

Abstract
Arsenic (As) is a known toxic element and carcinogen. Transcription factor nuclear factor-erythroid 2-related factor 2 (NRF2) controls cellular adaptation to oxidants and electrophiles by inducing antioxidant genes in response to redox stress. To explore associations between As level and NRF2-regulated cytoprotective genes expression, an observational study was conducted in a population of 61 occupationally exposed men with median (Me) age 50 years (interquartile range (IQR) 42–54) and in a control group of 52 men aged 40 (IQR 31–51.5) without occupational exposure. NRF2, KEAP1, GSTP1, HMOX1, NQO1, PRDX1, and TXNRD1 transcript levels were determined by means of quantitative real-time PCR along with the gene expression, methylation of NRF2 and KEAP1, as well as global DNA methylation were assessed. The median urine As tot. level in the exposed and control group was found to be 21.8 μg/g creat. (IQR 15.5–39.8 μg/g creat.) and 3.8 μg/g creat. (IQR 2.5–9.3) (p < 0.001). Global DNA methylation was significantly higher in occupationally exposed workers than in controls (Me 14.1 (IQR 9.5–18.1) vs Me 8.5 (IQR 5.9–12.6) p < 0.0001). NRF2 mRNA level was positively correlated with expression of all investigated NRF2-target genes in both groups (0.37 > R < 0.76, all p values < 0.0001). The multivariate linear regression adjusting for global methylation showed that As(III) level was significantly associated with expression of TXNRD1, GSTP1, HMOX1, and PRDX1. The results of this study indicate that arsenic occupational exposure is positively associated with global DNA methylation. The findings provide evidence for rather inactivation of NRF2-KEAP1 pathway in response to chronic arsenic exposure.
Keywords: Arsenic, NRF2, KEAP1, Cytoprotective genes, Gene expression, Methylation
Introduction
The factors present in the human environment significantly affect the integrity of the genetic material. Many of them have mutagenic and even carcinogenic effects. An important group of compounds with carcinogenic effect, which occur both in the living and occupational environment, are metals such as arsenic. This metaloid is classified by the International Agency for Research on Cancer [1], the Agency for Toxic Substances and Disease Registry [2], as a human carcinogen. The classification is based on epidemiological studies confirming the increase in the incidence of the lung cancer risk for inhalation exposure and skin and bladder cancers in people exposed to arsenic ingestion [3–5]. The main theory underlying the mechanisms of carcinogenesis of this factor is the role of oxidative stress [6–8]. Oxidative stress is the imbalance between prooxidant and antioxidant compounds/mechanisms in a cell resulting in an increase of prooxidative events [9]. This initiates a pathogenetic process which may ultimately lead to carcinogenesis. Abnormalities in the cells caused by oxidative stress are closely associated with the increased risk of cancer as a result of DNA damage, genomic DNA hypomethylation and hypermethylation, and changes in the regulatory mechanisms of cell proliferation and apoptosis. [6, 7, 10–12]. The antioxidant response is one of the most effective cellular defense mechanisms. Recent studies indicate that the main role in balancing the redox cell disorders and related consequences of exposure to inorganic arsenic (iAs) is played by transcription factor NRF2 (nuclear erythroid 2-related factor) linked to the antioxidant response element (ARE) of the nuclear DNA through a leucine zipper motif (bZip) [13]. The conducted genetic and biochemical studies confirm the importance of NRF2 in the transcriptional regulation of the ARE-dependent genes. NRF2 activates proteins with cytoprotective action deactivating electrophilic metabolites and reactive oxygen species stabilizing the redox potential. These include inter alia, glutathione S-transferase (GST), NAD(P)H-quinone 1 (NQO1) reductase, epoxide hydrolase, glutathione reductase, catalase, and superoxide dismutase [6, 7, 10, 11]. The main path that is responsible for the response to oxidative stress and the regulation of the activity of the NRF2 factor is that of KEAP1-NRF2-ARE. KEAP1 (Kelch-like ECH-associated protein) is responsible for cytoplasm-nuclear transport and proteasome destruction of NRF2. Under the conditions of homeostasis, the transcription factor NRF2 remains in the cytoplasm through forming a complex with KEAP1. When redox disorders occur, in response to the activity of the redox balance disrupters, NRF2 protein is released from the NRF2-KEAP1 complex and moves from the cytoplasm to the nucleus by activating the proper genes encoding proteins with cytoprotective action. The final result is an increased duration of NRF2 and increased expression of the ARE-dependent genes. Therefore, NRF2 is a protein referred to as necessary in the process aimed at protecting the cells against genetic damage under the effects of carcinogens [6, 7, 10, 11]. However, recent research attempts showed the “dark side” of the transcription factor NRF2. Independent studies indicated the responsibility of NRF2 for the processes of increased survival of tumor cells and stimulation of the carcinogenesis process [14, 15]. Mutations and epigenetic modifications affect the regulation and fate of NRF2, inter alia by disrupting the interactions between NRF2-KEAP1 and affecting NRF2 protein stabilization—these disturbances may ultimately enhance/reveal tumor phenotypes [14, 15]. Furthermore, transcription factor NRF2 over-expression caused by intense induction by xenobiotics can be a carcinogenic factor. Ikeda et al. [16], for the first time, described the negative impact of NRF2, pointing to its role in the development of liver cancer in animal experiments. Also, a significant impact is indicated on the part of the NRF2-dependent genes on the process of carcinogenesis. Heme oxygenese 1 enzyme (HO1 coded by HMOX1 gene), similar to NRF2 in normal homeostasis, exhibits antioxidant effects, while over-expression has been observed in different types of cancer changes [17]. In both cases, NRF2-dependent antioxidant response is indicated as a factor supporting the neoplastic processes by excessive induction mechanism NRF2-KEAP1 and epigenetic modifications. It seems interesting to assess the impact of exposure to inorganic arsenic in the conditions of occupational exposure on the expression of the transcription factor NRF2, NRF2-dependent genes, and ARE-dependent genes. The evaluation of these factors in a specific exposure related to specific lifestyle factors and professions will make it possible to better understand the potentially increased risk of tumors associated with the exposure to these metals. Therefore, the aim of the study was to investigate the association between iAs level in a population with occupational exposure to As and the link between iAs levels and NRF2-dependent genes (GSTP1, HMOX1, NQO1, PRDX1, TXNRD1) and others associated with redox homeostasis and epigenetic modification.
