

Neuroinvasive alphaherpesviruses produce diseases of clinical and economic significance in humans and veterinary animals but are predominantly associated with less serious recurrent disease. Like all viruses, herpesviruses assemble a metastable particle that selectively dismantles during initial infection. This process is made more complex by the presence of a tegument layer that resides between the capsid surface and envelope. Components of the tegument are essential for particle assembly and also serve as critical effectors that promote infection upon entry into cells. How this dynamic network of protein interactions is arranged within virions is largely unknown. We present a molecular approach to dissect the tegument, and with it we begin to tease apart the protein interactions that underlie this complex layer of the virion architecture.
KEYWORDS: herpesvirus, pseudorabies virus, PRV, virion, capsid, tegument, TEV protease, selective disassembly, structure
ABSTRACT
Herpesvirus particles have a complex architecture consisting of an icosahedral capsid that is surrounded by a lipid envelope. Connecting these two components is a layer of tegument that consists of various amounts of 20 or more proteins. The arrangement of proteins within the tegument cannot easily be assessed and instead is inferred from tegument interactions identified in reductionist models. To better understand the tegument architecture, we have developed an approach to probe capsid-tegument interactions of extracellular viral particles by encoding tobacco etch virus (TEV) protease sites in viral structural proteins, along with distinct fluorescent tags in capsid and tegument components. In this study, TEV sites were engineered within the pUL36 large tegument protein, a critical structural element that is anchored directly on the capsid surface. Purified pseudorabies virus extracellular particles were permeabilized, and TEV protease was added to selectively cleave the exposed pUL36 backbone. Interactions with the capsid were assessed in situ by monitoring the fate of the fluorescent signals following cleavage. Although several regions of pUL36 are proposed to bind capsids, pUL36 was found stably anchored to the capsid exclusively at its carboxyl terminus. Two additional tegument proteins, pUL37 and pUS3, were tethered to the capsid via pUL36, whereas the pUL16, pUL47, pUL48, and pUL49 tegument proteins were not stably bound to the capsid.
IMPORTANCE Neuroinvasive alphaherpesviruses produce diseases of clinical and economic significance in humans and veterinary animals but are predominantly associated with less serious recurrent disease. Like all viruses, herpesviruses assemble a metastable particle that selectively dismantles during initial infection. This process is made more complex by the presence of a tegument layer that resides between the capsid surface and envelope. Components of the tegument are essential for particle assembly and also serve as critical effectors that promote infection upon entry into cells. How this dynamic network of protein interactions is arranged within virions is largely unknown. We present a molecular approach to dissect the tegument, and with it we begin to tease apart the protein interactions that underlie this complex layer of the virion architecture.
INTRODUCTION
Viruses from the order Herpesvirales infect animals ranging from invertebrates to humans, with over 300 herpesviruses identified to date (1). Despite their wide prevalence, these viruses share a particle morphology and the ability to persist throughout the lifetime of their hosts (2). Neurotropic members of the Alphaherpesvirinae subfamily of the Herpesviridae family include clinical (e.g., herpes simplex virus 1; HSV-1) and veterinary (e.g., pseudorabies virus; PRV) viruses that infect mucosal-epithelial surfaces before transmitting to innervating neurons of the peripheral nervous system (3).
Herpes virions are ∼225 nm in diameter and consist of a linear double-stranded DNA genome, an icosahedral protein capsid, a protein tegument layer, and a lipid envelope containing viral glycoproteins (2, 4). Virion production begins in the nucleus, where capsid assembly and genome encapsidation occurs, and concludes in the cytoplasm with the envelopment of the capsid-tegument complex (5). While the structure of the capsid is solved to 3.1 Å for HSV-1 and 4.9 Å for PRV (6–8), the majority of the tegument layer departs from the radial symmetry of the capsid shell and cannot be visualized in great detail by composite averaging methods such as cryo-electron microscopy (cryo-EM) (9). This technical limitation is overcome by the application of cryo-electron tomography, but this method has yet to achieve resolutions necessary to infer protein distributions within the herpesvirus virion (10, 11). Despite this lack of architectural information, the constituents of the tegument layer are defined along with many pairwise interactions between them (2, 5, 12, 13). In addition, single-particle fluorescence analysis has revealed tegument protein localization to specific subregions of the viral particle (14, 15). Despite these advances, the question of how the tegument is organized remains poorly defined. Most of the inner tegument density is localized to capsid vertices, and to date, three tegument proteins have been proposed to contact the capsid directly: pUL16, pUL36, and pUL47 (16–19). The best-established capsid-tegument interaction is that of the pUL36 protein, which is directly anchored to the pUL25 capsid protein as part of a five-helix bundle around capsid vertices (6, 7, 18, 20–22).
The pUL36 protein is a large (330 kDa in PRV) and conserved tegument constituent (5). Since the initial finding that the C terminus of pUL36 binds to capsids via pUL25 in HSV-1 and PRV (18), other regions of this large tegument protein have also been proposed to bind capsids (21, 23–25). In this study, we sought to identify how pUL36 and other tegument proteins interact with the capsid in PRV. Because pUL36 is essential for viral replication, it was necessary to use an approach that was conditional and did not disrupt the formation of mature viral particles (26–32). To this end, we developed a selective on-particle disassembly assay to dissect capsid-tegument interactions from extracellular viral particles by targeted proteolysis. We demonstrate that the pUL36 C-terminal assembly site is capsid bound in dissected extracellular particles and was solely responsible for the stable attachment of pUL36 to the capsids. Two additional tegument proteins, pUS3 and pUL37, were retained on capsids in a pUL36-dependent manner following extracellular particle disassembly but did not form stable capsid interactions of their own accord. Four other tegument proteins, including pUL16 and pUL47, were not stably anchored to capsids from extracellular PRV particles, indicating that capsid interactions with these proteins is more relevant to particle assembly than as components of the mature architecture. The results provide a view into the protein interactions that contribute to the architecture of the metastable herpesvirus particle, and we expect that the selective on-particle disassembly assay will have broad application to the dissection of the architectures of many viruses.
(This article was submitted to an online preprint archive [33].)
RESULTS
Precision cleavage of pUL36 in viral particles.
The large tegument protein, pUL36, is a conserved virion component that is directly bound to the capsid surface (18, 21) and is essential for virus propagation (34–36). Although several mutagenesis studies have provided insights into pUL36 function (27, 28, 30, 37–40), the role of pUL36 in virion assembly has largely been refractory to investigation because of its presumably essential role and the challenge of avoiding spontaneous genetic repair when trans-complementing mutant viruses with the 9-kb UL36 open reading frame. Therefore, to dissect the PRV architecture, tobacco etch virus (TEV) protease cleavage sites were introduced into the pUL36 tegument protein. TEV protease recognizes a seven-amino-acid sequence, ENLYFQG, that is commonly included in recombinant proteins to allow for proteolytic removal of affinity tags following purification (41). The TEV protease recognition sequence is not predicted to be present in any PRV proteins based on the viral genomic sequence (42). Recombinant PRV strains were produced encoding red fluorescent capsids (pUL25/mCherry), green fluorescent pUL36, and a single TEV protease site within pUL36 (Fig. 1A) (43). Regions of pUL36 that exhibit low conservation were chosen as sites of TEV insertion, with the rationale that these areas would be tolerant of the small insertions and accessible for cleavage in the mature protein. The recombinant viruses produced plaques and titers that approximated those of the wild type (Fig. 1B and Table 1). Because pUL36 is critical for virion assembly and infectivity, these results indicated that TEV site insertion did not substantially impair pUL36 function (34–36).
FIG 1.

Recombinant PRV strains used in this study. (A) The degree of conservation across the pUL36 predicted amino acid sequence from seven alphaherpesviruses is shown as purple peaks (eShadow). The locations of TEV site insertions are indicated by vertical lines and labeled with flanking amino acid (aa) positions. Solid lines were cleavable with exogenous TEV protease, and dotted lines were refractory to cleavage. Regions of pUL36 suggested or shown to interact with capsids are marked by horizontal lines under the conservation plot. (B and C) Plaque diameters of recombinant viruses encoding a pUL25/mCherry capsid fusion, a GFP fusion to a designated tegument protein, and a TEV site within pUL36 were compared to a parental virus containing only the pUL25/mCherry tag. Diameters are presented as a percentage of the parental virus. Error bars indicate standard deviations (***, P < 0.05 compared to wild type).
