fluorescence signal per infected cell was plotted relative to that at time zero for each individual channel. In panels G and H, the error bars represent the standard deviation from the mean from 3 independent experiments.
HCoV OC43 undergoes cell-to-cell propagation.
We hypothesized that the dynamic viral platforms along axons may enable cell-to-cell propagation. Systems were herein designed to appreciate both the impact of free virus diffusion and cell-to-cell propagation. First, HCoV OC43 propagation was monitored in a lateral coculture system. Cells of interest were seeded on two distinct sets of coverslips. Susceptible cells on effector coverslips were infected at a low multiplicity of infection (MOI) of 0.01 with the recombinant reference HCoV OC43 (rOC) from the American Type Culture Collection (ATCC) (rOC/ATCC) for 2 h, washed extensively, and then combined in a small petri dish with a second coverslip with naive target cells (Fig. 6A). In this dish, both effectors and targets shared the same fluidic environment but did not have any direct cell contacts. Supernatants were harvested and cells were fixed at different time points, as indicated in Fig. 6B and C. The amount of released infectious virus particles and the percentage of infected cells were determined, respectively, by virus titration and immunofluorescence. Virus propagation was monitored in both human neuronal (LA-N-5) and nonneuronal epithelial (HRT-18) cell lines (51, 52). In both LA-N-5 (Fig. 6B) and HRT-18 (Fig. 6C) cells, the early 2.4% (LA-N-5 cells) and 0.3% (HRT-18 cells) of infected effector (indicative of proximal propagation) cells at 24 h rapidly progressed to reach 79% in LA-N-5 cells and 9% in HRT-18 cells by 72 h of incubation, which represented a 26- to 33-fold increase. Virus titers followed a similar trend. Colonization of distal target cells occurred but was delayed compared to the spread among effectors and coincided with the emergence of high virus titers. These data support the notion of the passive long-range diffusion of virus particles in both neuronal and epithelial cells, but also that cell proximity may somehow compensate for the low viral titers.
FIG 6.

HCoV OC43 sustains cell-to-cell propagation. (A) Schematic representation of the coculture system. (B, C) Propagation of HCoV OC43 in a coculture system in which two coverslips seeded with either infected cells (effector, MOI = 0.01) or naive target LA-N-5 cells (B) or HRT-18 cells (C) are placed side by side in a dish. Effector and target cells were cultured for up to 72 h postinfection. At the indicated time interval, supernatants were collected for titration of infectious virus (dotted black lines; refer to the y axis to the right) and cells were fixed and processed for immunofluorescence (colored lines; refer to the left y axis). The propagation of the infection, defined as the percentage of infected cells, was plotted separately for effector and target cells. (D to I) Cell-to-cell propagation assays. (D) HRT-18 cells were incubated with a fluid or semifluid inoculum (MOI = 1 for both) for 16 h, and infected cells were revealed by immunofluorescence. The resulting infectivity, defined as the percentage of infected cells, was normalized according to the fluid condition, which represented 100%. LA-N-5 cell cultures (E), primary mouse neuronal cell (PMNC) cultures (F), and HRT-18 cell cultures (G) were infected at an MOI of 0.01 and then overlaid with fluid or semifluid medium for 16 to 72 h. Cells were then fixed and immunostained. The propagation efficiency was plotted as the ratio between the percentage of infected cells at 72 h and the percentage of infected cells at 16 h. (H) A representative example of a wide field of infected LA-N-5 cells cultured in fluid and semifluid media. Colonies of infected cells are delimited by yellow dashed lines. (I) The percentages of infected cells in the whole field versus within colonies were compared. Error bars represent the standard deviation for the mean from 3 independent experiments (B, C, F) and 13 independent experiments (E), the range from the mean from 2 independent experiments (G), and the range from the mean for >12 fields or >40 colonies from 3 independent experiments (I).
To further characterize cell-to-cell propagation, methylcellulose was used to increase the density of the culture medium. A similar approach is commonly used to limit diffusion of particles when performing titration by plaque-forming assays of a broad array of viruses (53–56). A single-cycle assay over a period of 16 h in the semifluid medium resulted in an ≈99% decrease in the inoculum infectivity (Fig. 6D), thus confirming that methylcellulose successfully hinders particle diffusion. Next, propagation of HCoV OC43 under such restrictive conditions was assessed for 72 h and quantified by immunofluorescence from several low-magnification fields. Semifluid medium decreased substantially the propagation in LA-N-5 cell (Fig. 6E) and PMNC (Fig. 6F) cultures, yet residual propagation attributed to cell-to-cell propagation endured nonetheless. A similar trend was observed for HRT-18 cells (Fig. 6G), indicating that cell-to-cell propagation is not solely observed in neuronal cells. A closer look revealed virus propagation in the vicinity of colonized areas (Fig. 6H). Plotting the percentage of infected cells within these colonies compared to that within whole fields indicated that semifluid medium has, in fact, little (although a significant) effect on virus propagation locally while preventing the distal spread (Fig. 6I). Overall, these data suggest that HCoV OC43 indeed undergoes virus particle diffusion but that some productive cell-to-cell propagation occurs as well.
HCoV OC43 neuron-to-neuron propagation can be modulated.
The sensitivity of cell-to-cell propagation versus that of free diffusion of particles to various cues was compared in this system. Given that sialic acid has been shown to influence HCoV OC43 infection (57–59), the effect of various recombinant sialic acid binding lectins was compared (Fig. 7A). CCA (from Cancer antennarius) did not affect either mode of propagation, whereas MAA (from Maackia amurensis) impacted both types of propagation very similarly. SNA-1 (from Sambucus nigra) had an exacerbated negative impact on cell-to-cell propagation compared to free virus diffusion. Inversely, free virus diffusion was more affected by the wheat germ agglutinin (WGA). We also tested the effect of recombinant HCoV OC43 hemagglutinin-esterase (HE) protein (Fig. 7B), which removes acetyl groups from O-acetylated sialic acid to presumably free aggregated particles at the surface, thus promoting both virus entry and exit (51, 60–63). The recombinant wild-type (WT) HE protein severely impaired propagation under both conditions, whereas the HE S40T recombinant protein defective for acetylesterase activity did not. Similarly, both types of propagation were sensitive to the clone 4-3E.4 blocking antibody, recognizing the hypervariable region of spike (spanning approximately between amino acids 450 and 600) (Fig. 7C). The impact of the neutralizing antibody was less drastic in semifluid medium; however, we cannot exclude the possibility that the semifluid medium per se interfered with the neutralizing process. Doubling of the cell density tended to stimulate propagation but did so only under semifluid conditions, as no benefit was seen under fluid conditions (Fig. 7D), presumably because of the higher frequency of cell-to-cell contacts.
FIG 7.

Modulation of cell-to-cell and free virus infection. All experiments were performed using the same general procedures. LA-N-5 cells were infected, overlaid with semifluid or fluid medium with the indicated supplements, cultured for 72 h in fluid or semifluid medium, fixed, and immunostained to score the infection efficiency by immunofluorescence. Different cues were applied. (A, B) Various sialic acid-binding lectins (A) or recombinant WT HE or mutant HE S40T protein (devoid of acetylesterase function) (B) was added to the overlay medium. (C) The overlay media were supplemented with a nonneutralizing (3-2B.2) or a neutralizing (4-3E.4) monoclonal antibody, a control isotype, or preimmune rabbit serum, which served as a control for antispike serum. (D) LA-N-5 cells were seeded at 7,000 or 14,000 cells/cm2 before infection. (E) LA-N-5 cells were infected (MOI = 0.01) with various HCoV OC43 variants encoding the indicated spike mutants before the overlay medium was applied. Data were plotted as either the ratio between the infectivity for infected cells and the infectivity for the untreated controls (A to C) or between the percentage of infection at 72 h and the percentage of infection at 16 h (D and E). The error bars represent the range from the mean from two experiments (D) or the standard deviation from 3 (A to C) or 3 or 4 (E) experiments. *, P < 0.05; ns, not significant.
