

BoHV-1 transcription is rapidly activated during stress-induced reactivation from latency. The immediate early transcription unit 1 (IEtu1) promoter is regulated by the GR via two GREs. The IEtu1 promoter regulates expression of two viral transcriptional regulatory proteins, infected cell proteins 0 and 4 (bICP0 and bICP4), and thus must be stimulated during reactivation. This study demonstrates that activation of the IEtu1 promoter by the synthetic corticosteroid dexamethasone requires HCF-1. Interestingly, the GRE sites, but not the IE enhancer core element (TAATGARAT), were required for HCF-1-mediated GR promoter activation. The GR and HCF-1 were recruited to the IEtu1 promoter in transfected and infected cells. Collectively, these studies indicate that HCF-1 is critical for GR activation of the viral IE genes and suggest that an HCF-1–GR complex can stimulate the IEtu1 promoter in the absence of the viral IE activator VP16.
KEYWORDS: BoHV-1, HCF-1, glucocorticoid receptor, latency, transcriptional regulation
ABSTRACT
Following productive infection, bovine herpesvirus 1 (BoHV-1) establishes latency in sensory neurons. As in other alphaherpesviruses, expression of BoHV-1 immediate early (IE) genes is regulated by an enhancer complex containing the viral IE activator VP16, the cellular transcription factor Oct-1, and transcriptional coactivator HCF-1, which is assembled on an IE enhancer core element (TAATGARAT). Expression of the IE transcription unit that encodes the viral IE activators bICP0 and bICP4 may also be induced by the activated glucocorticoid receptor (GR) via two glucocorticoid response elements (GREs) located upstream of the enhancer core. Strikingly, lytic infection and reactivation from latency are consistently enhanced by glucocorticoid treatment in vivo. As the coactivator HCF-1 is essential for IE gene expression of alphaherpesviruses and recruited by multiple transcription factors, we tested whether HCF-1 is required for glucocorticoid-induced IE gene expression. Depletion of HCF-1 reduced GR-mediated activation of the IE promoter in mouse neuroblastoma cells (Neuro-2A). More importantly, HCF-1-mediated GR activation of the promoter was dependent on the presence of GRE sites but independent of the TAATGARAT enhancer core element. HCF-1 was also recruited to the GRE region of a promoter lacking the enhancer core, consistent with a direct role of the coactivator in mediating GR-induced transcription. Similarly, during productive lytic infection, HCF-1 and GR occupied the IE region containing the GREs. These studies indicate HCF-1 is critical for GR activation of the viral IE genes and suggests that glucocorticoid induction of viral reactivation proceeds via an HCF-1–GR mechanism in the absence of the viral IE activator VP16.
IMPORTANCE BoHV-1 transcription is rapidly activated during stress-induced reactivation from latency. The immediate early transcription unit 1 (IEtu1) promoter is regulated by the GR via two GREs. The IEtu1 promoter regulates expression of two viral transcriptional regulatory proteins, infected cell proteins 0 and 4 (bICP0 and bICP4), and thus must be stimulated during reactivation. This study demonstrates that activation of the IEtu1 promoter by the synthetic corticosteroid dexamethasone requires HCF-1. Interestingly, the GRE sites, but not the IE enhancer core element (TAATGARAT), were required for HCF-1-mediated GR promoter activation. The GR and HCF-1 were recruited to the IEtu1 promoter in transfected and infected cells. Collectively, these studies indicate that HCF-1 is critical for GR activation of the viral IE genes and suggest that an HCF-1–GR complex can stimulate the IEtu1 promoter in the absence of the viral IE activator VP16.
INTRODUCTION
Bovine herpesvirus 1 (BoHV-1) is an important risk factor for the polymicrobial disease bovine respiratory disease complex (BRDC). Acute BoHV-1 infection erodes mucosal surfaces in the upper respiratory tract, induces conjunctivitis, induces transient immune suppression, and promotes secondary infections by pathogenic bacteria, including Mannheimia haemolytica (1–3). BoHV-1 enhances interactions between the M. haemolytica leukotoxin and peripheral mononuclear cells, including neutrophils, leading to cell death and inflammation in the lung (4, 5). Life-threatening pneumonia frequently develops in calves coinfected with BoHV-1 and MH (6). In addition, the BoHV-1 entry receptor, poliovirus receptor related 1 (PVRL1), is a BRDC susceptibility gene for Holstein calves (7), further linking BoHV-1 to BRDC syndromes.
Like other alphaherpesvirus family members, BoHV-1 genes are expressed in three distinct phases during productive infection: immediate early (IE), early (E), and late (L) (8, 9). Transcription of IE genes is induced by cellular and viral transcription factors, including the viral IE activator VP16. This protein is packaged in the virion tegument, released upon infection, and assembled into the IE enhancer core (TAATGARAT) complexes to drive high-level expression of IE genes (10, 11). An essential cellular component of this enhancer complex is the transcriptional coactivator HCF-1 (host cell factor-1). HCF-1 mediates induced expression of the IE genes of several alphaherpesviruses, including herpes simplex virus 1 (HSV-1) and varicella-zoster virus (VZV), during initiation of lytic infection (12). It is recruited to IE enhancers via an interaction with VP16, where it forms stable multiprotein complexes on the IE enhancer core elements (13, 14). However, HCF-1 may also be recruited to IE promoters and enhancers through interactions with other cellular transcription factors (e.g., Sp1 and GA-binding protein [GABP]) that can promote IE gene expression (15, 16).
Functionally, HCF-1 is a component of multiple chromatin modification complexes. For HSV, an HCF-1 complex containing histone methyltransferases (SET1A or MLLs) and histone demethylases (LSD1 and JMJD2 family members) are critical to modulate the chromatin state of the infecting viral genome in order to drive the expression of viral IE genes (17–19). More recently, HCF-1 has been linked to the regulation of both transcription initiation and elongation stages of viral IE expression (20).
In addition to its roles in promoting lytic infection, HCF-1 also appears to be required during HSV-1 reactivation from latency. Upon exposure to stimuli that induce reactivation, HCF-1 is transported from a sequestered location in the cytoplasm to the neuronal nucleus (21), where it associates with viral IE enhancer-promoter domains (22). In addition, the HCF-1-associated histone demethylases are also required to promote IE gene expression during the initiation of viral reactivation. Thus, this coactivator and its associated components mediate IE expression during both lytic infection and viral reactivation.