TABLE 1.
Recombinant PRV used in this study
| Strain | Capsid tag | Tegument tag | TEV insertion site in pUL36 | Titer (PFU/ml) | Source |
|---|---|---|---|---|---|
| PRV-GS4848 | mRFP1-GFP-VP26 | 0.2 × 109 | 14 | ||
| PRV-GS4284 | pUL25/mCherry | 1.0 × 109 | 43 | ||
| PRV-GS5507 | pUL25/mCherry | GFP-pUL36 | 1.5 × 109 | 44 | |
| PRV-GS5962 | pUL25/mCherry | GFP-pUL36 | aa 317/318 (A) | 1.2 × 109 | This study |
| PRV-GS6046 | pUL25/mCherry | GFP-pUL36 | aa 482/483 (B) | 1.4 × 109 | This study |
| PRV-GS6047 | pUL25/mCherry | GFP-pUL36 | aa 599/600 | 0.7 × 109 | This study |
| PRV-GS6048 | pUL25/mCherry | GFP-pUL36 | aa 2021/2022 | 0.4 × 109 | This study |
| PRV-GS6210 | pUL25/mCherry | GFP-pUL36 | aa 2622/2623 (C) | 1.6 × 109 | This study |
| PRV-GS6220 | pUL25/mCherry | aa 2622/2623 (C) | 2.4 × 109 | This study | |
| PRV-GS6325 | pUL25/mCherry | pUL36-GFP | aa 2622/2623 (C) | 0.9 × 109 | This study |
| PRV-GS6267 | pUL25/mCherry | pUL37-GFP | aa 2622/2623 (C) | 0.5 × 109 | This study |
| PRV-GS6269 | pUL25/mCherry | pUL49-GFP | aa 2622/2623 (C) | 0.7 × 109 | This study |
| PRV-GS6292 | pUL25/mCherry | pUL48-GFP | aa 2622/2623 (C) | 0.7 × 109 | This study |
| PRV-GS6294 | pUL25/mCherry | pUS3-GFP | aa 2622/2623 (C) | 1.7 × 109 | This study |
| PRV-GS6372 | pUL25/mCherry | pUL16-GFP | aa 2622/2623 (C) | 1.1 × 109 | This study |
| PRV-GS6374 | pUL25/mCherry | pUL47-GFP | aa 2622/2623 (C) | 1.2 × 109 | This study |
Virions of each recombinant virus were collected from the supernatant of infected PK15 cells at 24 h postinfection. After a clarifying step to remove cell debris, viral particles were pelleted from the supernatant through a Nycodenz cushion by ultracentrifugation. The envelopes of the recovered particles were extracted using nonionic detergent (2% Triton X-100) by following a previously described procedure (20). The particles were then pelleted through a 35% sucrose cushion to remove detergent and solubilized material and were incubated in a reaction mixture with or without TEV protease (Fig. 2A). Treatment with TEV protease resulted in cleavage of pUL36 when the TEV site was placed at position A, B, or C, as determined by Western blot analysis (Fig. 2B). The N-terminal fragments of green fluorescent protein (GFP)-pUL36 that were produced were consistent with molecular weight predictions based on the locations of these sites. TEV protease did not cleave GFP-pUL36 in the remaining two viruses, suggesting the TEV sites at these positions were inaccessible to the protease (Fig. 1A, dashed vertical lines). Importantly, GFP-pUL36 cleavage was not observed when TEV protease was omitted (mock treatment) or in a virus that lacked a TEV site (Fig. 2B). These results demonstrated that TEV cleavage was specific and that cleavage occurred efficiently at TEV sites in position A, B, or C within pUL36.
FIG 2.

Recombinant TEV sites provide selective cleavage of pUL36. (A) Experimental workflow of extracellular virus particle harvesting and processing. (B) On-particle cleavage of GFP-pUL36 using viruses encoding a TEV site at position A, B, or C (Fig. 1A) was monitored by Western blot analysis using an anti-pUL36 antibody that recognizes an N-terminal epitope and an anti-VP5 antibody as a loading control.
Dissecting the capsid-pUL36 interface.
To better understand how pUL36 contacts the capsid, a quantitative approach was developed that paired single-particle fluorescence imaging with TEV processing of dual-color-tagged viral particles. These experiments were designed to determine if pUL36 makes multiple contacts with capsids in extracellular viral particles (Fig. 1A) (18, 21, 23–25). Using a virus that contains a TEV site and two fluorophores (an mCherry-tagged capsid and GFP fused to the N terminus of pUL36), the interface between pUL36 and capsids was probed. Specifically, if pUL36 contacts the capsid only through its C terminus, then TEV cleavage should result in loss of GFP from particles; if instead pUL36 forms multiple contacts, GFP should be retained. As a control, a virus encoding GFP fused to the C terminus of pUL36 was included, which would not be expected to release GFP from the capsid following TEV protease treatment (18). A virus that lacked a TEV site but contained a tandem red fluorescent protein (RFP)-GFP fusion to the pUL35 hexon tip capsid protein (RG-UL35) was included as an additional control (14). Capsids recovered from extracted extracellular viral particles were purified and reacted with TEV protease (Fig. 2A), which were then diluted and imaged to assay for retention of GFP fluorescence. In the absence of TEV, particles from all recombinant viruses displayed bright red and green fluorescence (14), and TEV treatment of the two control viruses did not reduce GFP emissions from the viral particles (Fig. 3). In contrast, particles containing a TEV site at position A or C in the GFP-pUL36 fusion emitted reduced green fluorescence following protease treatment. The loss of GFP emissions following cleavage at position C is noteworthy, as the result indicates that the three putative N-terminal capsid-binding sites together are insufficient for capsid binding under the conditions of the experiment. Given these results, the inability to release the GFP-UL36 N terminus following cleavage at position B was unexpected. An in silico analysis of pUL36 with the ExPASy COILS software predicts that the region of pUL36 around site B is a hydrophobic coiled coil. We suspect that cleavage at this site exposes hydrophobic residues, because viral particles cleaved at position B had a tendency to form heterogeneously sized clumps, consistent with aggregation. Despite the unexpected effect of particle clumping incurred by cleavage at position B, cleavage at positions A and C demonstrated the efficacy of the TEV selective disassembly approach for studying viral particle architecture and revealed that pUL36 is stably anchored to capsids solely at its C terminus.
FIG 3.
