

Abstract
Pancreatic cancer requires many genetic mutations. Combinations of underlying germline variants and environmental factors may increase the risk of cancer and accelerate the oncogenic process. We systematically reviewed, annotated and classified previously reported pancreatic cancer-associated germline variants in established risk genes. Variants were scored using multiple criteria and binned by evidence for pathogenicity, then annotated with published functional studies and associated biological systems/pathways. Twenty-two previously identified pancreatic cancer risk genes and 337 germline variants were identified from 97 informative studies that met our inclusion criteria. Fifteen of these genes contained 66 variants predicted to be pathogenic (APC, ATM, BRCA1, BRCA2, CDKN2A, CFTR, CHEK2, MLH1, MSH2, NBN, PALB2, PALLD, PRSS1, SPINK1, TP53). Pancreatic cancer risk genes were organized into key biological mechanisms that promote pancreatic oncogenesis within an oncogenic model. Development of precision medicine approaches requires updated variant information within the framework of an oncogenic progression model. Complex risk modeling may improve interpretation of early biomarkers and guide pathway-specific treatment for pancreatic cancer in the near future. Precision medicine is within reach.
Keywords: pancreatic cancer, germline variants, hereditary cancer syndrome, hereditary pancreatitis
Introduction
Pancreatic cancer remains an uncommon but highly lethal cancer, with an overall five-year survival of less than 8%.1 Lifetime risk for pancreatic cancer is 1.5%, with an estimated 53,670 new cases in the United States in 2017.1 While it is relatively rare, comprising about 3.1% of all cancer cases, pancreatic cancer is now ranked the third leading cause of death among all cancers in the United States. Its most common form is pancreatic ductal adenocarcinoma (PDAC), which also has the worst prognosis. As a recalcitrant cancer (cancer with a 5-year relative survival rate below 50%), the interest and research into the prediction, detection, and management of pancreatic cancer continues to intensify.
Multiple challenges confront physicians and scientists who specialize in pancreatic cancer diagnosis and treatment.2 First, most of the known risk factors are common, such as alcohol consumption, smoking, diet and obesity, and have only small independent effect sizes. Secondly, though genome wide association studies (GWAS) identified some risk loci, the effect sizes of these variants are too low to be of clinical value in current paradigms. Third, there are no easily identifiable and manageable pre-malignant lesions, with the exception of some cystic lesions. Fourth, the location of the pancreas limits accessibility for collection of biomarkers. Furthermore, biopsies are invasive and associated with risk of acute pancreatitis or other complications. Fifth, PDAC metastasizes early, typically before the original tumor is identified. Sixth, PDAC is resistant to cure from standard chemotherapy and radiation. Finally, PDAC strongly affects metabolism and the immune system, leading to rapid demise despite a relatively small tumor burden.
Pancreatic cancer is both an inherited and acquired genetic disorder. The pathobiology is complex, as no dominant germline mutation or environmental factor accounts for the majority of disease burden, suggesting that multiple factors and random events interact over a number of years to eventually cause pancreatic cancer later in life, typically PDAC.3 This process is represented by our Whitcomb-Shelton-Brand Progression Model of PDAC Oncogenesis illustrated in Figure 1.3 The inherited cancer susceptibility genes (left box) are shown to influence different steps in oncogenesis. In a subset of patients, injury and inflammation of the pancreas manifest as chronic pancreatitis or diabetes mellitus. When this process is aggravated by environmental factors such as alcohol and smoking, it may promote somatic mutagenesis and dedifferentiation of parenchymal cells into pancreatic cancer stem cells. As expected, those subjects who have an inherited pathogenic germline mutation are at even a greater risk since fewer critical steps are required.2,3
FIGURE 1.
