

Abstract
Purpose
AZD1775, a first-in-class, small-molecule inhibitor of the Wee1 tyrosine kinase, is under evaluation as a potential chemo- and radiosensitizer for treating glioblastoma. This study was to prospectively, quantitatively, and mechanistically investigate the penetration of AZD1775 across the human blood–brain barrier (BBB).
Experimental Design
AZD1775 plasma and tumor pharmacokinetics were evaluated in 20 patients with glioblastoma. The drug metabolism, transcellular passive permeability, and interactions with efflux and uptake transporters were determined using human derived in vitro systems. A whole-body physiologically based pharmacokinetic (PBPK) model integrated with a four-compartment permeability-limited brain model was developed for predicting the kinetics of AZD1775 BBB penetration and assessing the factors modulating this process.
Results
AZD1775 exhibited good tumor penetration in patients with glioblastoma, with the unbound tumor-to-plasma concentration ratio ranging from 1.3 to 24.4 (median, 3.2). It was a substrate for ABCB1, ABCG2, and OATP1A2, but not for OATP2B1 or OAT3. AZD1775 transcellular passive permeability and active efflux clearance across MDCKII–ABCB1 or MDCKII–ABCG2 cell monolayers were dependent on the basolateral pH. The PBPK model well predicted observed drug plasma and tumor concentrations in patients. The extent and rate of drug BBB penetration were influenced by BBB integrity, efflux and uptake active transporter activity, and drug binding to brain tissue.
Conclusions
In the relatively acidic tumor microenvironment where ABCB1/ABCG2 transporter-mediated efflux clearance is reduced, OATP1A2-mediated active uptake becomes dominant, driving AZD1775 penetration into brain tumor. Variations in the brain tumor regional pH, transporter expression/activity, and BBB integrity collectively contribute to the heterogeneity of AZD1775 penetration into brain tumors.
Introduction
Insufficient penetration of potentially effective chemotherapeutic agents across the blood–brain barrier (BBB) is a huge hurdle to the successful treatment of brain cancer (1). The BBB is composed of a continuous layer of brain endothelial cells connected by tight junctions. The tight junctions limit paracellular permeability, thus directly restricting the diffusional transport of hydrophilic substances and the entry of proteins and immune cells into the brain. Small lipophilic molecules may cross the BBB by transcellular passive diffusion, but the efficiency of this process is dependent on drug physicochemical properties (2). In addition, the BBB expresses a broad range of efflux and uptake transporters that actively modulate the transport of substrate drugs out of and into the brain (3, 4).
The pharmacokinetics of a drug in the brain, including the target site (e.g., brain tumor), are determined by multiple dynamic, interactive processes governed by both drug physicochemical properties (e.g., molecular weight, lipophilicity, charge, binding to plasma proteins/brain tissues, and affinity to uptake/efflux transporters) and brain pathophysiology (e.g., BBB integrity, transporter abundance/activity, cerebral blood flow, and cerebrospinal fluid bulk flow; refs. 5–7). Mechanistic understanding of the role of these interactive processes in drug BBB penetration and early, accurate prediction of drug exposure in the brain or brain tumors are of paramount importance to guide the rational drug development and treatment for brain cancer. However, the pharmacokinetics of many new and existing anticancer agents in human brain or brain tumors remain understudied, underreported, and misunderstood because of the difficulty in accessing human brain specimens and the lack of in vitro assays or animal models that reliably predict human BBB permeability. Clearly, it is imperative that novel translational approaches are developed to better understand drug penetration across human BBB. One such approach is the in vitro–in vivo extrapolation (IVIVE), physiologically based pharmacokinetic (PBPK) modeling that allows integration of drug-specific parameters (e.g., in vitro enzyme/transporter kinetic data) and system-specific parameters (e.g., brain pathophysiology) into a mechanistic pharmacokinetic model to predict the systemic and brain pharmacokinetics of a drug. Because PBPK models can delineate the fractional role of individual disposition pathways, they also provide an invaluable tool for mechanistically and quantitatively assessing the sources of intraindividual, interindividual, or interspecies variability in plasma/brain pharmacokinetics.
AZD1775 is a first-in-class, highly selective, potent, ATP-competitive, small-molecule inhibitor of the Wee1 tyrosine kinase. It shows an enzyme IC50 of 5.2 nmol/L (2.6 ng/mL) for inhibiting Wee1 activity and an IC50 of ~80 nmol/L (40 ng/mL) for inducingDNAdamage andG2-checkpoint escape in cell-based assays (https://ncats.nih.gov/files/AZD1775.pdf). The Wee1 kinase is a critical regulator of the S and G2–M checkpoint (8). Wee1 phosphorylates the Tyr 15 residue of the cyclin-dependent kinase 1 (CDK1) and inhibits its activity. Inhibition of Wee1 thereby activates CDK1, facilitating cells to enter the mitotic phase (9, 10). Additionally, inhibition of Wee1 causes aberrantly high CDK2 activity in S-phase cells, leading to unstable DNA replication structures and, ultimately, DNA damage (11). Thus, Wee1 inhibition offers a chemo- and radiosensitizing strategy by forcing cancer cells with damaged DNA to enter unscheduled mitosis and to undergo cell death (12). Because the tumor suppressor protein p53 regulates the G1 checkpoint, cancer cells harboring abnormalities in the p53 pathway become more dependent on G2 and S checkpoints in cell-cycle control (9, 13), and, therefore, p53-deficient cancer cells treated with Wee1 inhibitors would be particularly susceptible to DNA damage induction due to genetic and pharmacologic abrogation of both G1 and G2 checkpoints (14). Given the high frequency of p53 mutations and the pivotal role of the G2–M checkpoint in preventing the programmed cell death in glioblastoma (14), AZD1775 is under evaluation as a potential chemo- and radiosensitizer for the treatment of glioblastoma, the most common malignant primary brain cancer that universally develops resistance to radiation and chemotherapy and unrelentingly results in mortality.
As the first step in the clinical development, we prospectively assessed the tumor penetration of AZD1775 in patients with glioblastoma in a phase 0 clinical trial. Furthermore, leveraging in vitro studies and clinical data, we developed a whole-body PBPK model integrated with a four-compartment permeability-limited brain (4Brain) model to predict the extent and rate of AZD1775 penetration across the human BBB and assess the factors modulating this process.
Methods
Clinical pharmacokinetic study
The plasma and tumor pharmacokinetics of AZD1775 were evaluated in 20 patients with first-recurrence, de novo glioblastoma in a phase 0 trial (ClinicalTrials.gov identifier: NCT02207010). The protocol was approved by the Institutional Review Board of Barrow Neurological Institute, St. Joseph’s Hospital and Medical Center (Phoenix, AZ), and written informed consent was obtained from each patient. Patient characteristics (shown as the median and range) include age (59, 28–81 years), weight (79, 54–127 kg), height (172, 154–184 cm), body surface area (1.9, 1.52–2.43 m2), total bilirubin (0.5, 0.2–1.7 mg/dL), aspartate aminotransferase (16, 8–62 IU/L), alanine aminotransferase (24, 14–133 IU/L), serum albumin (4.0, 2.1–4.6 mg/dL), and serum creatine (0.8, 0.6–1.0 mg/dL).
AZD1775 was provided by AstraZeneca (Wilmington, DE) as 100-mg gelatin capsules. Patients were treated with a single oral dose of AZD1775 at the dose of 100 (4 patients), 200 (4 patients), or 400 mg (12 patients) prior to the surgical tumor resection. Patients receiving a dose of 100 or 200 mg had their tumor resection at 8 to 10 hours following dosing. Patients receiving a dose of 400 mg had their tumor resection at 2 to 4, 8 to 10, or 24 to 26 hours after dosing. For patients with the tumor resection scheduled at 2 to 4 hours following dosing, blood samples were collected at before dosing and at 2 to 4 (at the time of tumor resection), 8, and 24 hours after dosing. For patients with the tumor resection scheduled at 8 to 10 hours following dosing, blood samples were collected at before dosing and at 8 to 10 (at the time of tumor resection), 12, and 16 hours after dosing. For the patients with the tumor resection scheduled at 24 to 26 hours following dosing, blood samples were collected at before dosing and at 2, 8, and 24 to 26 hours (at the time of tumor resection) after dosing. The total and unbound concentrations of AZD1775 in plasma and tumor samples were determined using a validated LC/MS-MS method (15).