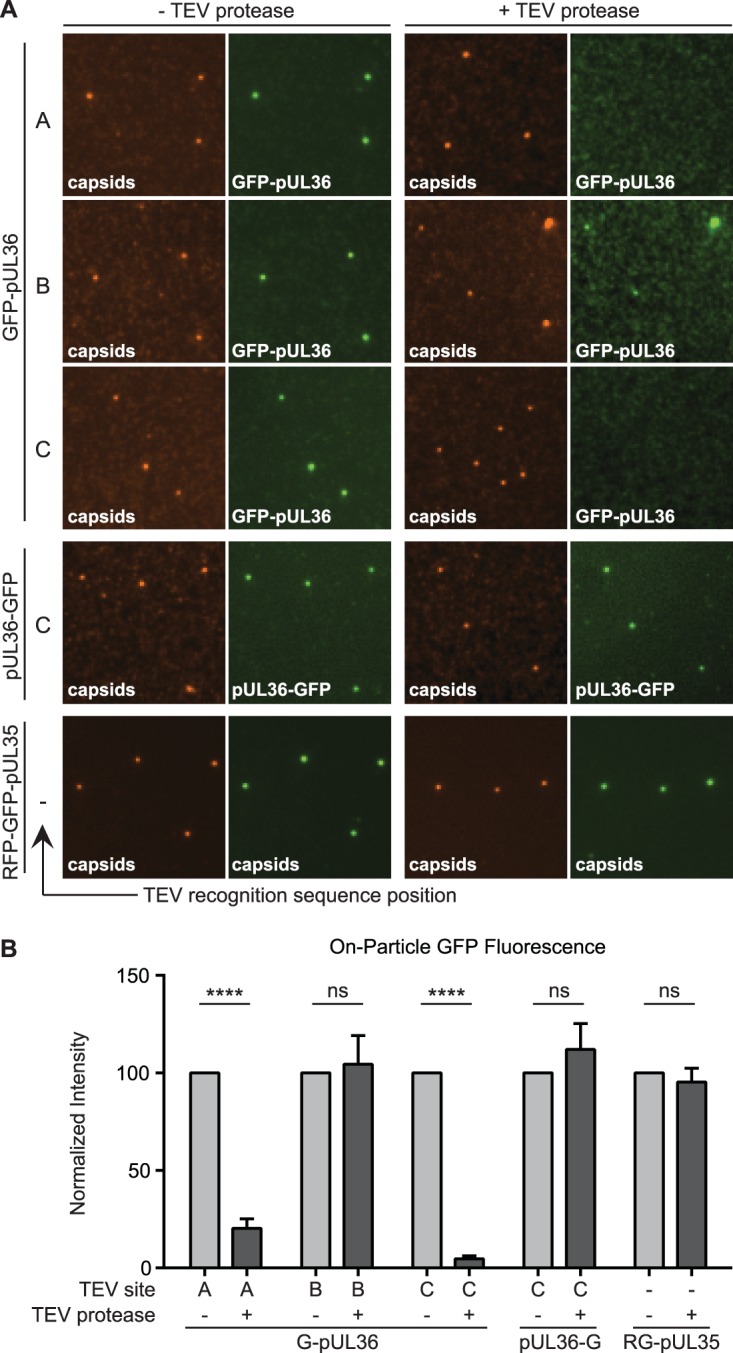
Mapping regions of pUL36 that maintain stable attachment to capsids. (A) Representative images of dual-fluorescent extracellular virus particles that were detergent extracted and incubated in the absence (left two columns) or presence (right two columns) of TEV protease. The position of the TEV site within pUL36 (position A, B, or C) is denoted, with a dash indicating no TEV site. All viruses encode the pUL25/mCherry capsid tag, with the exception of the RFP-GFP-pUL35 dual-capsid-tagged virus. (B) Fluorescence intensities from single particles represented in panel A. Values are normalized to the cognate “− TEV protease” control and represent data from n ≥ 3 experiments. Error bars indicate standard errors of the means (****, P < 0.001).
Recombinant PRV strains for the analysis of additional tegument-capsid interactions.
In addition to pUL36, other tegument proteins are proposed to interact with the capsid (16, 17, 45). Therefore, we extended the on-particle disassembly assay to determine whether six additional tegument proteins (pUS3, pUL16, pUL37, pUL47, pUL48, and pUL49) require pUL36 for stable capsid association in extracellular particles. A panel of dual-fluorescent viruses was constructed, each containing a TEV site within pUL36 at position C, an mCherry tag on the pUL25 capsid protein, and a GFP fusion to a tegument protein of interest. The position of the GFP fusion to each tegument protein was based on previous work, and analysis of released extracellular viral particles indicated that GFP-fused tegument proteins were incorporated at detectable levels in over 85% of particles (14). Recombinant viruses encoding GFP-tegument fusions grew to titers approximating those of wild-type PRV and were only moderately reduced for propagation in PK15 cells (Fig. 1C and Table 1).
Differential susceptibility of tegument proteins to detergent.
Many tegument proteins are involved in protein networks that connect the capsid to the membrane envelope (5). Because these interactions may be labile to detergent extraction, extracellular particles were exposed to 2% Triton X-100 to determine the degree to which each GFP-tegument fusion was solubilized from capsids on an individual particle level. Following purification from infected cell supernatants, viral particles were extracted and the extracted capsid-containing particles were isolated by ultracentrifugation through a sucrose cushion. The particles were resuspended, spotted onto coverslips, and imaged by fluorescence microscopy by the method outlined above (Fig. 2A). Representative images of mock- or detergent-extracted particles are presented in Fig. 4A. A previously described image analysis algorithm was used to quantify red and green emissions from individual viral particles, which were first identified based on red fluorescence intensities consistent with individual capsids (14). Representative GFP emission profiles in the presence and absence of detergent are shown for each virus in Fig. 4B. In the absence of detergent, GFP emissions from each tegument fusion were variable from particle to particle such that the distribution of tegument incorporation was Gaussian, which was consistent with previous work examining tegument incorporation into individual PRV virions (14). Following detergent extraction, capsid-associated GFP emissions varied greatly depending on the tegument protein to which GFP was fused. The preservation of capsid-associated emissions from the GFP-pUL36, pUL37-GFP, and pUS3-GFP fusions indicated that the association of these proteins with capsids was resistant to the extraction conditions and furthermore that GFP fluorescence was not inactivated by the procedure. Affirming the latter point, red and green fluorescence signals from RFP-GFP-pUL35 particles were unchanged by detergent extraction (Fig. 4C), indicating that reductions of GFP signal observed with pUL16-GFP, pUL47-GFP, pUL48-GFP, and pUL49-GFP could be ascribed to protein loss from the particles (46). Although not critical for the subsequent analysis, we note that following extraction, a fraction of either GFP-pUL36 or pUL37-GFP particles lost all detectable GFP emissions (25.5% and 21.7%, respectively) (Fig. 4B, inset percentages). Whether this observation hints at a population of particles that lack pUL36-capsid interactions or is due to the labile nature of these proteins under some conditions will require further investigation (20, 21).
FIG 4.

Tegument-capsid susceptibility to detergent extraction. (A) Dual-fluorescent extracellular viral particles containing the pUL25/mCherry capsid tag and GFP fused to a designated tegument protein were purified from the supernatant of infected cells and treated with buffer lacking (−) or containing (+) Triton X-100. Representative images of capsid and tegument emissions are shown, with each channel scaled equally across all samples. (B) GFP emission intensities from individual particles are show as histograms. The percentages in the upper right corner of each histogram indicate the proportion of particles with detectable GFP signal above threshold following detergent extraction. The data were fit to either Gaussian or decaying exponential distributions. All distributions had goodness-of-fit (R2) values of >0.95. (C) The red and green fluorescent signals of tandem-tagged mRFP-GFP-pUL35 particles were measured both before and after Triton X-100 extraction, and a with/without Triton X-100 detergent ratio was calculated for each fluorescence channel (n = 3). (D) Average red capsid (mCherry/pUL25) fluorescence intensities from purified particles following mock or Triton X-100 extraction (n = 3 per condition). Error bars indicate standard errors of the means. The solid horizontal line is the average intensity (intact plus mock extracted) across the seven capsid-tegument dual-fluorescent viruses. The dotted horizontal line is the average intensity following detergent extraction. (E) Estimated protein copy numbers based on average green fluorescence intensities (see Materials and Methods). Error bars indicate standard errors of the means.