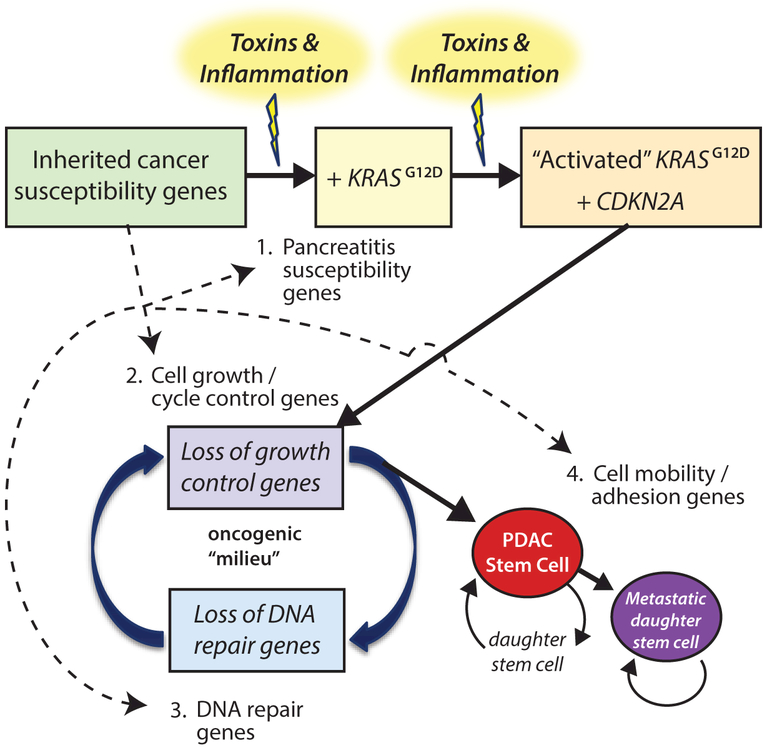
Progression model of PDAC oncogenesis. The probability that a person develops pancreatic cancer is dependent on progression through multiple stages, each of which requires changes to different robust biological systems. Prior knowledge of the biological function of key genes and pathogenic stresses allows for organization and integration of risk factors and their effects over time. Adapted from Whitcomb et al 2015.3
Germline mutations clearly increase risk for pancreatic cancer, and multiple familial pancreatic cancer kindreds have been described. While early estimations indicated that up to 10% of pancreatic cancer cases have a familial basis, only a limited number of susceptibility genes have been identified.4 The germline mutations within specific genes define rare hereditary cancer syndromes associated with high risks for pancreatic cancer. Examples include the ataxia telangiectasia gene (ATM), familial atypical multiple mole melanoma (FAMMM) syndrome linked to the CDKN2A gene, and Hereditary Breast and Ovarian Cancer syndrome caused by mutations in BRCA1 and BRCA2. The association of pancreatic cancer with familial syndromes provides insight into early mechanisms of oncogenesis and opportunities to identify high-risk patients for surveillance and prevention programs.
Despite current knowledge, interpretation and application of genetic risk of pancreatic cancer to manage patients remains challenging. Genetic testing and elucidation of targetable molecular pathways will be critical to improve and facilitate risk estimation, early diagnosis and personalized treatment. One approach involves the development of conceptual pancreatic cancer models, which allow complex information to be organized and predictive features tested in rational ways. Improvements in sequencing technology have facilitated the discovery of many germline variants associated with pancreatic cancer susceptibility in recent years. Furthermore, more complex combinations of low-penetrance variants that together markedly increase risk may be identified in some patients – a concept that has not yet been integrated into clinical interpretation and care in standardized ways.
Here, we review and summarize published germline variants associated with increased risks for pancreatic cancer, including new findings since our 2012 report.4 We list and describe multiple risk factors using an organizational approach adapted from our Progression Model of PDAC Oncogenesis (Fig. 1).
MATERIALS AND METHODS
Query Building
Germline variants associated with pancreatic cancer risk were identified through systematic review. First, reviews that discussed genes with germline variants identified in pancreatic cancer cases were retrieved from the PubMed database (http://www.ncbi.nlm.nih.gov/pubmed) from 2012 to June 2017. The following query (Query 1) was used:
("pancreatic cancer"[All Fields] OR "pancreatic neoplasms"[Mesh]) AND ("genes"[All Fields] OR "gene"[All Fields]) AND "germline"[All Fields] AND Review[ptyp] AND ("2012/01/01"[PDAT] : "2017/06/30"[PDAT]).