Materials and Methods
Study Population
This study was performed on copper mill workers (males n = 61) from the southwestern part of Poland with a median age of 50 years (interquartile range (IQR) 42–54 years). The median body mass index (BMI) was 28.1 kg/m2 (IQR 25.0–30.7 kg/m2). Non-occupationally exposed healthy controls (n = 52)—males with median age 40 years (IQR 32–51.5 years)—were recruited. The median body mass index in controls equaled 26.6 kg/m2 (IQR 24.4–28.4 kg/m2) (Table 1). The Institutional Ethics Committee for Scientific Research approved the study protocol and a written consent was obtained from each participant of the study.
Table 1.
Characteristics of examined subjects
| Exposed group | Control group | p values | |
|---|---|---|---|
| Median (IQR) | Median (IQR) | ||
| Age [years] | 50.00 (42.00–54.00) | 40.00 (32.00–51.50) | 0.0032 |
| BMI [kg/m2] | 28.09 (25.01–30.72) | 26.59 (24.42–28.35) | 0.0286 |
| Job seniority [years] | 27.00 (18.00–35.00) | NA | NA |
| N/N total (%) | N/N total (%) | ||
| Smoking | 19/61 (31.15%) | 15/52 (28.85%) | 0.7904 |
| Fish | 14/57 (24.56%) | 15/52 (28.85%) | 0.7729 |
NA not applicable
Arsenic Determination
Arsenic in the air and arsenic in urine were determined using inductively coupled plasma mass spectrometry (ICP-MS) as previously described by Janasik et al. [18]. Air samples were collected via the individual dosimetry method in the breathing zone of each worker continuously throughout a 6–7-h period of time. To determine arsenic and its compounds in the air of the workplace, ICP-MS was applied (version offered by NIOSH Manual of Analytical Methods, Fourth Edition Method 7301, Issue 1, 2003, Elements by ICP (Aqua Regia Ashing) and Method 7901, Issue 2, 1994, Arsenic Trioxide, as As) [19].
Analyses of As in Urine (As-U)
Urine samples were collected from the exposed group as well as from the controls. Workers provided spot urine samples immediately after the shift-end on the second day after exposure. The control subjects provided spot urine samples collected on the morning of arrival to the laboratory. Prior to dilution, the samples were centrifuged at 4000 rpm for 10 min and then the supernatant was diluted tenfold with 1.0% HNO3 for the total arsenic and the mobile phase for the speciation analyses. ELAN DRC-e ICP-MS with a dynamic reaction cell (Perkin Elmer, SCIEX, USA) was used for arsenic determination. The instrument Series 200 HPLC (Perkin Elmer, SCIEX, USA) was applied to separate arsenic chemical forms. The precision of the method for determination the individual forms of arsenic was as follows: AsB, 3.4%; As(III), 4.2%; DMA, 1.9%; MMA, 1.8%; and As(V), 2.3%. Accuracy measurement as the difference between the measurement and the accepted value for NIST 2669 was as follows: AsB, 1.1%; As(III), 1.5%; DMA, 2.2%; MMA, 3.9%; and As(V), 3.5%.
Certified reference material SRM 2669 (human urine) from the National Institute of Standard and Technology (NIST) was examined at the beginning and at the end of the analysis. The laboratory participates in the external quality program for the total arsenic determination organized by the Institute of Occupational Social and Environmental Medicine of the University of Erlangen, Nuremberg (G-EQUAS).
Gene Expression Analysis
NRF2, KEAP1, and five NRF2-targets selection: GSTP1, HMOX1, NQO1, PRDX1, and TXNRD1 were made according to studies data where associations between arsenic and NRF2-regulated genes were observed [20]. Total RNA was isolated from venous blood using PAXgene RNA Blood Mini Kit (PreAnalytiX GmbH, Hombrechtikon, Switzerland). Transcript levels in peripheral blood leukocytes were determined by means of quantitative real-time PCR (qPCR) with GAPDH and RPLP0 reference genes. Primer sequences for target genes were presented in a previous study [21]. The cDNA was synthesized with Transcriptor First Strand cDNA Synthesis Kit (Roche, Basel, Switzerland). All the samples were amplified in duplicate. Expression was quantified with FastStart Essential SYBR Green Master (Roche, Basel, Switzerland) using the LightCycler® 96 System (Roche, Basel, Switzerland). Gene expression data were evaluated by dCt method with reference genes-normalized relative quantification.
NRF2, KEAP1 Promoter Methylation
Promoter regions with transcriptional start site and first exon of NRF2 and KEAP1 were analyzed with transcriptional start sites DNA sequence: DBTSS database (http://dbtss.hgc.jp) for further CpG islands identification. Chemical modification of isolated genomic DNA was performed with Cells-to-CpG™ Bisulfite Conversion Kit (Thermo Fisher Scientific). DNA methylation of NRF2 and KEAP1 was analyzed using quantitative methylation-specific real-time PCR assay (qMSP) with FastStart Essential SYBR Green Master (Roche, Basel, Switzerland). Two sets of qMSP primers were designed using Methyl Primer Express ® Software v1.0 (Thermo Fisher Scientific, Carlsbad, CA, USA). The methylated (M) primers are 5′ TAACGTTTTTTCGGGGTTTC 3′(NRF2 F), 5′ CTCCGTTAACTCCCCGATAC 3′ (NRF2 R), 5′ TTTTTTTTAGATTTTGCGGTC 3′(KEAP1 F), and 5′ ATCTCCCGATTTCGTTACTAA 3′(KEAP1 R). The unmethylated (U) primers are 5′ TATTAATGTTTTTTTGGGGTTTT 3′(NRF2 F), 5′ CTCCATTAACTCCCCAATACCAA 3′(NRF2 R), 5′ TAATTTTTTTTAGATTTTGTGGTT 3′ (KEAP1 F), and 5′ CCATCTCCCAATTTCATTACTAAA 3′(KEAP1 R). A methylation index (MI) expressed as percentage of gene methylation was calculated for each sample using the following formula: MI = [1 / (1 + 2−(CtU−CtM)] × 100% [22].