In vitro studies
To allow IVIVE in the PBPK modeling, human-derived in vitro systems were used to determine AZD1775 drug-specific parameters, including (i) in vitro metabolic intrinsic clearance (CLint) by human liver and intestinal microsomes; (ii) apparent transcellular passive permeability in the apical-to-basolateral (Papp,A-B) and basolateral-to-apical (Papp,B-A) directions across MDCKII cell monolayers; (iii) interactions with efflux transporters ABCB1 and ABCG2 using MDCKII cells with overexpression of human ABCB1 or ABCG2 (named MDCKII-ABCB1 and MDCKII-ABCG2 cells); (iv) interactions with major BBB uptake transporters using HEK293 cells with overexpression of human OATP1A2, OATP2B1, or OAT3; and (v) fraction unbound in plasma (fu,p) and fraction unbound in brain tissue (fu,br) using patient plasma and brain tumor specimens. The details for in vitro experiments are presented in Supplementary Methods.
PBPK modeling and simulation
Simcyp Simulator V16 (Simcyp Ltd.) was used for PBPK modeling and simulations. A whole-body PBPK model integrated with a 4Brain model as implemented in the Simcyp Simulator was developed to predict the plasma and brain concentration-time profiles of total and unbound AZD1775. The model structure and assumptions are illustrated in Fig. 1 (16). System-specific parameters were derived based on the existing virtual Caucasian population within the Simcyp Simulator unless stated otherwise. Details on AZD1775 drug-specific parameters for the whole-body PBPK model and 4Brain model are presented in Table 1. In brief, physicochemical parameters were obtained from literature. The oral absorption was predicted using the Simcyp Advanced Dissolution, Absorption, and Metabolism model, where the dissolution rate was estimated using the built-in diffusion layer model and the human intestinal effective permeability was estimated by IVIVE scaling from the passive permeability determined from MDCKII cell monolayer. The drug distribution to all organs/tissues except for brain was perfusion rate limited. The hepatic and intestinal metabolic clearance was estimated by IVIVE scaling of the in vitro metabolic intrinsic clearance determined from human liver and intestinal microsomes, respectively. The renal clearance was assigned based on sensitivity analysis. Drug-dependent parameters for the 4Brain model were derived based on experimentally determined in vitro data on passive permeability and interaction with efflux and uptake transporter, as described below:
Figure 1.

A, Schematic illustration of the BBB, blood–cerebrospinal fluid (CSF) barrier, and brain–CSF barrier in human brain. Drug transport across the BBB (1) was governed by the passive bidirectional permeability and transporter-mediated active efflux and active uptake. Drug transport across the blood–CSF barrier (2) was controlled by the passive bidirectional permeability and transporter-mediated active efflux and active uptake. Drug transport across the brain–CSF barrier (3) was governed by the passive bidirectional permeability only. B, The structure of the 4Brain model as implemented in Simcyp Simulator V16. The passive bidirectional permeability across the BBB, blood–CSF barrier, and brain–CSF barrier are parameterized as PSB, PSC, and PSE, respectively. Active efflux and uptake at the BBB are parameterized as CLefflux,BBB and CLuptake,BBB, respectively. Active efflux and uptake at the blood–CSF barrier are parameterized as CLefflux,CSF and CLuptake,CSF, respectively. The model assumes that (i) only unbound and unionized drug can passively pass through all barriers, whereas transporters act upon unbound drug (including both unionized and ionized species); (ii) fluid balance is maintained by the circulation of CSF between spinal and cranial compartments and reabsorbed into the brain blood. Flow rates are described by the CSF secretion rate (QBCSFB), bulk flow rate from brain mass to cranial CSF (Qbulk), CSF flow rate out of cranial and spinal compartments (Qsink), CSF shuttle flow rate between cranial and spinal compartments (QSin and QSout), water transfer rate from the brain blood to brain mass (QBBB); (iii) the cerebral blood flow rate (QBrain) links the brain model to the whole-body PBPK model; (iv) metabolism in brain mass is negligible; and (v) all compartments are well stirred with defined volumes.
Table 1.
AZD1775 drug-specific parameters for the whole-body PBPK model integrated with a 4Brain model
| Parameter | Value | Comments/reference |
|---|---|---|
| Physicochemical | ||
| MW (g/mol) | 500.6 | ChEMBL database |
| LogP | 3.37 | ChEMBL database |
| PKa (monoprotic base) | 6.0 | Investigator brochure |
| B/P | 1.29 | Investigator brochure |
| fu,p | 0.2 | Experimental determined from pooled human plasma |
| Absorption: Advanced Dissolution, Absorption, and Metabolism (ADAM) model | ||
| MDCKII Papp (10−6 cm/s) for AZD1775 | 20 | Experimental determined from MDCKII cell monolayers |
| MDCKII Papp (10−6 cm/s) for midazolam | 62 | Experimental determined from MDCKII cell monolayers |
| Lag time (h) | 0 | Observed |
| fugut | 0.7 | Experimental determined from human liver microsomes |
| Qgut (L/h) | 10.2 | Simcyp predicted |
| Immediate release—diffusion layer model | ||
| Intrinsic aqueous solubility (mg/mL) | 0.034 | Simcyp predicted |
| Monodispersed radius (μm) | 10 | Assigned |
| Distribution: full PBPK | ||
| Vss (L/kg) | 19.2 | Predicted by Rodgers and Roland method (Method 2), Kp scalar of 1 |
| Elimination: whole-organ metabolic clearance | ||
| Human liver microsome (HLM): | ||
| Vmax (pmol/min/mg) | 197 | Experimental determined from HLM |
| Km (μmol/L) | 4.94 | Experimental determined from HLM |
| fuinc | 0.7 | Experimental determined from HLM |
| Human intestinal microsome (HIM): | ||
| CLint (μL/min) | 17 | Experimental determined from HIM |
| fumic | 0.7 | Experimental determined from HIM |
| CLR (L/h) | 6 | Optimized based on sensitivity analysis |
| 4Brain model | ||
| BBB | ||
| PSB (L/h) | 12.6 | IVIVE scaling of MDCKII Papp,A→B using Eq. 1a |
| fu,br | 0.07 | Experimental determined from patient brain tumor tissue |
| CLABCB1,vitro (μL/min/mg) | 152 and 43 | Estimated using Eq. 2 at the basolateral pH 7.4 and 6.5, respectivelyb |
| CLABCG2,vitro (μL/min/mg) | 76 and 33 | Estimated using Eq. 2 at the basolateral pH 7.4 and 6.5, respectivelyb |
| RAF for ABCB1 | 295 | Estimated using Eq. 3c |
| RAF for ABCG2 | 651 | Estimated using Eq. 3c |
| CLuptake,vivo (L/h) | 44.0 | Assigned based on sensitivity analysis |
| Blood–cranial CSF barrierd: | ||
| PSC (L/h) | 6.3 | Assumed to be half of PSB |
| fucsf | 1 | Assigned based on low protein concentration in CSF |
| Brain–cranial CSF barrier: | ||
| PSE (L/h) | 300 | Assigned given the high permeability of this barrier |
Abbreviations: B/P, blood-to-plasma partition ratio; CLABCB1,vitro and CLABCB1,vitro, ABCB1- and ABCG2-mediated in vitro efflux clearance, respectively; CLint, in vitro intrinsic metabolic clearance; CLR, renal clearance; CLuptake,vitro, uptake transporter-mediated in vitro clearance; CSF, cerebrospinal fluid; Eq., equation; fu,br, fraction unbound drug in brain tissue; fu,p, fraction of unbound drug in plasma; fucsf, fraction unbound drug in CSF; fugut, fraction of unbound drug in enterocytes; fumic, fraction of unbound drug in in vitro microsomal incubation; Km, substrate concentration at which half of Vmax is achieved; logP, logarithm of the neutral species octanol-to-buffer partition ratio; MW, molecule weight; Papp, apparent passive permeability; PKa, acid dissociation constant; PSB, passive permeability-surface area product on the BBB; Qgut, gut blood flow; RAF, in vivo–in vitro relative activity factor; Vmax, maximum metabolic rate; Vss, volume of distribution at steady-state using tissue volumes for a population representative of healthy volunteers population.