In Query 1, 24 reviews were identified. The genes identified from these pancreatic cancer reviews were compiled into a list for Query 2 – to acquire detailed information on previously identified germline variants in these genes. We also added the KRAS proto-oncogene to our query out of recognition that somatic KRAS activating mutations drive early oncogenesis and are found in >90% of PDAC cases5 (see discussion). Studies were then identified with the following query (Query 2):
("PANCREATIC CANCER"[ALL FIELDS] OR "PANCREATIC NEOPLASMS"[MESH] OR (("PANCREATIC"[ALL FIELDS] OR "PANCREAS"[ALL FIELDS]) AND "CANCER"[ALL FIELDS])) AND (("HEREDITARY PANCREATITIS"[ALL FIELDS] OR "PRSS1"[ALL FIELDS] OR "SPINK1"[ALL FIELDS] OR "SPINK2"[ALL FIELDS] OR "GGT1"[ALL FIELDS] OR "CFTR"[ALL FIELDS] OR "CTRC"[ALL FIELDS]) OR "KRAS"[ALL FIELDS] OR (("LI-FRAUMENI SYNDROME"[ALL FIELDS] OR "TP53"[ALL FIELDS]) OR "SMAD4"[ALL FIELDS] OR ("ATAXIA-TELANGIECTASIA"[ALL FIELDS] OR "ATM"[ALL FIELDS]) OR "CHEK2"[ALL FIELDS] OR ("FAMILIAL ATYPICAL MULTIPLE MOLE MELANOMA"[ALL FIELDS] OR "CDKN2A"[ALL FIELDS]) OR ("PEUTZ-JEGHERS"[ALL FIELDS] OR "STK11"[ALL FIELDS])) OR (("HEREDITARY BREAST AND OVARIAN CANCER"[ALL FIELDS] OR "BRCA1"[ALL FIELDS] OR "BRCA2"[ALL FIELDS] OR "BARD1"[ALL FIELDS]) OR ("FANCONI ANEMIA"[ALL FIELDS] OR "PALB2"[ALL FIELDS] OR "FANCA"[ALL FIELDS] OR "FANCC"[ALL FIELDS] OR "FANCG"[ALL FIELDS] OR "FANCM"[ALL FIELDS]) OR ("LYNCH SYNDROME"[ALL FIELDS] OR "MLH1"[ALL FIELDS] OR "MSH2"[ALL FIELDS] OR "MSH6"[ALL FIELDS] OR "PMS2"[ALL FIELDS] OR "EPCAM"[ALL FIELDS]) OR "POLN"[ALL FIELDS] OR "POLQ"[ALL FIELDS] OR "NBN"[ALL FIELDS] OR "PTEN"[ALL FIELDS]) OR ("PALLD"[ALL FIELDS] OR ("FAMILIAL ADENOMATOUS POLYPOSIS"[ALL FIELDS] OR "APC"[ALL FIELDS]))) AND ("0001/01/01"[PDAT] : "2017/06/30"[PDAT])
Relevant studies published before June 30, 2017 and available in English were retrieved, filtered and reviewed.
Literature Selection
Studies from Query 2 were first filtered by publication type and abstract keywords, then selected by the following inclusion criteria: (1) authors evaluated possible associations between germline variant(s) and risk for pancreatic cancer, or authors identified germline variant(s) in pancreatic cancer patients. Studies on non-adenocarcinoma types of pancreatic cancer were excluded; (2) the type of identified variant(s) is expected to alter protein structure (e.g. frameshift, missense, alternative splicing); and (3) identified variant(s) can be mapped onto the hg19 (human genome 19) reference genome with information provided in studies.
Data Collection
The following information was extracted from the filtered studies: First author, publication year, PubMed ID, gene name, germline variant (variant coding DNA nucleotide change with referenced transcript and amino acid change), mutation type, variant pathogenicity indicated in the study, if the variant is described in a hereditary cancer family and co-segregation with disease. Other information, including study population, number of cases/controls reported, and significance, was collected if available. Germline variants were mapped onto the human reference genome GRCh37 (UCSC hg19) and annotated with ExAC (Exome Aggregation Consortium) minor allele frequency (MAF),6 Combined Annotation Dependent Depletion (CADD) score,7 variant rsID (reference single nucleotide polymorphism [SNP] ID) (dbSNP [The Single Nucleotide Polymorphism database] build 147) and ClinVar clinical significance. Nucleotide changes and transcripts were formatted in HGVS (Human Genome Variation Society) and RefSeq (National Center for Biotechnology Information [NCBI] Reference Sequence Database) nomenclature, respectively.
Variant Evaluation and Categorization
Variants were scored across multiple criteria and binned according to the level of evidence for pathogenicity. The criteria and scoring algorithm used to evaluate variants was adapted from the American College of Medical Genetics and Genomics (ACMG) guidelines for variant interpretation (Table 1).8 Scores were summed across the seven criteria groups for each variant and binned into pathogenic categories according to aggregate scores: ≥5 – pathogenic (bin 1); 3–4.5 – likely pathogenic (bin 2); 0.5–2.5 – uncertain significance (bin 3); ≤0 – likely benign (bin 4). If a variant met more than one subcategory within a criteria group, the highest score was selected for that criterion. The criteria met from Table 1 and respective binning category were summarized and appended for each variant.
TABLE 1.