Fluorescence intensity measurements made from three independent experiments per virus were compiled to extrapolate estimated protein copy numbers before and after detergent extraction (Fig. 4D and E). As a technical point, these results demonstrated that the secondary ultracentrifugation step through a sucrose cushion did not alter red or green fluorescent intensities compared with virions isolated by a single purification step alone (compare intact and mock-extracted samples). However, detergent extraction was noted to consistently produce a slight reduction in red fluorescence from the pUL25/mCherry fusion encoded by any of the seven capsid/tegument dual-tagged viruses. The red fluorescence intensity measurements in the seven mock-extracted samples were averaged (Fig. 4D, solid horizontal line), and this intensity value was assigned a copy number of 70.5 based on the pUL25 copy number reported in a previous study (14). By comparing the mock-extracted average red fluorescent intensity to a similar average across the detergent-extracted conditions (Fig. 4D, dashed horizontal line), we extrapolated that the pUL25/mCherry copy number was reduced to 60.2 ± 5.4 following exposure to detergent. Interestingly, this number is consistent with five copies of pUL25/mCherry at each of the 12 capsid vertices (47, 48). While this value could be coincidental, it is possible that 60 copies of the pUL25/mCherry fusion are stably bound to capsids around the vertices, while a smaller number of pUL25 copies incorporate into virions by another means. We also note that a recent cryo-EM reconstruction of PRV reveals that there are 10 copies of pUL25 per vertex, indicating that fusing mCherry to pUL25 reduces its copy number at capsid vertices (6). Green fluorescence intensity measurements were converted to copy numbers by normalizing the average intensities to the reported copy number for GFP-pUL36, which is 107.1 copies/virion (Fig. 4E and Table 2) (14). For several of the tegument-tagged viruses, GFP emissions were significantly reduced in both overall intensity per particle and in terms of the number of particles with GFP signal above threshold following detergent extraction (Fig. 4B and E and Table 2). This precluded the inclusion of the dual-fluorescent TEV site containing viruses encoding GFP fusions to pUL16, pUL47, pUL48, and pUL49 in the subsequent on-particle disassembly assay.
TABLE 2.
Protein copies per extracellular viral particle
| Protein | Copy no. ± SD |
|
|---|---|---|
| Unextracted | Extracted | |
| GFP-pUL36 | 107.1 ± 1.4 | 79.1 ± 2.2 |
| pUL37-GFP | 78.8 ± 0.9 | 61.1 ± 3.5 |
| pUS3-GFP | 6.7 ± 0.1 | 10.3 ± 1.6 |
| pUL16-GFP | 79.6 ± 4.2 | 7.4 ± 0.5 |
| pUL47-GFP | 802.0 ± 46.6 | 37.0 ± 2.2 |
| pUL48-GFP | 574.0 ± 19.8 | 32.6 ± 12.5 |
| pUL49-GFP | 229.4 ± 9.8 | 55.7 ± 6.1 |
pUS3 and pUL37 connectivity to capsids in extracellular particles occurs through pUL36.
The nature of the attachment of pUS3 and pUL37 to capsids was explored by testing if these proteins were tethered to capsids via pUL36. Viral particles were isolated from the supernatant of cells infected with dual-fluorescent viruses encoding a TEV site at position C within pUL36, extracted, and incubated in the presence or absence of TEV protease (Fig. 5). Fluorescent intensities from individual viral particles were measured as described above. TEV protease treatment resulted in significant loss of both proteins from capsids, indicating that the pUS3 and pUL37 tegument proteins are bound to capsids indirectly via pUL36 and make no additional stable contacts with capsids.
FIG 5.

pUS3 and pUL37 tegument proteins are tethered to capsids by pUL36. (A) Extracted dual-fluorescent extracellular virus particles encoding a TEV site at position C in pUL36 were incubated in the absence or presence of TEV protease, and representative images of fluorescence emissions are shown. (B) Fluorescent intensities from single particles are presented normalized to the cognate “− TEV protease” control (n ≥ 3). Error bars indicate standard errors of the means (****, P < 0.001).
DISCUSSION
The herpesvirus tegument is notable both for its contribution to particle architecture and for effector functions that commence immediately upon entry into cells and subsequently act throughout the infectious cycle (5). Whereas tegument proteins are targeted for assembly into nascent virions late in infection, upon entry into cells they selectively disassemble such that only a subset remains capsid bound during cytoplasmic transit and nuclear delivery (49–52). These properties attest to the dynamic nature of the tegument, which is further highlighted by the finding that at least one tegument protein reconfigures within the intact virion upon cell contact (45). Alphaherpesviruses incorporate >20 types of tegument proteins (12, 13), some represented in hundreds of copies, with the total tegument mass constituting approximately half of the virion. While many pairwise interactions between viral tegument proteins have been described (5), it is not clear how these proteins coalesce in a mature particle. In this study, we developed a selective disassembly assay based on the TEV site-specific protease to investigate how several tegument proteins anchor onto the capsid in extracellular virus particles. Whereas traditional mutagenesis approaches have advanced our understanding of virion morphogenesis, this new approach allowed for precise dissection of the fully assembled viral particle. In this initial application of the method, we identified that PRV pUL36 is stably bound to capsids solely by its C terminus, and that tegument proteins pUS3 and pUL37 make no additional stable contacts with the capsid but are instead dependent upon pUL36 for stable tethering to the capsid (Fig. 6).
FIG 6.

Model of pUL36 topology on capsids from extracellular particles. Simplified illustration of a single copy of pUL36 shown bound to a vertex of the herpesvirus capsid by its C-terminal capsid-binding domain (CBD). The N terminus of pUL36 up to position A, housing the deubiquitinase catalytic domain (DUB), makes no stable contacts with the remainder of pUL36 or the capsid surface. Position B is located in a region of predicted coiled coils, and cleavage at this site does not release the N terminus of pUL36 from the capsid. Cleavage at position C releases the amino-terminal peptide (which constitutes the bulk of the pUL36 protein) from the capsid, indicating that only the sequence C terminal of position C affords stable attachment to capsids under the conditions of these experiments. Two capsid-associated tegument proteins, pUL37 and pUS3, are also released upon cleavage at position C, indicating that these proteins are dependent upon pUL36 for stable capsid association.
We previously used quantitative fluorescence imaging to localize tegument protein populations within intact extracellular PRV particles, resulting in an intravirion map of tegument distributions (14). That prior study also provided copy numbers of GFP-tagged tegument proteins and their variance from particle to particle. Building on that quantitative imaging approach, the current investigation examined how tegument proteins from individual PRV extracellular particles are affected by detergent extraction. We note that in addition to detergent extraction, this study differed from our prior analysis in that capsids were tagged with a pUL25/mCherry fusion, which better preserves the properties of wild-type virus than the earlier mRFP1-VP26 capsid reporter (43). Nevertheless, the estimated copy numbers of GFP-tegument fusion proteins in intact viral particles was on par between the studies, with the exception of pUL47 and pUL48, which were present in higher copies in the current report. Four tegument proteins (pUL16, pUL47, pUL48, and pUL49) were susceptible to extraction in 2% Triton X-100 and 0.5 M NaCl and were not retained uniformly on the capsid surface following solubilization. The histogram profiles of these proteins following extraction (i.e., decaying exponentials) suggested that the remaining residual proteins were retained in a nonspecific or stochastic fashion. The loss of pUL48 is notable, as this tegument protein interacts with pUL36 in HSV-1 to promote particle assembly (53–55). This finding highlights that interactions important for virion assembly are not necessarily preserved in mature particles, as is the case for the pUL16 tegument protein (16, 45). Three GFP-tagged tegument proteins (pUL36, pUL37, and pUS3) maintained Gaussian fluorescence emission profiles, indicating that these proteins remained capsid bound following detergent extraction (14). The results from the present study are largely consistent with prior studies that have examined differential extraction of bulk populations of extracellular herpesvirus particles using salt, nonionic detergent, reducing agents, or phosphorylation (20, 56–60). The differences observed in tegument extractability between studies indicate that the isolation, handling, and extraction conditions affect experimental outcomes. Nevertheless, the results are consistent with pUL36, pUL37, and pUS3 remaining capsid bound upon entering cells (49–51, 61, 62). The results obtained from the on-particle disassembly assay indicate that the retention of these proteins on capsids upon cell entry is due to their collective attachment to the capsid surface by the C terminus of pUL36.