PSB = Papp,A→B × SA (Eq. 1), where Papp,A→B is the apparent permeability determined from MDCKII cell monolayer (mean, 17.5 × 10−6 cm/s) and SA is the human brain microvasculature surface area (mean, 20 m2).
(Eq. 2), where CLefflux,vitro (μL/min/mg) is the efflux transporter-mediated in vitro clearance; ER is the efflux ratio determined from MDCKII-ABCB1 or MDCKII–ABCG2; Papp,A-B is the apparent passive permeability determined from MDCKII; SA is the filter surface area (0.143 cm2) in a 96-well transwell; and Procell is the protein amount of MDCKII-ABCB1 or –ABCG2 cells in a 96-well transwell.
(Eq. 3), where BMvPGB is the milligrams of brain microvessels per gram brain; BW is the average human brain weight; abundance in vivo or in vitro represents the ABCB1/ABCG2 transporter protein expression level in human brain microvessels or in MDCKII-ABCB1 and -ABCG2 cells, respectively.
Transporter-mediated clearance at the blood–cranial CSF barrier was not incorporated in the 4Brain model because little information is available on the transporters at this barrier, and also because the drug disposition in CSF was not the focus of this study.
The passive permeability-surface area product on the BBB (PSB) was predicted using Equation (Eq.) 1 (17), where Papp,A→B is the apparent permeability determined from MDCKII cell monolayer and SA is the human brain microvasculature surface area (SA; 15–25 m2).
| (1) |
The passive permeability-surface area product on the blood–cranial cerebrospinal fluid (CSF) barrier (PSC) was assumed to be half of the PSB (16). The passive permeability-surface area product on the brain–CSF barrier (PSE) was fixed at 300 L/h, assuming a high permeability of this barrier (16).
In vitro efflux transporter-mediated clearance (CLefflux,vitro) (μL/min/mg) was estimated using Eq. 2 (18), where ER is the efflux ratio determined from MDCKII-ABCB1 or -ABCG2 cells, Papp,A-B is the apparent passive permeability (cm/s) determined from MDCKII, SA is the filter surface area (0.143 cm2) in a 96-well transwell, and Procell is the protein amount (mg) of MDCKII-ABCB1 (mean, 0.019 mg/well) or -ABCG2 cells (mean, 0.014 mg/well) in a 96-well transwell.
| (2) |
CLefflux,vitro was scaled to the whole-brain in vivo efflux transporter-mediated clearance (CLefflux,vivo; μL/min/mg) by multiplying a relative activity factor (RAF; Eq. 3).
| (3) |
where abundance in vivo or in vitro represents the ABCB1/ABCG2 transporter protein expression level in brain microvessels (pmol/mg microvessels) or in cellular models (pmol/mg cells), respectively; BMvPGB is the milligrams of brain microvessels per gram brain, and BW is the brain weight (gram). Given an average BMvPGB of 0.244 mg protein/g brain (19), average brain weight of 1400 g, ABCB1 abundance in vivo of 6.06 pmol/mg and ABCG2 abundance in vivo of 8.14 pmol/mg in human brain microvessels (19), and ABCB1 abundance in vitro of 7.02 pmol/mg in MDCKII-ABCB1 cells and ABCG2 abundance in vitro of 4.27 pmol/mg in MDCKII-ABCG2 cells (our unpublished data), the RAF was estimated to be 294 for ABCB1 and 651 for ABCG2.
In vivo uptake transporter-mediated clearance (CLuptake,vivo) at the BBB was assigned based on sensitivity analysis. Efflux and uptake transporter-mediated clearance at the blood–CSF barrier was not incorporated in the 4Brain model given the little information available on the transporters at this barrier, and also because the drug disposition in CSF was not the focus of this study.
Simulations of 10 trials with 10 subjects in each trial were performed in an existing virtual Caucasian population following a single oral dose of 400 mg. The PBPK model was verified by comparing the predicted total and unbound plasma and brain concentration–time profiles with observed data in patients with glioblastoma.
To better understand the sources of the variability in drug brain pharmacokinetics, sensitivity analyses using the developed PBPK model were performed to examine the impact of BBB integrity (as assessed by PSB), transporter activity (as assessed by CLuptake,vivo and CLefflux,vivo), and drug binding to brain tissues (fu,br) on the extent and rate of BBB penetration. The extent of drug BBB penetration is often assessed by the total or unbound drug brain-to-plasma partition coefficient (Kp or Kp,uu), which can be estimated as the brain-to-plasma drug concentration ratio at the steady state (or brain equilibrium) or area under concentration–time curve (AUC) ratio. Given the notion that unbound drug concentration drives the in vivo pharmacologic effect, the use of Kp,uu as a measure of the extent of brain penetration is more pharmacologically relevant and therefore was used in this study. Analogous to the concept of drug oral absorption, the rate of BBB penetration was assessed by the time (Tmax,br) to achieve the maximum drug brain concentration (Cmax,br).
Statistical analysis
Comparisons of the cellular uptake of AZD1775 between the cell line overexpressing a particular uptake transporter and its vector control or between the presence and absence of a transporter inhibitor were performed using two-sided independent samples t test. A P value < 0.05 was regarded as statistically significant.
Results
AZD1775 tumor penetration in patients with glioblastoma
Figure 2 shows the brain tumor concentrations of total and unbound AZD1775 as well as the extent of drug tumor penetration (as assessed by the Kp and Kp,uu) in patients with glioblastoma receiving a single oral dose of AZD1775 (100, 200, or 400 mg). The unbound (pharmacologically active) AZD1775 tumor concentrations varied from 6 to 315 ng/g (median, 24 ng/g) across three dose levels and three sampling time points, which exceeded the enzyme IC50 (2.6 ng/mL) for inhibition of Wee1 activity. The extent of AZD1775 tumor penetration (Kp or Kp,uu) appeared independent of the dose and sampling times. There was large interindividual variability in AZD1775 tumor penetration in patients with glioblastoma. Overall, the Kp varied 10.6-fold (median, 9.1; range, 3.8–40.4) and the Kp,uu varied 19-fold (median, 3.2; range, 1.3–24.4) in 20 patients.
Figure 2.

Observed brain tumor pharmacokinetics of AZD1775 in 20 patients with glioblastoma following a single oral dose (100, 200, or 400 mg). A, Total AZD1775 tumor concentrations. B, Unbound AZD1775 tumor concentrations. C, Total AZD1775 tumor-to-plasma concentration ratios. D, Unbound AZD1775 tumor-to-plasma concentration ratios. Symbols represent observed data from individual patients, and lines represent median values.
In vitro studies
In vitro metabolism
The overall metabolic profiles of AZD1775 in human liver and intestinal microsomes were well described by Michaelis–Menten kinetics (Supplementary Fig. S1). The estimated in vitro metabolic kinetic parameters were used for prediction of whole-organ metabolic clearance of the liver and intestine in the PBPK model (Table 1).
Passive transcellular permeability and interaction with efflux transporters
The apparent permeability and efflux ratios for AZD1775 and the positive controls, loperamide (a typical substrate of ABCB1) and gefitinib (a typical substrate of ABCG2), on MDCKII, MDCKII-ABCB1, and MDCKII-ABCG2 cell monolayers are summarized in Fig. 3A. The positive controls with loperamide and gefitinib confirmed the functional expression of ABCB1 and ABCG2 in MDCKII-ABCB1 and MDCKII-ABCG2 cell lines, respectively. The mass recovery for AZD1775, loperamide, and gefitinib from all permeability experiments was within 85% to 115%. AZD1775 was highly permeable, with a mean passive permeability of 17.5×10–6 cm/s in the apical to basolateral direction across MDCKII monolayer. It exhibited an efflux ratio of 10.6 and 4.5 across MDCKII-ABCB1 and MDCKII-ABCG2 monolayers, respectively; the presence of an inhibitor of ABCB1 (cyclosporine A, 10 μmol/L) or ABCG2 (Ko143, 1 μmol/L) abrogated the efflux effect (Fig. 3A). These data collectively indicated that AZD1775 was a substrate of ABCB1 and ABCG2.