Global DNA Methylation
Global DNA methylation levels were assessed by colorimetric ELISA method using Methylflash Methylated DNA Quantification Kit (Epigentek, Farmingdale, NY, USA), according to manufacturer’s instructions. One hundred nanograms of DNA isolated from blood was used for analyses. Each sample was analyzed in duplicates. The calculation of 5-methylcytosine amount was done with the use of standard curve created using defined dilutions of methylated genomic DNA. Methylation levels were calculated relative to the methylated control DNA (included in the kit) and expressed as a percentage of total methylated DNA. The intrassay CV was 8.8%.
Statistical Analysis
For measurable parameters, the authors calculated median values with interquartile ranges. For non-measurable values, a number of observations with respected percentage were given. The Chi-square test for independent pairs was used with Yates correction or Fisher’s exact test, which were used if necessary. Mann-Whitney’s U test was used for non-paired categorical and continuous data analysis. Correlation coefficients were calculated with Spearman rank test. Linear regression models were performed in order to evaluate the relationship between gene expression values and As(III) level standardized to creatinine level with adjustment to methylation level. p values lower than 0.05 were considered statistically significant. Statistical analyses were done with STATISTICA 12.5 PL (StatSoft, Tulsa, OK, USA).
Results
The airborne arsenic concentrations (As-A) in occupational settings, measured using individual sampling, varied from 2.8 to 34.4 μg/m3 (mean 9.5 μg/m3). The occupational and environmentally (control) exposed group differed significantly in concentrations of arsenic in urine expressed both in microgram per liter and microgram per gram creatinine (Table 2). The mean creatinine values in urine of the exposed and control group were respectively 1.3 ± 0.64 g/l and 1.2 ± 0.63 g/l. According to the World Health Organization and American Conference on Governmental and Industrial Hygienist (ACGIH), urine specimens that were highly dilute or highly concentrated (creatinine concentration: > 0.3 g/L and < 3.0 g/L) were discarded and another sample was collected [23]. There was no significant correlation between the age and BMI and between methylation and gene expression regardless of the group.
Table 2.
Comparison of arsenic urinary concentration between groups
| Exposed group | Control group | p | |
|---|---|---|---|
| Median (IQR) | Median (IQR) | ||
| As-total (μg/l) | 29.50 (14.70–54.70) | 4.60 (2.55–15.90) | < 0.0001 |
| As-total (μg/g creat.) | 21.83 (15.49–39.77) | 3.75 (2.52–9.26) | < 0.0001 |
| As(III) (μg/l) | 3.10 (1.90–6.40) | 0.50 (0.19–0.70) | < 0.0001 |
| As(III) (μg/g creat.) | 2.30 (1.50–4.90) | 0.36 (0.16–0.59) | < 0.0001 |
| As-A (μg/m3) | 9.45 (2.80–34.35) | NA | NA |
| iAs (μg/l) | 4.50 (2.20–9.20) | 0.50 (0.17–0.95) | < 0.0001 |
| iAs (μg/g creat.) | 3.50 (2.10–6.90) | 0.37 (0.13–0.69) | < 0.0001 |
NA not applicable
Gene Expression
No statistically significant differences in NRF2-KEAP1-ARE pathway associated genes between exposed workers and controls were found. However, in the occupationally exposed workers group, the statistically significant relationship between NNR2 target genes and As(III) concentrations was found. A similar relationship was found between expression of NRF2 target genes and the sum of inorganic arsenic concentration (Table 3). NRF2 mRNA level was positively correlated with expression of NRF2 targets (Table 3) and with KEAP 1 (R = 0.72, p < 0.0001). KEAP1 mRNA level was positively associated with investigated gene expression (Table 4). Multivariate linear regression adjusting for global methylation indicated significant relationships between As(III)-U level standardized to creatinine and expression of TRXR1, GSTP1, HMOX1, and PRDX1 at the margin of statistical significance (Table 5, Fig. 1).
Table 3.
