

The results of this study reconcile the controversial reports regarding the anti-HIV-1 function of alpha interferon-induced MxB protein. In addition to the different cell types that may have contributed to the different observations, our data also suggest that VSV G protein-pseudotyped HIV-1 is much less inhibited by alpha interferon-induced MxB than HIV-1 itself is. Our results clearly demonstrate an important contribution of MxB to alpha interferon-mediated inhibition of HIV-1 in CD4+ T cells, which calls for using HIV-1 target cells and wild-type virus to test the relevance of the anti-HIV-1 activity of endogenous MxB and other restriction factors.
KEYWORDS: HIV-1, MxB, host restriction, interferon
ABSTRACT
Type I interferon inhibits viruses through inducing the expression of antiviral proteins, including the myxovirus resistance (Mx) proteins. Compared to the human MxA protein, which inhibits a wide range of viruses, the MxB protein has been reported to specifically inhibit primate lentiviruses, including HIV-1, and herpesviruses. Further, the role of endogenous MxB in alpha interferon-mediated inhibition of HIV-1 infection was questioned by a recent study showing that MxB knockout did not increase the level of infection by HIV-1 which carried the G protein of vesicular stomatitis virus (VSV), allowing infection of CD4-negative HT1080 cells. In order to further examine the anti-HIV-1 activity of endogenous MxB, we have used CRISPR/Cas9 to deplete MxB in different cell lines and observed a substantial restoration of HIV-1 infection in the presence of alpha interferon treatment. However, this rescue effect of MxB knockout became much less pronounced when infection was performed with HIV-1 carrying the VSV G protein. Interestingly, a CRISPR/Cas9 knockout screen of alpha interferon-stimulated genes in U87-MG cells revealed that the genes for interferon-induced transmembrane protein 2 (IFITM2) and IFITM3 inhibited VSV G-pseudotyped HIV-1 much more strongly than the rest of the genes tested, including the gene for MxB. Therefore, our results demonstrate the importance of MxB in alpha interferon-mediated inhibition of HIV-1 infection, which, however, can be underestimated if infection is performed with VSV G protein-pseudotyped HIV-1, due to the high sensitivity of VSV G-mediated infection to inhibition by IFITM proteins.
IMPORTANCE The results of this study reconcile the controversial reports regarding the anti-HIV-1 function of alpha interferon-induced MxB protein. In addition to the different cell types that may have contributed to the different observations, our data also suggest that VSV G protein-pseudotyped HIV-1 is much less inhibited by alpha interferon-induced MxB than HIV-1 itself is. Our results clearly demonstrate an important contribution of MxB to alpha interferon-mediated inhibition of HIV-1 in CD4+ T cells, which calls for using HIV-1 target cells and wild-type virus to test the relevance of the anti-HIV-1 activity of endogenous MxB and other restriction factors.
INTRODUCTION
HIV-1 infection is inhibited by type I interferon (IFN) (reviewed in reference 1). Several genes that are stimulated by type I IFN, called IFN-stimulated genes (ISGs), have been reported to bear anti-HIV-1 activity (1–5), including the myxovirus resistance gene B (MxB) (6–8). Humans carry two Mx genes, MxA and MxB (also called Mx1 and Mx2, respectively), both of which respond to IFN stimulation (reviewed in reference 9). The antiviral function of MxA has been well received for its inhibition of a range of different viruses, including influenza viruses, human parainfluenza virus type 3, measles virus, hepatitis B virus, and others (10). Discovery of the antiviral activity of MxB came relatively late, which was made in 2013, when HIV-1 was reported to be subject to inhibition by MxB (6–8). Recently, human herpesviruses were also shown to be restricted by MxB (11).
MxA and MxB are large dynamin-like GTPases (reviewed in reference 9). Both proteins fold into similar structures, comprising a stalk that mediates oligomerization of the Mx protein and a globular domain that bears the GTPase activity (12–15). The formation of oligomers is indispensable for both MxA and MxB to restrict their target viruses. While MxA depends on its GTPase activity for its antiviral function, the GTPase activity of MxB is required to inhibit human herpesviruses but not HIV-1 (6, 8, 11–19). MxA is cytoplasmic, whereas MxB is seen more at the cytoplasmic face of the nuclear envelope. In contrast to MxA, which exploits its unstructured loop 4 to recognize viral nucleoproteins (20, 21), MxB uses its N-terminal region to achieve targeting of the HIV-1 capsid core (22–26).
Results of mutagenesis studies and viral escape experiments suggest that the HIV-1 capsid protein is the viral target of MxB (6–8, 22, 24), which is further supported by the association of MxB with HIV-1 capsid assembly in vitro (13, 27). MxB does not affect HIV-1 reverse transcription but, rather, reduces the levels of HIV-1 two-copy long terminal repeat (LTR) circles and the integrated viral DNA (6–8), as well as alters the distribution of HIV-1 DNA integration sites in cellular DNA (24).
Several studies indicate a role of MxB in suppressing HIV-1 acquisition and replication in patients. First, significantly higher levels of MxB mRNA were reported in highly exposed HIV-seronegative Kenyan sex workers, suggesting an association of MxB with reduced HIV-1 susceptibility in this cohort (28). Second, an MxB haplotype, rs2074560, showed a higher association with HIV-1-exposed seronegative individuals, likely as a result of the increased MxB expression associated with rs2074560 and lower HIV-1 replication (29). Equally important, MxB resistance mutations in the viral capsid protein, such as the H87Q and G116A mutations, have a high prevalence in circulating HIV-1 strains (30–32). Moreover, the CRF08 BC strain, a result of recombination of the A116 subtype C strain and the 116G subtype B strain, carries the MxB-sensitive G116 capsid but regains the MxB-resistant A116 mutation in more than half of its descendants after 13 years of circulating in China (31). All these findings support an inhibitory pressure of MxB on HIV-1 in patients.
However, the role of MxB in type I IFN-mediated inhibition of HIV-1 infection remains unsettled. When the anti-HIV-1 activity of MxB was reported in 2013, three labs used short hairpin RNA (shRNA) to knock down MxB in different cell lines, including SupT1, THP-1, U87/CD4/CXCR4, and HOS cells, and all observed a substantial restoration of HIV-1 infection in the presence of alpha interferon (IFN-α) (6–8). A subsequent study by Opp et al. utilized Cas9/guide RNA (gRNA) to knock out MxB in THP-1 cells and HT1080 cells and reported no role of MxB in IFN-α-mediated inhibition of HIV-1 infection (33). Subsequent to this report, the MxB knockout experiment in THP-1 cells was repeated by Bulli et al., and the data showed a significant alleviation of HIV-1 inhibition by IFN-α (34). With the aim to reconcile these inconsistent reports and further elucidate the importance of IFN-α-induced MxB in controlling HIV-1 infection, we used Cas9/gRNA to knock out MxB in different cell lines, including HT1080, U87, and SupT1 cells. The results suggest that the anti-HIV-1 potency of IFN-α-induced endogenous MxB can be cell type dependent, with the weakest anti-HIV-1 effect being found in HT1080 cells. The data also suggest that vesicular stomatitis virus (VSV) G protein-pseudotyped HIV-1 is less sensitive to inhibition by IFN-α-induced endogenous MxB. Overall, our results further support an important role of MxB in the IFN-α-mediated anti-HIV-1 response in HIV-1 target cells.