The major influence of the HCoV OC43 spike glycoprotein on virus propagation was demonstrated in previous studies (24, 26, 33, 47, 64). The fast propagation through the CNS of the spike amino acid 183 to 241-encoding (rOC/S183–241) protein coincided with exacerbated neurovirulence (47, 64), whereas the attenuated spike G758R-encoding (rOC/SG758R) protein was found to reach the brain stem with delayed kinetics (24, 33). In all these studies, the prototypical variant (herein termed WT) obtained from the American Type Culture Collection (ATCC) always appeared to be moderate in both propagation and neurovirulence, hence generally considered a convenient intermediate point of comparison. To verify if the two modes of propagation are influenced equivalently by the crucial viral factor spike, the propagation of these three variants was compared under both fluid and semifluid conditions (Fig. 7E). The rOC/S183–241 virus propagated 2 to 3 times faster than rOC/ATCC under both conditions, suggesting a more rapid and/or efficient replication cycle that benefits equivalently both free-particle diffusion and neuron-to-neuron transfer. In contrast, rOC/SG758R failed to propagate under semifluid conditions, while its propagation remained very comparable to that of rOC/ATCC under fluid conditions, indicating that a single mutation on the spike glycoprotein is sufficient to abrogate specifically cell-to-cell propagation. Therefore, although they were similar in several molecular aspects, cell-to-cell propagation and free virus diffusion remain two different processes that have distinct genetic determinants.
Axonal transport enables HCoV OC43 neuron-to-neuron propagation.
To verify if cell-to-cell propagation in neurons relies on axonal transport, we tested the effect of the microtubule-disturbing agents vinblastine (destabilizing) and paclitaxel (stabilizing) (Fig. 8A) on HCoV OC43 propagation under both fluid and semifluid conditions. Under fluid conditions, these drugs did not affect virus propagation in LA-N-5 cells over the range of concentrations tested during the 72 h of incubation, indicating a minimal effect of these drugs on the virus general egress. In sharp contrast, both drugs decreased virus propagation in a dose-dependent manner under semifluid conditions, indicating that microtubules are preferentially required for neuron-to-neuron propagation (Fig. 8B). None of the drugs affected propagation in the epithelial HRT-18 cell line (Fig. 8C), excluding a nonneuronal pleiotropic effect. Significantly fewer spike platforms could be observed on the axonal structures of infected LA-N-5 cells upon a 24-h treatment with paclitaxel (Fig. 8D and E), confirming interference with axonal transport. The neuron-specific requirement for microtubules under semifluid conditions indicates that axonal transport plays an important role during neuron-to-neuron HCoV OC43 propagation.
FIG 8.

Axons allow neuron-to-neuron propagation. (A) LA-N-5 cells were treated with the indicated concentration of vinblastine (Vin) or paclitaxel (Pac) in fluid medium for 72 h and then stained for spike glycoprotein (red), βIII-tubulin (green), and nuclei (blue). Representative pictures taken by confocal microscopy are shown. (B, C) LA-N-5 cells (B) and HRT-18 cells were infected at an MOI of 0.01 and cultured for 72 h in fluid or semifluid medium containing the specified concentration of vinblastine or paclitaxel. Infected cells were then fixed and immunostained to determine the percentage of infection. (D, E) Effect of paclitaxel on the axonal association of spike platforms. (D) Infected LA-N-5 cells (MOI = 0.2) were treated with 250 nM paclitaxel for 20 h, fixed, and immunostained. (E) Data plotted in the graph are the amount of spike platforms per micrometer of infected axons. The error bars represent the standard deviation from the mean from 3 independent experiments (B, C) or the standard deviation for >200 individual axonal structures (E). *, P < 0.05.
DISCUSSION
The widely circulating respiratory HCoV OC43 is naturally neuroinvasive and can disseminate within the CNS. In line with many other neuroinvasive viruses (40–42, 44), HCoV OC43 probably finds its way to the CNS through the olfactory tract, probably along the olfactory nerve. This is supported by our detection of HCoV OC43 in the OSN and the olfactory bulb early upon intranasal exposure (Fig. 1A to C and 2C) and the failed neuroinvasion in ZnSO4-treated mice (Fig. 2D). The rare presence of HCoV OC43 in the liver of intranasally inoculated mice and its undetectable level in the bloodstream at days 2 to 5 suggest that neuroinvasion occurs independently of the low level of systemic infection (Fig. 2B). In light of the weak RNA signal in the CNS upon intralingual infection, we cannot formally rule out the possibility of neuroinvasion by the sensory or autonomic circuitry. Indeed, the trigeminal nerve possesses nociceptive neurons, which are present in the respiratory epithelium of the nasal cavity (65) and can theoretically be infected by HCoV OC43 to invade the CNS, as suggested for mouse hepatitis virus (MHV) years ago (43). From the periphery (nasal cavity), the trigeminal nerve mainly follows a neuronal circuit which ends at the brain stem, which could explain our data related to intralingual inoculation of the virus. The stronger colonization of the brain stem than the cortex at day 3 is also consistent with this interpretation, although it is unclear if the lower RNA titers in the cortex are caused by a delayed infection or a reduced tropism. Nevertheless, neuroinvasion through the OSN appears to be dominant because (i) these cells are clearly the most susceptible to infection in the nasal cavity and the olfactory bulb is the first CNS tissue colonized upon intranasal inoculation (Fig. 1A and 2C); (ii) microscopy data show a clear hierarchal trend from the olfactory bulb toward the brain stem and spinal cord (Fig. 1); (iii) the magnitude of infection both in the amount of infected cells (Fig. 1) and in virus RNA titers (Fig. 2C) is systematically higher in this zone of the CNS than in the cortex, brain stem, and spinal cord; and (iv) ZnSO4, reported to destroy olfactory neuroepithelium (45), abrogates neuroinvasion.
Making use of our animal model and neuronal cell cultures, we herein characterized a new mechanism of HCoV OC43 propagation that appears to have in vivo relevance for neuropropagation. From the olfactory bulb, neuropropagation along the various axonal connections through the CNS is supported by key observations: first, the seemingly dynamic association of structural viral material along axons both ex vivo and in vitro (Fig. 3 to 5); second, the existence of a neuron-to-neuron mode of propagation particularly efficient at a high cellular density (Fig. 6 and 7); and third, the disruption of neuron-to-neuron propagation by microtubule-disrupting agents (Fig. 8). Nevertheless, the contribution of freely diffusing particles must not be overlooked. Isolated infected cells could indeed be observed scattered through the brain at 3 to 4 days post-intranasal inoculation. The latter type of propagation appeared to be dominant in our cell culture systems. However, in vitro systems do not necessarily reflect perfectly the situation prevailing in vivo. For instance, various physical barriers impossible to mimic in vitro are expected to limit diffusion of free particles in vivo. In culture, particles sediment on susceptible cells as a two-dimensional layer, which dos not reflect properly the dynamic heterogeneous three-dimensional environment found in vivo. As suggested by the positive effect of cell-cell contacts on HCoV OC43 propagation under semifluid conditions (Fig. 7D), the interneuronal connection within the CNS is likely to boost the neuron-to-neuron propagation efficiency compared to the free particle diffusion efficiency in a way difficult to predict in vitro, where neuronal connections are not necessarily fully mature. Despite these limitations, local neuron-to-neuron propagation in vitro appeared to be only twice less efficient. We hypothesize that freely diffusing founder viral particles initiate stochastic infections in naive regions of the brain, thus establishing a small viral colony around which to expand at the rhythm of the neuron-to-neuron propagation. Such local propagation combined with a subsequent round of distal colonization with new founder virus particles would indeed ensure a fast spread throughout the CNS.