Criteria and Scoring Algorithm to Evaluate Variant Pathogenicity
| Category | Number | Criteria | Score |
|---|---|---|---|
| Common Polymorphism | 0 | ExAC MAF for this variant is >5% | −2 |
| Null variant | 1 | Variant can be classified as a null variant (nonsense, frameshift, canonical +/−1 or 2 splice sites, initiation codon) | +1.5 |
| Function | 2(a) | Variant damaging effects are supported by a functional study | +2 |
| 2(b) | Variant is located within a functional domain but has not yet been confirmed by a functional assay | +1 | |
| 2(c) | Amino acid change has been previously established to have a damaging functional effect (regardless of nucleotide change) | +2 | |
| 2(d) | Variant is a novel missense mutation AND another missense mutation has previously reported at this residue with a damaging functional effect | +1.5 | |
| Computational prediction | 3(a) | CADD score for this variant is ≥30 | +1 |
| 3(b) | CADD score for this variant is ≥20 and <30 | +0.5 | |
| 3(c) | CADD score for this variant is <10 | −1 | |
| Significance | 4(a) | Statistical significance is observed, with odds ratio >5 (95% CI does not overlap with 1) | +2 |
| 4(b) | Statistical significance is observed | +1 | |
| Multiple reports | 5(a) | Variant is reported in 3 or more study cohorts as possibly disease causing | +1.5 |
| 5(b) | Variant is reported in 2 different study cohorts as possibly disease causing | +1 | |
| Variant co-segregation | 6(a) | Co-segregation of variant with disease in multiple affected family members is observed | +2 |
| 6(b) | Variant failed to co-segregate with disease (if examined) | −1 |
Pathogenicity score of each variant is calculated by adding up scores from highest scored criterion met in each category. Our criteria and scoring algorithm was adapted from the American College of Medical Genetics and Genomics (ACMG) guidelines for variant interpretation.8
Risk Factor Review
According to protein function/molecular pathway and The Progression Model of PDAC Oncogenesis,3 genetic risk factors identified in Query 1 were organized into four different biological systems of cellular processes and oncogenic steps – (1) cell injury; (2) cell growth and cycle control; (3) DNA repair, and (4) cell mobility and adhesion (Table 2). We separate cell growth/cycle regulation and DNA repair, but recognize the interaction and synergism of acquired mutations that progressively impair both of them.
TABLE 2.
Functional Organization of Germline Risk Factors
| Systems | Gene Symbol | Function in Model | Inherited Disorder | References |
|---|---|---|---|---|
|
Cell injury (pancreatitis susceptibility) |
PRSS1 | Protease, premature activation induces pancreatitis | Hereditary pancreatitis | 3,4,137–143 |
| SPINK1 | Trypsin inhibitor | Hereditary pancreatitis | 139,140,142,143 | |
| SPINK2 | Trypsin inhibitor | 137 | ||
| GGT1 | Metabolism of glutathione, anti-oxidative defense | 3 | ||
| CTRC | Degrades prematurely activated tyrpsin | Hereditary pancreatitis | 142 | |
| CFTR | Anion channel required for epithelial cell secretion/absorption | Cystic fibrosis, Hereditary pancreatitis | 3,137,140,143 | |
| Cell growth/cycle control | KRAS | Proliferation signaling | Cardiofaciocutaneous syndrome, Noonan syndrome | 3,138,139,141,142,144,145 |