During productive infection by BoHV-1, the IE transcription unit 1 (IEtu1) promoter drives expression of the bICP0 and bICP4 major viral transcriptional regulatory proteins (23–25) (Fig. 1A), while IEtu2 drives the expression of bICP22. In addition to VP16-mediated induction of IE gene expression, IEtu1 can also be trans-activated by the glucocorticoid receptor (GR) when cells are treated with the synthetic corticosteroid dexamethasone (DEX) (26). GR-mediated activation of this IE promoter requires functional GR binding sites located more than 700 bases upstream of the single enhancer core TAATGARAT motif (Fig. 1B).
FIG 1.
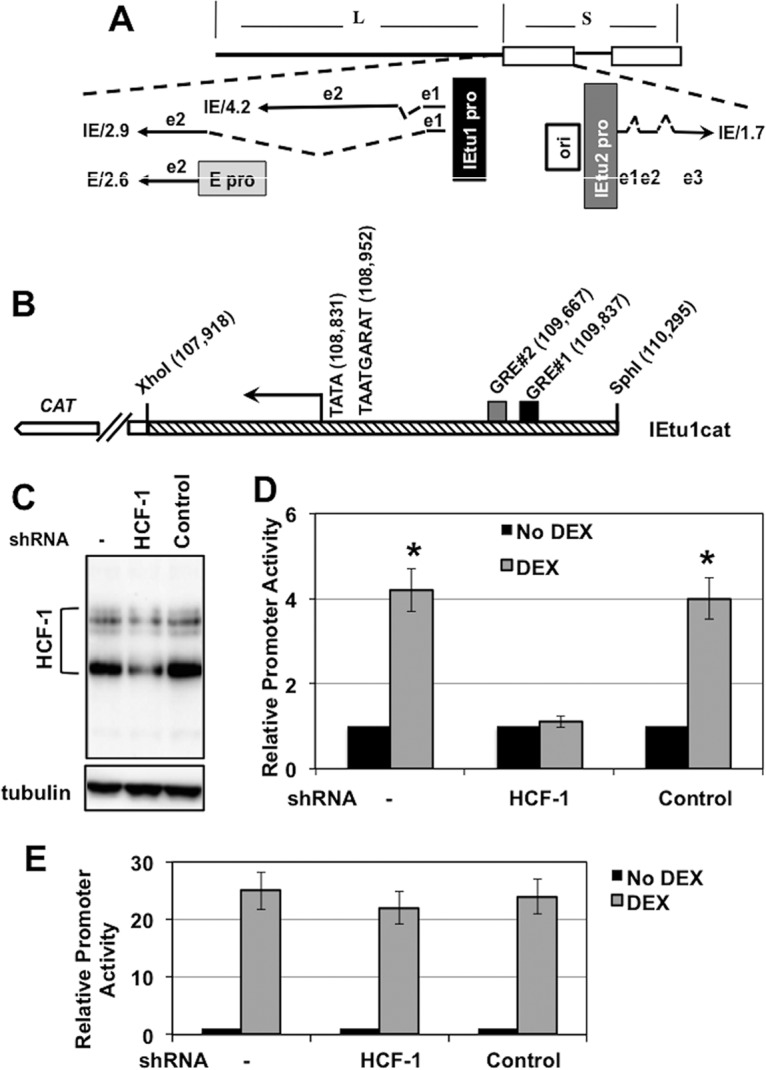
Regulation of IEtu1 promoter activity by DEX. (A) Location of IE transcripts and promoters that drive their expression during productive infection (23–25). IE/4.2 encodes the bICP4 protein, and IE/2.9 encodes the bICP0 protein. One IE promoter activates expression of IE/4.2 and IE/2.9 (IEtu1, black rectangle). A second bICP0 transcript (E/2.6) is expressed from an early promoter (E pro; gray rectangle). The IEtu2 promoter (IEtu2 pro) drives expression of the bICP22 protein. Solid lines in the transcript position map represent exons (e1, e2, or e3), and dashed lines denote introns. ORI, origin of replication. (B) Schematic of the IEtu1 promoter (IEtu1cat) with the transcription initiation site (arrow), TATA box, TAATGARAT core, and GRE sites (GRE#1 and GRE#2). The genomic coordinates of the first nucleotide of each respective motif are shown. (C) Neuro-2A cells were mock transfected (−) or transfected with plasmids that express an HCF1-specific shRNA or a control shRNA. At 24 h posttransfection, resolved cell lysates were probed for HCF-1 and control tubulin. (D) Neuro-2A cells were transfected with plasmids that express an HCF1-specific shRNA (shRNAc504) or a control shRNA. Twenty-four hours later, cells were cotransfected with the IEtu1 promoter-reporter and a plasmid expressing the mouse GR and subsequently treated with vehicle or 10 μM water-soluble DEX. Reporter activity is shown in the presence or absence of DEX treatment. The results are averages ± standard deviations (SD) from 3 independent experiments, and the asterisk denotes a significant difference relative to other samples (P < 0.05 by Student t test). (E) Neuro-2A cells were mock transfected (−) or cotransfected with the indicated plasmids expressing the HCF-1 shRNA (c504) or control shRNA. Twenty-four hours later, cells were transfected with an MMTV LTR reporter. The reporter activity is shown in the presence or absence of DEX treatment. The results are averages ± SD from 3 independent experiments.
Like other alphaherpesviruses, primary infection with BoHV-1 results in the establishment of lifelong latency in sensory ganglia (27–29). In contrast to productive infection, the only viral gene that is abundantly expressed in latently infected sensory neurons is the latency-related gene (9, 30). Importantly, stress stimuli, resulting in increased corticosteroid levels, enhance the incidence of BoHV-1 reactivation from latency (31). Furthermore, treatment with DEX consistently initiates BoHV-1 reactivation from latency in vivo (27–29). Thus, as corticosteroids bind and activate the GR and mineralocorticoid receptor (MR), the activated nuclear receptors may mediate early events during reactivation from latency, including induction of viral IE gene expression.
In this study, we demonstrate that the coactivator HCF-1 is required for GR-mediated stimulation of the viral IE (bICP0 and bICP4) expression in mouse neuroblastoma cells. The GREs, but not the enhancer core TAATGARAT motif, were required for HCF-1–GR-mediated induction of IE gene expression. In contrast, HCF-1 was not necessary for DEX-mediated activation of the mouse mammary tumor virus long terminal repeat (MMTV LTR), suggesting an inherent gene or context-dependent role for the coactivator in GR-mediated transcription. Together, these studies suggest that BoHV-1 IE genes can be directly induced via HCF-1-GR in the absence of the viral IE activator VP16.
RESULTS
HCF-1 stimulates GR-mediated trans-activation of BoHV-1 IE promoter.