Our microscopy approaches shed light on the mechanisms by which HCoV OC43 could leave the axon to promote neuron-to-neuron propagation. Accumulations of membrane-embedded spike, E, and HE structural proteins, herein termed spike platforms, were found to decorate axons in vitro. These platforms appeared to be spatially static, probably because they were anchored to elements of the cytoskeleton. Even if the platforms observed by live cell imaging were similar to those observed postfixation (Fig. 5A), we cannot rule out the possibility that some sort of motility was impaired by the bivalent antibody used during our labeling procedure. However, the two antibodies used (anti-OC43 spike serum and monoclonal antibody 3-2B.2) showed no neutralizing capabilities under either fluid or semifluid conditions (Fig. 7C), thus precluding any impact of such theoretical motility on neuron-to-neuron propagation. This observed staticity does not preclude dynamism, as new viral products compensate for the gradual loss of older proteins, which is indeed reminiscent of sites of assembly and virus release. The implied anterograde transport of virus material along axons toward exit sites could be explained by two generic models (reviewed in reference 66): (i) virus assembly at the cell bodies and then transport toward release sites (e.g., as in the Herpesviridae) or (ii) convergence along axons of structural subunits in discrete sites where assembly and release can be achieved (e.g., as in the Rhabdoviridae). Our inability to detect nucleocapsid in viral platforms may suggest that its recruitment to spike/E/HE assembled scaffolds is a terminal process quickly resolved by the release of the particle, therefore precluding its transient observation. This possibility is in line with the second model mentioned above. However, we cannot exclude the possibility that the strong nucleocapsid signal across the axon cytosol, as seen in other previous studies (23, 67, 68), masks a dimmer signal corresponding to a nucleocapsid coassembled with the other structural proteins. It is interesting to note that viral platforms are phenotypically very similar to those derived from the infection of neurons by the rabies virus (RABV), pseudorabies virus (PRV), and herpes simplex virus 1 (69–72), two alphaherpesviruses whose accumulation in varicosities enriched in presynaptic markers gave rise to the yet unconfirmed hypothesis of transsynaptic viral transfer (71, 73). Whether this transsynaptic propagation instead of a nonsynaptic stochastic secretion along the axons could apply for HCoV OC43 remains to be verified. To further examine these possibilities, the development of tools enabling more accurate and sensitive live monitoring of HCoV OC43 propagation will be of paramount importance.
Our data suggest that propagation via diffusion of free particles and neuron-to-neuron transfer share several mechanistic similarities, as seen for other viruses (reviewed in reference 74). For instance, the two tested modes of propagation tended to be similarly affected by antispike neutralizing antibody, recombinant HE, and sialic acid-binding lectins (Fig. 7A to C). Furthermore, spike amino acid 183 to 241-encoding virus (rOC/U183–241) showed a 2- to 3-fold increased propagation in both cases (Fig. 7E). Subtle divergences between the two modes of propagation were nevertheless noticed. The negative impact of SNA-I was significantly more pronounced in semifluid medium. Moreover, spike G758R-encoding virus (rOC/SG758R) failed to propagate from neuron to neuron. The reasons behind this incapacity are still unclear but may underline the privileged contribution of some still unknown attachment factor(s) to a specific mode of propagation. A precedent may exist, as the differential expression profile of MHV cellular receptors appeared to modulate its spread in a manner reminiscent of neuron-to-neuron propagation (75). Our previous work demonstrated that HCoV OC43 spike G758R and spike amino acid 183 to 241 mutants conferred attenuated and exacerbated neurovirulence in mice, respectively, despite equivalent virus loads in the brain at their onset of infection (24, 33, 64). A key observation was that HCoV OC43 spike G758R was affected in its capacity to reach the spinal cord, whereas the HCoV OC43 spike amino acid 183 to 241 mutant reached the spinal cord even more efficiently than the reference virus. Therefore, the efficiency of neuron-to-neuron propagation is associated with neuropathogenesis. It raises the intriguing possibility that neuron-to-neuron propagation efficiency defines the outcome of infection by enabling the colonization of areas of the CNS, such as the spinal cord, otherwise difficult for HCoV OC43 to reach by stochastic diffusion. A similar interpretation was drawn in a report that compared the propagation of the A59 strain of MHV to the more virulent JHM.SD strain, described as relying more on neuron-to-neuron transfer (75). The virus probably does not benefit from this exacerbated neurovirulence, as the HCoV OC43 spike amino acid 183 to 241 mutant was isolated in vitro and not in vivo (24). In fact, the spike G758R variant is predominantly found in primary isolates (33), suggesting, unsurprisingly, that low neurovirulence, although associated with limited propagation, ensures host survival and favors long-term persistence. Therefore, slowing the progression of HCoV OC43 toward the brain stem and spinal cord by hindering neuron-to-neuron propagation may represent a strategically valid approach to avoid neuronal damage. Indeed, additional studies are warranted to quantify the impact of neuron-to-neuron propagation during HCoV neuropathogenesis. In this context, the identification and characterization of the molecular determinants distinctly regulating the two aforementioned phenomena, including their receptor(s) and attachment factor(s), will be the key to a better understanding of their associated neuropathology and eventually to the elaboration of antiviral countermeasures.
MATERIALS AND METHODS
Ethics statement.
All mouse experiments were approved by the Institutional Animal Care and Use Ethics Committee (IACUC) of the Institut National de la Recherche Scientifique (INRS) and conform to the Canadian Council on Animal Care (CCAC). Animal care and use protocols numbers 1304-02 and 1205-03 were issued by the IACUC of INRS for the animal experiments described herein. All the experiments with both wild-type viruses and mutant viruses (viruses with an S protein with a potential gain of function) were approved by the institutional biosafety committee (IBC) at INRS (certificate 2013-07), as all biosafety level 2 safety level measures were applied to prevent infection of all laboratory workers and the potential spread of viruses.
Viruses and cell lines.
The recombinant HCoV OC43 virus (rOC/ATCC) (26) harbors the same sequence as the reference virus HCoV OC43 (VR-759), obtained in the 1980s from the American Type Culture Collection (ATCC). HCoV OC43 rOC/S183–241 and rOC/S G758R were previously described (33, 47, 64). Virus stocks were generated by transfecting each cDNA clone in BHK-21 cells, amplified by two passages in the HRT-18 cell line, and sequenced to confirm that no mutations were introduced during the process.
The human ileocecal colorectal adenocarcinoma HRT-18 cell line (a gift from the late David Brian, University of Tennessee) was cultured in minimal essential medium alpha (MEM-alpha; Life Technologies) supplemented with 10% (vol/vol) fetal bovine serum (FBS; Multicell) and was used to produce viral stocks and perform experiments. The human neuroblastoma LA-N-5 cell line (a kind gift of Stephan Ladisch, George Washington University School of Medicine) was maintained in RPMI medium supplemented with 15% (vol/vol) fetal bovine serum (FBS), 10 mM HEPES, 1 mM sodium pyruvate, and 100 μM nonessential amino acids (Gibco-Invitrogen). LA-N-5 cells were differentiated into human neurons as previously described (48). Briefly, 1.25 × 104 LA-N-5 cells were seeded in RPMI supplemented with 20% (vol/vol) FBS, 10 mM HEPES, 1 mM sodium pyruvate, and 100 μM nonessential amino acids on 24-well glass coverslips that had been coated beforehand with 1/20 Matrigel matrix basement membrane (Corning) following the manufacturer's procedures. On the next day and every 2 days for 8 days, the medium was replaced with fresh Dulbecco modified Eagle medium (DMEM) supplemented with 10% (vol/vol) FBS, 10 mM HEPES, 1 mM sodium pyruvate, 100 μM nonessential amino acids, 50 μg/ml gentamicin (Wisent), and 10 μM all-trans-retinoic acid (Sigma-Aldrich).