Association between NRF2-regulated gene expression and arsenic concentrations in urine
| Exposed | NRF2 | KEAP1 | HMOX1 | GSTP1 | NQO1 | PRDX1 | TRXR1 |
| As tot. (μg/l) | 0.17 | 0.08 | 0.13 | 0.06 | 0.01 | 0.11 | 0.18 |
| As(III) (μg/l) | 0.29 | 0.22 | 0.31 | 0.24 | 0.15 | 0.28 | 0.35 |
| As(V) (μg/l) | 0.16 | 0.15 | 0.16 | 0.14 | 0.06 | 0.27 | 0.18 |
| DMA (μg/l) | 0.11 | 0.04 | 0.07 | 0.07 | − 0.01 | 0.13 | 0.12 |
| MMA (μg/l) | 0.24 | − 0.01 | 0.01 | − 0.01 | − 0.01 | 0.13 | 0.19 |
| AsB (μg/l) | 0.19 | 0.10 | 0.16 | 0.12 | 0.02 | 0.07 | 0.18 |
| As-A (μg/m3) | − 0.03 | − 0.14 | − 0.01 | 0.00 | 0.07 | 0.06 | 0.02 |
| Sum iAs+MMA (μg/l) | 0.28 | 0.13 | 0.22 | 0.15 | 0.09 | 0.25 | 0.31 |
| Sum iAs (μg/l) | 0.27 | 0.21 | 0.29 | 0.22 | 0.13 | 0.30 | 0.32 |
| Sum iAs+MMA+DMA (μg/l) | 0.19 | 0.10 | 0.14 | 0.11 | 0.04 | 0.18 | 0.20 |
| Control | NRF2 | KEAP1 | HMOX1 | GSTP1 | NQO1 | PRDX1 | TRXR1 |
| As tot. (μg/l) | − 0.12 | − 0.19 | − 0.09 | − 0.14 | − 0.16 | − 0.16 | − 0.20 |
| As(III) (μg/l) | − 0.08 | − 0.15 | − 0.18 | − 0.12 | − 0.21 | − 0.17 | − 0.18 |
| As(V) (μg/l) | 0.31 | 0.17 | 0.11 | 0.38 | − 0.18 | 0.34 | 0.09 |
| DMA (μg/l) | − 0.10 | − 0.19 | − 0.08 | − 0.11 | − 0.18 | − 0.17 | − 0.17 |
| MMA (μg/l) | − 0.12 | − 0.22 | − 0.25 | − 0.23 | − 0.15 | − 0.24 | − 0.18 |
| AsB (μg/l) | − 0.13 | − 0.17 | − 0.06 | − 0.04 | − 0.10 | − 0.13 | − 0.19 |
| Sum iAs+MMA (μg/l) | − 0.12 | − 0.20 | − 0.20 | − 0.20 | − 0.20 | − 0.13 | − 0.23 |
| Sum iAs (μg/l) | − 0.10 | − 0.19 | − 0.19 | − 0.18 | − 0.22 | − 0.12 | − 0.23 |
| Sum iAs+MMA+DMA (μg/l) | − 0.11 | − 0.18 | − 0.09 | − 0.14 | − 0.16 | − 0.17 | − 0.19 |
Table 4.
Association between NRF2 and NRF2-regulated gene expression
| NRF2-dCT | KEAP1-dCT | |||
|---|---|---|---|---|
| R | p | R | p | |
| HMOX1-dCT | 0.66 | < 0.0001 | 0.75 | < 0.0001 |
| GSTP1-dCT | 0.51 | < 0.0001 | 0.69 | < 0.0001 |
| NQO1-dCT | 0.30 | 0.0013 | 0.45 | < 0.0001 |
| PRDX1-dCT | 0.48 | < 0.0001 | 0.62 | < 0.0001 |
| TRXR1-dCT | 0.76 | < 0.0001 | 0.69 | < 0.0001 |
R, correlation coefficient
Table 5.
Association between gene expression, methylation, and urine arsenic level in occupational exposure group
| As(III) (μg/g creat.) | 5-mC % | NRF2, KEAP1 promoter methylation | As(III) (μg/g creat.) adj. global methylation | As(III) (μg/g creat.) adj. NRF2, KEAP1 promoter methylation | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| R | p | R | p | R | p | B | p | B | p | |
| NRF2-dCT | 0.35 | 0.0061 | 0.20 | 0.1218 | − 0.21 | 0.1004 | 0.11 | 0.0067 | 0.08 | 0.0324 |
| KEAP1-dCT | 0.32 | 0.0133 | 0.25 | 0.0494 | − 0.03 | 0.8469 | 0.09 | 0.0217 | 0.10 | 0.0090 |
| HMOX1-dCT | 0.36 | 0.0051 | 0.28 | 0.0324 | 0.08 | 0.0211 | ||||
| GSTP1-dCT | 0.27 | 0.0348 | 0.06 | 0.6256 | 0.07 | 0.0288 | ||||
| NQO1-dCT | 0.22 | 0.0949 | 0.26 | 0.0449 | 0.06 | 0.2194 | ||||
| PRDX1-dCT | 0.30 | 0.0184 | 0.06 | 0.6689 | 0.08 | 0.0271 | ||||
| TRXR1-dCT | 0.39 | 0.0019 | 0.18 | 0.1728 | 0.10 | 0.0159 | ||||
R, correlation coefficient; B, interpretation as R
Fig. 1.
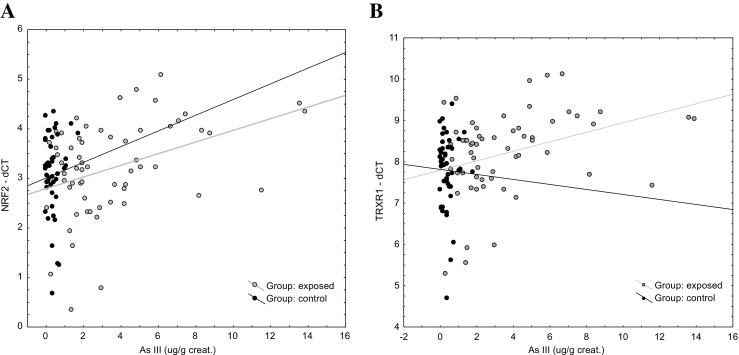
Example of relationship between As(III) concentration in urine and NRF2 (a) and TRXR1 (b) expression
DNA Methylation
The occupationally exposed group showed a significantly higher degree of NRF2 methylation (Me 0.47% (IQR 0.29–0.59%) vs Me 0.27% (IQR 0.13–0.47%), p = 0.0017), KEAP1 methylation (Me 0.13% (IQR 0.03–0.20%) vs Me 0.04% (IQR 0.001–0.18%), p = 0.0238), and overall 5-mC content (Me 1.41% (IQR 0.95–1.81%) vs Me 0.85% (IQR 0.59–1.26%) p < 0.0001) than the control group. Moreover, smokers in the occupational exposure group observed a higher degree of methylation of the 5-mC (Me 1.63% (IQR 1.41–1.99%) vs Me 1.35% (IQR 0.76–1.55%) p = 0.0039).