Despite these findings, several prior studies have implicated the presence of capsid binding sites in pUL36 in addition to the C-terminal capsid-binding domain (21, 23–25). Furthermore, pUL36 can assemble into extracellular particles when the C-terminal capsid-binding site is eliminated, although it is subsequently lost from capsids upon entry into cells (63). This indicates that pUL36 assembly into virions can occur by a mechanism that is distinct from its ability to form a stable attachment to the capsid surface, likely by the pUL48 tegument protein, as evidenced by its role in recruiting pUL36 into extracellular light particles that lack capsids (64). How the capsid is brought into the nascent forming virion in the absence of the UL36 C terminus is unclear but may involve additional interactions that promote assembly but are subsequently labile in virions. As already noted, there is compelling evidence that pUL16 behaves in this manner (16, 45), and it will be of interest to learn if the other putative capsid-binding regions of pUL36 do as well. Our results also provide evidence that dynamic alterations in intravirion protein interactions promote stable capsid association: a previous study identified the pUL21 tegument protein as being necessary for the efficient incorporation of pUS3 into PRV virions, whereas the current study indicates that pUS3 is stably anchored to capsids by pUL36 in PRV (65). More research will be needed to parse out how the tegument layer attaches to capsids, and we cannot rule out that there are additional tegument proteins that form stable contacts with capsids that were not included in this study. However, cryo-EM reconstruction of PRV and HSV-1 virions do no attest to capsid interactions apart from pUL36 (6, 7, 22).
This study provides new details about the alphaherpesvirus tegument network. The selective disassembly assay developed for these studies should prove useful for future investigations of the particle architectures of many viruses, as well as for deciphering changes in the herpesvirus tegument that can occur temporally or following glycoprotein-ligand interactions (16, 58). Because diverse herpesviruses share many architectural features and a common particle morphology, insights from this study may prove informative for viruses across the Herpesviridae family.
MATERIALS AND METHODS
Cells and virus strains.
Recombinant PRV strains used in this study were constructed by two-step, red-mediated recombination with the pBecker3 infectious clone (66, 67). A summary of the recombinant strains of PRV used in this study is provided in Table 1, and the primers used for their production are listed in Table 3. Tobacco etch virus (TEV) protease recognition sites and the GFP coding sequence were inserted into the pBecker3 derivative pGS4284, which encodes a pUL25/mCherry (red fluorescent capsid) fusion, and pGS5507, which additionally encodes a GFP-pUL36 fusion (43, 44). Recombinant clones were initially identified by restriction enzyme analysis and confirmed by sequencing. Infectious clones were transfected into PK15 epithelial cells as previously described to produce stocks of recombinant PRV (49). Working stocks were made by an additional passage through PK15 cells. PK15 cells were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% (vol/vol) bovine growth supplement (BGS; HyClone). During infection, the BGS concentration was reduced to 2%. Titers of virus stocks were determined on PK15 cells as previously described (34). Titer measurements were compiled from a minimum of 3 individual working stocks. Individual plaque diameters on PK15 cells were measured from the average of two orthogonal cross sections based on pUL25/mCherry fluorescence emissions. Greater than 30 plaques were measured per experiment. This process was repeated three times to arrive at the average plaque size for each virus, which was normalized as a percentage of the parental virus plaque diameter. Statistical significance was determined by one-way analysis of variance (ANOVA) with a multiple-comparison test.
TABLE 3.
Primers used to construct PRV recombinants unique to this study
| Strain | Template | Primer sequencea (5′–3′) |
|---|---|---|
| GS5962 | pEP-KanS2 | ACGGTCGGCGGGCGGCCGTCGCGCAAGTTCCGCCCGCGCACCCGCCAGCTGGAGAATTTGTACTTCCAAGGAGAGCCCATGGGCTTCAGGATGACGACGATAAGTAGGG |
| GAGCGGGGCCAGCTCCAGCTCCCGGACGGCGAAGCCCATGGGCTCCCCTTGGAAGTACAAATTCTCCAGCTGGCGGGTGCGCGGGCGCAACCAATTAACCAATTCTGATTAG | ||
| GS6046 | pEP-KanS2 | GAGGGCTCCTACGCGCAGCTCTCGGCGTGCTTCATGGACGCCGCGGAGAATTTGTACTTCCAAGGAGCGCGGGACCCCGGCAGGATGACGACGATAAGTAGGG |
| GCGCGCCACCACCGTCTGGTCGTAGACGCCGCCGGGGTCCCGCGCCCCTTGGAAGTACAAATTCTCCGCGGCGTCCATGAACAACCAATTAACCAATTCTGATTAG | ||
| GS6047 | pEP-KanS2 | GCGCAGTACAGCGCCAAGGCCATGCTGATGGACCGCAACGGCGCGGAGAATTTGTACTTCCAAGGACAGCGCTTCCGCGTGAGGATGACGACGATAAGTAGGG |
| GCGGCGCAGGCCCGCCACCGCGGCGTTGGCCACGCGGAAGCGCTGCCCTTGGAAGTACAAATTCTCCGCGCCGTTGCGGTCCAACCAATTAACCAATTCTGATTAG | ||
| GS6048 | pEP-KanS2 | GACTCGCAGCTGGCGGTGTCGCGCGTGCTCTGGGCGCCGCCCGAGAATTTGTACTTCCAAGGACCGCCGCGACGCCCGAGGATGACGACGATAAGTAGGG |
| CAGCAGCGGGTCGTCCTCGAGCGTGAAGCCCGGGCGTCGCGGCGGCCCTTGGAAGTACAAATTCTCGGGCGGCGCCCAGAGCAACCAATTAACCAATTCTGATTAG | ||
| GS6210GS6220GS6325 | pEP-KanS2 | CGAACGTCGGTGGGTATTAGAGACCACAGTCGCCGAAAATCCGAGAATTTGTACTTCCAAGGAATGGCTCTTAGGATGACGACGATAAGTAGGG |
| CTGGCGCGGGGCCGATACGGGCCTTCGCGGAACAAGAGCCATCCCTTGGAAGTACAAATTCTCGGATTTTCGCAACCAATTAACCAATTCTGATTAG | ||
| GS6267 | pEP-EGFP-in | CGTGGATCGGCTCCTGGCCGAGGTCGACGCCGTGTCCAAAGTGAGCAAGGGCGAGGAG |
| GGCTGAAATAACACACGCGCGTGGGGGAAAAATCTTTTTACTTGTACAGCTCGTCCATGC | ||
| GS6269 | pEP-EGFP-in | GGACGAGAGCACCCCCGGGCGGAAGGGAAAAGTGTATAAAGTGAGCAAGGGCGAGGAG |
| CAGCGGACGACTGAGCGGGGTGCCATTTGCAACGCCTTTACTTGTACAGCTCGTCCATGC | ||
| GS6292 | pEP-EGFP-in | CCTCATCCCCCGCGACGCGCTCAACCGGATGTTTGAGATGGTGAGCAAGGGCGAGGAG |
| AGGTGGTCGGGCGCCGCAGATCCGACCGCGCGCGGCGTCACTTGTACAGCTCGTCCATGC | ||
| GS6294 | pEP-EGFP-in | CGCCCTTCCGCTGATGAGATACTCAACTTTGGAATGTGGACCGTAGTGAGCAAGGGCGAGGAG |
| GCGCAAAGGTGTGTGTGTCCTACCGCTCGGAGCCGGGCCGTTTTACTTGTACAGCTCGTCCATGC | ||
| GS6372 | pEP-EGFP-in | CCCCGCCCGCGCCATCCCCGAGCTAATAAACGATTATGTGAGCAAGGGCGAG |
| GCCACAATACAAACGCAAGTACCCATTTTTTTCATTTCACTTGTACAGCTCGTC | ||
| GS6374 | pEP-EGFP-in | CCGAGCGCATTTATCGGCGCGCCGGCCGCCCCGGGGCCGCGGTGGCGGAGGTGGTGTGAGCAAGGGCGAGGAG |
| CTCGGGCGCGGCGGAGCATGGCGGAGGCAGAGGCGGGTTACTTGTACAGCTCGTCCATGC |
Template homologies are in boldface.
Western blot analysis.