Mixed primary cultures of mouse CNS cells were prepared as previously described (33). Briefly, the cortex and hippocampus from 14- to 16-day-old embryonic CD1 pup brains were harvested. The tissues were dissociated in Hanks balanced salt solution plus trypsin-EDTA (0.5%) (ratio, 10:1) for 15 min at 37°C and then by gentle up-and-down pipetting. The debris was decanted, and the supernatants were transferred to Neurobasal medium (Invitrogen) supplemented with 1 mM GlutaMAX-I (Life Technologies), 10 mM HEPES buffer, 1 mM sodium pyruvate, 2% (vol/vol) B27 supplement (Life Technologies), 50 μg/ml gentamicin, and 10% (vol/vol) horse serum (Life Technologies). Cells were then seeded at 2 × 105 cells/cm2 and grown on poly-d-lysine (final concentration, 50 μg/ml)-treated glass coverslips in the same medium. From the next day until they were ready for infection (6 to 7 days), the medium was replaced every 2 days with fresh Neurobasal medium without horse serum.
Evaluation of viral neuroinvasiveness.
Fifteen-day-old mice were subjected to intranasal inoculation (injection of 5 μl in each nostril), intralingual inoculation (single injection of 10 μl using a Hamilton syringe laterally at the center of the tongue), or intracranial inoculation (as previously described [24]) of rOC/ATCC using 104.25 50% tissue culture infective doses (TCID50) per 10 μl. Sham-infected mice received phosphate-buffered saline (PBS). To block neuroinvasion, 5 μl of ZnSO4 (0.17 M) was instilled in both nostrils of the mice 3 days before intranasal infection with the virus. Intralingual and intracerebral inoculations were performed under peritoneal anesthesia of ketamine-xylazine (ketamine at 200 mg/kg of body weight and xylazine at 10 mg/kg) (38). Whole infected brain, liver, and blood were harvested 5 days later (Fig. 2B) or the olfactory bulb, cortex, brain stem, spinal cord, and blood were harvested at between 1 and 5 days postinfection (Fig. 2C) from peritoneally anesthetized mice (ketamine at 200 mg/kg and xylazine at 10 mg/kg) and frozen at −80°C. Whole-brain tissue was shredded/lysed by extensive agitation in 1 ml QIAzol lysis reagent (Qiagen) supplemented with shredding beads. Total brain RNA was extracted by the QIAzol-chloroform-propanol manufacturer's procedure, the amount was evaluated using an ND1000 spectrophotometer (NanoDrop), and the extracted RNA was frozen at −80°C. Virus RNA copy numbers were quantified in triplicate by real-time RT-PCR using a TaqMan RNA-to-cycle threshold (CT) 1-step kit (Applied Biosystems/Life Technologies) in a 20-μl reaction mixture with 10 μl 2× TaqMan RT-PCR mix (containing carboxy-X-rhodamine as a passive reference dye), 900 nM forward and reverse primers targeting a 68-bp region of the HCoV OC43 M gene (76) (forward primer OC43-FP [5′-ATGTTAGGCCGATAATTGAGGACTAT-3′, nucleotides [nt] 433 to 458]; reverse primer OC43-RP [5′-AATGTAAAGATGGCCGCGTATT-3′, nt 479 to 500]), 200 nM 6-carboxyfluorescein (FAM)–black hole quencher 1 probe OC43-TP (FAM-5′-CATACTCTGACGGTCACAAT-3′, nt 459 to 478), 0.5 μl 40× TaqMan reverse transcriptase enzyme mix, and 800 ng extracted brain RNA. Serially diluted cRNA standards were used for the generation of a standard curve. Amplification and detection were performed in a StepOnePlus real-time PCR system apparatus and analyzed with StepOne software (version 2.3; Applied Biosystems). The limit of detection was defined as the average signal obtained for the corresponding negative controls for each organ.
Infection of cell lines and primary mouse CNS cultures.
The HRT-18 cell, LA-N-5 cell, and primary mouse neuronal cell (PMNC) cultures were infected with the indicated recombinant HCoV OC43 at the indicated multiplicity of infection (MOI) for 2 h in MEM-alpha supplemented with 1% (vol/vol) FBS (HRT-18 cells), Neurobasal medium with B27–GlutaMAX-I (PMNCs), or DMEM supplemented with 1% (vol/vol) FBS (LA-N-5 cells). The inoculum was then discarded and replaced with fresh MEM-alpha supplemented with 1% (vol/vol) FBS (HRT-18 cells), fresh Neurobasal medium with B27–GlutaMAX-I (PMNCs), or fresh DMEM supplemented with 2.5% (vol/vol) FBS (LA-N-5 cells) and incubated for up to 72 h, as indicated above. During infection and further incubation, LA-N-5 cells and PMNCs were kept at 37°C and HRT-18 cells were kept at 33°C.
Titration of infectious particles.
Virus titers from cell culture supernatants were determined by an indirect immunoperoxidase assay (IPA) on HRT-18 cells, as previously described (77). Briefly, HRT-18 cells were infected from serially diluted virus-containing supernatants for 2 h and incubated for 96 h. Cells were methanol fixed and incubated with the mouse primary antibody 4-3E.4 (dilution 1/50), which detects the S protein of HCoV OC43. Immunocomplexes were detected by incubating with a secondary horseradish peroxidase-conjugated goat anti-mouse immunoglobulin antibody diluted 1/500 (Kirkegaard & Perry Laboratories) and revealed with 0.025% (wt/vol) 3,3-diaminobenzidine tetrahydrochloride (Sigma-Aldrich) and 0.01% (vol/vol) hydrogen peroxide in 1× PBS. Infectious virus titers were calculated by the Karber method (77).
Immunofluorescence.
Confocal immunofluorescence was performed either after intracerebral infection of 22-day-old mice, for which perfusion with 4% (wt/vol) paraformaldehyde (PFA) was performed at 5 days postinfection, or after intranasal infection of 15-day-old mice, for which perfusion was performed every day for a 4-day period. For intracerebral infection, brain sections were sagitally sliced into 60-μm-thick sections with a Lancer Vibratome, and for intranasal infection, heads were harvested and the fur, skin, and lower jaw were removed. Whole heads were decalcified in 6% EDTA (pH 8) at 4°C for 5 days and then transferred to 30% sucrose solution, being processed 3 days later. Serial sections were collected in PBS, treated for 10 min with H2O2 to disrupt erythrocytes, washed twice for 5 min each time in PBS, and permeabilized for 2 h in PBS supplemented with 0.1% (vol/vol) Triton X-100. Sections were further incubated for 1 h in 0.1% Triton–PBS supplemented with 1% (vol/vol) horse serum and then overnight at 4°C in 0.05% Triton–PBS containing 1% horse serum and OC43-cross-reacting ascetic anti-hepatitis E virus (anti-HEV) N (1/500) (33), βIII-tubulin (1/1,000; Abcam), and/or a polyclonal anti-OC43 spike serum (1/250) generated from rabbit immunization with a fragment of the spike glycoprotein encompassing amino acids 16 to 334. Sections were washed 3 times (for 15 min each time) in 0.05% Triton–PBS and incubated for 2 h in 0.05% Triton–PBS supplemented with 1% horse serum and 1/500 adequate Alexa Fluor 488- or Alexa Fluor 568-coupled secondary antibodies (Life Technologies). Immunostained sections were washed twice (5 min each time) in 0.05% Triton–PBS, counterstained for nuclei with 10 μg/ml Hoechst (Invitrogen), and then washed again 4 times (for 15 min each time) in 0.05% Triton–PBS. Sections were then mounted in Prolong Diamond antifade mounting medium (Molecular Probes) on glass slides and imaged on a Zeiss LSM780 confocal microscope equipped with a 30-mW 405-nm diode laser, a 25-mW 458/488/514-nm argon multiline laser, and 20-mM DPSS 561-nm laser mounted on a Zeiss Axio Observer microscope at a magnification of ×630 (Carl Zeiss Microimaging). All immunostaining steps were carried out at room temperature with agitation, unless otherwise stipulated.