Discussion
As-Mediated Activation of NRF2-KEAP1 Antioxidant Pathway
Oxidative stress is the most widely accepted and studied mechanism of arsenic toxicity. Many researchers nowadays have speculated that the activation of the NRF2-KEAP1-ARE pathway by arsenic is beneficial and is likely to be an attempt of cells to counteract the damage effects of the metalloid. Arsenic was reported to induce the NRF2-dependent antioxidant response, although the detailed mechanism of NRF2 induction by arsenic has to be further explored [12–15]. Arsenic elicited both a beneficial NRF2-dependent antioxidant response and a cell damaging effect. The net outcome in response to arsenic may be dictated by arsenic species, dose, and duration of arsenic exposure. Nevertheless, activation of the NRF2-KEAP1-ARE pathway represented the initial attempt to counteract detrimental effects induced by arsenic and to maintain cellular homeostasis, which is the first barrier against the effects of exposure to arsenic. In experimental studies, increased NRF2 activity was observed in osteoblasts under the impact of arsenic through the transcriptional activation of the genes encoding HO-1, Prx I. Inorganic arsenic(III) strengthens the cellular expression of NRF2 on the transcription level and activates the expression of the NRF2-dependent genes in cell lines of human keratinocytes (HaCaT) [20, 24]. Exposure to inorganic arsenic is a stimulus that causes increased accumulation of nuclear NRF2. Zhao et al. [25] found that interactions between NRF2, KEAP1, and NRF1 coordinated the regulation of cellular antioxidant reactions in response to acute exposure to arsenic. The role of NRF2 in prevention of damage resulting from oxidative stress and prevention of pathogenetic processes is due to the effect of arsenic on the interactions between KEAP1 and E3 subunit of ubiquitine ligase (Cul 3) for NRF2. By decreasing Cul3 activity, NRF2 ultimately inhibits the degradation of the transcription factor in the cytoplasm and increases the lifetime of NRF2 from about 20 to 200 min [20]. Dinkova-Kostova et al. [26] as well as Eggler et al. [27] revealed that a number of inducers interacted with cysteine thiols of KEAP1 to inhibit NRF2 turnover and activate NRF2. He and Ma [13] showed that NRF2 contains evolutionarily conserved cysteine residues critical for response to antioxidant and electrophile inducers including arsenic. Furthermore, NRF2 cysteines are required for suppression of KEAP1-dependent ubiquitination and transcription activation of NRF2. Therefore, a dual sensor mechanism, in which both KEAP1 and NRF2 recognize inducers, has evolved to ensure a wide range of ligand recognition by NRF2. We found that NRF2 mRNA level was positively correlated with expression of investigated genes and with KEAP1 mRNA level which may confirm the dual mechanism of inducer recognition, especially because we obtained also positive correlations between KEAP1 mRNA level and studies genes expression.
Activation of the antioxidant response element in cytoprotective genes is the first barrier that is responsible for detoxification of arsenic. These genes include redox-balancing proteins (heme oxygenase, tioredoxin reductase), both I and II phase detoxification enzymes (NQO1, GST) and another stress response protein. The conducted studies demonstrate that occupational arsenic exposure rather inactivates NRF2 and expression of cytoprotective genes, TXNRD1 GSTP1, HMOX1, PRDX1 and expression is of interest and was statistically associated with inorganic arsenic urine level (Table 4, Fig. 1). Although NRF2 pathway activation by iAs has been reported in various cell types [28–30], however, the experimental data in vivo are very limited and not fully elucidated in humans. Several in vitro experiments have indicated that the TRX system is involved in As-induced cell responses. Hansen et al. and Myers et al. [31, 32] reported that the mRNA and protein expression of TXNRD1 are induced in As(III)-treated cells. Rea et al. [33] confirmed that the mRNA level of the selenoenzyme thioredoxin reductase was increased up to 13-fold under analysis of primary keratinocytes treated with arsenite; however, enzyme activities or protein levels of TRXR1 were not analyzed. Another study, monitoring global gene expression in keratinocytes treated with arsenite, revealed increases in mRNA encoding thioredoxin and TRXR1 [33]. These studies confirm that increased ROS generated in long-term exposure to arsenic is the primary mechanism behind the increased rates of cancer in populations with elevated arsenic exposure. Dodmane et al. [34] reported increased gene expression (HMOX1, GPX2, TXNRD1) in 1T1 urotherial cells, HEK001 keratinocytes, and HBE (human bronchial epithelial) cells. However, similar to Bailey et al. [35, 36] emphasize the differences in the results of the analysis of gene expression in vitro and in vivo. Clewell et al. [37] explain these differences, among others suppression of adaptive responses to oxidative stress with chronic exposure. The results of studies of gene expression in the current project with an average working time of exposure to inorganic arsenic 27 years (range 18–40), can justify the thesis of inhibition/suppression of adaptive responses as a result of chronic exposure. To our knowledge, this is the first work on the study of gene expression in the case of occupational exposure to inorganic arsenic. The observed dependence due to the relatively small and specific group should be interpreted with caution and necessarily confirmed by other studies in occupational exposure conditions.
As and Global DNA Methylation
DNA methylation is one of the most commonly studied epigenetic regulation mechanisms and is involved in the regulation of many biological processes through the regulation of gene expression. Global genomic DNA methylation is a hallmark of many types of cancers. The underlying mechanisms and physiologic consequences of As-induced alterations in genomic DNA methylation are unknown, probably its mechanism of SAM (S-adenosyl methionine) insufficiency and DNMT (DNA methyltransferase) gene expression [38]. Global DNA hypomethylation was found to be a causative factor for arsenic-induced carcinogenicity, which causes aberrant gene expression, in steroid-, apoptosis-, and cell cycle-related genes. The majority of animal and in vitro studies report that arsenic induces global DNA hypomethylation. Animal studies have suggested that As induces hepatic genomic hypomethylation of DNA [25, 39]. Genomic DNA hypomethylation commonly occurs in tumors and transformed cells and is thought to constitute an early event in some cancers [40]. Benbrahim-Tallaa et al. [41] exposed for 29 weeks human prostatic epithelial cells to As(III) in 5 μM concentration and observed genomic DNA hypomethylation, which was accompanied by reduced DNA methyltransferase activity.