RESULTS
Knockout of MxB in HT1080 cells marginally alleviates the inhibition of HIV-1 infection by IFN-α2b.
We first knocked out MxB in HT1080 cells and measured the effect on HIV-1 infection. Two gRNAs were accordingly designed (Fig. 1A). gRNA1 targets exon II, which is downstream of the ATG start codon and which overlaps one of the gRNAs used by Opp et al. in their study (33). gRNA2 targets exon IV. Both gRNAs were cloned into the lentiCRISPRv2 vector, which carries the U6 promoter cassette to express gRNA and the elongation factor 1α promoter to express Cas9 (35). HT1080 cells were transduced with lentiviruses that packaged the Cas9/gRNA sequence and then subjected to selection with puromycin, to generate stably transduced cell lines. The lentiCRISPRv2 vector that did not have a gRNA inserted was used to create the control cell line. We also designed one gRNA to target the STAT1 gene. STAT1, together with STAT2, is phosphorylated by IFN-α-activated JAK1/TYK2 and activates the expression of ISGs (36). Therefore, depletion of STAT1 is expected to block IFN-α signaling transduction and prevent the expression of ISGs. The results of Western blotting of the generated cell lines showed that both MxB gRNAs efficiently depleted the expression of MxB upon IFN-α2b stimulation but did not affect the expression of STAT1, suggesting that MxB knockout does not exert a general effect on the cellular response to IFN-α stimulation (Fig. 1B). The STAT1 gRNA depleted the STAT1 expression that was enhanced by IFN-α2b and dramatically reduced MxB expression, which confirmed the impaired IFN-α response due to STAT1 depletion (Fig. 1B). As HT1080 cells do not express HIV-1 receptors CD4 and CXCR4/CCR5, we challenged these cells with VSV G-pseudotyped HIV-1 (HIV-1/VSV G). Two doses of IFN-α2b, 500 U/ml and 1,000 U/ml, were used to treat HT1080 cells prior to exposure to HIV-1/VSV G. HIV-1 infectivity was determined by infecting TZM-bl indicator cells (37). IFN-α2b treatment decreased the level of HIV-1 infection in control cells to 15% (500 U/ml IFN-α2b) and 6% (1,000 U/ml IFN-α2b) of the level of infection without IFN-α2b treatment (Fig. 1C). When STAT1 was depleted, the level of HIV-1 infection in the presence of 1,000 U/ml IFN-α2b rose from 6% to 40%, indicating a significant loss of IFN-α2b inhibition (Fig. 1C). In contrast, IFN-α2b diminished HIV-1 infection in MxB knockout cells as strongly as it did in the control cells (Fig. 1C), which suggests that in HT1080 cells, IFN-α2b-induced MxB does not contribute to the maximal inhibition of VSV G-pseudotyped HIV-1 by IFN-α2b, in agreement with the results obtained by Opp et al. (33).
FIG 1.
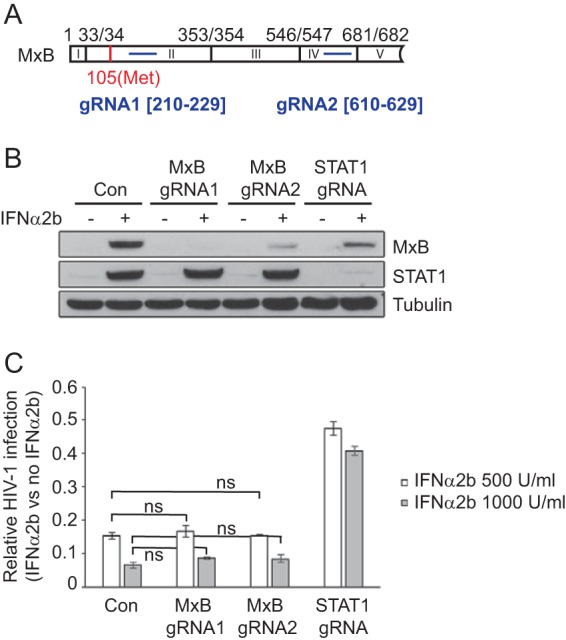
Effect of MxB depletion on IFN-α2b inhibition of HIV-1/VSV G infection in HT1080 cells. (A) Illustration of the gRNA target sites (in blue) in the MxB gene. The first 5 exons (I to V) of MxB are shown. The boundary of adjacent exons is specified by the nucleotide positions in MxB mRNA (GenBank accession number NM_002463.1). The ATG start codon (at nucleotide position 105) is highlighted in red. (B) Levels of MxB and STAT1 in control cells (Con) and cells stably transduced with Cas9/gRNA in the presence and absence of IFN-α2b (500 U/ml). The levels of tubulin were also determined as the internal control. (C) Inhibition of HIV-1/VSV G infection by IFN-α2b (500 U/ml and 1,000 U/ml). The levels of infectious HIV-1 were determined by infecting TZM-bl indicator cells. Suppression of HIV-1 infection in the control and knockout cell lines was calculated by dividing the levels of HIV-1 production with IFN-α2b treatment by those without IFN-α2b treatment. The data shown are the averages from three independent infections of HT1080 cells. Error bars indicate standard deviations. ns, not significant.
Knockout of MxB in SupT1 cells and U87/CD4 cells significantly restores HIV-1 infection in the presence of IFN-α2b.