Confocal microscopy and live cell imaging.
The presence of spike platforms on LA-N-5 axons was evaluated by fixing the cells on ice using 4% PFA (Electron Microscopy Sciences). Fixed cells were washed with PBS and then incubated with 1/200 polyclonal anti-OC43 spike for 1 h at 37°C, washed twice, and reincubated for 45 min at room temperature with adequate 1/500 Alexa Fluor 568 antibody. Following 3 PBS washes, surface-stained cells were permeabilized for 5 min with 0.1% Triton–PBS and sequentially stained for 1 h at room temperature for total βIII-tubulin using specific antibodies (1/1,000; Abcam) and the appropriate 1/500 Alexa Fluor 488 secondary antibody along with 2 μg/ml Hoechst. For PMNCs, surface staining was conducted at 37°C for 15 in 2% FBS–Neurobasal medium supplemented with 1/200 anti-OC43 spike polyclonal serum, washed twice for 2 min each time in medium, and then fixed for 5 min with 4% PFA. Fixed cells were incubated for 30 min at room temperature with the appropriate Alexa Fluor 568 antibody diluted 1/1,000 in PBS. Cells were washed twice, fixed again for 5 min, and permeabilized with 0.1% Triton–PBS for 5 min at room temperature. Cells were then stained for total βIII-tubulin for 1 h at 37°C in 0.1% (vol/vol) Triton–PBS, washed twice in PBS, and incubated for 1 h at room temperature with the appropriate 1/1,000 Alexa Fluor 488 antibody and 2 μg/ml Hoechst.
To assess the colocalization of surface HCoV OC43 spike glycoprotein with other virus structural proteins, infected LA-N-5 cells were incubated at 37°C for 15 min with either 1/200 polyclonal anti-OC43 spike serum (generated from rabbit immunization with a fragment of the spike glycoprotein encompassing amino acids 16 to 334), 1/2 monoclonal 4-3E.4 anti-OC43 spike antibody, and/or 1/200 monoclonal KD9-40 anti-bovine coronavirus HE protein antibody (kindly provided by Sylvia van Drunen Littel-van den Hurk, University of Saskatchewan, Saskatchewan, Canada), reportedly recognizing OC43 HE (51), in 2.5% FBS–DMEM. Cells were washed twice for 2 min each time in medium and then reincubated for 15 min at room temperature in 2.5% FBS–DMEM supplemented with 1/500 Alexa Fluor 568 secondary antibody. Cells were fixed in 4% PFA after 3 2-min washes in medium. Where total costainings were needed, surface-stained cells were next permeabilized in 0.1% (vol/vol) Triton–PBS for 5 min at room temperature and incubated for 2 h at 37°C in 5% (vol/vol) FBS–PBS supplemented with 1/500 polyclonal anti-OC43-E protein, 1/2 monoclonal 4E11.3 anti-HEV N protein hybridoma supernatant, 1/2 4-3E.4 anti-OC43 spike hybridoma supernatant, or 1/500 polyclonal anti-OC4 spike serum. Cells were then washed twice and reincubated with adequate Alexa Fluor 488 antibodies diluted 1/500 in PBS. Nuclei were counterstained with 2 μg/ml Hoechst.
Immunostained cells (LA-N-5 cells or PMNCs) were all mounted on glass slides with Prolong Diamond antifade mounting medium after 3 final PBS washes. The images shown in the figures are nonoverlapping maximal projections generated by ImageJ software from single z-stacks exported from Zen black software (Carl Zeiss Microimaging).
LA-N-5 cells and PMNCs were seeded on Matrigel or a collagen–poly-d-lysine-coated live cell imaging dish (MatTek Laboratories), differentiated, and infected as indicated above. Real-time imaging was performed by live surface staining of spike platforms at 37°C for 15 min with either 1/200 polyclonal anti-OC43 spike serum or 1/4 monoclonal 3-2B.2 anti-OC43 spike antibody as indicated above and immediately imaged at 37°C for up to 35 min at ≈0.125 frame/s using Zen (version 2.1) software. The presented images and videos are thick acquisitions from a maximal pinhole aperture obtained and processed using Zen black software (Carl Zeiss Microimaging).
All images and videos were acquired with a Zeiss LSM780 confocal microscope equipped with a 30-mW 405-nm diode laser, a 25-mW 458/488/514-nm argon multiline laser, and a 20-mM DPSS 561-nm laser mounted on a Zeiss Axio Observer microscope at a magnification of ×630 (Carl Zeiss Microimaging). For real-time imaging, the Axio Observer microscope was further equipped with temperature-, humidity-, and CO2-controlling stage-top incubators.
For ratiometric measurements, 20-h-infected LA-N-5 cells were initially surface stained live as described above first using the polyclonal antispike serum and Alexa Fluor 568 antibodies. Cells were then chased (or not) for up to 3 h at 37°C before being subjected to a similar second surface staining using instead the monoclonal 4-3E.4 spike antibody and Alexa Fluor 488 antibodies. To account for the loss of immunocomplexes during the biologically permissive chase period, parallel samples were fixed with 4% PFA before the initial staining. The same acquisition settings were used for all samples. Images were exported, axons and bodies were separated in distinct files using Photoshop CS5.1 software (Adobe), and quantification from maximal z-stack projections was generated by ImageJ software. The absolute signal per infected cell was calculated by dividing the bulk signal in each field by the number of nuclei. The resulting values were normalized to the corresponding value at time zero. Brightness and contrast values remained unchanged through the process to avoid biased quantification.
Virus propagation assay.
For virus propagation assays, cells were always seeded on glass coverslips, coated or not following the seeding procedures (see above). Immediately after infection of HRT-18 cells, LA-N-5 cells, or PMNCs (as described above), the inocula were replaced with propagation medium. Propagation medium was prepared as follows: 2× DMEM (Multicell) was supplemented with 2 mM GlutaMAX-I, 20 mM HEPES buffer, 100 μg/ml gentamicin, 2 mM sodium pyruvate, and 200 μM (vol/vol) nonessential amino acids. FBS at 2% (vol/vol), 5% (vol/vol), or 4% (vol/vol) B27 plus cobalamin at 0.013698 mg/ml was also added, respectively, for HRT-18 cells, LA-N-5 cells, and PMNCs. Supplemented 2× DMEM was then diluted 1/2 in either 4% (wt/vol) methylcellulose (semifluid medium) or H2O (fluid medium). Cells were overlaid with semifluid or fluid medium and incubated at 37°C for the time periods indicated above, after which the supernatants were discarded and cells were directly fixed for 15 min in 4% PFA. Where specified, the following compounds were also added to fluid and semifluid medium following a previously described procedure (the final concentrations are indicated): 2 to 250 nM vinblastine or paclitaxel (Sigma-Aldrich); 1/200 anti-OC43 spike polyclonal serum, 1/4 hybridoma supernatant containing anti-OC43 spike nonneutralizing 3-2B.2, and neutralizing 4-3E.4 monoclonal antibodies (51); 10 μg/ml lectin SNA-I (from Sambucus nigra), WGA (from Triticum vulgare), CCA (from Cancer antennarius), or MAA (from Maackia amurensis) (EY Laboratories); and 10 μg/ml HE and HE S40T recombinant proteins (78).