However, instances have been reported of arsenic inducing global hypermethylation. Mass and Wang [42] and Davis et al. [43] observed global DNA hypermethylation on the cell lines exposed to inorganic arsenic. In contrast to the majority of animal and in vitro studies, human studies reported global hypo- and hypermethylation-induced arsenic exposure. Tellez-Plaza et al. [44] observed decreased global DNA methylation associated with a higher arsenic exposure level. Pilsner et al. [45] reported genomic methylation of PBL (peripheral blood leukocytes) DNA positively associated with plasma folate concentrations. They found that individuals with genomic hypomethylation of PBL DNA had a 1.8-fold increase risk for skin lesions, suggesting that changes in genomic methylation of PBL DNA may serve as an early biomarker of molecular events associated with the initiation and/or progression of As-induced skin lesions. Furthermore, these findings suggest that methylation of PBL DNA may serve as a functional biomarker to identify individuals at risk for future skin lesion development. In contrast, Majumdar et al. [46] indicated that arsenic levels in drinking water were associated with global DNA hypermethylation. In a case-control study, Pilsner et al. [47] showed an association between increasing global DNA methylation and urinary arsenic. Our findings confirmed that inorganic arsenic exposure in occupational settings (by inhalation) induces DNA hypermethylation.
Similarly to Niedzwiecki et al. [48], we did not find any significant correlation between age and 5-mC methylation, probably for the same reason, i.e., too small group in respect of age, in contrast to Pilsner et al. [45] observations. In our study, we found statistically significant differences between 5-mC % methylation in occupationally exposed group and controls, moreover smokers in occupational group showed a higher degree of 5-mC%. Generally, cigarette smoke impedes tracheobronchial clearance, possibly prolonging the exposure of bronchial epithelium to arsenic-containing dust particles, thus increasing the dose to the target pulmonary tissue [49]. Cigarette smoke is considered to be one of the most powerful modifiers of DNA methylation. In the present work, we observed increased hypermethylation in smokers, in contrast to another study where smoking is a proven factor limiting methylation, which is important in carcinogenesis [50]. A limited number of studies related to human exposure to arsenic and epigenetic modifications makes it difficult to explain this observation. Evidence from studies on populations exposed to As suggests a trend for an association between increasing arsenic exposure with increased DNA methylation although additional studies are needed to confirm those findings.
Conclusions
The findings provide evidence showing inactivation of NRF2-KEAP1 pathway in response to chronic arsenic exposure rather, but results should be interpreted with caution and necessarily confirmed by other studies in occupational exposure conditions. Moreover, occupational arsenic exposure is positively associated with global DNA methylation but the mechanisms are not clear yet. For this reason, further studies are necessary to determine the impact of exposure to arsenic on methylation and consequently the possibility to apply changes in DNA methylation as biomarkers of adverse health effects associated with iAs exposure.
Funding Information
This work was supported by the Grant for Scientific Research (2012/07/B/NZ7/04257) of the National Center of Sciences and the internal grant IMP 1.20 from the statutory activity resources of the Nofer Institute of Occupational Medicine in Lodz.
Compliance with Ethical Standards
The Institutional Ethics Committee for Scientific Research approved the study protocol and a written consent was obtained from each participant of the study.
Competing Interests
The authors declare that they have no competing interests.
References
- 1.International Agency for Research on Cancer (1987) Overall evaluation of carcinogenicity: an updating of IARC monographs volumes 1 to 42. Carcinogenicity in humans and in experimental animals, and summaries of other relevant data, for agents for which there are data on carcinogenicity in humans. IARC Monogr Eval Carcinog Risk Hum Suppl [PubMed]
- 2.Agency for Toxic Substances and Disease Registry (1990) Arsenic toxicity. Case studies in environmental medicine. Atlanta, GA: Agency for Toxic Substances and Disease Registry, U.S. Department of Health & Human Services, Public Health Services. HE20.7917:5 504-U
- 3.FOWLER BRUCE A., CHOU C.-H. SELENE J., JONES ROBERT L., CHEN C.-J. Handbook on the Toxicology of Metals. 2007. Arsenic; pp. 367–406. [Google Scholar]
- 4.Järup L, Pershagen G, Wall S. Cumulative arsenic exposure and lung cancer in smelter workers: a dose-response study. Am J Ind Med. 1989;15(1):31–41. doi: 10.1002/ajim.4700150105. [DOI] [PubMed] [Google Scholar]
- 5.Lauwerys R, Hoet P. Industrial chemical exposure—guidelines for biological monitoring. Boca Raton: Lewis Publishers, CRC Press; 2001. [Google Scholar]
- 6.De Vizcaya-Ruiz A, Barbier O, Ruiz-Ramos R, Cebrian ME. Biomarkers of oxidative stress and damage in human populations exposed to arsenic. Mutat Res. 2009;674(1-2):85–92. doi: 10.1016/j.mrgentox.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 7.Swaran JS. Arsenic-induced oxidative stress and its reversibility. Free Radic Biol Med. 2001;51:257–281. doi: 10.1016/j.freeradbiomed.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 8.Liu QJ, Kadiiska MB. Role of oxidative stress in cadmium toxicity and carcinogenesis. Toxicol Appl Pharmacol. 2009;238(3):209–214. doi: 10.1016/j.taap.2009.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sies H. Oxidative stress: oxidants and antioxidants. Exp Physiol. 1997;1:291–295. doi: 10.1113/expphysiol.1997.sp004024. [DOI] [PubMed] [Google Scholar]