Previous studies demonstrated that DEX stimulates the BoHV-1 IE (IEtu1) promoter activity in mouse neuroblastoma cells (Neuro-2A) (26). This IE promoter contains two consensus GR binding sites (Fig. 1B) that are required for DEX-mediated trans-activation (26). Based on the requirement for the cellular coactivator HCF-1 for HSV IE expression during lytic infection and during viral reactivation (13), we tested whether HCF-1 was required for DEX induction of BoHV-1 IE gene expression. As shown in Fig. 1C, HCF-1 was specifically depleted in Neuro-2A cells expressing an HCF-1 short hairpin RNA (shRNA) compared to cells transfected with a control shRNA construct. Strikingly, in HCF-1-depleted cells, DEX-mediated induction of the IE promoter construct was reduced (Fig. 1D). Interestingly, in contrast to the regulation of the BoHV-1 IE gene, DEX-induced transcription of the MMTV LTR was unaffected by HCF-1 depletion (Fig. 1E). Thus, the role of HCF-1 in DEX-mediated transcriptional activation is gene and context dependent.
GREs but not the enhancer core elements are required for HCF-1-GR induction.
A region of the IE promoter encompassing the two GREs (DEX responsive region [DRR]) but lacking the IE enhancer core element (TAATGARAT) was important for stimulation by DEX (26) (DRR and 3′-DRR) (Fig. 2A). In contrast, constructs containing the simian virus 40 (SV40) promoter alone or fused to IE sequences upstream of the GREs (5′-DRR) were unresponsive. Importantly, DEX-mediated transcription of constructs containing the IE GREs was dependent on HCF-1 (Fig. 2B). Thus, IE promoter sequences that contain both GREs but lack the IE enhancer core element were sufficient for HCF-1-mediated DEX induction.
FIG 2.

HCF-1 is required for DEX stimulation of the IEtu1 promoter. (A) Schematic of IEtu1 promoter and location of DEX responsive region (DRR). GRE#1 (black rectangle) and GRE#2 (gray rectangle) are shown in the IEtu1 promoter fragment. (B) Neuro-2A cells were transfected with plasmids expressing the HCF-1 shRNA (c504) or control shRNA. Twenty-four hours later, cells were cotransfected with the DRR, 3′-DRR, or 5′-DRR reporter construct and a plasmid expressing the mouse GR. DEX-induced reporter promoter activity was measured 3 h posttreatment. The results are averages ± SD from 3 independent experiments. *, P < 0.05, Student t test.
To further confirm that the enhancer core element was not required for DEX-mediated induction of the IE gene, this element was deleted from a minimal IEtu1 promoter in which the sequences between the GRE sites and the TAATGARAT enhancer core element had been removed (IEtu1 Collapsed). In transfected Neuro-2A cells, the IEtu1 Collapsed promoter and IEtu1ΔTAATGARAT were trans-activated with similar efficiency by the GR when DEX was added to cultures (Fig. 3B). This study supported the finding that GR-mediated activation of the IEtu1 promoter was independent of the IE enhancer core element.
FIG 3.
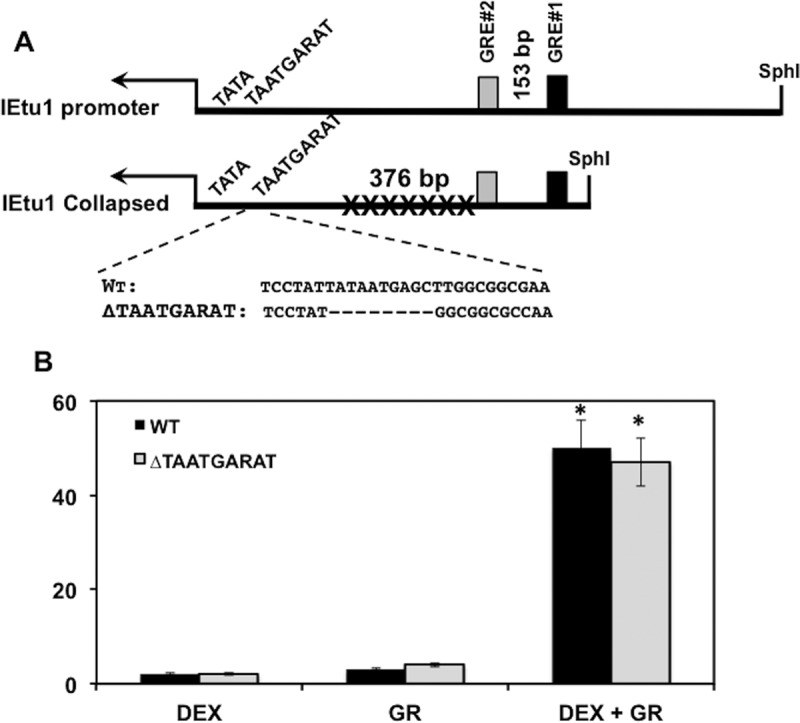
Mutation of TAATGARAT motif has no effect on DEX-mediated activation of IEtu1 promoter. (A) Schematic of IEtu1 promoter and IEtu1 Collapsed construct. The IEtu1 TAATGARAT sequence (10) and ΔTAATGARAT mutant used for this study are shown. (B) Neuro-2A cells were transfected with IEtu1 Collapsed or ΔTAATGARAT luciferase constructs (0.5 μg DNA) or cotransfected with GR expression construct (1.0 μg) using Lipofectamine 3000. Where indicated, DEX was added at 16 h after transfection. Dual-Luciferase activity was performed as previously described at 40 h after transfection (34). The results are averages ± SD from 3 independent experiments. *, P < 0.05 by Student t test.
HCF-1 is recruited to sequences that contain the GREs.
To test whether HCF-1 was recruited to the promoter region containing the GREs, chromatin immunoprecipitation (ChIP) assays were performed in transfected Neuro-2A cells. Cells were transfected with the 3′-DRR followed by treatment with vehicle or DEX. As shown in Fig. 4B, ChIP studies demonstrated HCF-1 and GR cooccupied the GRE region of the 3′-DRR plasmid. No specific PCR product was amplified from ChIPs of cells transfected with the 3′-DRR plasmid when IPs were performed using the control IgG. Interestingly, treatment with DEX had little effect on HCF-1 or GR occupancy of the 3′-DRR (Fig. 4B and C). Although this result could be due to low levels of corticosteroids in media containing 2% stripped fetal bovine serum, studies have demonstrated that the GR can associate with GREs in the absence of corticosteroids (32, 33). Thus, HCF-1 and GR were recruited to the IE GRE region in the absence of a discernible enhancer core motif.
FIG 4.