To quantify virus propagation in HRT-18 or LA-N-5 cells at the time points indicated above, fixed cells were washed once in PBS and permeabilized in chilled 100% methanol for 5 min at −20°C. Samples were rehydrated for 5 min in PBS and stained for 2 h at 37°C using a 1/2 dilution of mixed hybridoma supernatants containing equivalent volumes of 4E11.3 anti-HEV N protein and 4-3E.4 anti-OC43 spike monoclonal antibodies. Cells were washed twice for 5 min each time in PBS and then probed for 45 min at room temperature with anti-mouse immunoglobulin Alexa Fluor 488 secondary fluorescent antibodies (1/500; Life Technologies) along with 2 μg/ml Hoechst to counterstain the nuclei. Following 3 PBS washes, the coverslips were either directly imaged at a magnification of ×100 on a Zeiss LSM780 confocal microscope using the 30-mW 405-nm diode laser and 25-mW 458/488/514-nm argon multiline laser or mounted with Immuno Mount mounting medium (Fisher Scientific) on glass slides to be analyzed later on. For each sample, ≥6 4-by-4 tiles of full fields were taken and exported from Zen black software (Carl Zeiss Microimaging), and cells were quantified using the cell image analysis software CellProfiler (www.cellprofiler.org/).
Minor modifications to this protocol were applied to assess propagation in PMNCs. For instance, methanol-permeabilized cells were incubated for 1 h at room temperature with 2% (wt/vol) PBS and then for 1 h at 37°C in the 1/2 dilution of our mixed hybridoma supernatants supplemented with 0.1% Triton. Cells were incubated with the secondary antibodies for 1 h at room temperature.
Statistical tests.
For propagation and platform dynamism kinetics, statistical analyses were conducted by two-way analysis of variance (ANOVA) followed by a Sidak test. Statistics for other propagation assays were assessed using a ratio paired Student t test when comparing fluid and semifluid media and an unpaired Student t test for mutant comparisons. The statistical validation of the effect of paclitaxel on axonal spike association was done using an unpaired Student t test. Statistical significance was defined as P values of <0.05 and <0.01.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jessie Tremblay for excellent technical assistance.
This work was supported by grant MT-9203 from the CIHR's Institute of infection and Immunity (III) to Pierre J. Talbot. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.00404-18.
REFERENCES
- 1.Talbot P, Jacomy H, Desforges M. 2008. Pathogenesis of human coronaviruses other than severe acute respiratory syndrome coronavirus, p 313–324. In Perlman S, Gallagher T, Snijder EJ (ed.), Nidoviruses. ASM Press, Washington, DC. [Google Scholar]
- 2.Forgie S, Marrie TJ. 2009. Healthcare-associated atypical pneumonia. Semin Respir Crit Care Med 30:67–85. doi: 10.1055/s-0028-1119811. [DOI] [PubMed] [Google Scholar]
- 3.Freymuth F, Vabret A, Dina J, Cuvillon-Nimal D, Lubin C, Vaudecrane A, Guillois B, Gouarin S, Petitjean J, Lafaix-Delaire F, Brouard J. 2010. Bronchiolitis viruses. Arch Pediatr 17:1192–1201. doi: 10.1016/j.arcped.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vabret A, Dina J, Brison E, Brouard J, Freymuth F. 2009. Human coronaviruses. Pathol Biol (Paris) 57:149–160. (In French.) doi: 10.1016/j.patbio.2008.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drosten C, Gunther S, Preiser W, van der Werf S, Brodt HR, Becker S, Rabenau H, Panning M, Kolesnikova L, Fouchier RA, Berger A, Burguiere AM, Cinatl J, Eickmann M, Escriou N, Grywna K, Kramme S, Manuguerra JC, Muller S, Rickerts V, Sturmer M, Vieth S, Klenk HD, Osterhaus AD, Schmitz H, Doerr HW. 2003. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med 348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 6.Kuiken T, Fouchier RA, Schutten M, Rimmelzwaan GF, van Amerongen G, van Riel D, Laman JD, de Jong T, van Doornum G, Lim W, Ling AE, Chan PK, Tam JS, Zambon MC, Gopal R, Drosten C, van der Werf S, Escriou N, Manuguerra JC, Stohr K, Peiris JS, Osterhaus AD. 2003. Newly discovered coronavirus as the primary cause of severe acute respiratory syndrome. Lancet 362:263–270. doi: 10.1016/S0140-6736(03)13967-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, Penaranda S, Bankamp B, Maher K, Chen MH, Tong S, Tamin A, Lowe L, Frace M, DeRisi JL, Chen Q, Wang D, Erdman DD, Peret TC, Burns C, Ksiazek TG, Rollin PE, Sanchez A, Liffick S, Holloway B, Limor J, McCaustland K, Olsen-Rasmussen M, Fouchier R, Gunther S, Osterhaus AD, Drosten C, Pallansch MA, Anderson LJ, Bellini WJ. 2003. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 8.Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. 2012. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 367:1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- 9.Gu J, Gong E, Zhang B, Zheng J, Gao Z, Zhong Y, Zou W, Zhan J, Wang S, Xie Z, Zhuang H, Wu B, Zhong H, Shao H, Fang W, Gao D, Pei F, Li X, He Z, Xu D, Shi X, Anderson VM, Leong AS. 2005. Multiple organ infection and the pathogenesis of SARS. J Exp Med 202:415–424. doi: 10.1084/jem.20050828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morfopoulou S, Brown JR, Davies EG, Anderson G, Virasami A, Qasim W, Chong WK, Hubank M, Plagnol V, Desforges M, Jacques TS, Talbot PJ, Breuer J. 2016. Human coronavirus OC43 associated with fatal encephalitis. N Engl J Med 375:497–498. doi: 10.1056/NEJMc1509458. [DOI] [PubMed] [Google Scholar]
- 11.Arbour N, Day R, Newcombe J, Talbot PJ. 2000. Neuroinvasion by human respiratory coronaviruses. J Virol 74:8913–8921. doi: 10.1128/JVI.74.19.8913-8921.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stewart JN, Mounir S, Talbot PJ. 1992. Human coronavirus gene expression in the brains of multiple sclerosis patients. Virology 191:502–505. doi: 10.1016/0042-6822(92)90220-J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turgay C, Emine T, Ozlem K, Muhammet SP, Haydar AT. 2015. A rare cause of acute flaccid paralysis: human coronaviruses. J Pediatr Neurosci 10:280–281. doi: 10.4103/1817-1745.165716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Algahtani H, Subahi A, Shirah B. 2016. Neurological complications of Middle East respiratory syndrome coronavirus: a report of two cases and review of the literature. Case Rep Neurol Med 2016:3502683. doi: 10.1155/2016/3502683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arabi YM, Harthi A, Hussein J, Bouchama A, Johani S, Hajeer AH, Saeed BT, Wahbi A, Saedy A, AlDabbagh T, Okaili R, Sadat M, Balkhy H. 2015. Severe neurologic syndrome associated with Middle East respiratory syndrome corona virus (MERS-CoV). Infection 43:495–501. doi: 10.1007/s15010-015-0720-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gu J, Korteweg C. 2007. Pathology and pathogenesis of severe acute respiratory syndrome. Am J Pathol 170:1136–1147. doi: 10.2353/ajpath.2007.061088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lau KK, Yu WC, Chu CM, Lau ST, Sheng B, Yuen KY. 2004. Possible central nervous system infection by SARS coronavirus. Emerg Infect Dis 10:342–344. doi: 10.3201/eid1002.030638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y, Li H, Fan R, Wen B, Zhang J, Cao X, Wang C, Song Z, Li S, Li X, Lv X, Qu X, Huang R, Liu W. 2016. Coronavirus infections in the central nervous system and respiratory tract show distinct features in hospitalized children. Intervirology 59:163–169. doi: 10.1159/000453066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Principi N, Bosis S, Esposito S. 2010. Effects of coronavirus infections in children. Emerg Infect Dis 16:183–188. doi: 10.3201/eid1602.090469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsai LK, Hsieh ST, Chang YC. 2005. Neurological manifestations in severe acute respiratory syndrome. Acta Neurol Taiwan 14:113–119. [PubMed] [Google Scholar]