Figure 3.

Apparent passive permeability in the apical-to-basolateral (Papp,A-B) and basolateral-to-apical (Papp,B-A) directions and efflux ratio (ER) across MDCKII, MDCKII-ABCB1, and MDCKII-ABCG2 cell monolayers. A, The Papp,A-B, Papp,B-A, and ER of AZD1775 (5 μmol/L) and the positive controls, loperamide (a typical substrate of ABCB1, 5 μmol/L) and gefitinib (a typical substrate of ABCG2, 1 μmol/L), in the absence or presence of an inhibitor of ABCB1 (cyclosporine A, 10 μmol/L) or ABCG2 (Ko143, 1 μmol/L). B, The Papp,A-B, Papp,B-A, and ER of AZD1775 (5 μmol/L) at a fixed apical pH (7.4) and varying basolateral pH (7.4, 7.0, 6.5, and 6.0). Data are expressed as the mean from three independent experiments (triplicate in each experiment), with the relative SD <20%.
Notably, AZD1775 exhibited pH-dependent passive permeability and active efflux across MDCKII, MDCKII-ABCB1, and MDCKII-ABCG2 cell monolayers (Fig. 3B). At a fixed pH (7.4) in the apical chamber (mimicking blood circulation) and with varying pH (7.4, 7.0, 6.5, or 6.0) in the basolateral chamber (mimicking brain interstitium), the passive permeability of AZD1775 across MDCKII decreased in the basolateral to apical direction while increasing in the apical to basolateral direction, thereby resulting in a net influx at a relatively acidic basolateral pH (e.g., efflux ratio of 0.8 at pH 6.5; Fig. 3B). This observation was in accordance with the pH partition theory, that is, for weak base or acid drugs, the extent to which they are ionized is exponentially related to the pH of their milieu. As ionization of a molecule substantially decreases its lipophilicity, slight differences in pH may markedly influence the ability of the drug to traverse across biological membranes. At a relative acidic basolateral pH, the ionization of AZD1775, a weak base drug, was increased, and as a result, the basolateral-to-apical permeability decreased and the drug was trapped in the basolateral compartment. Similar findings were also observed on MDCKII-ABCB1 or MDCKII-ABCG2 monolayers, and of particular note, efflux ratios were markedly reduced at a relatively acidic basolateral pH (Fig. 3B). For example, as the basolateral pH decreased from 7.4 to 6.5, the efflux ratio was reduced by 60% (from 9.2 to 3.7) on MDCKII-ABCB1 and by 45% (from 4.7 to 2.6) on MDCKII-ABCG2. Because the transport of AZD1775 across MDCKII-ABCB1 and MDCKII-ABCG2 monolayers occurred via both passive diffusion and active efflux, the observed reduction in efflux ratios at a relatively acidic basolateral pH was likely attributable to the decrease of both passive outward diffusion rate and active efflux rate. Since the transporter expression level or activity of ABCB1 and ABCG2 was not regulated by pH changes (20–22), it is plausible that the decreased active efflux rate was due to less drug remaining within the apical membrane of cell monolayer to interact with efflux transporters at a relatively acidic basolateral pH, a condition favorable to trapping a weak base drug into the basolateral compartment.
Interaction with uptake transporters
The cellular uptake of AZD1775 and the positive control, estrone-3-sulfate (a typical substrate for OATP1A2, OATP2B1, and OAT3), in HEK293 cells with overexpression of OATP1A2, OATP2B1, or OAT3 and their respective vector control cells is summarized in Fig. 4. The cellular uptake of estrone-3-sulfate was significantly higher in HEK293 cells overexpressing OATP1A2, OATP2B1, or OAT3 compared with their respective vector control (P < 0.001), and the presence of a specific transporter inhibitor (i.e., 100 μmol/L sulfobromophthalein for OATP1A2, 10 μmol/L fluvastatin A for OATP2B1, or 50 μmol/L probenecid for OAT3) reduced estrone-3-sulfate uptake to a level similar to that in the vector control (Fig. 4). These data confirmed the functional activity of the studied transporters in the present in vitro systems. The cellular uptake of AZD1775 in cells overexpressing OATP1A2 was significantly higher than that in the vector control (P = 0.001), and the presence of an OATP1A2 inhibitor (i.e., 100 μmol/L sulfobromophthalein) reduced the drug uptake to the level similar to that in the vector control (P = 0.005; Fig. 4A), collectively suggesting that AZD1775 was a substrate of OATP1A2. On the contrary, the cellular uptake of AZD1775 was similar between the cells overexpressing OATP2B1 or OAT3 and their respective vector control or between the absence and presence of a specific transporter inhibitor (Fig. 4B and C), suggesting that AZD1775 was not a substrate of OATP2B1 or OAT3.
Figure 4.
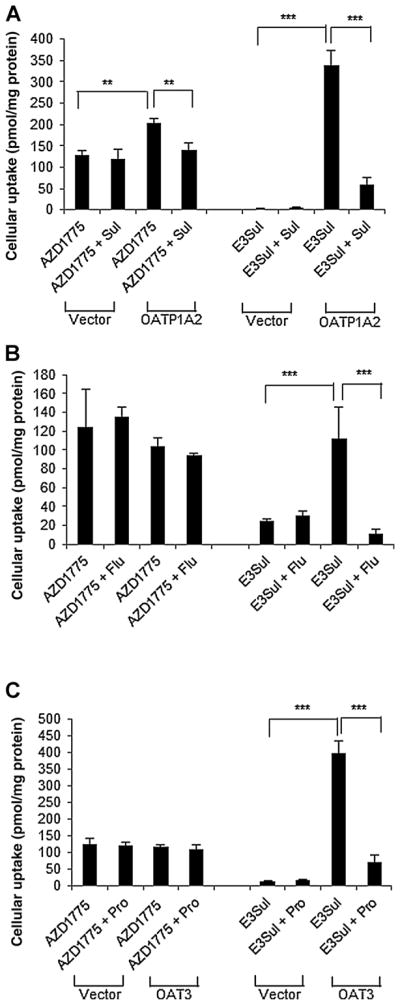
Cellular uptake of AZD1775 and the positive control, estrone-3-sulfate (E3Sul; a typical substrate for OATP1A2, OATP2B1, and OAT3), in the absence or presence of a specific transporter inhibitor [i.e., 100 μmol/L sulfobromophthalein (Sul) for OATP1A2, 10 μmol/L fluvastatin A (Flu) for OATP2B1, or 50 μmol/L probenecid (Pro) for OAT3] in HEK293 cells with overexpression of (A) OATP1A2, (B) OATP2B1, or (C) OAT3 and their respective vector control cells. Data are expressed as the mean ± SD from three independent experiments, with triplicate in each experiment. **, P < 0.01; ***, P < 0.001.
Binding to plasma proteins and brain tissue
In 20 patients with glioblastoma, AZD1775 exhibited a higher binding to brain tissues (fu,br: 0.067 ± 0.013; range, 0.042–0.10) than to plasma proteins (fu,p: 0.182 ± 0.024; range, 0.15–0.24).
PBPK modeling and simulation
PBPK modeling
A whole-body PBPK model integrated with a 4Brain model (Fig. 1) was developed to predict the kinetics of AZD1775 penetration across the BBB. Drug penetration across the BBB was controlled by the passive permeability, active efflux, and active uptake at the BBB. Given an average passive permeability of 17.5 × 10−6 cm/s determined from MDCKII cell monolayer and an average human brain microvascular surface area of 20 m2, the passive diffusion clearance (PSB) at the BBB was estimated to be 12.6 L/h (Eq. 1 and Table 1). Notably, AZD1775 exhibited pH-dependent efflux across MDCKII-ABCB1 or MDCKII-ABCG2 cell monolayers (Fig. 3B). As the basolateral pH varied from 7.4 to 6.5, ABCB1-mediated in vitro clearance decreased from 152 to 43 μL/min/mg, and similarly ABCG2-mediated in vitro clearance decreased from 76 to 33 μL/min/mg. Therefore, using the IVIVE scaling strategy based on the relative in vivo–in vitro activity factor of 295 for ABCB1 and 651 for ABCG2 (Eq. 3), it was estimated that ABCB1- and ABCG2-mediated efflux clearance at the BBB would reduce from 5.7 to 2.1 L/h as the brain interstitial pH decreased from 7.4 to 6.5.