| TP53 | Cell cycle regulation and arrest after DNA damage, apoptosis | Li-Fraumeni syndrome | 3,137,139,141,143,146 | |
| SMAD4 | Tumor suppressor, growth inhibition | Hereditary hemorrhagic telangiectasia, Juvenile polyposis syndrome, Myhre syndrome | 139 | |
| ATM | DNA damage response at DSBs (double-strand breaks) | Ataxia-telangiectasia | 3,4,137–143,145–147 | |
| CHEK2 | Cell cycle checkpoint regulator, DSB response | Li-Fraumeni syndrome | 147 | |
| CDKN2A | Cell arrest at G1 and G2 checkpoints, apoptosis | Familial atypical multiple mole melanoma (FAMMM) syndrome | 3,4,137–143,146 | |
| STK11 | Regulates cell growth, proliferation and DNA damage response | Peutz-Jeghers Syndrome |
3,4,137–139,141–143 | |
| PTEN | Regulates cell cycle, survival and metabolism | PTEN hamartoma tumor syndrome | 141 | |
| DNA repair | BRCA1 | DSB DNA repair | Hereditary Breast and Ovarian cancer syndrome | 3,4,117,137–141,143,146–150 |
| BRCA2 | DSB DNA repair | Hereditary Breast and Ovarian cancer syndrome | 3,4,137–147,149,151,152 | |
| BARD1 | DSB DNA repair | Hereditary Breast and Ovarian cancer syndrome | 147 | |
| PALB2 | DSB DNA repair | Fanconi anemia | 3,4,117,137–143,145,146,149 | |
| FANCA | DNA repair | Fanconi anemia | 137 | |
| FANCC | DNA repair | Fanconi anemia | 137,141,143,146,147 | |
| FANCG | DNA repair | Fanconi anemia | 137,141,143 | |
| FANCM | DNA repair | Fanconi anemia | 147 | |
| MLH1 | Mismatch repair | Lynch Syndrome | 3,4,137,139,141,143,146 | |
| MSH2 | Mismatch repair | Lynch Syndrome | 3,4,137,139,141,143,146 | |
| MSH6 | Mismatch repair | Lynch Syndrome | 3,4,137,141,146 | |
| PMS2 | Mismatch repair | Lynch Syndrome | 3,4,137,147 | |
| EPCAM | Mismatch repair* | Lynch Syndrome | 3 | |
| POLN | DNA damage repair | 147 | ||
| POLQ | DNA damage repair | 147 | ||
| NBN | DNA damage repair | Nijmegen breakage syndrome | 147 | |
| Cell mobility/adhesion | PALLD | Cytoskeletal component | 3,140,143,146 | |
| APC | Regulates cell migration and adhesion, Wnt pathway antagonist | Familial adenomatous polyposis | 3,4,137,141,143 |
Deletions in the last few exons of EPCAM inactivate MSH2 to indirectly cause defects in mismatch repair.
A critical review and discussion of each risk gene was conducted in the context of this model. These organizational themes represent a framework for constructing rigorous disease models for risk assessment and therapeutic decision-making.
RESULTS
Search Results
In Query 2, we identified 3463 potentially relevant studies published on or before June 30, 2017 in PubMed (Fig. 2). A total of 578 (16.7%) studies were excluded according to their publication type (i.e., they were review articles). Of the remaining 2885 studies, 2182 (75.6%) were identified to be irrelevant upon keyword screening of their title and abstract (if available). An additional 495 (70.4%) were excluded because they did not provide information on pancreatic cancer inherited risk genes or germline variants from pancreatic cancer patients. Finally, we examined the 208 remaining studies, 97 (46.6%) of which reported on germline variants associated with pancreatic cancer that passed our variant filtering criteria. The characteristics of these 97 studies are summarized in Supplementary Table 1.
FIGURE 2.
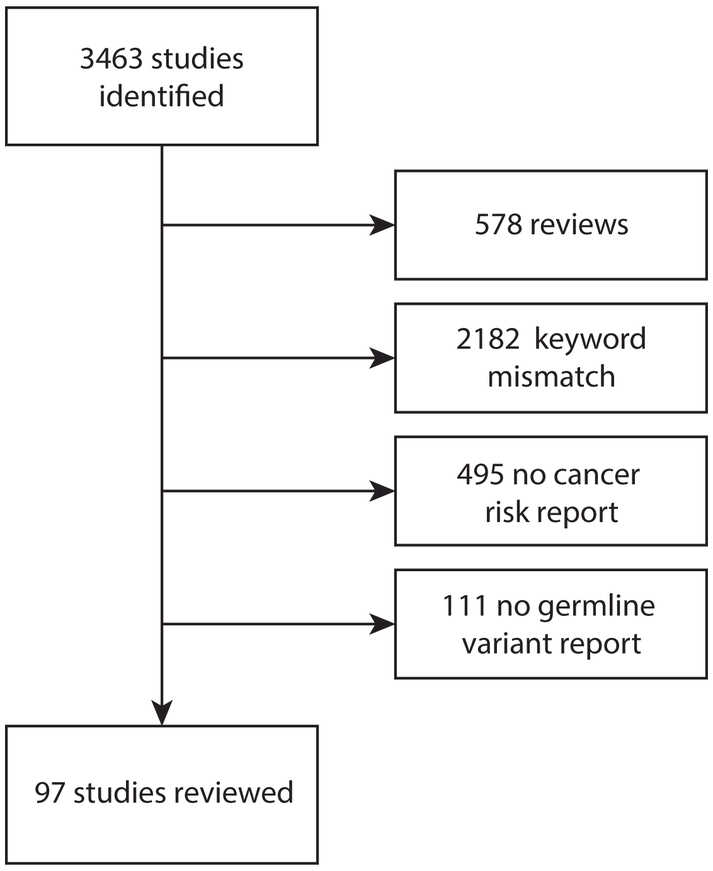
Flow chart describing the search and filter results of Query 2.