- 10.Baird L, Dinkova-Kostova AT. The cytoprotective role of the Keap1–Nrf2 pathway. Arch Toxicol. 2011;85(4):241–272. doi: 10.1007/s00204-011-0674-5. [DOI] [PubMed] [Google Scholar]
- 11.Motohashi H, Yamamoto M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol Med. 2004;10(11):549–557. doi: 10.1016/j.molmed.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 12.He X, Chen M, Lin G, Ma Q. Arsenic induces NAD(P)H-quinone oxidoreductase I by disrupting the Nrf2-Keap1-Cul3 complex and recruiting Nrf2-Maf to the antioxidant response element enhancer. J Biol Chem. 2012;281:23620–23631. doi: 10.1074/jbc.M604120200. [DOI] [PubMed] [Google Scholar]
- 13.He X, Ma Q. Critical cysteine residues of Kelch-like ECH-associated protein 1 in arsenic sensing and suppression of nuclear factor erythroid 2-related factor 2. JPET. 2010;332(1):66–75. doi: 10.1124/jpet.109.160465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kensler TW, Wakabayashi N. Nrf2: friend or foe for chemoprevention? Carcinogenesis. 2010;31(1):90–99. doi: 10.1093/carcin/bgp231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Nicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, et al. Oncogene induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–109. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ikeda H, Nishi S, Sakai M. Transcription factor Nrf2/MafK regulates rat placental glutathione S-transferase gene during hepatocarcinogenesis. Biochem J. 2004;380(2):515–521. doi: 10.1042/bj20031948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jozkowicz A, Was H, Dulak J. Heme oxygenase-1 in tumors: is it a false friend? Antioxid Redox Signal. 2007;9(12):2099–2118. doi: 10.1089/ars.2007.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Janasik B, Reszka E, Stanislawska M, Wieczorek E, Fendler W, Wasowicz W. Biological monitoring and the influence of genetic polymorphism of As3MT and GSTs on distribution of urinary arsenic species in occupational exposure workers. Int Arch Occup Environ Health. 2015;88(6):807–818. doi: 10.1007/s00420-014-1009-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.NIOSH Manual of analytical methods, fourth edition method 7301, issue 1, (2003) Elements by ICP (Aqua Regia Ashing) and method 7901, issue 2, 1994, arsenic trioxide, as As)
- 20.Sinha D, Biswas J, Bishayee A. Nrf2-mediated redox signaling in arsenic carcinogenesis a review. Arch Toxicol. 2012;87:383–396. doi: 10.1007/s00204-012-0920-5. [DOI] [PubMed] [Google Scholar]
- 21.Reszka E, Wieczorek E, Jablonska E, Janasik B, Fendler W, Wasowicz W. Association between plasma selenium level and NRF2 target genes expression in humans. J Trace Elem Med Biol. 2015;30:102–106. doi: 10.1016/j.jtemb.2014.11.008. [DOI] [PubMed] [Google Scholar]
- 22.Lu L, Katsaros D, De la Longrais IA, Sochirca O, Yu H. Hypermethylation of let-7a-3 in epithelial ovarian cancer is associated with low insulin-like growth factor-II expression and favorable prognosis. Cancer Res. 2007;67(21):10117–10122. doi: 10.1158/0008-5472.CAN-07-2544. [DOI] [PubMed] [Google Scholar]
- 23.ACGIH . TLV® and BEIs® based on the documentation of the threshold limit values for chemical substances and physical agents and biological exposure indices. Cincinnati: ACGIH®; 2014. [Google Scholar]
- 24.Pi J, Diwan BA, Sun Y, Liu J, Qu W, He Y, Styblo M, Waalkes MP. Arsenic-induced malignant transformation of human keratinocytes: involvement of Nrf2. Free Radic Biol Med. 2008;45(5):651–658. doi: 10.1016/j.freeradbiomed.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao R, Hou Y, Zhang Q, Woods CG, Xue P, Fu J, Yarborough K, Guan D, Andersen ME, Pi J. Cross-regulations among NRFs and KEAP1 and effects of their silencing on arsenic-induced antioxidant response and cytotoxicity in human keratinocytes. Environ Health Perspect. 2012;120(4):583–589. doi: 10.1289/ehp.1104580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. PNAS. 2002;99(18):11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eggler AL, Liu G, Pezzuto JM, Breemen RB, Mesecar AD. Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. PNAS. 2005;102(29):10070–10075. doi: 10.1073/pnas.0502402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shah Palak, Trinh Elaine, Qiang Lei, Xie Lishi, Hu Wen-Yang, Prins Gail, Pi Jingbo, He Yu-Ying. Arsenic Induces p62 Expression to Form a Positive Feedback Loop with Nrf2 in Human Epidermal Keratinocytes: Implications for Preventing Arsenic-Induced Skin Cancer. Molecules. 2017;22(2):194. doi: 10.3390/molecules22020194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chiu PR, YC H, Hsieh BS, Huang TC, Cheng HL, Huang LW, Chang KL. Osteoblast activate the Nrf2 signalling pathway in response to arsenic trioxide treatment. Int J Biochem Cell Biol. 2016;79:327–336. doi: 10.1016/j.biocel.2016.08.036. [DOI] [PubMed] [Google Scholar]
- 30.Nishimoto S, Suzuki T, Koike S, Yuan B, Takagi N, Ogasawara Y. Nrf2 activation ameliorates cytotoxic effects of arsenic trioxide in acute promyelocytic leukemia cells through increased glutathione levels and arsenic efflux from cells. Toxicol Appl Pharmacol. 2016;305:161–168. doi: 10.1016/j.taap.2016.06.017. [DOI] [PubMed] [Google Scholar]