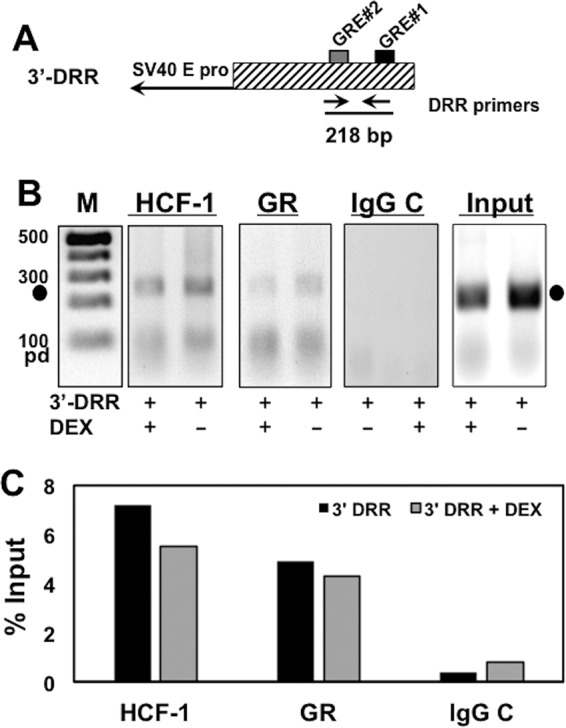
HCF-1 and GR occupy GRE sites of IEtu1 promoter in transfected cells. (A) Schematic of the 3′-DRR promoter construct. Location of the 3′-DRR primer pair and expected PCR fragment size are shown. (B) Neuro-2A cells were cotransfected with the plasmid expressing the mouse GR and the 3′-DRR reporter construct. Cultures were subsequently treated with 10 μM DEX (+) for 3 h. HCF-1 and GR occupancy were assessed by ChIP assays using the DRR primer set. IgG, control; input, 10% of total DNA; M, DNA size markers. The results are representative of three independent studies. Closed circles denote 217-bp PCR product. pd, primer dimer. (C) The intensity of bands in panel B was quantified using a Bio-Rad ChemiDoc system.
To investigate if HCF-1 and GR bound the BoHV-1 IE GRE region during productive infection, bovine kidney cells (CRIB) were mock infected or infected with BoHV-1 and treated with control vehicle or DEX for 7 h, and ChIP assays performed using control IgG, HCF-1, and GR antibodies. PCRs of ChIP DNAs were done using a primer set that amplifies a 107-bp fragment containing the two IE GREs (Fig. 5A) (34). As shown in Fig. 5B, both HCF-1 and GR occupied the IE gene region containing the GREs in BoHV-1-infected CRIB cells (lanes 2 and 3). No product was seen in immunoprecipitates from mock-infected cells (lanes 1) or in control IgG immunoprecipitates. Similar to the results using transfected promoter constructs, DEX treatment had no significant impact on the HCF-1 or GR occupancy of the viral IE GRE region (Fig. 5B, compare lanes 2 and 3 and C). Taken together, the data indicate that GR and HCF-1 occupied the IE promoter proximal to the GRE elements in both transfected IE promoter reporters and BoHV-1-infected cells.
FIG 5.
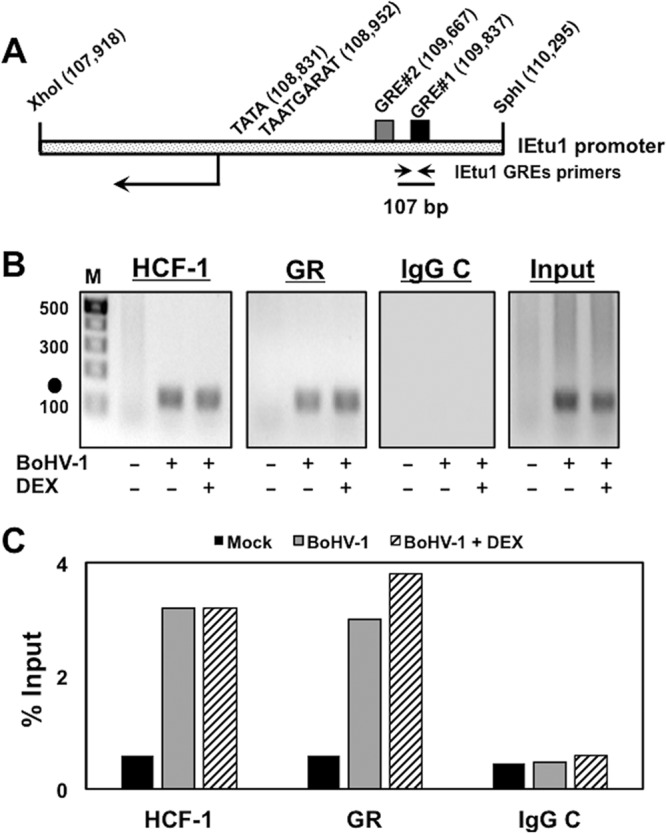
HCF-1 and GR occupy the BoHV-1 IEtu1 promoter in BoHV-1-infected cells. (A) Schematic of the IEtu1 promoter with the genomic locations of the TATA box, TAATGARAT core, GREs, and IEtu1 GRE ChIP primer pair. (B) CRIB cells were mock infected (−) or infected with BoHV-1 (+, MOI of 1) and treated with vehicle (−) or DEX (+) for 7 h. CRIB cells were transfected with the GR expression plasmid (1 μg plasmid) for 12 h prior to infection. HCF-1 and GR occupancy were assessed by ChIP assays using the DRR primer set. IgG, control; input, 10% of total DNA; M, DNA size markers (bp). The results are representative of three independent studies. Closed circles denote 107-bp PCR product. (C) The intensity of bands in panel B was quantified using a Bio-Rad ChemiDoc system.
Interactions between GR and 3′-DRR requires the GREs and ½ GREs.