By incorporating a passive diffusion clearance of 12.6 L/h and active efflux clearance of 5.7 or 2.1 L/h at the BBB, the PBPK model predicted the Kp,uu to be 0.78 and 0.92, respectively, at the brain interstitial pH of 7.4 and 6.5. Further sensitivity analysis with varying passive diffusion clearance (1.26–126 L/h) and active efflux clearance (0.57–57 L/h) suggested that the Kp,uu ranged from 0.11 to 1.03 at two extreme scenarios. These results suggested that active uptake must be involved in the BBB penetration of AZD1775 to achieve the observed unbound tumor-to-plasma ratios (median, 3.2; range, 1.6–24.4) in patients with glioblastoma. Subsequent sensitivity analysis with fixed passive diffusion clearance (12.6 L/h) and active efflux clearance (2.1 L/h) indicated the Kp,uu increased from 1.2 to 8.0 as the active uptake clearance at the BBB increased from 2.1 to 105 L/h. Therefore, by incorporating a passive clearance of 12.6 L/h, active efflux clearance of 2.1 L/h, and active uptake clearance of 44.0 L/h at the BBB, the final PBPK model predicted the Kp,uu to be 3.2, which was in good agreement with clinically observed data.
The developed PBPK model adequately predicted the plasma and brain concentration–time profiles of total and unbound AZD1775 in patients with glioblastoma. As shown in Fig. 5, the model-predicted total and unbound AZD1775 plasma and brain concentration–time profiles following a single oral dose of 400 mg in a virtual Caucasian population (n = 100) well recovered observed concentration data in patients with glioblastoma, with ~97% of observed data falling between the 5th and 95th percentile of the predicted mean concentration–time profile.
Figure 5.

Physiologically based pharmacokinetic modeling. A and B, Model-predicted and clinically observed AZD1775 total and unbound plasma concentration–time profiles. C and D, Model-predicted and clinically observed AZD1775 total and unbound brain tumor concentration–time profiles. Simulations of 10 virtual trials with 10 subjects in each were performed in a virtual Caucasian population following a single oral dose of 400 mg. Observed data were obtained from 12 Caucasian patients with glioblastoma receiving a single oral dose of 400 mg. The symbols represent observed data. The thick black line represents overall mean concentration–time profile for the virtual population (n = 100); dotted and dashed lines represent the 5th and 95th percentiles of the mean concentration, respectively.
Factors modulating drug BBB penetration
The impact of BBB integrity (assessed by passive diffusion clearance), transporter activity (assessed by active efflux/uptake clearance), and drug binding to brain tissue on the extent and rate of drug BBB penetration is illustrated in Fig. 6 and Supplementary Fig. S2. The impact of BBB integrity on the extent of BBB penetration was modulated by active transport process. When insignificant active efflux or uptake transport was present, variations in BBB integrity had a negligible influence on the extent of penetration, as indicated by a constant Kp,uu (~1.0) when the passive diffusion clearance varied 100-fold (1.26–126 L/h; Fig. 6A and B; Supplementary Fig. S2A and S2B). When active efflux was dominant, compromised BBB integrity (i.e., increasing passive diffusion clearance) led to a progressive increase in BBB penetration (Fig. 6A and Supplementary Fig. S2A), whereas when active uptake was dominant, compromised BBB integrity led to a progressive decrease in BBB penetration (Fig. 6B and Supplementary Fig. S2B). However, irrespective of whether active efflux or uptake is present, compromised BBB integrity generally resulted in quicker BBB penetration (i.e., shorter time to reach the maximum drug concentration in the brain; Fig. 6C and D; Supplementary Fig. S2C and S2D).
Figure 6.

Sensitivity analyses using the developed PBPK model. A, Impact of BBB integrity, as assessed by passive diffusion clearance (PSB) and active efflux clearance at the BBB (CLefflux,BBB) on the extent of BBB penetration (assessed by Kp,uu). B, Impact of BBB integrity (PSB) and active uptake clearance at the BBB (CLuptake,BBB) on the extent of BBB penetration (Kp,uu). C, Impact of BBB integrity (PSB) and active efflux clearance at the BBB (CLefflux,BBB) on the rate of BBB penetration, as assessed by the time to achieve the maximum drug brain concentration (Tmax,br). D, Impact of BBB integrity (PSB) and active uptake clearance at the BBB (CLuptake,BBB) on the rate of BBB penetration (Tmax,br). Different colors of surface areas represent different values of the Kp,uu or Tmax,br.
Increasing active efflux clearance led to a decrease in the extent of BBB penetration, whereas the opposite was observed as active uptake clearance increased (Fig. 6A and B). Of note, the degree of the impact of active transporter activity on drug BBB penetration was dependent on the magnitude of passive permeability. Specifically, the extent of BBB penetration (i.e., Kp,uu) was more sensitive to the change of active transporter activity when the passive permeability was relatively lower (Fig. 6A and B; Supplementary Fig. S2E and S2F). The rate of BBB penetration was insensitive to the change of active uptake, whereas quicker BBB penetration was expected as active efflux increased (Fig. 6C and D; Supplementary Fig. S2G and S2H).
Irrespective of BBB integrity or transporter activity, variations in drug binding to brain tissue influenced the rate of BBB penetration and total drug brain exposure but did not influence the unbound drug brain exposure. Specifically, the decrease of drug binding to brain tissue resulted in quicker BBB penetration while having no influence on the extent of BBB penetration (i.e., Kp,uu; Supplementary Fig. S2I and S2J).
Discussion
This study shows that the Wee1 kinase inhibitor AZD1775 extensively accumulates in human glioblastoma tumor tissue and achieves pharmacologically active and potentially therapeutic concentrations in patients. This is unexpected for a dual substrate of ABCB1 and ABCG2, two main efflux transporters expressed at human BBB to restrict the penetration of substrate drugs into the brain (4). Our in vitro cellular studies suggest that the ABCB1/ABCG2–mediated efflux clearance of AZD1775 is significantly reduced at a relatively acidic basolateral pH (which mimics acidic brain tumor microenvironment). In addition, AZD1775 is a substrate of OATP1A2, an uptake transporter expressed at human BBB to facilitate the transport of substrate drugs from the blood to brain (3, 24). Our mechanistic PBPK model further indicates that the extent of AZD1775 penetration into brain tumors is mainly dependent on the relative transporting efficiency of ABCB1/ABCG2 and OATP1A2. In acidic tumor microenvironment where ABCB1/ABCG2–mediated active efflux clearance is reduced, OATP1A2-mediated active uptake becomes dominant to drive AZD1775 penetration into brain tumors. These findings not only have direct clinical relevance for the treatment of glioblastomas with AZD1775 but also emphasize the need to consider the unique tumor microenvironment and transporter-mediated active uptake mechanisms in the evaluation of brain/tumor pharmacokinetics of agents in the treatment of brain cancer. In addition, the PBPK model we developed offers a mechanistic tool for the prediction of plasma and brain tumor pharmacokinetics, and provides the basis for the design of future prospective investigations for evaluating alternative and improved dosing regimens of AZD1775 and related agents.
Although the present data support further clinical development of AZD1775 for treating glioblastoma, it is noteworthy that our clinical observations are at odds with preclinical findings in an orthotopic glioblastoma xenograft mouse model, which indicated a limited distribution of AZD1775 in mouse normal brain and tumor tissues (23). Although the exact underlying mechanism is yet to be determined, the observed interspecies variability in AZD1775 BBB penetration underscores the critical value of early prospective evaluation of drug penetration across human BBB to inform decision-making regarding future clinical development and trial design.