Twenty-two genes were we reported in multiple studies, strengthening the evidence for pathogenicity (Table 3).
TABLE 3.
Summary of Reviewed Genes
| Gene Symbol* | No. of Studies | Country | No. of Variants |
|---|---|---|---|
| APC | 4 | Canada, Germany, Israel, US | 3 |
| ATM | 9 | Canada, Germany, Japan, US | 22 |
| BARD1 | 1 | US | 1 |
| BRCA1 | 19 | Canada, Israel, Italy, Poland, Sweden, US | 26 |
| BRCA2 | 31 | Canada, Germany, Iceland, Israel, Italy, Japan, Korea, Spain, UK, US | 87 |
| CDKN2A | 16 | Canada, Germany, Italy, Netherlands, Poland, US | 46 |
| CFTR | 10 | Belgium, Germany, Italian, Spain, UK, US | 15 |
| CHEK2 | 5 | Czech Republic, Germany, Poland, US | 4 |
| FANCA | 2 | US | 13 |
| FANCC | 3 | US | 13 |
| FANCM | 1 | US | 1 |
| MLH1 | 6 | Canada, Italy, Japan, US | 8 |
| MSH2 | 6 | Canada, Ireland, Italy, US | 12 |
| MSH6 | 3 | Canada, US | 8 |
| NBN | 2 | Czech Republic, US | 1 |
| PALB2 | 14 | Canada, Czech Republic, Germany, Italy, Japan, Poland, Spain, US | 52 |
| PALLD | 3 | Canada, US | 3 |
| PMS2 | 2 | US | 7 |
| PRSS1 | 4 | France, Spain, UK | 4 |
| SPINK1 | 5 | Finland, Germany, Italy, Japan, US | 3 |
| STK11 | 2 | Italy, US | 2 |
| TP53 | 6 | Canada, Czech Republic, Japan, US | 6 |
Gene symbols follow the Hugo Gene Nomenclature Committee [HGNC] Gene ID.
US indicates, United States; UK, United Kingdom.
Germline Variants Associated With Pancreatic Cancer
Hundreds to thousands of germline variants are expected to contribute to pancreatic cancer susceptibility, many of which are rare or private variants. Germline variants that were identified in multiple pancreatic cancer cases and found to be both common and either pathogenic or likely pathogenic by multiple criteria were summarized. Twenty variants were identified in multiple studies and reported here as pathogenic (Table 4). An additional 46 variants were categorized as likely pathogenic (Supplementary Table 2), and the remaining variants were categorized as variants of uncertain significance (VUS) (see Supplementary Table 3 for all variants).
TABLE 4.
‘Pathogenic’ Germline Variants (Bin 1)
| Gene | cDNA Nucleotide Change | rsID | Population | Reference |
|---|---|---|---|---|
| ATM | c.5932G>T | rs587779852 | US, Europe | 153,154 |
| ATM | c.8266A>T | rs371638537 | US | 58 |
| BRCA1 | c.5263_5264insC | rs80357906 | (Ashkenazi Jewish) US, Canada, Poland, Israel | 65,82,83,155–159 |
| BRCA1 | c.68_69delAG* | rs386833395 | (Ashkenazi Jewish) US | 69,160 |
| BRCA1 | c.66_67delAG* | rs796856605 | (Ashkenazi Jewish) US, Israel, Canada | 82,83,155,158,159,161,162 |
| BRCA2 | c.5946delT | rs80359550 | (Ashkenazi Jewish) US, Israel, Canada | 69,81–83,156,158,159,161–166 |
| BRCA2 | c.6591_6592delTG | rs80359605 | Germany, UK | 80,167,168 |
| BRCA2 | c.9227G>A | rs80359187 | Germany, UK | 80,168 |
| BRCA2 | c.10095delCinsGAATTATATCT | rs276174803 | Germany, UK | 80,168 |
| CDKN2A | c.457G>T | rs45476696 | US | 60,69,169,170 |
| CDKN2A | c.377T>A | rs104894098 | US | 60,68,69,169,171 |
| CDKN2A | c.301G>T | rs104894094 | US, Italy | 60,68,69,111,169 |
| CDKN2A | c.225_243del19 | rs730881674 | Netherlands, US | 68,69,169,172–174 |
| CDKN2A | c.148C>T | rs864622636 | Germany, US | 69,175 |
| CDKN2A | c.−34G>T | rs1800586 | US | 68,69,169,170 |
| CHEK2 | c.1100delC | rs555607708 | Germany, Poland, US | 153,157,176 |
| PALB2 | c.509_510delGA† | rs515726124 | Czech Republic, Poland, US | 60,89,157 |
| PALB2 | c.508_509delAG† | - | Europe | 88 |
| PALB2 | c.172_175delTTGT | rs180177143 | Poland, US | 65,157,177 |
| SPINK1 | c.194+2T>C‡ | rs148954387 | (Japanese) Japan, Germany | 26,178 |
All germline variants are reported in patients with pancreatic cancer in multiple studies, and all variants except one SPINK1 variant are reported in familial pancreatic cancer cases.