- 21.Yeh EA, Collins A, Cohen ME, Duffner PK, Faden H. 2004. Detection of coronavirus in the central nervous system of a child with acute disseminated encephalomyelitis. Pediatrics 113:e73–e76. doi: 10.1542/peds.113.1.e73. [DOI] [PubMed] [Google Scholar]
- 22.Cristallo A, Gambaro F, Biamonti G, Ferrante P, Battaglia M, Cereda PM. 1997. Human coronavirus polyadenylated RNA sequences in cerebrospinal fluid from multiple sclerosis patients. New Microbiol 20:105–114. [PubMed] [Google Scholar]
- 23.Jacomy H, Fragoso G, Almazan G, Mushynski WE, Talbot PJ. 2006. Human coronavirus OC43 infection induces chronic encephalitis leading to disabilities in BALB/C mice. Virology 349:335–346. doi: 10.1016/j.virol.2006.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jacomy H, St-Jean JR, Brison E, Marceau G, Desforges M, Talbot PJ. 2010. Mutations in the spike glycoprotein of human coronavirus OC43 modulate disease in BALB/c mice from encephalitis to flaccid paralysis and demyelination. J Neurovirol 16:279–293. doi: 10.3109/13550284.2010.497806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arbour N, Cote G, Lachance C, Tardieu M, Cashman NR, Talbot PJ. 1999. Acute and persistent infection of human neural cell lines by human coronavirus OC43. J Virol 73:3338–3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.St-Jean JR, Desforges M, Talbot PJ. 2006. Genetic evolution of human coronavirus OC43 in neural cell culture. Adv Exp Med Biol 581:499–502. doi: 10.1007/978-0-387-33012-9_88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fazzini E, Fleming J, Fahn S. 1992. Cerebrospinal fluid antibodies to coronavirus in patients with Parkinson's disease. Mov Disord 7:153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson-Lussenburg CM, Zheng Q. 1987. Coronavirus and multiple sclerosis: results of a case/control longitudinal serological study. Adv Exp Med Biol 218:421–429. doi: 10.1007/978-1-4684-1280-2_51. [DOI] [PubMed] [Google Scholar]
- 29.Sibley WA, Bamford CR, Clark K. 1985. Clinical viral infections and multiple sclerosis. Lancet i:1313–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brison E, Jacomy H, Desforges M, Talbot PJ. 2011. Glutamate excitotoxicity is involved in the induction of paralysis in mice after infection by a human coronavirus with a single point mutation in its spike protein. J Virol 85:12464–12473. doi: 10.1128/JVI.05576-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brison E, Jacomy H, Desforges M, Talbot PJ. 2014. Novel treatment with neuroprotective and antiviral properties against a neuroinvasive human respiratory virus. J Virol 88:1548–1563. doi: 10.1128/JVI.02972-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Do Carmo S, Jacomy H, Talbot PJ, Rassart E. 2008. Neuroprotective effect of apolipoprotein D against human coronavirus OC43-induced encephalitis in mice. J Neurosci 28:10330–10338. doi: 10.1523/JNEUROSCI.2644-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Le Coupanec A, Desforges M, Meessen-Pinard M, Dube M, Day R, Seidah NG, Talbot PJ. 2015. Cleavage of a neuroinvasive human respiratory virus spike glycoprotein by proprotein convertases modulates neurovirulence and virus spread within the central nervous system. PLoS Pathog 11:e1005261. doi: 10.1371/journal.ppat.1005261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jean A, Quach C, Yung A, Semret M. 2013. Severity and outcome associated with human coronavirus OC43 infections among children. Pediatr Infect Dis J 32:325–329. doi: 10.1097/INF.0b013e3182812787. [DOI] [PubMed] [Google Scholar]
- 35.Lee J, Storch GA. 2014. Characterization of human coronavirus OC43 and human coronavirus NL63 infections among hospitalized children <5 years of age. Pediatr Infect Dis J 33:814–820. doi: 10.1097/INF.0000000000000292. [DOI] [PubMed] [Google Scholar]
- 36.Charles PC, Walters E, Margolis F, Johnston RE. 1995. Mechanism of neuroinvasion of Venezuelan equine encephalitis virus in the mouse. Virology 208:662–671. doi: 10.1006/viro.1995.1197. [DOI] [PubMed] [Google Scholar]
- 37.Rochel S, Margolis FL. 1980. The response of ornithine decarboxylase during neuronal degeneration and regeneration in olfactory epithelium. J Neurochem 35:850–860. doi: 10.1111/j.1471-4159.1980.tb07082.x. [DOI] [PubMed] [Google Scholar]
- 38.Jacomy H, Talbot PJ. 2003. Vacuolating encephalitis in mice infected by human coronavirus OC43. Virology 315:20–33. doi: 10.1016/S0042-6822(03)00323-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.St-Jean JR, Jacomy H, Desforges M, Vabret A, Freymuth F, Talbot PJ. 2004. Human respiratory coronavirus OC43: genetic stability and neuroinvasion. J Virol 78:8824–8834. doi: 10.1128/JVI.78.16.8824-8834.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barnett EM, Perlman S. 1993. The olfactory nerve and not the trigeminal nerve is the major site of CNS entry for mouse hepatitis virus, strain JHM. Virology 194:185–191. doi: 10.1006/viro.1993.1248. [DOI] [PubMed] [Google Scholar]
- 41.Mori I. 2015. Transolfactory neuroinvasion by viruses threatens the human brain. Acta Virol 59:338–349. doi: 10.4149/av_2015_04_338. [DOI] [PubMed] [Google Scholar]
- 42.Perlman S, Evans G, Afifi A. 1990. Effect of olfactory bulb ablation on spread of a neurotropic coronavirus into the mouse brain. J Exp Med 172:1127–1132. doi: 10.1084/jem.172.4.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perlman S, Jacobsen G, Afifi A. 1989. Spread of a neurotropic murine coronavirus into the CNS via the trigeminal and olfactory nerves. Virology 170:556–560. doi: 10.1016/0042-6822(89)90446-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Riel D, Verdijk R, Kuiken T. 2015. The olfactory nerve: a shortcut for influenza and other viral diseases into the central nervous system. J Pathol 235:277–287. doi: 10.1002/path.4461. [DOI] [PubMed] [Google Scholar]
- 45.Hansen LF, Hammer M, Petersen SH, Nielsen GD. 1994. Effects of intranasal ZnSO4 irrigation on olfactory and trigeminal cues. Physiol Behav 55:699–704. [DOI] [PubMed] [Google Scholar]
- 46.Katsetos CD, Legido A, Perentes E, Mork SJ. 2003. Class III beta-tubulin isotype: a key cytoskeletal protein at the crossroads of developmental neurobiology and tumor neuropathology. J Child Neurol 18:851–866. doi: 10.1177/088307380301801205. [DOI] [PubMed] [Google Scholar]
- 47.Favreau DJ, Desforges M, St-Jean JR, Talbot PJ. 2009. A human coronavirus OC43 variant harboring persistence-associated mutations in the S glycoprotein differentially induces the unfolded protein response in human neurons as compared to wild-type virus. Virology 395:255–267. doi: 10.1016/j.virol.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hill DP, Robertson KA. 1998. Differentiation of LA-N-5 neuroblastoma cells into cholinergic neurons: methods for differentiation, immunohistochemistry and reporter gene introduction. Brain Res Brain Res Protoc 2:183–190. doi: 10.1016/S1385-299X(97)00041-X. [DOI] [PubMed] [Google Scholar]