AZD1775 belongs to the Biopharmaceutics Classification System (BCS) class II compound characterized by high permeability and low solubility. It is a good substrate of ABCB1 and ABCG2 (Fig. 3), two major efflux transporters that are highly expressed in the apical side of the BBB responsible for extruding substrate drugs from the brain to blood (4). Surprisingly, the Kp,uu of AZD1775 ranges from 1.3 to 24.4 (median, 3.2) in patients with glioblastoma. The PBPK modeling and simulation suggest that to achieve this degree of brain tumor penetration (i.e., Kp,uu > 1), a dominant active drug uptake has to present at the BBB. Several drug uptake transporters including OATP1A2, OATP2B1, and OAT3 have been identified in human brain microvessels (3). In particular, OATP1A2 is present at high abundance in the luminal membrane of capillary endothelial cells of human BBB (3, 24), implicating a critical role of this transporter in facilitating the entry of substrate drugs from the blood to brain. Our cellular uptake experiments suggest that AZD1775 is a substrate of OATP1A2, but not for OATP2B1 or OAT3 (Fig. 4), supporting the premise that OATP1A2-mediated uptake is likely involved in AZD1775 brain penetration. Notably, AZD1775 and other OATP1A2 substrate drugs (e.g., imatinib, methotrexate, paclitaxel, fentanyl, and pentazocine) are also the substrates of ABCB1 and/or ABCG2 (25–29). Similar to AZD1775, fentanyl and pentazocine show good BBB penetration (30, 31). It is known that the Kp,uu is determined by the ratio of net influx and net efflux clearances that can occur via both passive diffusion and transporter-mediated active transport (5). Active efflux increases the net efflux clearance, whereas active influx increases the net influx clearance. For a dual substrate of efflux and uptake transporters, it is plausible that under certain circumstances, active efflux and/or outward passive diffusion at the BBB is reduced, and, consequently, net efflux clearance is diminished and net influx clearance becomes dominant, thereby leading to increased drug brain penetration.
One circumstance that could lead to diminished net efflux clearance at the BBB is the acidic tumor microenvironment. There is firm evidence that the interstitial pH of many solid tumors, including glioblastoma, is more acidic (pH, 5.8–7.2) than normal tissue interstitial pH (7.0–7.8; refs. 32–34). The relatively acidic tumor microenvironment not only plays an important role in tumor pathophysiology but also can influence the tumor penetration and distribution of weak base or weak acid chemotherapeutic agents (32, 33). As supported by the pH partition theory and demonstrated by our in vitro cellular permeability experiments, at a relatively acidic basolateral pH, AZD1775 permeability decreased in the basolateral-to-apical direction while increasing in the apical-to-basolateral direction, thus resulting in a pronounced reduction in the efflux ratios on MDCKII-ABCB1 and MDCKII-ABCG2 monolayers (Fig. 3B). These in vitro findings can be extrapolated to an in vivo situation. By IVIVE scaling of the passive permeability determined from MDCKII cells (Eq. 1) and efflux ratios determined from MDCKII-ABCB1 and -ABCG2 cells (Eq. 2), it is predicted that when brain tumor interstitial pH varies from 7.4 to 6.5, the outward passive diffusion clearance at the BBB would decrease by ~26% (from 14.4 to 10.6 L/h), whereas there would be no apparent change in the inward passive diffusion clearance (from 12.2 to 12.9 L/h); meanwhile, the active efflux clearance would decrease by 63% (from 5.66 to 2.07 L/h). Hence, assuming the uptake transporter-mediated clearance remains the same (44 L/h), the overall decrease of the net efflux clearance would lead to a 1.6-fold increase in the Kp,uu (from 2.8 to 4.5). Collectively, these data provide mechanistic and quantitative evidence supporting the notion that the relative acidic tumor environment is favorable to the penetration and trapping of a weak base drug that is a substrate of ABCB1/ABCG2 into brain tumors due to reduced net efflux clearance at the BBB. These data also suggest that variations in the brain tumor regional pH can contribute to the heterogeneous penetration of weak base drugs such as AZD1775 into brain tumors.
In addition, the variation in BBB transporter expression/activity is an important factor accounting for the interspecies, interindividual, and intraindividual variability in drug penetration into the brain or brain tumors. The impact of efflux or uptake transporter activity on the extent of BBB penetration (Kp,uu) is illustrated by the PBPK simulation and sensitivity analyses. Of note, this impact is more significant for drugs with low passive permeability compared to those with high passive permeability (Fig. 6A and B; Supplementary Fig. S2E and S2F). The transporter function of ABCB1, ABCG2, or OATP1A2 can be impaired due to genetic polymorphisms or pharmacologic inhibition. A recent study using positron emission tomography (PET) clearly demonstrated that in people with a common non-synonymous SNP of the ABCG2 gene (421C>A), which occurs in ~20% of the population, pharmacologic inhibition of ABCB1 caused a significant increase in brain distribution of dual ABCB1/ABCG2 substrates (35). Three common synonymous SNPs of the ABCB1 gene (3435C>T, 2677G>T/A, and 1236C>T) have been associated with the susceptibility to fentanyl-induced respiration suppression, indicating the impact of ABCB1 polymorphisms on the brain penetration of the ABCB1 substrate fentanyl, a synthetic opioid receptor agonist (28). A number of SNPs have been identified in the OATP1A2 gene, and a few functional variants have been characterized in vitro (24, 36, 37). For example, the 516A>C, 559G>A, and 833A>—deletion variants have been associated with a reduced uptake function of OATP1A2 (24, 38), whereas the 38T>C variant increases the uptake of estrone sulfate and methotrexate (38). In light of the key role of ABCB1, ABCG2, and OATP1A2 in the BBB penetration of AZD1775, it is plausible that functional genetic polymorphisms of these transporters may account for the interindividual variability in the brain pharmacokinetics of AZD1775. Further investigation in a larger patient population is needed to test this hypothesis.
Another brain pathophysiology factor that may contribute to heterogeneous drug penetration into brain tumors is the variation in BBB integrity (39, 40). The magnitude of brain tumor vascular permeability varies both spatially and temporally, with the greatest permeability elevation in tumor core and a largely or completely intact BBB at the proliferating tumor edge or infiltrating tumor regions (41). One conventional way to measure BBB integrity is by gadolinium-enhanced magnetic resonance imaging (MRI; ref. 42). However, it is important to note that given the complex physiology of the BBB permeability affected by active transporters, molecule charge, and binding to plasma protein or brain tissue, contrast enhancement by water-soluble, non-ionic gadolinium agents may not accurately reflect the BBB penetration of lipophilic, charged, efflux/uptake transporter substrate drugs (40). The interplay of BBB integrity and active transporter activity on the extent of drug BBB penetration (i.e., Kp,uu) can be illustrated by PBPK simulations (Fig. 6 and Supplementary Fig. S2). When insignificant active transport is present, variation in BBB integrity has negligible impact on the Kp,uu (Fig. 6A and B; Supplementary Fig. S2A and S2B). This may explain, at least in part, the observation that permeability can differ by more than 100-fold among compounds capable of penetrating into the brain (43). When active efflux is dominant, compromised BBB integrity results in increased Kp,uu, whereas an opposite change is observed when active uptake is dominant (Fig. 6A and B; Supplementary Fig. S2A and S2B). Hence, given the fact that plasma protein-bound drugs generally do not cross the intact or partially impaired BBB, the general belief that the extent of drug penetration into contrast-enhanced tumor area is higher than that in non-enhanced area may be valid only for the drugs that undergo dominant active efflux transport at the BBB (e.g., topotecan and methotrexate; refs. 44, 45) or those that are BSC class III compounds characterized with high water solubility and low passive permeability (e.g., gadolinium imaging agents) but may not be applied to the drugs that undergo dominant active uptake at the BBB (e.g., AZD1775) or those that are not the substrates of efflux/uptake transporters. The PBPK simulations suggest that the penetration of AZD1775 into the invasive edge or infiltrative region of glioblastoma where the BBB remains largely intact would be better than, or at least similar to, that in bulky tumor areas where the BBB is “leaky,” assuming other factors (e.g., regional pH and transporter activity) remain the same in these regions. Further studies are needed to verify this prediction by comparisons of the Kp,uu measured from paired tumor samples (enhanced vs. non-enhanced region) of each individual patient. Because invasive, infiltrative glioblastoma regions are often unresectable and give rise to recurrent disease, a drug with good penetration into these regions would provide a huge therapeutic advantage.