HT1080 cells are epithelial cells in nature and are not target cells for HIV-1. We therefore asked whether IFN-α-induced MxB can suppress HIV-1 infection in CD4+ T cells and whether HIV-1 without the VSV G protein is more susceptible to inhibition by IFN-α2b-induced MxB. We chose to knock out MxB in a CD4+ T cell line, SupT1 (Fig. 2A), and then compared the response of HIV-1 to IFN-α2b treatment in the control and the MxB knockout SupT1 cell lines. We also depleted STAT1 using Cas9/gRNA and observed that depletion of STAT1, which impairs the cellular response to IFN-α, restored the level of HIV-1 infection under IFN-α2b treatment from 10% in the control cells to 60% in STAT1 knockout cells (Fig. 2B). Depletion of MxB by either gRNA1 or gRNA2 elevated the level of HIV-1 infection in the presence of IFN-α2b from 10% to 40% (Fig. 2B), which indicates a marked attenuation of IFN-α2b-mediated HIV-1 inhibition as a result of MxB knockout. Interestingly, when the infection was performed with HIV-1 carrying the VSV G protein, although depletion of STAT1 still effectively rescued HIV-1 infection, knockout of MxB increased the level of HIV-1 infection from 10% to only 20%, which is significantly lower than the 40% restoration when infection was performed with HIV-1 itself (Fig. 2B). These results suggest that MxB plays a major role in IFN-α-mediated inhibition of HIV-1 in T cells. This inhibitory role of MxB becomes less pronounced against HIV-1 that is pseudotyped with the VSV G protein.
FIG 2.

Depletion of MxB in SupT1 and U87/CD4 cells rescues HIV-1 infection under IFN-α2b treatment. (A) Levels of MxB and STAT1 in the control SupT1 cells and in SupT1 cells that were stably transduced with Cas9/gRNA in the absence and presence of IFN-α2b (500 U/ml). The levels of tubulin served as the internal control. (B) IFN-α2b (500 U/ml) suppression of HIV-1 infection, without or with VSV G protein pseudotyping, in the control and knockout SupT1 cells. Relative HIV-1 infection was calculated with the values in the absence IFN-α2b arbitrarily set as 1. (C) Levels of MxB and STAT1 in control and knockout U87/CD4/CXCR4 cells in the absence and presence of IFN-α2b. (D) IFN-α2b (100 U/ml) suppression of HIV-1 NL4-3 or VSV-G-pseudotyped NL4-3 (NL4-3/VSV G) in control and knockout U87/CD4/CXCR4 cells. (E) Results of Western blotting to show the depletion of MxB and STAT1 in U87/CD4/CCR5 cell lines that were stably transduced with Cas9/gRNA. (F) IFN-α2b (100 U/ml) suppression of HIV-1 AD8-1 or VSV-G-pseudotyped AD8-1 (AD8-1/VSV G) in the control and MxB knockout U87/CD4/CCR5 cell lines. The results shown are the averages from three independent infections of the knockout cell lines. (G) IFN-α2b (500 U/ml) suppression of HIV-1 H87Q and G208A mutants in the control SupT1 cells or in SupT1 cells with MxB or STAT1 knockout. The level of wild-type HIV-1 NL4-3 infection in the control SupT1 cells in the absence of IFN-α2b was arbitrarily set as 1. The infection levels of the H87Q and G208A mutants were calculated accordingly. The results shown are the averages from three independent infection experiments. Error bars indicate standard deviations. *, P < 0.05; **, P < 0.005; ns, not significant.
We further investigated the anti-HIV-1 activity of MxB in U87 cells, a human glioblastoma cell line. When MxB was knocked out in U87/CD4/CXCR4 cells (Fig. 2C), the level of HIV-1 infection in the presence of IFN-α2b rose from 10% (control cells) to 40% (MxB gRNA1) and 30% (MxB gRNA2), an increase similar to that seen when STAT1 was knocked out (Fig. 2D). However, marginal increases were observed in MxB knockout U87/CD4/CXCR4 cells when infection was performed with HIV-1 carrying VSV G, in contrast to an increase from 5% (control cells) to 40% as a result of STAT1 knockout. Similar results were obtained when MxB was knocked out in U87/CD4/CCR5 cells and infections were performed with either the R5-tropic HIV-1 strain AD8-1 or AD8-1 carrying the VSV G protein (Fig. 2E and F). These data further demonstrate a strong inhibition of HIV-1 by IFN-α-induced MxB, but this inhibition is weakened against VSV G-pseudotyped HIV-1.
The MxB-resistant capsid mutation H87Q is less inhibited by IFN-α2b.
Mutations in HIV-1 capsid mediate resistance to MxB (6–8). We therefore chose to test two mutants with such capsid mutations, H87Q and G208A (22, 30), for their ability to resist IFN-α2b inhibition. The results showed that the G208A mutant was inhibited by IFN-α2b in SupT1 cells to similar degrees as wild-type HIV-1 (Fig. 2G). In contrast, IFN-α2b decreased the level of infection by the H87Q mutant to only 40% of that without exposure to IFN-α2b, which is significantly higher than the 15% observed for wild-type HIV-1 (Fig. 2G). Again, knockout of MxB significantly increased the level of infection by wild-type HIV-1 and the G208A mutant in the presence of IFN-α2b treatment (Fig. 2G). These results suggest that some MxB resistance capsid mutations render HIV-1 resistant to IFN-α inhibition.
Knockout of MxB alleviates the inhibition of HIV-1 reporter viruses by both IFN-α and IFN-β.
Given that MxB impairs the nuclear import of HIV-1 DNA (6, 8), we further investigated the effect of MxB knockout on IFN-α2b-mediated inhibition of HIV-1 using reporter viruses NLENY1 and NLENY1-ES/VSV G, which express yellow fluorescent protein (YFP) and allow quantification of the infected cells by flow cytometry (38). Virus NLENY1 carries the HIV-1 Env protein, whereas NLENY1-ES/VSV-G does not express HIV-1 Env protein and is pseudotyped with the VSV G protein. The results in Fig. 3 show that IFN-α2b treatment led to a 2-fold reduction of SupT1 cells that were infected either by NLENY1 or by NLENY1-ES/VSV G. Knockout of STAT1 ablated the IFN-α2b-mediated inhibition of both viruses. As opposed to a nearly complete rescue of NLENY1 virus infection from IFN-α2b inhibition by MxB knockout, the rescue effect on NLENY1-ES/VSV G was much less pronounced (Fig. 3). We further tested the effect of IFN-β on these two HIV-1 reporter viruses. As expected, the MxB protein was induced in the control SupT1 cells by IFN-β but was not expressed in the MxB knockout or STAT1 knockout SupT1 cell lines (Fig. 4A). Infection with both NLENY1 and NLENY1-ES/VSV G decreased 2-fold with IFN-β treatment (Fig. 4B and C). This inhibition was lost in MxB knockout cells for the NLENY1 virus but persisted for NLENY1-ES/VSV G. These data further demonstrate that MxB has an important role in type I interferon-mediated control of HIV-1 infection, including that by both IFN-α and IFN-β, albeit the role of MxB is less prominent when HIV-1 is pseudotyped with the VSV G protein.
FIG 3.