To investigate if HCF-1 and GR bound to the 3′-DRR when both GREs were mutated, a mutant lacking both GREs (Δ2xGRE) was cotransfected with the GR and ChIP performed using control IgG, HCF-1, and GR antibodies. PCRs of ChIP DNAs were done using the primer set flanking the GR#1 and GR#2 sites as described for Fig. 4. As expected, GR and HCF-1 were bound to the wild-type (wt) 3′-DRR (Fig. 6A). Mutating both GREs in the 3′-DRR fragment (Δ2xGRE; Fig. 6C) reduced GR binding but did not impact HCF-1 binding. Interestingly, although binding of the GR to the ΔGRE2 fragment was reduced compared to that of the wt 3′-DRR, we consistently detected an increase in GR binding to the Δ2xGRE2 construct when DEX was added, suggesting that additional GR-responsive sequences are present in this construct. Therefore, the 3′-DRR fragment was examined for potential GRE half-sites (½ GREs), as half-sites have been shown to bind GR monomers in the presence of DEX and to regulate specific target genes (35) (Fig. 6D). Four ½ GRE binding sites (A to D) were identified in the 3′-DRR adjacent to GRE#1 and GRE#2. To investigate if these sites played a role in binding HCF-1 and GR, a 3′-DRR construct with mutations in both GREs and all 4 ½ GRE binding sites (GRE Null) was constructed and transfected into Neuro-2A cells. As shown in Fig. 6A, mutation of the 4 ½ sites abrogated the DEX-responsive binding of GR compared to mutation of the two consensus GREs (Δ2xGRE2). In contrast, HCF-1 bound to the GRE Null fragment as efficiently as the wt 3′-DRR and the Δ2xGRE constructs. Thus, GR binding to the 3′-DRR fragment was mediated by both the two whole GREs and the ½ GREs. However, binding of HCF-1 to the 3′-DRR was independent of this complex GR binding domain, suggesting that the protein could be recruited by additional unknown factors.
FIG 6.
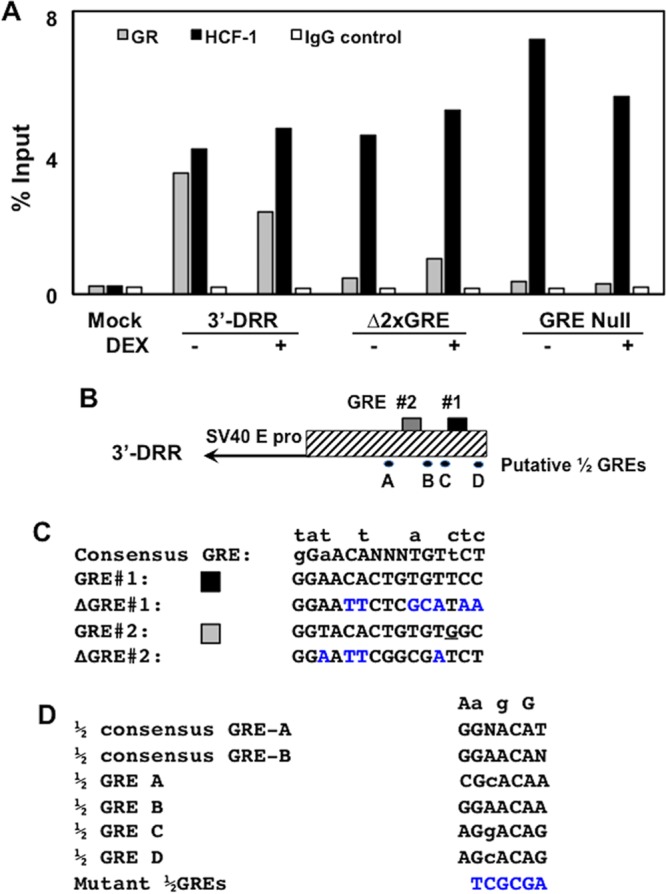
HCF-1, but not GR, occupies sequences in 3′-DRR when GRE sites are mutated. (A) Neuro-2A cells were cotransfected with the plasmid expressing the mouse GR and the 3′-DRR construct. Cultures were subsequently treated with 10 μM DEX (+) for 3 h at 45 h after transfection. HCF-1 and GR occupancy were assessed by ChIP assays using the DRR primer set. IgG, control; input, 10% of total DNA; M, DNA size markers. The intensity of bands was quantified using a Bio-Rad ChemiDoc system. The trends in this ChIP assay are consistent with results from three independent studies. (B) Schematic of the 3′-DRR promoter construct and location of the two GREs and four ½ GREs. (C) Sequence of the consensus GRE, the two GREs in the IEtu1 promoter, and mutants. Nucleotides above the consensus GRE denote changes that still result in GR binding and transactivation. Blue nucleotides denote mutations introduced into GRE#1 and GRE#2 (26). The underlined G in GRE #2 is a mismatch to the consensus GRE. (D) The two consensus ½ GREs (GRE-A and GRE-B) were previously described (35), and nucleotides listed above the sequences are differences observed in some ½ GREs that are capable of trans-activation by a GR monomer. Sequences of the four ½ GREs were identified in the 3′-DRR. A NruI restriction enzyme site (TCGCGA) was used to mutate each of the four ½ GRE sites (blue nucleotides).
To examine the effect that the ½ GREs have on DEX-induced promoter activation, Neuro-2A cells were cotransfected with the wt 3′-DRR construct or those lacking the GREs and/or the 4 ½ GREs (Fig. 7A). Mutating all 4 ½ GREs (Δ½GREs) decreased DEX-mediated promoter activity approximately 2-fold, while mutating both GREs (Δ2xGRE) reduced DEX-mediated activation to nearly basal levels (Fig. 7B). When all 4 ½ GREs and both GREs were mutated (GRE Null), DEX-mediated activation was similar to that of the Δ2xGRE mutant. Thus, the 4 ½ GRE sites contribute to DEX-induced stimulation of promoter activity but are dependent on the presence of full GREs.
FIG 7.
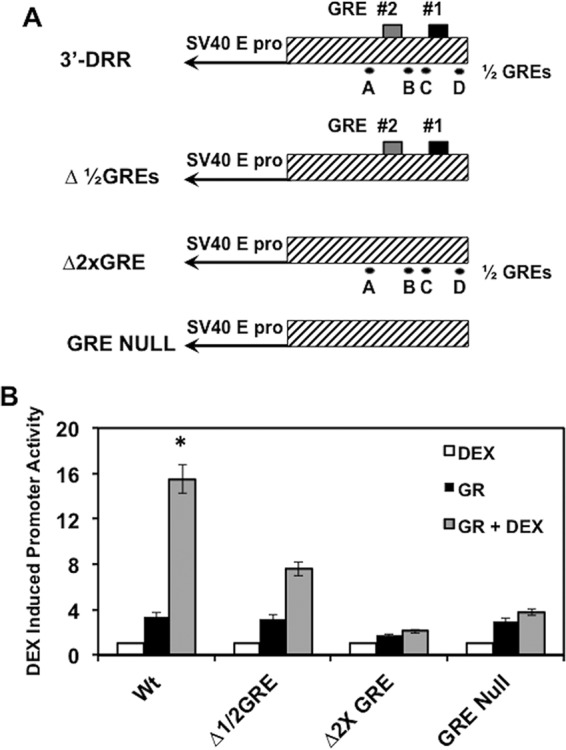
½ GREs modulate DEX-mediated activation of 3′-DRR sequences. (A) Luciferase constructs used to examine the effects of GREs and ½ GREs on DEX-mediated activation. (B) Neuro-2A cells were transfected with the designated luciferase construct (0.25 μg DNA) using Lipofectamine 3000 and were designated the GR expression construct (1.0 μg), as described in Materials and Methods. Empty vector was included to maintain the same DNA concentrations for each sample. Where indicated, DEX was added at 16 h after transfection. Dual-Luciferase activity was performed at 40 h after transfection as previously described (34). The results are averages ± SD from 3 independent experiments, and an asterisk denotes significant differences between other samples (P < 0.05 by Student t test).