- 49.Bosch BJ, de Haan CA, Rottier PJ. 2004. Coronavirus spike glycoprotein, extended at the carboxy terminus with green fluorescent protein, is assembly competent. J Virol 78:7369–7378. doi: 10.1128/JVI.78.14.7369-7378.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Verheije MH, Hagemeijer MC, Ulasli M, Reggiori F, Rottier PJ, Masters PS, de Haan CA. 2010. The coronavirus nucleocapsid protein is dynamically associated with the replication-transcription complexes. J Virol 84:11575–11579. doi: 10.1128/JVI.00569-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Desforges M, Desjardins J, Zhang C, Talbot PJ. 2013. The acetyl-esterase activity of the hemagglutinin-esterase protein of human coronavirus OC43 strongly enhances the production of infectious virus. J Virol 87:3097–3107. doi: 10.1128/JVI.02699-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mounir S, Talbot PJ. 1992. Sequence analysis of the membrane protein gene of human coronavirus OC43 and evidence for O-glycosylation. J Gen Virol 73(Pt 10):2731–2736. [DOI] [PubMed] [Google Scholar]
- 53.Alfson KJ, Avena LE, Beadles MW, Staples H, Nunneley JW, Ticer A, Dick EJ Jr, Owston MA, Reed C, Patterson JL, Carrion R Jr, Griffiths A. 2015. Particle-to-PFU ratio of Ebola virus influences disease course and survival in cynomolgus macaques. J Virol 89:6773–6781. doi: 10.1128/JVI.00649-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Juarez D, Long KC, Aguilar P, Kochel TJ, Halsey ES. 2013. Assessment of plaque assay methods for alphaviruses. J Virol Methods 187:185–189. doi: 10.1016/j.jviromet.2012.09.026. [DOI] [PubMed] [Google Scholar]
- 55.Watanabe T, Horikawa Y, Sato K, Saito H. 1982. Methylcellulose media for plaque assay of murine leukemia virus. J Clin Microbiol 16:542–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zurbach KA, Moghbeli T, Snyder CM. 2014. Resolving the titer of murine cytomegalovirus by plaque assay using the M2-10B4 cell line and a low viscosity overlay. Virol J 11:71. doi: 10.1186/1743-422X-11-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krempl C, Schultze B, Herrler G. 1995. Analysis of cellular receptors for human coronavirus OC43. Adv Exp Med Biol 380:371–374. doi: 10.1007/978-1-4615-1899-0_60. [DOI] [PubMed] [Google Scholar]
- 58.Kunkel F, Herrler G. 1996. Structural and functional analysis of the S proteins of two human coronavirus OC43 strains adapted to growth in different cells. Arch Virol 141:1123–1131. doi: 10.1007/BF01718615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vlasak R, Luytjes W, Spaan W, Palese P. 1988. Human and bovine coronaviruses recognize sialic acid-containing receptors similar to those of influenza C viruses. Proc Natl Acad Sci U S A 85:4526–4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.de Groot RJ. 2006. Structure, function and evolution of the hemagglutinin-esterase proteins of corona- and toroviruses. Glycoconj J 23:59–72. doi: 10.1007/s10719-006-5438-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rottier PJ. 1990. Background paper. Coronavirus M and HE: two peculiar glycoproteins. Adv Exp Med Biol 276:91–94. [DOI] [PubMed] [Google Scholar]
- 62.Smits SL, Gerwig GJ, van Vliet AL, Lissenberg A, Briza P, Kamerling JP, Vlasak R, de Groot RJ. 2005. Nidovirus sialate-O-acetylesterases: evolution and substrate specificity of coronaviral and toroviral receptor-destroying enzymes. J Biol Chem 280:6933–6941. doi: 10.1074/jbc.M409683200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vlasak R, Luytjes W, Leider J, Spaan W, Palese P. 1988. The E3 protein of bovine coronavirus is a receptor-destroying enzyme with acetylesterase activity. J Virol 62:4686–4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meessen-Pinard M, Le Coupanec A, Desforges M, Talbot PJ. 2017. Pivotal role of receptor-interacting protein kinase 1 and mined lineage kinase domain-like in neuronal cell death induced by the human neuroinvasive coronavirus OC43. J Virol 91:e01513-. doi: 10.1128/JVI.01513-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lochhead JJ, Thorne RG. 2012. Intranasal delivery of biologics to the central nervous system. Adv Drug Deliv Rev 64:614–628. doi: 10.1016/j.addr.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 66.Taylor MP, Enquist LW. 2015. Axonal spread of neuroinvasive viral infections. Trends Microbiol 23:283–288. doi: 10.1016/j.tim.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Frana MF, Behnke JN, Sturman LS, Holmes KV. 1985. Proteolytic cleavage of the E2 glycoprotein of murine coronavirus: host-dependent differences in proteolytic cleavage and cell fusion. J Virol 56:912–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pasick JM, Kalicharran K, Dales S. 1994. Distribution and trafficking of JHM coronavirus structural proteins and virions in primary neurons and the OBL-21 neuronal cell line. J Virol 68:2915–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bauer A, Nolden T, Schroter J, Romer-Oberdorfer A, Gluska S, Perlson E, Finke S. 2014. Anterograde glycoprotein-dependent transport of newly generated rabies virus in dorsal root ganglion neurons. J Virol 88:14172–14183. doi: 10.1128/JVI.02254-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Howard PW, Wright CC, Howard T, Johnson DC. 2014. Herpes simplex virus gE/gI extracellular domains promote axonal transport and spread from neurons to epithelial cells. J Virol 88:11178–11186. doi: 10.1128/JVI.01627-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Saksena MM, Wakisaka H, Tijono B, Boadle RA, Rixon F, Takahashi H, Cunningham AL. 2006. Herpes simplex virus type 1 accumulation, envelopment, and exit in growth cones and varicosities in mid-distal regions of axons. J Virol 80:3592–3606. doi: 10.1128/JVI.80.7.3592-3606.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Taylor MP, Kramer T, Lyman MG, Kratchmarov R, Enquist LW. 2012. Visualization of an alphaherpesvirus membrane protein that is essential for anterograde axonal spread of infection in neurons. mBio 3:e00063-12. doi: 10.1128/mBio.00063-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.De Regge N, Nauwynck HJ, Geenen K, Krummenacher C, Cohen GH, Eisenberg RJ, Mettenleiter TC, Favoreel HW. 2006. Alpha-herpesvirus glycoprotein D interaction with sensory neurons triggers formation of varicosities that serve as virus exit sites. J Cell Biol 174:267–275. doi: 10.1083/jcb.200510156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sattentau QJ. 2011. The direct passage of animal viruses between cells. Curr Opin Virol 1:396–402. doi: 10.1016/j.coviro.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 75.Bender SJ, Phillips JM, Scott EP, Weiss SR. 2010. Murine coronavirus receptors are differentially expressed in the central nervous system and play virus strain-dependent roles in neuronal spread. J Virol 84:11030–11044. doi: 10.1128/JVI.02688-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vijgen L, Keyaerts E, Moes E, Maes P, Duson G, Van Ranst M. 2005. Development of one-step, real-time, quantitative reverse transcriptase PCR assays for absolute quantitation of human coronaviruses OC43 and 229E. J Clin Microbiol 43:5452–5456. doi: 10.1128/JCM.43.11.5452-5456.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lambert F, Jacomy H, Marceau G, Talbot PJ. 2008. Titration of human coronaviruses using an immunoperoxidase assay. J Vis Exp 2008:751. doi: 10.3791/751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li Z, Michael IP, Zhou D, Nagy A, Rini JM. 2013. Simple piggyBac transposon-based mammalian cell expression system for inducible protein production. Proc Natl Acad Sci U S A 110:5004–5009. doi: 10.1073/pnas.1218620110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.