Each symbol represents a group of variants that result in same cDNA and protein change.
Absent in familial pancreatic cancer study.
cDNA indicates complementary DNA; US, United States; UK, United Kingdom.
A review of the pancreatic cancer susceptibility genes indicated that they are each subsets of four different biological systems – cell injury; cell growth and cycle control; DNA repair; and cell mobility and adhesion. It was not possible to determine if individuals had mutations in multiple biological systems from the available studies.
DISCUSSION
Pancreatic cancer (OMIM #260350) is an inherited and acquired genetic disorder, for which an increased risk is associated with a number of different genes in different biological systems. Predictive genetic testing can be used to estimate patient risk for pancreatic cancer, particularly if there is a known pathogenic risk variant in the family or another high pre-existing risk, such as chronic pancreatitis. However, understanding overall risk of pancreatic cancer and other complex disorders remains a challenge for clinicians and scientists. Epidemiology and family studies demonstrate a small overall increased risk (standardized incidence ratio (SIR) of 1.88; 95% confidence interval (CI), 1.27–2.68) of pancreatic cancer among first-degree relatives of patients with pancreatic cancer.9 Since most environmental factors also confer relatively small increased risk (2–3 fold), we hypothesize that independent pathogenic germline variants in cancer susceptibility genes are small because they only affect one step in a complex process. However, a combination of inherited and acquired pathogenic genetic factors, affecting multiple steps in a pathogenic pathway and driven by environmental stressors,10 will have a large combined effect. For example, smoking is known to approximately double the risk of pancreatic cancer. This relative risk may appear low, but for patients with hereditary pancreatitis, doubling a 20% risk of pancreatic cancer to 40% is clinically important, especially as smoking also decreases the age of onset up to 20 years.11,12 Knowledge of the effect of smoking in hereditary pancreatitis resulted in a drastic reduction in smoking in these patients over the past 20 years, with a marked decrease in the overall rate of pancreatic cancer by age 70 years.13 Thus, understanding the combination of risk factors in patients with high pre-existing risk should influence clinical management decisions.
Still, the identification of genetic variants in an individual poses several challenges with interpretation and subsequent determination of beneficial actions. One approach, used here, continuously updates and expands the list of reported variants, with an expectation that, with sufficient numbers, pathogenic variants will be enriched in subjects with pancreatic cancer compared to control populations. Databases such as ClinVar, which aggregate information on the relationships between genomic variants and human phenotypes, are an important resource of variants and their proposed clinical significance identified through clinical testing laboratories. However, the data is organized on a model of rare germline single disease susceptibility loci with strong genetic effects, without a framework to recognize the contributions of additive or interacting variants. A more sophisticated solution may require a paradigm shift from traditional Mendelian genetics to disease modeling with outcome simulation to anticipate the efficacy of possible interventions. Application of the new paradigm results in true personalized medicine.
Fortunately, knowledge of PDAC continues to grow through discoveries from complementary approaches. Genetically engineered mouse models (GEMMs) have demonstrated the unequivocal interrelationship between pancreatic inflammation, specific genetic variants and PDAC development.14–19 Likewise, next generation sequencing of PDAC tumors and early lesions provides insights into the complex but stereotypic pathways of oncogenesis.20 The clinical application of this knowledge naturally lags behind by a number of years, but continued organization and interpretation of accumulated clinical and translational data has the potential to rapidly accelerate the development of clinically useful tools. The ACMG guidelines for variant interpretation8 represents an important step for harmonizing reporting practices across clinical laboratory reports, noting that in clinical practice “pathogenic” (e.g. bin 1) and “likely pathogenic” (e.g. bin 2) are often combined.