DISCUSSION
It is well established that, upon infection, alphaherpesvirus IE genes are induced by the assembly of a potent enhancer complex containing cellular transcription factors, the viral IE activator VP16, and the cellular coactivator HCF-1. In this context, HCF-1 plays a critical role in mediating transitions in the chromatin state of the infecting virus that promote viral IE gene expression. Similarly, HCF-1 also appears to play a significant role in mediating the initiation of viral reactivation from latency. However, the factors and mechanisms involved in stress-mediated induction of reactivation from latency remain unclear. In BoHV-1 latently infected calves, treatment with the synthetic corticosteroid DEX efficiently induces viral reactivation in sensory neurons. Here, we show that HCF-1 is required for glucocorticoid receptor-mediated induction of viral IE genes in the absence of VP16.
In reporter assays, HCF-1 was required for DEX-mediated stimulation of the BoHV-1 IE promoter. Furthermore, both HCF-1 and GR occupied the IE GRE sites of the reporters and of the native IE promoter during lytic infection. However, HCF-1 binding was not affected by mutating the GREs or ½ GREs, suggesting that the recruitment of HCF-1 was independent of the GR and likely mediated by additional factors. Importantly, depletion of HCF-1 had no effect on the basal-level activity of the IEtu1 promoter or of constructs containing the minimal SV40 promoter fused to the IE GRE sequences. Thus, HCF-1 was only required to potentiate GR-mediated induction of the IEtu1 promoter. Interestingly, in contrast to the BoHV-1 IE gene, HCF-1 does not play a role in MMTV LTR promoter activity following dexamethasone treatment in Neuro-2A cells. This specificity is not unexpected, as GR interacts with many transcription factors and transcriptional coactivators, and there is promoter-specific usage of coactivators (36, 37) at GR target genes.
Importantly, the single TAATGARAT motif in the IEtu1 promoter was not required for DEX-mediated activation, indicating that GR induction was independent of the assembly of the VP16 enhancer complex. Conversely, the IE GRE sites are not important for VP16-mediated induction of the IEtu1 promoter, as a deletion mutant lacking these sites is activated by VP16 with efficiency similar to that of the wild-type promoter (10). Thus, the GREs within the IEtu1 promoter may not play a critical role during productive infection of cultured cells where VP16 is abundantly present. Rather, HCF-1-GR-mediated induction of viral IE expression may be highly significant during the initial stages of viral reactivation from latency when the viral IE activator is not expressed.
Strikingly, HCF-1 is primarily detected in the cytoplasm of sensory neurons during HSV-1 latency. Following stimulation that promotes viral reactivation, HCF-1 is rapidly transported to the nucleus (21) and associates with viral IE enhancer domains (22). In addition, HCF-1-associated histone demethylases are required to mediate IE gene expression and stimulation of reactivation (18, 19). Similarly, HCF-1 is detected in the nucleus of bovine TG neurons within 30 min posteuthanasia (38). In vivo, DEX-mediated induction of the IEtu1 promoter is rapid and expression of bICP0 is readily detected within 1.5 h posttreatment of latently infected calves (39, 40). Thus, the regulated transport of HCF-1 and the induction of the viral IE genes by HCF-1–GR could provide a mechanism by which stress signals promote the initiation of reactivation (in the absence of the viral IE activator VP16).
MATERIALS AND METHODS
Cells and virus.
Murine neuroblastoma cells (Neuro-2A) were grown in Eagle's minimal essential medium (EMEM) supplemented with 10% fetal calf serum, penicillin (10 U/ml), and streptomycin (100 μg/ml). CRIB (bovine kidney cells) were propagated as previously described (41).
Plasmids.
pIE1 (IEtu1cat; gift of V. Misra, University of Saskatchewan) was previously described (10, 11). The IEtu1 DRR enhancer constructs were previously described (26) and are summarized in Fig. 2A. pCAT3 contains a minimal SV40 early promoter driving chloramphenicol acetyltransferase (CAT) expression. The respective IEtu1 promoter sequences were cloned upstream of the SV40 early promoter. The ΔGRE, Δ2xGRE, and GRE null constructs shown in Fig. 7A were synthesized by GenScript and were inserted upstream at unique KpnI and XhoI restriction sites of the minimal SV40 early promoter driving luciferase (pGL3-Promoter Vector). IEtu1 Collapsed and the GenScript-synthesized ΔTAATGARAT construct, and these fragments were inserted at unique KpnI and HindIII restriction sites of the pGL3-Basic vector. A mouse GR expression vector was a gift of J. Cidlowski, NIH. Control (ACTACCGTTGTTATAGGTG) and HCF-1 (GTACCTGAATGACTTATAT) shRNAs were cloned in pSilencer 2.1-U6 (Ambion).
Promoter activity measurements.
Neuro-2A cells were cotransfected with the indicated plasmids, as detailed in the respective figure legends, using NeuroTransIt (MIR2145; Mirus) or Lipofectamine 3000 (Invitrogen) according to the manufacturer's instructions. At 5 h posttransfection, cells were incubated in MEM supplemented with 2% charcoal stripped fetal bovine serum (Gibco) and treated with control vehicle or 10 μM water-soluble DEX (Sigma) for 24 h. At 48 h posttransfection, cell lysates were prepared by freeze/thaw cycles in 0.25 M Tris-HCl, pH 7.4. Cell debris was pelleted by centrifugation, and protein concentrations were determined. CAT activity was measured by incubating with 0.1 uCi [14C]chloramphenicol (CFA754; Amersham Biosciences) and 0.5 mM acetyl-coenzyme A (A2181; Sigma) as described previously (26). Following thin-layer chromatography, CAT activity was quantitated using a Bio-Rad molecular imager FX (Molecular Dynamics, CA). Data are expressed as fold induction of samples containing DEX relative to control vehicle. Dual-Luciferase assays were performed as described previously (34).
ChIP assay.