PK15 cells were infected at a multiplicity of infection (MOI) of 10 with recombinant PRV encoding GFP-pUL36 fusions or GFP fusions to other tegument proteins (Table 1). A 5× final concentration of sample buffer (FSB; 60 mM Tris [pH 6.8], 10% [vol/vol] glycerol, 0.01% bromophenol blue, 2% [wt/vol] SDS, 5% [vol/vol] β-mercaptoethanol) was added to either purified extracellular virions or extracted and TEV-treated particles. Samples were boiled for 5 min and separated on 4 to 20% SDS polyacrylamide gradient gels (Mini-PROTEAN TGX gel; Bio-Rad). Proteins were transferred to 0.45-μm-pore-size Immobilon-FL membranes (Millipore) and blocked in 5% (wt/vol) powdered milk in PBS (13.7 mM NaCl, 0.27 mM KCl, 1 mM Na2HPO4, 0.2 mM KH2PO4) for 80 min at room temperature. PRV pUL36 was detected using a previously described antipeptide rabbit antibody at a dilution of 1:2,500 (68). PRV VP5 was detected using the mouse monoclonal anti-VP5 clone 3C10 (a gift from Lynn Enquist, Princeton University) at a dilution of 1:1,000. The mouse monoclonal GFP clone B-2 (sc-9996; Santa Cruz) was used to detect GFP-tagged proteins at a dilution of 1:1,000. Antibodies were prepared in 1% milk–PBST (13.7 mM NaCl, 0.27 mM KCl, 1 mM Na2HPO4, 0.2 mM KH2PO4, 0.1% Tween 20) and incubated with membranes overnight at 4°C. Secondary antibodies were prepared in 1% milk–PBST and incubated with membranes for 60 min at room temperature in the dark. Detection was performed using a LI-COR Odyssey Fc imaging system with donkey anti-rabbit IRDye 680RD or goat anti-mouse IRDye 800CW secondary antibodies at a dilution of 1:10,000 (LI-COR Biosciences).
Preparation of extracted viral particles.
Approximately 4.6 × 107 cells in six 15-cm plates were infected with PRV strains at an MOI of 10 PFU/cell and incubated for 24 h in DMEM (Gibco) supplemented with 2% (vol/vol) BGS. Supernatants (approximately 15 ml per plate) were cleared of cell debris by a 20-min, 5,000 × g, fixed-angle centrifugation in two 50-ml conical tubes at 4°C. The cleared supernatant was split into three SW28 centrifugation tubes, and each was underlaid with 3 ml of 10% (wt/vol) Nycodenz in TNE buffer (20 mM Tris [pH 7.6], 500 mM NaCl, 1 mM EDTA). Viral particles were pelleted through the Nycodenz cushions by centrifuging for 1 h at 13,000 rpm at 4°C in an SW28 rotor. The pellets were resuspended and combined in TNE buffer (20 mM Tris [pH 7.6], 500 mM NaCl, 1 mM EDTA) and dispersed by gentle pipetting, resulting in the intact viral particle preparation. In some instances the viral particle preparation was extracted in 1 ml of buffer containing 2% Triton X-100 in TNE supplemented with 2 μl of protease inhibitor cocktail (P8340; Sigma) for 20 min on ice. The extracted particles were separated from solubilized membrane and proteins by diluting the 1-ml preparation with 4 ml TNE, overlaying onto a 5-ml 35% sucrose cushion in TNE buffer, and centrifuging for 1 h at 25,000 rpm in an SW41 rotor at 4°C. The pellet was resuspended in PBS and dispersed by gentle pipetting, resulting in the extracted viral particle preparation. For light microscopy, resuspended intact or extracted particles were diluted with PBS and imaged on a plasma-treated no. 1.5, 22- by 22-mm coverslip in a wax-sealed microscope slide chamber as previously described (14, 69).
TEV treatment of extracted viral particles.
Approximately 2 × 106 extracted viral particles in PBS were incubated in a ProTEV plus (Promega) reaction mix containing 1× ProTEV buffer (50 mM HEPES [pH 7.0], 0.5 mM EDTA) supplemented with 1 mM dithiothreitol (DTT) and either 5 to 10 U of ProTEV or an equal volume of ProTEV storage buffer lacking enzyme (50 mM HEPES [pH 7.5], 300 mM NaCl, 1 mM DTT, 1 mM EDTA, 50% glycerol, 0.1% Triton X-100) for 18 h at 30°C in the dark. Following the incubation, particles were diluted in 5× FSB for Western blot analysis or were diluted and dispersed in PBS for imaging by fluorescence microscopy.
Quantitative fluorescence microscopy.
Particles were imaged on an inverted wide-field Nikon Eclipse TE2000-E microscope fitted with a 60×, 1.4-numeric-aperture objective, a Cascade II:512 camera, and a Lumen PRO fluorescence illumination system set at 100% light output. mRFP1 and mCherry fluorescence was imaged using a 572/35-nm excitation filter and a 632/60-nm emission filter; enhanced GFP was imaged using a 470/40-nm excitation filter and a 525/50-nm emission filter (Chroma Technology Corporation). All fluorophores were imaged with the same dual-band-pass dichroic mirror. With one exception, red and green emissions were sequentially captured using 1.7-s exposures, with a 1-MHz digitizer and zero electron-multiplying gain. The exception was images captured of PRV-GS6374 particles, which had green emissions approaching saturation and were therefore captured with 1.0-s exposures that were then linearly scaled by a factor of 1.7. Fluorescent intensities from individual particles were enumerated with an automated, custom algorithm as described previously (14). Briefly, the algorithm identified diffraction-limited red fluorescent punctae consistent with individual capsids. Red fluorescent capsids outside ±3 standard deviations were postfiltered from the data sets to remove noise and virion clusters. Red and spatially correlated green intensity measurements from the extracellular viral particles were compiled from three independent experiments, with 800 to 1,300 particles measured per experiment. Nonlinear Gaussian regression was performed in GraphPad Prism. Red intensity measurements were converted to protein copy number by normalizing to the reported copy number for PRV pUL25/GFP (70.5 copies/virion), and green intensity measurements were converted to copy numbers by normalizing to the reported copy number for PRV GFP-pUL36 (107.1 copies/virion) (14). Protein copy numbers were plotted as bar graphs from three independent experiments (two-way ANOVA with a multiple-comparison test).
ACKNOWLEDGMENTS
We thank Sarah Antinone for helping edit the manuscript. The monoclonal antibody directed against the PRV VP5 protein was generously provided by Lynn Enquist. Sequencing services were performed at the Northwestern University Genomics Core Facility.
This work, including the efforts of Gregory A. Smith, was funded by National Institute of Allergy and Infectious Diseases (NIAID) (AI056346). The Cellular and Molecular Basis of Disease from the National Institutes of Health (T32 GM08061) provided support for Gina R. Daniel and Caitlin E. Pegg, and a Graduate Research Fellowship from the National Science Foundation (DGE-1324585) provided support for Caitlin E. Pegg.