- 31.Hansen JM, Zhang H, Jones DP. Differential oxidation of thioredoxin-1, thioredoxin-2 and glutathione by metal ions. Free Radic Biol Med. 2006;40(1):138–145. doi: 10.1016/j.freeradbiomed.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 32.Myers JM, Antholine WE, Myers CR. Hexavalent chromium causes the oxidation of thioredoxin in human bronchial epithelial cells. Toxicology. 2008;246(2-3):222–233. doi: 10.1016/j.tox.2008.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rea MA, Gregg JP, Qin Q, Phillips MA, Rice RH. Global alteration of gene expression in human keratinocytes by inorganic arsenic. Carcinogenesis. 2003;24(4):747–756. doi: 10.1093/carcin/bgg010. [DOI] [PubMed] [Google Scholar]
- 34.Dodmane PR, Arnold LL, Kakiuchi-Kiyota S, Qiu F, Liu X, Rennard SI, Cohen SM. Cytotoxicity and gene expression changes induced by inorganic and organic trivalent arsenicals in human cells. Toxicology. 2013;312:18–29. doi: 10.1016/j.tox.2013.07.008. [DOI] [PubMed] [Google Scholar]
- 35.Bailey K, Xia Y, Ward WO, Knapp GW, Owen RD, Thai SF. Global gene expression profiling of hyperkeratotic skin lesions from inner Mongolians chronically exposed to arsenic. Toxicol Pathol. 2009;37(7):849–859. doi: 10.1177/0192623309351725. [DOI] [PubMed] [Google Scholar]
- 36.Bailey KA, Hester SD, Knapp GW, Owen RD, Thai SF. Gene expression of normal human epidermal keratinocytes modulated by trivalent arsenicals. Mol Carcinog. 2010;49(12):981–998. doi: 10.1002/mc.20677. [DOI] [PubMed] [Google Scholar]
- 37.Clewell HJ, Thomas RS, Kenyon EM, Hughes MF, Adair BM, Gentry PR, Yager JW. Concentration- and time-dependent genomic changes in the mouse urinary bladder following exposure to arsenate in drinking water for up to 12 weeks. Toxicol Sci. 2011;123(2):421–432. doi: 10.1093/toxsci/kfr199. [DOI] [PubMed] [Google Scholar]
- 38.Reichard JF, Schnekenburger M, Puga A. Long term low-dose arsenic exposure induces loss of DNA methylation. Biochem Biophys Res Commun. 2007;352(1):188–192. doi: 10.1016/j.bbrc.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen H, Li S, Liu J, Diwan BA, Barrett JC, Waalkes MP. Chronic inorganic arsenic exposure induces hepatic global and individual gene hypomethylation: implication for arsenic hepatocarcinogenesis. Carcinogenesis. 2004;25(9):1779–1786. doi: 10.1093/carcin/bgh161. [DOI] [PubMed] [Google Scholar]
- 40.Robertson KD, Wolffe AP. DNA methylation in health and disease. Nat Rev Genet. 2000;1(1):11–19. doi: 10.1038/35049533. [DOI] [PubMed] [Google Scholar]
- 41.Benbrahim-Tallaa L, Waterland RA, Styblo M, Achanzar WE, Weber MM, Waalkes MP. Molecular events associated with arsenic-induced malignant transformation of human prostatic epithelial cells: abberant genomic DNA methylation and K-ras oncogene activation. Toxicol Appl Pharmacol. 2005;206(3):288–298. doi: 10.1016/j.taap.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 42.Mass MJ, Wang L. Arsenic alters cytosine methylation patterns of the promoter of the tumor suppressor gene p53 in human lung cells: a model for a mechanism of carcinogenesis. Mutat Res – Rev Res. 1997;386(3):263–277. doi: 10.1016/S1383-5742(97)00008-2. [DOI] [PubMed] [Google Scholar]
- 43.Davis CD, Uthus EO, Finley JW. Dietary selenium and arsenic affect DNA methylation in vitro in Caco-2 cells and in vivo in rat liver and colon. J Nutr. 2000;130(12):2903–2909. doi: 10.1093/jn/130.12.2903. [DOI] [PubMed] [Google Scholar]
- 44.Tellez-Plaza M, Wan-yee T, Yan S, Umans JG, Francesconi KA, Goessler W, et al. Association of global DNA methylation and global DNA hydroxymethylation with metals and other exposures in human blood DNA samples. Environ Health Perspect. 2014;122(9):946–954. doi: 10.1289/ehp.1306674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pilsner JR, Liu X, Ahsan H, Ilievski V, Slavkovich V, Levy D, Factor-Litvak P, Graziano JH, Gamble MV. Folate deficiency, hyperhomocysteinemia, low urinary creatinine and hypomethylation of leukocyte DNA are risk factors for arsenic-induced skin lesions. Environ Health Perspect. 2009;117(2):254–260. doi: 10.1289/ehp.11872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Majumdar S, Chanda S, Ganguli B, Guha Mazumder DN, Lahiri S, Dasgupta UB. Arsenic exposure induces genomic hypermethylation. Environ Toxicol. 2010;25(3):315–318. doi: 10.1002/tox.20497. [DOI] [PubMed] [Google Scholar]
- 47.Pilsner JR, Liu X, Ahsan H, Ilievski V, Slavkovich V, Levy D, Factor-Litvak P, Graziano JH, Gamble MV. Genomic methylation of peripheral blood leukocyte DNA: influences of arsenic and folate in Bangladeshi adults. Am J Clin Nutr. 2007;86(4):1179–1186. doi: 10.1093/ajcn/86.4.1179. [DOI] [PubMed] [Google Scholar]
- 48.Niedzwiecki MM, Liu X, Hall MN, Thomas T, Slavkovich V, Ilievski V, Levy D, Alam S, Siddique AB, Parvez F, Graziano JH, Gamble MV. Sex-specific association of arsenic exposure with global DNA methylation and hydroxymethylation in leukocytes: results from two studies in Bangladesh. Cancer Epidemiol Biomark. 2015;24(11):1748–1757. doi: 10.1158/1055-9965.EPI-15-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hertz-Picciotto I, Smith AH, Holtzman D, Lipsett M, Alexeeff G. Synergism between occupational arsenic exposure and smoking in the induction of lung cancer. Epidemiology. 1992;3(1):23–31. doi: 10.1097/00001648-199201000-00006. [DOI] [PubMed] [Google Scholar]
- 50.Ken WK, Pausova Z. Cigarette smoking and DNA methylation. Front Genet. 2013;4:1–11. doi: 10.3389/fgene.2013.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]