ChIP studies were performed as previously described (26). Neuro-2A cells were grown in 100-mm dishes and were cotransfected with the designated IE promoter constructs (4 μg DNA) and a plasmid that expresses GR (1 μg DNA). For these studies, cells were transfected with the indicated plasmids using Lipofectamine 3000 (Invitrogen) according to the manufacturer's instructions. Twenty-four hours after transfection, Neuro-2A cells were cultured in MEM containing 2% charcoal-stripped fetal calf serum. The cultures were treated with vehicle or DEX (10 μM; Sigma) for 4 h. For ChIP assays of BoHV-1-infected cells, CRIB cells were mock infected or infected with BoHV-1 (multiplicity of infection [MOI] of 1), followed by DEX treatment for 7 h. Formaldehyde cross-linked cells were lysed in buffer A (50 mM HEPES, pH 7.5, 140 mM NaCl, 1 mM EDTA [pH 8.0], 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS) containing protease inhibitors. Following sonication, lysates were precleared using salmon sperm DNA-agarose (Millipore). Cleared lysate was incubated with 2 μg of anti-GR (3660S; Cell Signaling), anti-HCF-1 (A301-400A; Bethyl Laboratories), or control rabbit IgG (18140; Sigma) in buffer B (50 mM Tris HCl [pH 8.0], 150 mM NaCl, 2 mM EDTA [pH 8.0], 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) for 12 h at 4°C. Immunoprecipitates were collected using Dynabeads protein A beads (Life Technologies) and washed extensively with buffer C (20 mM Tris-HCl, pH 8.0, 500 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% SDS). PCR analyses of isolated DNAs were done using previously described primers (26, 34) that amplify the 3′-DRR (5′-TAGCCGCTCCATTCTCTC-3′ and 5′-AAAAGTGGGGAAGCAGGG-3′) to yield a 218-bp fragment or the IEtu1 GREs (5′-CCCACTTTTGCCTGTGTG-3′ and 5′-TTTTCCTCCTCCTTCCCC-3′) to yield a 107-bp fragment.
ACKNOWLEDGMENTS
This research was supported by funding to C.J. (USDA-NIFA Competitive Grants Program [13-01041 and 16-09370], Sitlington Endowment, National Institute of Neurological Disorders and Stroke of the NIH [R21NS102290], and Oklahoma Center for Respiratory and Infectious Diseases [NIH Centers for Biomedical Research Excellence grant P20GM103648]) and by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, NIH (T.M.K.).
REFERENCES
- 1.Highlander SK, Fedorova ND, Dusek DM, Panciera R, Alvarez LE, Renehart C. 2000. Inactivation of Pasteurella (Mannheimia) haemolytica leukotoxin causes partial attenuation of virulence in a calf challenge model. Infect Immun 68:3916–3922. doi: 10.1128/IAI.68.7.3916-3922.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Highlander SK. 2001. Molecular genetic analysis of virulence in Mannheimia (Pasteurella) haemolytica. Front Biosci 6:D1128–D1150. [DOI] [PubMed] [Google Scholar]
- 3.Zecchinon L, Fett T, Desmecht D. 2005. How Mannheimia haemolytica defeats host defense through a kiss of death mechanism. Vet Res 36:133–156. doi: 10.1051/vetres:2004065. [DOI] [PubMed] [Google Scholar]
- 4.Rivera-Rivas JJ, Kisiela D, Czuprynski CJ. 2009. Bovine herpesvirus type 1 infection of bovine bronchial epithelial cells increases neutrophil adhesion and activation. Vet Immunol Immunopathol 131:167–176. doi: 10.1016/j.vetimm.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 5.Leite F, Kuckleburg C, Atapattu D, Schulz R, Czuprynski CJ. 2004. BHV-1 infection and inflammatory cytokines amplify the interaction between Mannheimia haemolytica leukotoxin with bovine peripheral blood mononuclear cells in vitro. Vet Immunol Immunopathol 99:193–202. doi: 10.1016/j.vetimm.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 6.Yates WD, Babiuk LA, Jericho KW. 1983. Viral-bacterial pneumonia in calves: duration of the interaction between bovine herpesvirus 1 and Pasteurella haemolytica. Can J Comp Med 47:257–264. [PMC free article] [PubMed] [Google Scholar]
- 7.Neibergs HL, Seabury CM, Wojtowicz AJ, Wang Z, Scraggs E, Kiser JN, Neupane M, Womack JE, Van Eenennaam A, Hagevortm GR, Lehenbauer TW, Aly S, Davis J, Taylor JF, Bovine Respiratory Disease Complex Coordinated Agricultural Research Team. 2014. Susceptibility loci revealed for bovine respiratory disease complex in pre-weaned Holstein calves. BMC Genomics 15:1–19. doi: 10.1186/1471-2164-15-1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones C. 1998. Alphaherpesvirus latency: its role in disease and survival of the virus in nature. Adv Virus Res 51:81–133. doi: 10.1016/S0065-3527(08)60784-8. [DOI] [PubMed] [Google Scholar]
- 9.Jones C. 2003. Herpes simplex virus type 1 and bovine herpesvirus 1 latency. Clin Microbiol Rev 16:79–95. doi: 10.1128/CMR.16.1.79-95.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Misra V, Bratanich AC, Carpenter D, O'Hare P. 1994. Protein and DNA elements involved in transactivation of the promoter of the bovine herpesvirus (BHV) 1 IE-1 transcription unit by the BHV alpha gene trans-inducing factor. J Virol 68:4898–4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Misra V, Walker S, Hayes S, O'Hare P. 1995. The bovine herpesvirus alpha gene trans-inducing factor activates transcription by mechanisms different from those of its herpes simplex virus type 1 counterpart VP16. J Virol 69:5209–5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Narayanan A, Nogueira ML, Ruyechan WT, Kristie TM. 2005. Combinatorial transcription of herpes simplex virus and varicella zoster virus immediate early genes is strictly determined by the cellular coactivator HCF-1. J Biol Chem 280:1369–1375. doi: 10.1074/jbc.M410178200. [DOI] [PubMed] [Google Scholar]
- 13.Kristie TM. 2015. Dynamic modulation of HSV chromatin drives initiation of infection and proides targets for epigenetic therapies. Virology 479-480:555–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vogel JL, Kristie TM. 2013. The dynamics of HCF-1 modulation of herpes simplex virus chromatin during initiation of infection. Viruses 5:1272–1291. doi: 10.3390/v5051272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vogel JL, Kristie TM. 2009. The novel coactivator C1 (HCF) coordinates multiprotein enhancer formation and mediates transcription activation by GABP. EMBO J 19:683–690. doi: 10.1093/emboj/19.4.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gunther M, Laithier M, Brison O. 2000. A set of proteins interacting with transcription factor Sp1 identified in a two-hybrid screening. Mol Cell Biochem 210:131–142. doi: 10.1023/A:1007177623283. [DOI] [PubMed] [Google Scholar]