Knockout of MxB alleviates inhibition of HIV-1 reporter virus by IFN-α2b. IFN-α2b (500 U/ml) was used to treat SupT1 cells, including control and MxB/gRNA1, MxB/gRNA2, or STAT1/gRNA knockout SupT1 cells. Sixteen hours later, cells were infected with HIV-1 reporter viruses NLENY1 or NLENY1-ES/VSV G. The number of infected (YFP-positive) cells was scored with flow cytometry. (A) The results of one representative infection experiment are shown. (B) The averages from three independent experiments are summarized in the bar graph. The values of IFN-α2b inhibition were calculated as described in the legend to Fig. 1C. Error bars indicate standard deviations. *, P < 0.05; **, P < 0.005. SSC-A, side scatter area.
FIG 4.

IFN-β inhibition of HIV-1 is mitigated by MxB knockout. The control, MxB knockout, or STAT1 knockout SupT1 cell lines were treated with IFN-β (100 U/ml) for 16 h before being infected with NLENY1 or NLENY1-ES/VSV G. (A) Expression of MxB and STAT1 with or without IFN-β treatment was measured by Western blotting. (B) The number of infected (YFP-positive) cells was scored with flow cytometry. The results of one representative infection experiment are shown. (C) The values of IFN-β inhibition were determined as described in the legend to Fig. 1C. The averages from three independent infection experiments were calculated. Error bars indicate standard deviations. *, P < 0.05; **, P < 0.005. SSC-A, side scatter area.
Ectopically expressed MxB inhibits HIV-1 carrying the VSV G protein.
We next asked whether MxB inhibits HIV-1 to a lesser degree when the virus carries the VSV G protein. This may explain why VSV G-pseudotyped HIV-1 responded poorly to the rescue by MxB depletion in the presence of IFN-α treatment. To this end, we used a SupT1 cell line expressing exogenous MxB protein upon induction by doxycycline (7). We treated the MxB SupT1 cell lines with a range of doxycycline concentrations and induced MxB expression from low (induced by 5 ng/ml doxycycline) to high (induced by 100 ng/ml) levels (Fig. 5A). When the treated cells were infected with either NL4-3 or NL4-3 carrying the VSV G protein, expression of the viral Gag and p24/CA proteins diminished gradually with an increase in MxB expression, indicating suppression of viral infection (Fig. 5A and B). We also performed Western blotting to measure the level of endogenous MxB in SupT1 cells upon treatment with IFN-α2b (500 U/ml) and found that its level was equivalent to the MxB amount that was induced by doxycycline at 5 ng/ml to 10 ng/ml in the MxB-expressing SupT1 cell line (Fig. 5C). We then measured the levels of infectious HIV-1 particles that were produced by the infected SupT1 cells. Doxycycline did not markedly affect HIV-1 production from control SupT1 cells that did not express exogenous MxB (Fig. 5D). The doxycycline-induced MxB inhibited both HIV-1 NL4-3 and HIV-1 NL4-3 carrying VSV G to similar degrees across all doxycycline concentrations tested (Fig. 5D). The inhibition of HIV-1 by exogenous MxB expression was slightly greater than the inhibition of HIV-1 carrying VSV G, but the differences were not significant. These data suggest that MxB, when ectopically expressed in the absence of other IFN-α-induced ISGs, inhibits HIV-1 and VSV G-pseudotyped HIV-1 equally well.
FIG 5.
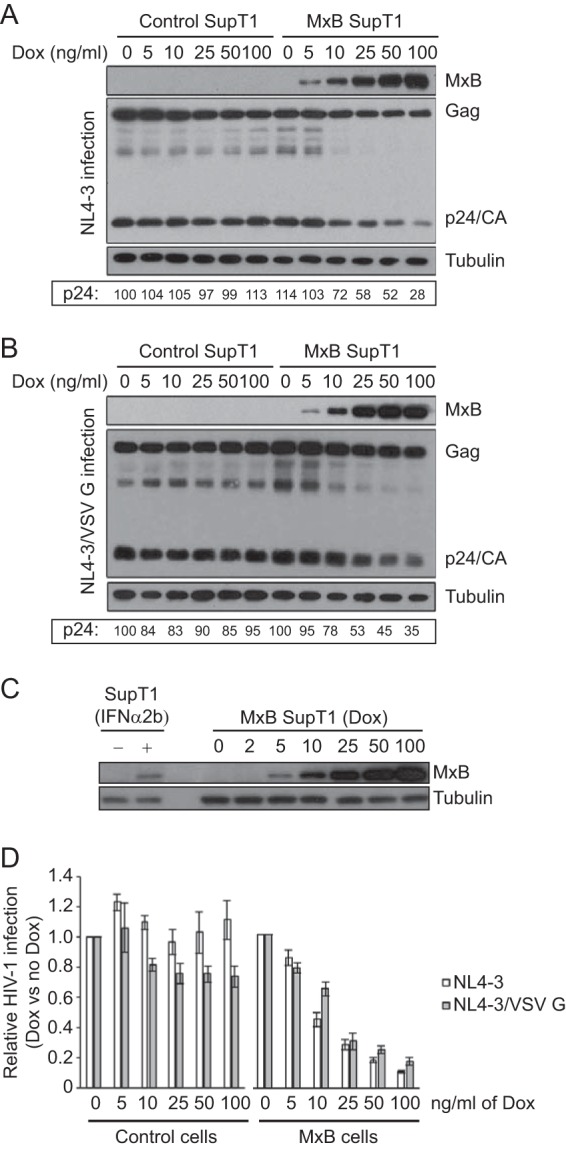
Suppression of HIV-1 NL4-3 or NL4-3/VSV G by ectopically expressed MxB. (A, B) SupT1 cells that were stably transduced with a tetracycline-inducible MxB expression retroviral vector were treated with increasing doses of doxycycline (Dox), followed by challenge with NL4-3 (equivalent to 100 ng p24) or NL4-3/VSV G (equivalent to 10 ng p24) viruses. The levels of MxB and viral Gag/p24 proteins were measured by Western blotting, and representative blots from one of the three independent infection experiments are shown for NL4-3-infected cells (A) and for NL4-3/VSV G-infected cells (B). The p24 signals were quantified using ImageJ software and are presented with the value of p24 for NL4-3 in control cells (0 ng/ml doxycycline treatment) arbitrarily set as 100. (C) SupT1 cells were treated with 500 U/ml IFN-α2b for 16 h to induce the expression of endogenous MxB. The MxB-expressing SupT1 cell line was treated with different doses of doxycycline (in ng/ml) for 16 h. Cell lysates were examined by Western blotting using anti-MxB antibody. The levels of tubulin were also probed as the internal control. (D) The levels of viruses in the supernatants were determined by infecting TZM-bl indicator cells. The virus amount from the infection without doxycycline treatment was arbitrarily set as 1. The data shown are averages from three independent transfections. Error bars indicate standard deviations.