REFERENCES
- 1.Davison AJ. 2011. Evolution of sexually transmitted and sexually transmissible human herpesviruses. Ann N Y Acad Sci 1230:E37–E49. doi: 10.1111/j.1749-6632.2011.06358.x. [DOI] [PubMed] [Google Scholar]
- 2.Mettenleiter TC, Klupp BG, Granzow H. 2009. Herpesvirus assembly: an update. Virus Res 143:222–234. doi: 10.1016/j.virusres.2009.03.018. [DOI] [PubMed] [Google Scholar]
- 3.Smith G. 2012. Herpesvirus transport to the nervous system and back again. Annu Rev Microbiol 66:153–176. doi: 10.1146/annurev-micro-092611-150051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knipe DM, Howley PM. 2013. Fields virology, 6th ed Wolters Kluwer Health/Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 5.Owen DJ, Crump CM, Graham SC. 2015. Tegument assembly and secondary envelopment of alphaherpesviruses. Viruses 7:5084–5114. doi: 10.3390/v7092861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu Y-T, Jiang J, Bohannon KP, Dai X, Luxton GW, Hui WH, Bi G-Q, Smith GA, Zhou ZH. 2017. A pUL25 dimer interfaces the pseudorabies virus capsid and tegument. J Gen Virol 98:2837–2849. doi: 10.1099/jgv.0.000903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dai X, Zhou ZH. 2018. Structure of the herpes simplex virus 1 capsid with associated tegument protein complexes. Science 360:eaao7298. doi: 10.1126/science.aao7298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yuan S, Wang J, Zhu D, Wang N, Gao Q, Chen W, Tang H, Wang J, Zhang X, Liu H, Rao Z, Wang X. 2018. Cryo-EM structure of a herpesvirus capsid at 3.1 A. Science 360:eaao7283. doi: 10.1126/science.aao7283. [DOI] [PubMed] [Google Scholar]
- 9.Smith GA. 2017. Assembly and egress of an alphaherpesvirus clockwork. Adv Anat Embryol Cell Biol 223:171–193. doi: 10.1007/978-3-319-53168-7_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grunewald K, Desai P, Winkler DC, Heymann JB, Belnap DM, Baumeister W, Steven AC. 2003. Three-dimensional structure of herpes simplex virus from cryo-electron tomography. Science 302:1396–1398. doi: 10.1126/science.1090284. [DOI] [PubMed] [Google Scholar]
- 11.Schmid MF, Hecksel CW, Rochat RH, Bhella D, Chiu W, Rixon FJ. 2012. A tail-like assembly at the portal vertex in intact herpes simplex type-1 virions. PLoS Pathog 8:e1002961. doi: 10.1371/journal.ppat.1002961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kramer T, Greco TM, Enquist LW, Cristea IM. 2011. Proteomic characterization of pseudorabies virus extracellular virions. J Virol 85:6427–6441. doi: 10.1128/JVI.02253-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loret S, Guay G, Lippe R. 2008. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J Virol 82:8605–8618. doi: 10.1128/JVI.00904-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bohannon KP, Jun Y, Gross SP, Smith GA. 2013. Differential protein partitioning within the herpesvirus tegument and envelope underlies a complex and variable virion architecture. Proc Natl Acad Sci U S A 110:E1613–E1620. doi: 10.1073/pnas.1221896110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laine RF, Albecka A, van de Linde S, Rees EJ, Crump CM, Kaminski CF. 2015. Structural analysis of herpes simplex virus by optical super-resolution imaging. Nat Commun 6:5980. doi: 10.1038/ncomms6980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meckes DG Jr, Wills JW. 2007. Dynamic interactions of the UL16 tegument protein with the capsid of herpes simplex virus. J Virol 81:13028–13036. doi: 10.1128/JVI.01306-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scholtes LD, Yang K, Li LX, Baines JD. 2010. The capsid protein encoded by U(L)17 of herpes simplex virus 1 interacts with tegument protein VP13/14. J Virol 84:7642–7650. doi: 10.1128/JVI.00277-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coller KE, Lee JI, Ueda A, Smith GA. 2007. The capsid and tegument of the alphaherpesviruses are linked by an interaction between the UL25 and VP1/2 proteins. J Virol 81:11790–11797. doi: 10.1128/JVI.01113-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou ZH, Chen DH, Jakana J, Rixon FJ, Chiu W. 1999. Visualization of tegument-capsid interactions and DNA in intact herpes simplex virus type 1 virions. J Virol 73:3210–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Newcomb WW, Brown JC. 2010. Structure and capsid association of the herpesvirus large tegument protein UL36. J Virol 84:9408–9414. doi: 10.1128/JVI.00361-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cardone G, Newcomb WW, Cheng N, Wingfield PT, Trus BL, Brown JC, Steven AC. 2012. The UL36 tegument protein of herpes simplex virus 1 has a composite binding site at the capsid vertices. J Virol 86:4058–4064. doi: 10.1128/JVI.00012-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fan WH, Roberts AP, McElwee M, Bhella D, Rixon FJ, Lauder R. 2015. The large tegument protein pUL36 is essential for formation of the capsid vertex-specific component at the capsid-tegument interface of herpes simplex virus 1. J Virol 89:1502–1511. doi: 10.1128/JVI.02887-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baker ML, Jiang W, Wedemeyer WJ, Rixon FJ, Baker D, Chiu W. 2006. Ab initio modeling of the herpesvirus VP26 core domain assessed by cryoEM density. PLoS Comput Biol 2:e146. doi: 10.1371/journal.pcbi.0020146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pasdeloup D, Blondel D, Isidro AL, Rixon FJ. 2009. Herpesvirus capsid association with the nuclear pore complex and viral DNA release involve the nucleoporin CAN/Nup214 and the capsid protein pUL25. J Virol 83:6610–6623. doi: 10.1128/JVI.02655-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roberts AP, Abaitua F, O'Hare P, McNab D, Rixon FJ, Pasdeloup D. 2009. Differing roles of inner tegument proteins pUL36 and pUL37 during entry of herpes simplex virus type 1. J Virol 83:105–116. doi: 10.1128/JVI.01032-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bottcher S, Granzow H, Maresch C, Mohl B, Klupp BG, Mettenleiter TC. 2007. Identification of functional domains within the essential large tegument protein pUL36 of pseudorabies virus. J Virol 81:13403–13411. doi: 10.1128/JVI.01643-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mohl BS, Bottcher S, Granzow H, Fuchs W, Klupp BG, Mettenleiter TC. 2010. Random transposon-mediated mutagenesis of the essential large tegument protein pUL36 of pseudorabies virus. J Virol 84:8153–8162. doi: 10.1128/JVI.00953-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee JI, Luxton GW, Smith GA. 2006. Identification of an essential domain in the herpesvirus VP1/2 tegument protein: the carboxy terminus directs incorporation into capsid assemblons. J Virol 80:12086–12094. doi: 10.1128/JVI.01184-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee JI, Sollars PJ, Baver SB, Pickard GE, Leelawong M, Smith GA. 2009. A herpesvirus encoded deubiquitinase is a novel neuroinvasive determinant. PLoS Pathog 5:e1000387. doi: 10.1371/journal.ppat.1000387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zaichick SV, Bohannon KP, Hughes A, Sollars PJ, Pickard GE, Smith GA. 2013. The herpesvirus VP1/2 protein is an effector of dynein-mediated capsid transport and neuroinvasion. Cell Host Microbe 13:193–203. doi: 10.1016/j.chom.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bottcher S, Maresch C, Granzow H, Klupp BG, Teifke JP, Mettenleiter TC. 2008. Mutagenesis of the active-site cysteine in the ubiquitin-specific protease contained in large tegument protein pUL36 of pseudorabies virus impairs viral replication in vitro and neuroinvasion in vivo. J Virol 82:6009–6016. doi: 10.1128/JVI.00280-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Batterson W, Furlong D, Roizman B. 1983. Molecular genetics of herpes simplex virus. VIII. Further characterization of a temperature-sensitive mutant defective in release of viral DNA and in other stages of the viral reproductive cycle. J Virol 45:397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Daniel GR, Pegg CE, Smith GA. 2018. Dissecting the herpesvirus architecture by targeted proteolysis. bioRxiv doi: 10.1101/311894. [DOI] [PMC free article] [PubMed]