- 17.Narayanan A, Ruyechan WT, Kristie TM. 2007. The cofactor host cell factor-1 mediates Set1 and MLL1 H3K4 trimethylation at herpesvirus immediate early promoters for initiation of infection. Proc Natl Acad Sci U S A 104:10835–10840. doi: 10.1073/pnas.0704351104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liang Y, Vogel J, Narayanan A, Peng H, Kristie TM. 2009. Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat Med 15:1312–1318. doi: 10.1038/nm.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liand Y, Vogel JL, Arbuckle JH, Rai G, Jadhav A, Simeonov A, Maloney DJ, Kristie TM. 2013. Targeting the JMJD2 histone demethylases to epigenitically control herpesvirus infection and reactivation from latency. Sci Transl Med 5:167ra165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alfonso-Dunn R, Turner AW, Jean Beltran PM, Arbuckle JH, Budayeva HG, Cristea IM, Kristie TM. 2017. Transcriptional elongation of HSV immediate early genes by the super elongation complex drives lytic infection and reactivation from latency. Cell Host Microbe 21:507–515. doi: 10.1016/j.chom.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kristie TM, Vogel JL, Sears AE. 1999. Nuclear localization of the C1 factor (host cell factor) in sensory neurons correlates with reactivation of herpes simplex virus from latency. Proc Natl Acad Sci U S A 96:1229–1233. doi: 10.1073/pnas.96.4.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Whitlow ZW, Kristie TM. 2009. Recruitment of the transcriptional coactivator HCF-1 to viral immediate-early promoters during initiation of reactivation from latency of herpes simplex virus type 1. J Virol 83:9591–9595. doi: 10.1128/JVI.01115-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wirth UV, Fraefel C, Vogt B, Vlcek C, Paces V, Schwyzer M. 1992. Immediate-early RNA 2.9 and early RNA 2.6 of bovine herpesvirus 1 are 3′ coterminal and encode a putative zinc finger transactivator protein. J Virol 66:2763–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wirth UV, Gunkel K, Engels M, Schwyzer M. 1989. Spatial and temporal distribution of bovine herpesvirus 1 transcripts. J Virol 63:4882–4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wirth UV, Vogt B, Schwyzer M. 1991. The three major immediate-early transcripts of bovine herpesvirus 1 arise from two divergent and spliced transcription units. J Virol 65:195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kook I, Henley C, Meyer F, Hoffmann F, Jones C. 2015. Bovine herpesvirus 1 productive infection and the immediate early transcription unit 1 are stimulated by the synthetic corticosteroid dexamethasone. Virology 484:377–385. doi: 10.1016/j.virol.2015.06.010. [DOI] [PubMed] [Google Scholar]
- 27.Homan EJ, Easterday BC. 1983. Experimental latent and recrudescent bovine herpesvirus-1 infections in calves. Am J Vet Res 44:309–313. [PubMed] [Google Scholar]
- 28.Inman M, Lovato L, Doster A, Jones C. 2002. A mutation in the latency-related gene of bovine herpesvirus 1 disrupts the latency reactivation cycle in calves. J Virol 76:6771–6779. doi: 10.1128/JVI.76.13.6771-6779.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rock D, Lokensgard J, Lewis T, Kutish G. 1992. Characterization of dexamethasone-induced reactivation of latent bovine herpesvirus 1. J Virol 66:2484–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jones C, da Silva LF, Sinani D. 2011. Regulation of the latency-reactivation cycle by products encoded by the bovine herpesvirus 1 (BHV-1) latency-related gene. J Neurovirol 17:535–545. doi: 10.1007/s13365-011-0060-3. [DOI] [PubMed] [Google Scholar]
- 31.Jones C. 2014. Reactivation from latency by alpha-herpesvirinae subfamily members: a stressful situation. Curr Topics Virol 12:99–118. [Google Scholar]
- 32.Pandit S, Geissler W, Harris G, Sitlani A. 2002. Allosteric effects of dexamethasone and RU486 on glucocorticoid receptor-DNA interactions. J Biol Chem 277:1538–1543. doi: 10.1074/jbc.M105438200. [DOI] [PubMed] [Google Scholar]
- 33.Schmitt J, Stunnenberg HG. 1993. The glucocorticoid receptor hormone binding domain mediates transcriptional activation in vitro in the absence of ligand. Nucleic Acids Res 21:2673–2681. doi: 10.1093/nar/21.11.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.El-Mayet FS, Sawant L, Thungunutla P, Jones C. 2017. Combinatorial effects of the glucocorticoid receptor and Krüppel-like transcription factor 15 on bovine herpesvirus 1 transcription and productive infection. J Virol 91:e00904-17. doi: 10.1128/JVI.00904-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schiller B, Chodankar R, Watson LC, Stallcup MR, Yamamoto KR. 2014. Glucocorticoid receptor binds half sites as a monomer and regulates specific target genes. Genome Biol 15:418. doi: 10.1186/s13059-014-0418-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oakley RH, Cidlowski JA. 2013. The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J Allergy Clin Immunol 132:1033–1044. doi: 10.1016/j.jaci.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ratman D, Vanden Berghe W, Dejager L, Libert C, Tavernier J, Beck IM, De Bosscher K. 2013. How glucocorticoid receptors modulate the activity of other transcription factors: a scope beyond tethering. Mol Cell Endocrinol 380:41–54. doi: 10.1016/j.mce.2012.12.014. [DOI] [PubMed] [Google Scholar]
- 38.Lu R, Misra V. 2000. Potential role for luman, the cellular homologue of herpes simplex virus VP16 (alpha gene trans-inducing factor), in herpesvirus latency. J Virol 74:934–943. doi: 10.1128/JVI.74.2.934-943.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frizzo da Silva L, Kook I, Doster A, Jones C. 2013. Bovine herpesvirus 1 regulatory proteins, bICP0 and VP16, are readily detected in trigeminal ganglionic neurons expressing the glucocorticoid receptor during the early stages of reactivation from latency. J Virol 87:11214–11222. doi: 10.1128/JVI.01737-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kook I, Doster A, Jones C. 2015. Bovine herpesvirus 1 regulatory proteins are detected in trigeminal ganglionic neurons during the early stages of stress-induced escape from latency. J Neurovirol 21:585–591. doi: 10.1007/s13365-015-0339-x. [DOI] [PubMed] [Google Scholar]
- 41.Flores EF, Kreutz LC, Donis RO. 1996. Swine and ruminant pestiviruses require the same cellular factor to enter bovine cells. J Gen Virol 77:1295–1303. doi: 10.1099/0022-1317-77-6-1295. [DOI] [PubMed] [Google Scholar]