VSV G-mediated infection is strongly suppressed by IFN-α-induced IFITM proteins.
Since VSV G-pseudotyped HIV-1 was strongly suppressed by IFN-α and knockout of MxB only slightly reduced this inhibition, we were interested in knowing which IFN-α-induced protein(s) plays the major inhibitory role. To this end, we designed gRNAs to target 53 ISGs which were reported to be upregulated by IFN-α in U87-MG cells by more than 3-fold (8). In order to achieve efficient knockout, three gRNAs were designed for each ISG (see Table S1 in the supplemental material). The three gRNAs of each ISG were pooled to transduce U87-MG cells. The stably transduced cell lines, selected with puromycin, were treated with IFN-α2b to induce the expression of ISGs, prior to challenge with VSV G-pseudotyped HIV-1. HIV-1 infection was monitored by measuring the levels of infectious HIV-1 in the culture supernatants through infection of TZM-bl indicator cells. We calculated the IFN-α2b inhibition of HIV-1 in each ISG knockout cell line by dividing the level of HIV-1 production in the presence of IFN-α2b treatment by the level of HIV-1 production in the absence of IFN-α2b treatment. We set this value from the control cells as 1 and present the data for all ISG knockout cell lines in Fig. 6A. As the positive control of this screen study, STAT1 was knocked out, which led to a 25- to 30-fold restoration of HIV-1 infection in the presence of IFN-α2b (Fig. 6A). In agreement with the data in Fig. 2, MxB knockout only moderately relieved the IFN-α2b inhibition of HIV-1/VSV G (Fig. 6A). Remarkably, knockout of interferon-induced transmembrane protein 3 (IFITM3) increased HIV-1/VSV G infection in the presence of IFN-α by 10-fold; knockout of IFITM2 led to a 5-fold increase (Fig. 6A), an enhancement of HIV-1/VSV G infection that was markedly higher than that achieved by knocking out any of the other ISGs. Knockout of the majority of the ISGs did not alleviate the inhibition of HIV-1 infection by IFN-α2b. In addition, knockout of some ISGs, including ISG15, USP18, DHX58, and IFI44L, moderately enhanced IFN-α-mediated inhibition, which suggests a possible negative regulation of the interferon response by these ISGs.
FIG 6.

Cas9/gRNA knockout screen in U87-MG cells for ISGs that alleviate IFN-α2b inhibition of HIV-1 NL4-3/VSV G. (A) Effects of ISG knockout (KO) on IFN-α2b (100 U/ml) suppression of HIV-1 NL4-3/VSV G infection. Fifty-three ISGs were selected for the knockout study. For each ISG, a pool of three gRNAs was used to stably transduce U87-MG cells. NL4-3/VSV G infection was monitored by measuring the levels of viruses produced through infection of TZM-bl cells. IFN-α2b suppression of NL4-3/VSV G infection was calculated by dividing the amount of viruses produced in the presence of IFN-α2b by the amount of viruses produced without IFN-α2b treatment. The value from control cells in each screening experiment is arbitrarily set as 1, so that all the data from multiple experiments can be summarized in the bar graph. If the value from an ISG knockout cell line is greater than 1, then the knockout of this ISG leads to an increase in HIV-1/VSV G infection in the presence of IFN-α2b. The results shown are the averages from two independent screens of all 53 ISGs. For clarity, data are presented in two graphs. (B) Results of Western blotting to show the levels of IFITM2 or IFITM3 in the U87-MG knockout cell lines. Three gRNAs were tested for IFITM2 or IFITM3. One cell line was created using all three gRNAs (1/2/3) to achieve maximal knockout efficiency. (C) IFN-α2b (100 U/ml) inhibition of NL4-3/VSV G infection in IFITM2 or IFITM3 knockout U87-MG cell lines. The levels of viruses that were produced in the absence or presence of IFN-α2b were determined by infecting TZM-bl indicator cells. The results shown are the averages from three independent infections. RLU, relative luciferase units. Error bars indicate standard deviations.
To confirm the dominant contribution of IFITM2 and IFITM3 to IFN-α2b-mediated inhibition of HIV-1/VSV G infection, we generated cell lines which were stably transduced with one gRNA for IFITM2 or IFITM3. The IFITM2 gRNA1 and gRNA3 efficiently depleted IFITM2 (Fig. 6B) and increased the level of HIV-1/VSV G infection in the presence of IFN-α2b by 5-fold (Fig. 6C). For IFITM3, gRNA2 diminished IFITM3 greater than the other two gRNAs (Fig. 6B) and elevated the level of HIV-1/VSV G infection by 8-fold (Fig. 6C). When all three gRNAs for IFITM2 or IFITM3 were used, maximal depletion of each gene was observed (Fig. 6B), which consequently generated the highest rescue effect on HIV-1/VSV G infection in the presence of IFN-α2b compared to that generated when an individual gRNA was used for knockout (Fig. 6C). Together, these data support the dominant role of IFITM3 and IFITM2 in IFN-α2b-mediated inhibition of VSV G-pseudotyped HIV-1.
DISCUSSION
The results of this study demonstrate that depletion of MxB with Cas9/gRNA markedly alleviates IFN-α-mediated inhibition of HIV-1 infection. This significant role of MxB in the anti-HIV-1 response by IFN-α is also supported by several studies that used shRNA or Cas9/gRNA to deplete endogenous MxB (6–8). In particular, Bulli et al. recently used two gRNAs to knock out MxB in THP-1 cells and observed a significant increase in the level of HIV-1 infection in the presence of IFN-α treatment (34), which is in agreement with the results observed using shRNA to deplete MxB in THP-1 cells (6, 8). Further, we observed that this role of MxB can be underestimated when the VSV G protein is used to pseudotype HIV-1, with the possibility of no effect of MxB depletion on IFN-α inhibition of HIV-1 infection in some cell lines, including HT1080 cells. The inhibitory pressure of endogenous MxB in the context of IFN-α stimulation, as observed in tissue culture studies by multiple groups, is further supported by the association of high MxB levels with low HIV-1 acquisition levels in vivo (28, 29) and the prevalence of MxB resistance capsid mutations in circulating HIV-1 strains (30–32). The role of MxB in type I interferon-mediated antiviral defense is also supported by a recent report showing that knockdown of MxB markedly rescues infections by herpes simplex virus 1 (HSV-1) in the presence of IFN-α treatment (11). Further study is warranted to investigate the contribution of MxB to interferon-mediated inhibition of viruses in primary cells, including knockout of MxB in human peripheral blood mononuclear cells (PBMCs) and primary CD4+ T cells. Technical challenges include the relatively low transfection and transduction efficiency of primary cells and the difficulty to select for and enrich knockout primary cells, as is often practiced with transformed cell lines.