In conclusion, using an integrated quantitative, clinical pharmacology approach that leverages clinical trial, IVIVE, and PBPK modeling, we provided the first clinical evidence of good tumor penetration of AZD1775 in patients with glioblastoma, and we further elucidated the underlying mechanism. Specifically, in the relatively acidic tumor microenvironment where ABCB1/ABCG2–mediated efflux clearance is reduced, OATP1A2-mediated active uptake becomes dominant to drive AZD1775 penetration into brain tumors. Variations in the brain/tumor regional pH, transporter expression/activity, and BBB integrity collectively attribute to the heterogeneity of drug penetration into brain tumors. Our study not only provides quantitative and mechanistic insights into the penetration of AZD1775 into brain tumors to inform decision-making regarding further clinical development of this novel agent for the treatment of glioblastoma but also composes a framework for prospective, quantitative, and mechanistic investigating the penetration of novel anticancer drugs across human BBB in general.
Supplementary Material
Translational Relevance.
Early, accurate prediction and mechanistic understanding of penetration of anticancer drugs across the human blood–brain barrier (BBB) are of paramount importance to the rational drug development and therapy for brain cancer. In this study, using an integrated, quantitative clinical pharmacology approach that leverages clinical trial, in vitro–in vivo extrapolation (IVIVE), and physiologically based pharmacokinetic (PBPK) modeling, we provided the first clinical evidence of good tumor penetration of AZD1775 in patients with glioblastoma, and we further elucidated the underlying mechanism. Our study provided quantitative and mechanistic insights into the heterogeneous brain/brain tumor penetration of AZD1775 to inform decision-making regarding further clinical development. The developed mechanistic PBPK model, verified by clinical observed data, can be generalized to other drugs that undergo similar disposition pathways. This study composes a framework for prospective, quantitative, and mechanistic investigation of the penetration of novel anticancer drugs across the human BBB in early-phase clinical development.
Acknowledgments
The authors thank AstraZeneca for providing the drug (AZD1775) for this study. The authors particularly thank the patients enrolled in the study.
This study was supported by the United States Public Health Service Cancer Center Support Grant P30 CA022453, the American Society of Clinical Oncology Career Development Award, and the Ben and Catherine Ivy Foundation.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Authors’ Contributions
Conception and design: J. Li, A. Sparreboom, N. Sanai
Development of methodology: J. Li, J. Wu, Y. Xie
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): J. Li, J. Wu, X. Bao, N. Honea, Y. Xie, A. Sparreboom
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): J. Li, J. Wu, S. Kim
Writing, review, and/or revision of the manuscript: J. Li, J. Wu, N. Honea, Y. Xie, A. Sparreboom, N. Sanai
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): J. Li, J. Wu, X. Bao, N. Honea
Study supervision: J. Li, N. Sanai
References
- 1.Henderson JT, Piquette-Miller M. Blood-brain barrier: an impediment to neuropharmaceuticals. Clin Pharmacol Ther. 2015;97:308–13. doi: 10.1002/cpt.77. [DOI] [PubMed] [Google Scholar]
- 2.Pardridge WM. Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab. 2012;32:1959–72. doi: 10.1038/jcbfm.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bronger H, Konig J, Kopplow K, Steiner HH, Ahmadi R, Herold-Mende C, et al. ABCC drug efflux pumps and organic anion uptake transporters in human gliomas and the blood-tumor barrier. Cancer Res. 2005;65:11419–28. doi: 10.1158/0008-5472.CAN-05-1271. [DOI] [PubMed] [Google Scholar]
- 4.Miller DS. Regulation of ABC transporters at the blood-brain barrier. Clin Pharmacol Ther. 2015;97:395–403. doi: 10.1002/cpt.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hammarlund-Udenaes M, Friden M, Syvanen S, Gupta A. On the rate and extent of drug delivery to the brain. Pharm Res. 2008;25:1737–50. doi: 10.1007/s11095-007-9502-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Lange EC. The mastermind approach to CNS drug therapy: translational prediction of human brain distribution, target site kinetics, and therapeutic effects. Fluids Barriers CNS. 2013;10:12. doi: 10.1186/2045-8118-10-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamamoto Y, Danhof M, de Lange EC. Microdialysis: the key to physiologically based model prediction of human CNS target site concentrations. AAPS J. 2017;19:891–909. doi: 10.1208/s12248-017-0050-3. [DOI] [PubMed] [Google Scholar]
- 8.Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol Cancer Ther. 2009;8:2992–3000. doi: 10.1158/1535-7163.MCT-09-0463. [DOI] [PubMed] [Google Scholar]
- 9.Parker LL, Piwnica-Worms H. Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science. 1992;257:1955–7. doi: 10.1126/science.1384126. [DOI] [PubMed] [Google Scholar]
- 10.Watanabe N, Broome M, Hunter T. Regulation of the human WEE1Hu CDK tyrosine 15-kinase during the cell cycle. EMBO J. 1995;14:1878–91. doi: 10.1002/j.1460-2075.1995.tb07180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beck H, Nahse V, Larsen MS, Groth P, Clancy T, Lees M, et al. Regulators of cyclin-dependent kinases are crucial for maintaining genome integrity in S phase. J Cell Biol. 2010;188:629–38. doi: 10.1083/jcb.200905059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Witt Hamer PC, Mir SE, Noske D, Van Noorden CJ, Wurdinger T. WEE1 kinase targeting combined with DNA-damaging cancer therapy catalyzes mitotic catastrophe. Clin Cancer Res. 2011;17:4200–7. doi: 10.1158/1078-0432.CCR-10-2537. [DOI] [PubMed] [Google Scholar]
- 13.Kuerbitz SJ, Plunkett BS, Walsh WV, Kastan MB. Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Proc Natl Acad Sci U S A. 1992;89:7491–5. doi: 10.1073/pnas.89.16.7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leijen S, Beijnen JH, Schellens JH. Abrogation of the G2 checkpoint by inhibition of Wee-1 kinase results in sensitization of p53-deficient tumor cells to DNA-damaging agents. Curr Clin Pharmacol. 2010;5:186–91. doi: 10.2174/157488410791498824. [DOI] [PubMed] [Google Scholar]
- 15.Wu J, Sanai N, Bao X, LoRusso P, Li J. An aqueous normal-phase chromatography coupled with tandem mass spectrometry method for determining unbound brain-to-plasma concentration ratio of AZD1775, a Wee1 kinase inhibitor, in patients with glioblastoma. J Chromatogr B Anal Technol Biomed Life Sci. 2016;1028:25–32. doi: 10.1016/j.jchromb.2016.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gaohua L, Neuhoff S, Johnson TN, Rostami-Hodjegan A, Jamei M. Development of a permeability-limited model of the human brain and cerebrospinal fluid (CSF) to integrate known physiological and biological knowledge: estimating time varying CSF drug concentrations and their variability using in vitro data. Drug Metab Pharmacokinet. 2016;31:224–33. doi: 10.1016/j.dmpk.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 17.Badhan RK, Chenel M, Penny JI. Development of a physiologically-based pharmacokinetic model of the rat central nervous system. Pharmaceutics. 2014;6:97–136. doi: 10.3390/pharmaceutics6010097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kalvass JC, Pollack GM. Kinetic considerations for the quantitative assessment of efflux activity and inhibition: implications for understanding and predicting the effects of efflux inhibition. Pharm Res. 2007;24:265–76. doi: 10.1007/s11095-006-9135-x. [DOI] [PubMed] [Google Scholar]