- 34.Smith GA, Enquist LW. 1999. Construction and transposon mutagenesis in Escherichia coli of a full-length infectious clone of pseudorabies virus, an alphaherpesvirus. J Virol 73:6405–6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Desai PJ. 2000. A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA-filled capsids in the cytoplasm of infected cells. J Virol 74:11608–11618. doi: 10.1128/JVI.74.24.11608-11618.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Z, Selariu A, Warden C, Huang G, Huang Y, Zaccheus O, Cheng T, Xia N, Zhu H. 2010. Genome-wide mutagenesis reveals that ORF7 is a novel VZV skin-tropic factor. PLoS Pathog 6:e1000971. doi: 10.1371/journal.ppat.1000971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fuchs W, Klupp BG, Granzow H, Mettenleiter TC. 2004. Essential function of the pseudorabies virus UL36 gene product is independent of its interaction with the UL37 protein. J Virol 78:11879–11889. doi: 10.1128/JVI.78.21.11879-11889.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Böttcher S, Klupp BG, Granzow H, Fuchs W, Michael K, Mettenleiter TC. 2006. Identification of a 709-amino-acid internal non-essential region within the essential conserved tegument protein(p) UL36 of pseudorabies virus. J Virol 80:9910–9915. doi: 10.1128/JVI.01247-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abaitua F, Daikoku T, Crump CM, Bolstad M, O'Hare P. 2011. A single mutation responsible for temperature-sensitive entry and assembly defects in the VP1-2 protein of herpes simplex virus. J Virol 85:2024–2036. doi: 10.1128/JVI.01895-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abaitua F, Hollinshead M, Bolstad M, Crump CM, O'Hare P. 2012. A nuclear localization signal in herpesvirus protein VP1-2 is essential for infection via capsid routing to the nuclear pore. J Virol 86:8998–9014. doi: 10.1128/JVI.01209-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kapust RB, Tozser J, Fox JD, Anderson DE, Cherry S, Copeland TD, Waugh DS. 2001. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng 14:993–1000. doi: 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]
- 42.Szpara ML, Tafuri YR, Parsons L, Shamim SR, Verstrepen KJ, Legendre M, Enquist LW. 2011. A wide extent of inter-strain diversity in virulent and vaccine strains of alphaherpesviruses. PLoS Pathog 7:e1002282. doi: 10.1371/journal.ppat.1002282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bohannon KP, Sollars PJ, Pickard GE, Smith GA. 2012. Fusion of a fluorescent protein to the pUL25 minor capsid protein of pseudorabies virus allows live-cell capsid imaging with negligible impact on infection. J Gen Virol 93:124–129. doi: 10.1099/vir.0.036145-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huffmaster NJ, Sollars PJ, Richards AL, Pickard GE, Smith GA. 2015. Dynamic ubiquitination drives herpesvirus neuroinvasion. Proc Natl Acad Sci U S A 112:12818–12823. doi: 10.1073/pnas.1512559112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meckes DG Jr, Wills JW. 2008. Structural rearrangement within an enveloped virus upon binding to the host cell. J Virol 82:10429–10435. doi: 10.1128/JVI.01223-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chalfie M, Kain S. 2006. Green fluorescent protein: properties, applications, and protocols, 2nd ed Wiley-Interscience, Hoboken, NJ. [Google Scholar]
- 47.Conway JF, Cockrell SK, Copeland AM, Newcomb WW, Brown JC, Homa FL. 2010. Labeling and localization of the herpes simplex virus capsid protein UL25 and its interaction with the two triplexes closest to the penton. J Mol Biol 397:575–586. doi: 10.1016/j.jmb.2010.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Toropova K, Huffman JB, Homa FL, Conway JF. 2011. The herpes simplex virus 1 UL17 protein is the second constituent of the capsid vertex-specific component required for DNA packaging and retention. J Virol 85:7513–7522. doi: 10.1128/JVI.00837-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luxton GW, Haverlock S, Coller KE, Antinone SE, Pincetic A, Smith GA. 2005. Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc Natl Acad Sci U S A 102:5832–5837. doi: 10.1073/pnas.0500803102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Granzow H, Klupp BG, Mettenleiter TC. 2005. Entry of pseudorabies virus: an immunogold-labeling study. J Virol 79:3200–3205. doi: 10.1128/JVI.79.5.3200-3205.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Antinone SE, Smith GA. 2010. Retrograde axon transport of herpes simplex virus and pseudorabies virus: a live-cell comparative analysis. J Virol 84:1504–1512. doi: 10.1128/JVI.02029-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aggarwal A, Miranda-Saksena M, Boadle RA, Kelly BJ, Diefenbach RJ, Alam W, Cunningham AL. 2012. Ultrastructural visualization of individual tegument protein dissociation during entry of herpes simplex virus 1 into human and rat dorsal root ganglion neurons. J Virol 86:6123–6137. doi: 10.1128/JVI.07016-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vittone V, Diefenbach E, Triffett D, Douglas MW, Cunningham AL, Diefenbach RJ. 2005. Determination of interactions between tegument proteins of herpes simplex virus type 1. J Virol 79:9566–9571. doi: 10.1128/JVI.79.15.9566-9571.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Svobodova S, Bell S, Crump CM. 2012. Analysis of the interaction between the essential herpes simplex virus 1 tegument proteins VP16 and VP1/2. J Virol 86:473–483. doi: 10.1128/JVI.05981-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ko DH, Cunningham AL, Diefenbach RJ. 2010. The major determinant for addition of tegument protein pUL48 (VP16) to capsids in herpes simplex virus type 1 is the presence of the major tegument protein pUL36 (VP1/2). J Virol 84:1397–1405. doi: 10.1128/JVI.01721-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morrison EE, Wang YF, Meredith DM. 1998. Phosphorylation of structural components promotes dissociation of the herpes simplex virus type 1 tegument. J Virol 72:7108–7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wolfstein A, Nagel CH, Radtke K, Dohner K, Allan VJ, Sodeik B. 2006. The inner tegument promotes herpes simplex virus capsid motility along microtubules in vitro. Traffic 7:227–237. doi: 10.1111/j.1600-0854.2005.00379.x. [DOI] [PubMed] [Google Scholar]
- 58.Newcomb WW, Brown JC. 2009. Time-dependent transformation of the herpesvirus tegument. J Virol 83:8082–8089. doi: 10.1128/JVI.00777-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Radtke K, Kieneke D, Wolfstein A, Michael K, Steffen W, Scholz T, Karger A, Sodeik B. 2010. Plus- and minus-end directed microtubule motors bind simultaneously to herpes simplex virus capsids using different inner tegument structures. PLoS Pathog 6:e1000991. doi: 10.1371/journal.ppat.1000991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Newcomb WW, Jones LM, Dee A, Chaudhry F, Brown JC. 2012. Role of a reducing environment in disassembly of the herpesvirus tegument. Virology 431:71–79. doi: 10.1016/j.virol.2012.05.017. [DOI] [PubMed] [Google Scholar]
- 61.Copeland AM, Newcomb WW, Brown JC. 2009. Herpes simplex virus replication: roles of viral proteins and nucleoporins in capsid-nucleus attachment. J Virol 83:1660–1668. doi: 10.1128/JVI.01139-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Coller KE, Smith GA. 2008. Two viral kinases are required for sustained long distance axon transport of a neuroinvasive herpesvirus. Traffic 9:1458–1470. doi: 10.1111/j.1600-0854.2008.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schipke J, Pohlmann A, Diestel R, Binz A, Rudolph K, Nagel CH, Bauerfeind R, Sodeik B. 2012. The C-terminus of the large tegument protein pUL36 contains multiple capsid binding sites that function differently during assembly and cell entry of herpes simplex virus. J Virol 86:3682–3700. doi: 10.1128/JVI.06432-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fuchs W, Granzow H, Klupp BG, Kopp M, Mettenleiter TC. 2002. The UL48 tegument protein of pseudorabies virus is critical for intracytoplasmic assembly of infectious virions. J Virol 76:6729–6742. doi: 10.1128/JVI.76.13.6729-6742.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Michael K, Klupp BG, Karger A, Mettenleiter TC. 2007. Efficient incorporation of tegument proteins pUL46, pUL49, and pUS3 into pseudorabies virus particles depends on the presence of pUL21. J Virol 81:1048–1051. doi: 10.1128/JVI.01801-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Smith GA, Enquist LW. 2000. A self-recombining bacterial artificial chromosome and its application for analysis of herpesvirus pathogenesis. Proc Natl Acad Sci U S A 97:4873–4878. doi: 10.1073/pnas.080502497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tischer BK, Smith GA, Osterrieder N. 2010. En passant mutagenesis: a two step markerless red recombination system. Methods Mol Biol 634:421–430. doi: 10.1007/978-1-60761-652-8_30. [DOI] [PubMed] [Google Scholar]
- 68.Leelawong M, Lee JI, Smith GA. 2012. Nuclear egress of pseudorabies virus capsids is enhanced by a subspecies of the large tegument protein that is lost upon cytoplasmic maturation. J Virol 86:6303–6314. doi: 10.1128/JVI.07051-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Smith GA, Gross SP, Enquist LW. 2001. Herpesviruses use bidirectional fast-axonal transport to spread in sensory neurons. Proc Natl Acad Sci U S A 98:3466–3470. doi: 10.1073/pnas.061029798. [DOI] [PMC free article] [PubMed] [Google Scholar]