MxB strongly suppresses VSV G-pseudotyped HIV-1 when ectopically expressed in the absence of other ISGs (33). In agreement with this, when we compared MxB inhibition of HIV-1 with or without VSV G pseudotyping, we observed only modestly less suppression of VSV G-pseudotyped HIV-1 than HIV-1 itself along a range of MxB expression levels that were induced with different doses of doxycycline (Fig. 5). We suspect that in the context of IFN-α stimulation, other ISGs might have influenced the anti-HIV-1 effect of the endogenous MxB. In support of this possibility, results from our knockout screen of ISGs in U87-MG cells showed a dominant inhibition of VSV G-pseudotyped HIV-1 by IFN-α-induced IFITM2 and IFITM3 proteins. This observation may explain the relatively moderate increase in HIV-1/VSV G infection when the other ISGs, including MxB, were knocked out (Fig. 6). This is likely because IFITM2 and IFITM3 are so potent in inhibiting VSV G-mediated infection that HIV-1/VSV G infection is still substantially suppressed when other anti-HIV-1 ISGs are depleted.
Mutations across the N- and C-terminal domains of the capsid protein have been reported to confer resistance to MxB to various degrees (6–8, 22, 24, 30). One group of mutations is located in the cyclophilin A binding loop in the N-terminal domain of capsid, including mutations at amino acid positions V86, H87, G89, and P90. A second cluster is found in a solvent-accessible loop between helices 10 and 11 in the C-terminal domain of capsid, including amino acid positions P207 and G208. Several more MxB-resistant capsid mutations, such as the cell cycle-dependent mutation N57S, were also identified and are predominantly located at the surface of the capsid hexamer. These MxB-resistant capsid mutations modulate HIV-1 sensitivity to IFN-α to different degrees. Some mutations, such as N57S, result in modest resistance to IFN-α (6), and some, such as the G208A mutation, do not confer any resistance in this regard (33), while other mutations, such as N74D and P90A, render HIV-1 more sensitive to IFN-α inhibition (34). The results of our study showed that the MxB resistance mutation H87Q is significantly refractory to IFN-α. These different responses of MxB-resistant HIV-1 mutations to IFN-α may result from the diverse effects of these mutations on the functions of capsid, ranging from capsid stability, core uncoating, routes of nuclear entry, and interaction with a plethora of host factors. Thus, it is expected that in spite of the marked inhibition of HIV-1 by MxB, resistance to MxB may not always lead to resistance to IFN-α. All the same, it is encouraging to observe that some of these MxB resistance mutations, such as N57S and H87Q, do confer resistance to IFN-α inhibition of HIV-1.
In summary, our data further support the important contribution of MxB to IFN-α-mediated inhibition of HIV-1 and other viruses, including human herpesviruses. The anti-HIV-1 activity of MxB in the context of IFN-α stimulation can be underestimated when infection is performed with VSV G-pseudotyped HIV-1. This is partially due to the high sensitivity of VSV G-mediated virus entry to IFN-α-induced IFITM proteins, which might have shadowed the anti-HIV-1 effect of other ISGs.
MATERIALS AND METHODS
Plasmid DNA.
The infectious HIV-1 DNA clone NL4-3 was obtained from the NIH AIDS Reagent Program; the AD8-1 clone was kindly provided by Eric O. Freed. Site-directed mutagenesis (Clontech) was performed to generate HIV-1 NL4-3 mutants that carry capsid mutation H87Q or G208A. The lentiCRISPRv2 plasmid was purchased from Addgene. The guide RNA primers were synthesized by Invitrogen and inserted into the BstBI site in lentiCRISPRv2. HIV-1 reporter virus DNAs NLENY1-IRES and NLENY1-IRES-ES were kindly provided by David Levy (38).
Cell culture.
HEK293T, HT1080, TZM-bl, U87-MG, U87/CD4/CXCR4, and U87/CD4/CCR5 cells were grown in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum (FBS; Invitrogen), 50 U/ml penicillin, and 50 μg/ml streptomycin (Invitrogen). SupT1 cells were grown in RPMI 160 medium supplemented with 10% FBS, 50 U/ml penicillin, and 50 μg/ml streptomycin (Invitrogen). TZM-bl is an HIV indicator cell line that carries the HIV-1 LTR-luciferase expression cassette, which responds to transactivation by HIV-1 Tat (37). HEK293T and HT1080 cells were purchased from ATCC. U87 cell lines were obtained from the NIH AIDS Reagent Program. Transfection of HEK293T cells was conducted with the Lipofectamine reagent (Invitrogen) in accordance with the manufacturer's instructions.
Generating MxB knockout cell lines.
Lentivirus particles were produced through the transfection of HEK293T cells using plasmid DNA clones lentiCRISPRv2 (expressing single guide RNA [sgRNA] targeting MxB), psPAX2 (expressing Gag and Gag-Pol), and pVSV-G, as described in reference 39. Next, HT1080, SupT1, and U87 cells were infected with these lentiviruses, followed by puromycin selection to generate stable cell lines. The control cell line was created using the lentiCRISPRv2 vector that does not express sgRNA. MxB knockout efficiency was determined by Western blotting, examining the levels of MxB in the created cell lines in which IFN-α2b (500 U/ml; Sigma) was used to induce MxB expression for 24 h. The anti-MxB antibodies were generated as we previously described (7). The sequences of the MxB sgRNAs are 5′-TGTGGTGGCACTGTGCCGAA-3′ for sgRNA1 and 5′-GTTCCGGTAGCTGATCCTTC-3′ for sgRNA2. The STAT1 knockout cell lines were generated using the same method. The sequence of STAT1 sgRNA is 5′-GCAGCTTGACTCAAAATTCC-3′.
HIV-1 infection.