- 19.Uchida Y, Ohtsuki S, Katsukura Y, Ikeda C, Suzuki T, Kamiie J, et al. Quantitative targeted absolute proteomics of human blood-brain barrier transporters and receptors. J Neurochem. 2011;117:333–45. doi: 10.1111/j.1471-4159.2011.07208.x. [DOI] [PubMed] [Google Scholar]
- 20.Neuhoff S, Ungell AL, Zamora I, Artursson P. pH-dependent bidirectional transport of weakly basic drugs across Caco-2 monolayers: implications for drug-drug interactions. Pharm Res. 2003;20:1141–8. doi: 10.1023/a:1025032511040. [DOI] [PubMed] [Google Scholar]
- 21.Goda K, Balkay L, Marian T, Tron L, Aszalos A, Szabo G., Jr Intracellular pH does not affect drug extrusion by P-glycoprotein. J Photochem Photobiol B Biol. 1996;34:177–82. doi: 10.1016/1011-1344(95)07282-9. [DOI] [PubMed] [Google Scholar]
- 22.Altenberg GA, Young G, Horton JK, Glass D, Belli JA, Reuss L. Changes in intra- or extracellular pH do not mediate P-glycoprotein-dependent multidrug resistance. Proc Natl Acad Sci U S A. 1993;90:9735–8. doi: 10.1073/pnas.90.20.9735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pokorny JL, Calligaris D, Gupta SK, Iyekegbe DO, Jr, Mueller D, Bakken KK, et al. The efficacy of the Wee1 inhibitor MK-1775 combined with temozolomide is limited by heterogeneous distribution across the blood-brain barrier in glioblastoma. Clin Cancer Res. 2015;21:1916–24. doi: 10.1158/1078-0432.CCR-14-2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee W, Glaeser H, Smith LH, Roberts RL, Moeckel GW, Gervasini G, et al. Polymorphisms in human organic anion-transporting polypeptide 1A2 (OATP1A2): implications for altered drug disposition and central nervous system drug entry. J Biol Chem. 2005;280:9610–7. doi: 10.1074/jbc.M411092200. [DOI] [PubMed] [Google Scholar]
- 25.Eechoute K, Franke RM, Loos WJ, Scherkenbach LA, Boere I, Verweij J, et al. Environmental and genetic factors affecting transport of imatinib by OATP1A2. Clin Pharmacol Ther. 2011;89:816–20. doi: 10.1038/clpt.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van de Steeg E, van Esch A, Wagenaar E, Kenworthy KE, Schinkel AH. Influence of human OATP1B1, OATP1B3, and OATP1A2 on the pharmacokinetics of methotrexate and paclitaxel in humanized transgenic mice. Clin Cancer Res. 2013;19:821–32. doi: 10.1158/1078-0432.CCR-12-2080. [DOI] [PubMed] [Google Scholar]
- 27.Shukla S, Ohnuma S, Ambudkar SV. Improving cancer chemotherapy with modulators of ABC drug transporters. Curr Drug Targets. 2011;12:621–30. doi: 10.2174/138945011795378540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park HJ, Shinn HK, Ryu SH, Lee HS, Park CS, Kang JH. Genetic polymorphisms in the ABCB1 gene and the effects of fentanyl in Koreans. Clin Pharmacol Ther. 2007;81:539–46. doi: 10.1038/sj.clpt.6100046. [DOI] [PubMed] [Google Scholar]
- 29.Moriki Y, Suzuki T, Fukami T, Hanano M, Tomono K, Watanabe J. Involvement of P-glycoprotein in blood-brain barrier transport of pentazocine in rats using brain uptake index method. Biol Pharm Bull. 2004;27:932–5. doi: 10.1248/bpb.27.932. [DOI] [PubMed] [Google Scholar]
- 30.Henthorn TK, Liu Y, Mahapatro M, Ng KY. Active transport of fentanyl by the blood-brain barrier. J Pharmacol Exp Ther. 1999;289:1084–9. [PubMed] [Google Scholar]
- 31.Suzuki T, Moriki Y, Goto H, Tomono K, Hanano M, Watanabe J. Investigation on the influx transport mechanism of pentazocine at the blood-brain barrier in rats using the carotid injection technique. Biol Pharm Bull. 2002;25:1351–5. doi: 10.1248/bpb.25.1351. [DOI] [PubMed] [Google Scholar]
- 32.Gerweck LE, Seetharaman K. Cellular pH gradient in tumor versus normal tissue: potential exploitation for the treatment of cancer. Cancer Res. 1996;56:1194–8. [PubMed] [Google Scholar]
- 33.Tannock IF, Rotin D. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res. 1989;49:4373–84. [PubMed] [Google Scholar]
- 34.Martin GR, Jain RK. Noninvasive measurement of interstitial pH profiles in normal and neoplastic tissue using fluorescence ratio imaging microscopy. Cancer Res. 1994;54:5670–4. [PubMed] [Google Scholar]
- 35.Bauer M, Romermann K, Karch R, Wulkersdorfer B, Stanek J, Philippe C, et al. Pilot PET study to assess the functional interplay between ABCB1 and ABCG2 at the human blood-brain barrier. Clin Pharmacol Ther. 2016;100:131–41. doi: 10.1002/cpt.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou Y, Yuan J, Li Z, Wang Z, Cheng D, Du Y, et al. Genetic polymorphisms and function of the organic anion-transporting polypeptide 1A2 and its clinical relevance in drug disposition. Pharmacology. 2015;95:201–8. doi: 10.1159/000381313. [DOI] [PubMed] [Google Scholar]
- 37.Iida A, Saito S, Sekine A, Mishima C, Kondo K, Kitamura Y, et al. Catalog of 258 single-nucleotide polymorphisms (SNPs) in genes encoding three organic anion transporters, three organic anion-transporting polypeptides, and three NADH:ubiquinone oxidoreductase flavoproteins. J Hum Genet. 2001;46:668–83. doi: 10.1007/s100380170019. [DOI] [PubMed] [Google Scholar]
- 38.Badagnani I, Castro RA, Taylor TR, Brett CM, Huang CC, Stryke D, et al. Interaction of methotrexate with organic-anion transporting polypeptide 1A2 and its genetic variants. J Pharmacol Exp Ther. 2006;318:521–9. doi: 10.1124/jpet.106.104364. [DOI] [PubMed] [Google Scholar]
- 39.Lockman PR, Mittapalli RK, Taskar KS, Rudraraju V, Gril B, Bohn KA, et al. Heterogeneous blood-tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clin Cancer Res. 2010;16:5664–78. doi: 10.1158/1078-0432.CCR-10-1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gerstner ER, Fine RL. Increased permeability of the blood-brain barrier to chemotherapy in metastatic brain tumors: establishing a treatment paradigm. J Clin Oncol. 2007;25:2306–12. doi: 10.1200/JCO.2006.10.0677. [DOI] [PubMed] [Google Scholar]
- 41.Ewing JR, Brown SL, Lu M, Panda S, Ding G, Knight RA, et al. Model selection in magnetic resonance imaging measurements of vascular permeability: Gadomer in a 9L model of rat cerebral tumor. J Cereb Blood Flow Metab. 2006;26:310–20. doi: 10.1038/sj.jcbfm.9600189. [DOI] [PubMed] [Google Scholar]
- 42.Heye AK, Culling RD, del Valdes Hernandez MC, Thrippleton MJ, Wardlaw JM. Assessment of blood-brain barrier disruption using dynamic contrast-enhanced MRI. A systematic review Neuroimage Clin. 2014;6:262–74. doi: 10.1016/j.nicl.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu X, Tu M, Kelly RS, Chen C, Smith BJ. Development of a computational approach to predict blood-brain barrier permeability. Drug Metab Dispos. 2004;32:132–9. doi: 10.1124/dmd.32.1.132. [DOI] [PubMed] [Google Scholar]
- 44.Leggas M, Adachi M, Scheffer GL, Sun D, Wielinga P, Du G, et al. Mrp4 confers resistance to topotecan and protects the brain from chemotherapy. Mol Cell Biol. 2004;24:7612–21. doi: 10.1128/MCB.24.17.7612-7621.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blakeley JO, Olson J, Grossman SA, He X, Weingart J, Supko JG. Effect of blood brain barrier permeability in recurrent high grade gliomas on the intratumoral pharmacokinetics of methotrexate: a microdialysis study. J Neurooncol. 2009;91:51–8. doi: 10.1007/s11060-008-9678-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.