HIV-1 was produced by transfecting HEK293T cells with proviral DNA clones NL4-3, AD8-1, NL4-3/H87Q, NL4-3/G208A, or NLENY1. The pVSV-G plasmid was cotransfected to produce HIV-1 particles that carry the VSV glycoprotein. The amounts of HIV-1 produced were determined by either measuring HIV-1 capsid/p24 by enzyme-linked immunosorbent assay or measuring HIV-1 reverse transcriptase activity, as previously described (40). An amount of HIV-1 equivalent to 100 ng p24 was used to infect different cell lines. For HIV-1 that was pseudotyped with the VSV G protein, an amount of virus equivalent to 10 ng p24 was used for infection because of the high entry efficiency of the VSV G protein. Prior to HIV-1 infection, cells were treated with either IFN-α2b (Sigma) or IFN-β (Sigma) for 24 h or left untreated. Each infection was performed in duplicate. Each experiment was independently repeated at least three times. The inoculated HIV-1 was removed 16 h after infection. After an additional 24-hour culture, 50 μl of the culture supernatants was used to infect the TZM-bl indicator cells in duplicate. Forty hours later, cells were collected, and the luciferase activity in the cell lysates was measured using a luciferase assay system (Promega). The results represent the levels of infectious HIV-1 that were produced. For infections by NLENY1 and NLENY1-ES/VSV-G, the infected cells were harvested and fixed in 4% paraformaldehyde. The number of YFP-positive cells was scored by flow cytometry. All HIV-1 infection experiments were performed in the certified biocontainment level 3 lab at the Lady Davis Institute.
ISG knockout screen in U87-MG cells.
We selected 53 ISGs whose levels increased in U87-MG cells by more than 3-fold after IFN-α2b treatment, as shown in the microarray data by Goujon et al. (8). Three sgRNAs were designed to target each of these 53 ISGs and were cloned into lentiCRISPRv2. The sequences of all sgRNAs are listed in Table S1 in the supplemental material. For each ISG, viruses that encode the individual sgRNA were pooled and used to transduce U87-MG cells. After selection with puromycin for stably transduced cell lines, HIV-1/VSV G (which was in an amount equivalent to 15 ng capsid/p24 and which carries the VSV glycoprotein) was used to infect these cell lines that had been treated with IFN-α2b (100 U/ml) or left untreated. At 40 h postinfection, the levels of infectious HIV-1 in the culture supernatants were determined by infecting TZM-bl cells. The knockout efficiency of STAT1, IFITM2, and IFITM3 was examined by Western blotting. The anti-STAT1 antibody was purchased from Santa Cruz Biotechnology (C-111), and the anti-IFITM2 (12769-1-AP) and anti-IFITM3 (11714-1-AP) antibodies were purchased from the Proteintech Group.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by funding from the Canadian Institutes of Health Research (HOP-143171).
We thank Kavita Raniga for critically reading the manuscript and Zhen Wang and Yimeng Wang for technical assistance.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.00422-18.
REFERENCES
- 1.Doyle T, Goujon C, Malim MH. 2015. HIV-1 and interferons: who's interfering with whom? Nat Rev Microbiol 13:403–413. doi: 10.1038/nrmicro3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kane M, Zang TM, Rihn SJ, Zhang F, Kueck T, Alim M, Schoggins J, Rice CM, Wilson SJ, Bieniasz PD. 2016. Identification of interferon-stimulated genes with antiretroviral activity. Cell Host Microbe 20:392–405. doi: 10.1016/j.chom.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu J, Pan Q, Rong L, He W, Liu SL, Liang C. 2011. The IFITM proteins inhibit HIV-1 infection. J Virol 85:2126–2137. doi: 10.1128/JVI.01531-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krapp C, Hotter D, Gawanbacht A, McLaren PJ, Kluge SF, Sturzel CM, Mack K, Reith E, Engelhart S, Ciuffi A, Hornung V, Sauter D, Telenti A, Kirchhoff F. 2016. Guanylate binding protein (GBP) 5 is an interferon-inducible inhibitor of HIV-1 infectivity. Cell Host Microbe 19:504–514. doi: 10.1016/j.chom.2016.02.019. [DOI] [PubMed] [Google Scholar]
- 6.Kane M, Yadav SS, Bitzegeio J, Kutluay SB, Zang T, Wilson SJ, Schoggins JW, Rice CM, Yamashita M, Hatziioannou T, Bieniasz PD. 2013. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature 502:563–566. doi: 10.1038/nature12653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu Z, Pan Q, Ding S, Qian J, Xu F, Zhou J, Cen S, Guo F, Liang C. 2013. The interferon-inducible MxB protein inhibits HIV-1 infection. Cell Host Microbe 14:398–410. doi: 10.1016/j.chom.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 8.Goujon C, Moncorge O, Bauby H, Doyle T, Ward CC, Schaller T, Hue S, Barclay WS, Schulz R, Malim MH. 2013. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 502:559–562. doi: 10.1038/nature12542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haller O, Staeheli P, Schwemmle M, Kochs G. 2015. Mx GTPases: dynamin-like antiviral machines of innate immunity. Trends Microbiol 23:154–163. doi: 10.1016/j.tim.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 10.Haller O, Kochs G. 2011. Human MxA protein: an interferon-induced dynamin-like GTPase with broad antiviral activity. J Interferon Cytokine Res 31:79–87. doi: 10.1089/jir.2010.0076. [DOI] [PubMed] [Google Scholar]
- 11.Crameri M, Bauer M, Caduff N, Walker R, Steiner F, Franzoso FD, Gujer C, Boucke K, Kucera T, Zbinden A, Munz C, Fraefel C, Greber UF, Pavlovic J. 2018. MxB is an interferon-induced restriction factor of human herpesviruses. Nat Commun 9:1980. doi: 10.1038/s41467-018-04379-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alvarez FJD, He S, Perilla JR, Jang S, Schulten K, Engelman AN, Scheres SHW, Zhang P. 2017. CryoEM structure of MxB reveals a novel oligomerization interface critical for HIV restriction. Sci Adv 3:e1701264. doi: 10.1126/sciadv.1701264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fribourgh JL, Nguyen HC, Matreyek KA, Alvarez FJ, Summers BJ, Dewdney TG, Aiken C, Zhang P, Engelman A, Xiong Y. 2014. Structural insight into HIV-1 restriction by MxB. Cell Host Microbe 16:627–638. doi: 10.1016/j.chom.2014.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao S, von der Malsburg A, Dick A, Faelber K, Schroder GF, Haller O, Kochs G, Daumke O. 2011. Structure of myxovirus resistance protein A reveals intra- and intermolecular domain interactions required for the antiviral function. Immunity 35:514–525. doi: 10.1016/j.immuni.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 15.Gao S, von der Malsburg A, Paeschke S, Behlke J, Haller O, Kochs G, Daumke O. 2010. Structural basis of oligomerization in the stalk region of dynamin-like MxA. Nature 465:502–506. doi: 10.1038/nature08972. [DOI] [PubMed] [Google Scholar]
- 16.Pitossi F, Blank A, Schroder A, Schwarz A, Hussi P, Schwemmle M, Pavlovic J, Staeheli P. 1993. A functional GTP-binding motif is necessary for antiviral activity of Mx proteins. J Virol 67:6726–6732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ponten A, Sick C, Weeber M, Haller O, Kochs G. 1997. Dominant-negative mutants of human MxA protein: domains in the carboxy-terminal moiety are important for oligomerization and antiviral activity. J Virol 71:2591–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dicks MD, Goujon C, Pollpeter D, Betancor G, Apolonia L, Bergeron JR, Malim MH. 2015. Oligomerization requirements for MX2-mediated suppression of HIV-1 infection. J Virol 90:22–32. doi: 10.1128/JVI.02247-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buffone C, Schulte B, Opp S, Diaz-Griffero F. 2015. Contribution of MxB oligomerization to HIV-1 capsid binding and restriction. J Virol 89:3285–3294. doi: 10.1128/JVI.03730-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitchell PS, Patzina C, Emerman M, Haller O, Malik HS, Kochs G. 2012. Evolution-guided identification of antiviral specificity determinants in the broadly acting interferon-induced innate immunity factor MxA. Cell Host Microbe 12:598–604. doi: 10.1016/j.chom.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patzina C, Haller O, Kochs G. 2014. Structural requirements for the antiviral activity of the human MxA protein against Thogoto and influenza A virus. J Biol Chem 289:6020–6027. doi: 10.1074/jbc.M113.543892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Busnadiego I, Kane M, Rihn SJ, Preugschas HF, Hughes J, Blanco-Melo D, Strouvelle VP, Zang TM, Willett BJ, Boutell C, Bieniasz PD, Wilson SJ. 2014. Host and viral determinants of Mx2 antiretroviral activity. J Virol 88:7738–7752. doi: 10.1128/JVI.00214-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kong J, Xu B, Wei W, Wang X, Xie W, Yu XF. 2014. Characterization of the amino-terminal domain of Mx2/MxB-dependent interaction with the HIV-1 capsid. Protein Cell 5:954–957. doi: 10.1007/s13238-014-0113-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matreyek KA, Wang W, Serrao E, Singh P, Levin HL, Engelman A. 2014. Host and viral determinants for MxB restriction of HIV-1 infection. Retrovirology 11:90. doi: 10.1186/s12977-014-0090-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goujon C, Moncorge O, Bauby H, Doyle T, Barclay WS, Malim MH. 2014. Transfer of the amino-terminal nuclear envelope targeting domain of human MX2 converts MX1 into an HIV-1 resistance factor. J Virol 88:9017–9026. doi: 10.1128/JVI.01269-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goujon C, Greenbury RA, Papaioannou S, Doyle T, Malim MH. 2015. A triple-arginine motif in the amino-terminal domain and oligomerization are required for HIV-1 inhibition by human MX2. J Virol 89:4676–4680. doi: 10.1128/JVI.00169-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fricke T, White TE, Schulte B, de Souza Aranha Vieira DA, Dharan A, Campbell EM, Brandariz-Nunez A, Diaz-Griffero F. 2014. MxB binds to the HIV-1 core and prevents the uncoating process of HIV-1. Retrovirology 11:68. doi: 10.1186/s12977-014-0068-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stein DR, Shaw SY, McKinnon LR, Abou M, McCorrister SJ, Westmacott GR, Fowke KR, Plummer FA, Ball TB. 2015. Mx2 expression is associated with reduced susceptibility to HIV infection in highly exposed HIV seronegative Kenyan sex workers. AIDS 29:35–41. doi: 10.1097/QAD.0000000000000490. [DOI] [PubMed] [Google Scholar]
- 29.Sironi M, Biasin M, Cagliani R, Gnudi F, Saulle I, Ibba S, Filippi G, Yahyaei S, Tresoldi C, Riva S, Trabattoni D, De Gioia L, Lo Caputo S, Mazzotta F, Forni D, Pontremoli C, Pineda JA, Pozzoli U, Rivero-Juarez A, Caruz A, Clerici M. 2014. Evolutionary analysis identifies an MX2 haplotype associated with natural resistance to HIV-1 infection. Mol Biol Evol 31:2402–2414. doi: 10.1093/molbev/msu193. [DOI] [PubMed] [Google Scholar]
- 30.Liu Z, Pan Q, Liang Z, Qiao W, Cen S, Liang C. 2015. The highly polymorphic cyclophilin A-binding loop in HIV-1 capsid modulates viral resistance to MxB. Retrovirology 12:1. doi: 10.1186/s12977-014-0129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei W, Guo H, Ma M, Markham R, Yu XF. 2016. Accumulation of MxB/Mx2-resistant HIV-1 capsid variants during expansion of the HIV-1 epidemic in human populations. EBioMedicine 8:230–236. doi: 10.1016/j.ebiom.2016.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakayama EE, Saito A, Sultana T, Jin Z, Nohata K, Shibata M, Hosoi M, Motomura K, Shioda T, Sangkitporn S, Loket R, Saeng-Aroon S. 2018. Naturally occurring mutations in HIV-1 CRF01_AE capsid affect viral sensitivity to restriction factors. AIDS Res Hum Retroviruses 34:382–392. doi: 10.1089/AID.2017.0212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Opp S, Vieira DA, Schulte B, Chanda SK, Diaz-Griffero F. 2015. MxB is not responsible for the blocking of HIV-1 infection observed in alpha interferon-treated cells. J Virol 90:3056–3064. doi: 10.1128/JVI.03146-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bulli L, Apolonia L, Kutzner J, Pollpeter D, Goujon C, Herold N, Schwarz SM, Giernat Y, Keppler OT, Malim MH, Schaller T. 2016. Complex interplay between HIV-1 capsid and MX2-independent alpha interferon-induced antiviral factors. J Virol 90:7469–7480. doi: 10.1128/JVI.00458-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanjana NE, Shalem O, Zhang F. 2014. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rauch I, Muller M, Decker T. 2013. The regulation of inflammation by interferons and their STATs. JAKSTAT 2:e23820. doi: 10.4161/jkst.23820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC. 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob Agents Chemother 46:1896–1905. doi: 10.1128/AAC.46.6.1896-1905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levy DN, Aldrovandi GM, Kutsch O, Shaw GM. 2004. Dynamics of HIV-1 recombination in its natural target cells. Proc Natl Acad Sci U S A 101:4204–4209. doi: 10.1073/pnas.0306764101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F. 2014. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liang C, Rong L, Quan Y, Laughrea M, Kleiman L, Wainberg MA. 1999. Mutations within four distinct Gag proteins are required to restore replication of human immunodeficiency virus type 1 after deletion mutagenesis within the dimerization initiation site. J Virol 73:7014–7020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.