Cell Injury Risk – PRSS1, SPINK1, and CFTR
Chronic pancreatitis (CP) has been long established to be a strong risk factor for pancreatic cancer,21 and pancreatic inflammation is an early event in the carcinogenic process. Recurring or persistent inflammation is a known driver of cellular turnover, which inevitably increases the chance of somatic mutagenesis and promotes a microenvironment that selects for malignant cell properties.22,23 Indeed, KRAS mutations, which are found in precursor lesions (PanINs), can be found in CP pancreata with disease duration of ≥ 3 years.24 An association between acute pancreatitis and risk for pancreatic cancer was also recently identified.25 The risk of pancreatic cancer appears to be high with long-standing chronic pancreatitis, especially when genetic mutations in pancreatitis susceptibility genes cause early onset chronic pancreatitis. While these mutations increase the risk of pancreatic cancer in patients who develop chronic pancreatitis, most pancreatic cancer patients do not have commonly recognized mutations in PRSS1, SPINK1 or CFTR.26
PRSS1 (Gene ID: 5644) codes for protease, serine 1, also known as trypsin-1 and cationic trypsinogen. Cationic trypsinogen is a digestive enzyme precursor and the most highly expressed isotype of three trypsinogens that are expressed in the pancreas. Trypsinogen is activated to trypsin, serving as an endoprotease to cleave peptide chains at arginine or lysine. Trypsin is also a master regulator that activates trypsinogen and other pancreatic zymogens and inactivates other trypsin molecules. Cationic trypsin (the active form of trypsinogen) is the primary driver of cell injury and inflammation in acute pancreatitis, which can lead to CP. Gain-of-function mutations in PRSS1 result in susceptibility to premature activation and/or resistance to degradation. Other mechanisms, described below, impair trypsin inhibitors or the ability of duct cells to quickly flush trypsinogen out of the pancreatic duct and into the duodenum.
Patients with PRSS1 gain-of-function variants (e.g. p.Asn29Ile [p.N29I], p.Arg122His [p.R122H]) are at high risk of hereditary pancreatitis (OMIM: 167800), which is associated with a first attack around age 10–12 years and a high risk for progression to CP in the 2nd or 3rd decade of life. Estimates for cumulative risk for pancreatic cancer at age 70 range from 7.2–40%,11,13,27,28 which is believed to be a consequence of lifetime exposure of the pancreas to inflammation. However, PRSS1 mutations are an uncommon cause of pancreatitis (~1%), and only 5% of patients with CP (all etiologies) will develop pancreatic cancer over a 20 year period.29
SPINK1 (Gene ID: 6690) codes for serine peptidase inhibitor, Kazal type 1, also known as pancreatic secretory trypsin inhibitor. It is a suicide trypsin inhibitor that is upregulated with inflammation to protect the pancreas from autodigestion by trypsin and other pancreatic digestive enzymes that might be activated by trypsin. Loss-of-function mutations in SPINK1 indirectly increase the risk for trypsin-related injury. Pathogenic variants in SPINK1 and its regulatory elements are common in the general population (~2%) and are associated with CP.30 While pathogenic SPINK1 germline variants can cause an autosomal recessive form of pancreatitis, they more commonly act as disease modifiers in combination with other genetic variants.31 Studies have also confirmed that SPINK1 mutations are associated with a higher risk for pancreatic cancer, particularly in patients with chronic CP.32–34 The association between SPINK1 variants and pancreatic cancer appears small, since SPINK1 variants are only important in the context of recurrent, premature trypsin activation, and of the patients who do develop recurrent acute and chronic pancreatitis, only a small fraction progress to pancreatic cancer over time. Finally, of the patients who do develop pancreatic cancer, only a subset have pre-existing CP. Nevertheless, there is a real risk within the right context.
CFTR (Gene ID: 1080) codes for the cystic fibrosis transmembrane conductance regulator protein. CFTR is an anion channel expressed in secretory or absorptive epithelial cells of the respiratory, digestive, and reproductive systems and the skin. It can change conformations to be primarily a chloride-conducting or bicarbonate-conducing channel. Several organs utilize the bicarbonate conducting function, including the pancreas, vas deferens and sinuses.35 Severe mutations in CFTR cause classic cystic fibrosis, an autosomal recessive disorder (OMIM: 602421) by impairing both chloride and bicarbonate conductance. Lack of ion conductance results in lack of fluid secretion and, therefore, pancreatic inflammation from retained trypsin. More recently, CFTR variants have been discovered that impair bicarbonate conductance only, leaving chloride conductance intact.35 While both forms are associated with pancreatitis, heterozygous CFTR carriers also have an increased risk of recurrent acute and chronic pancreatitis,36,37 particularly in the presence of SPINK1 or CTRC mutations,38,39 or pancreas divisum.40,41
Patients with CFTR-associated chronic pancreatitis are at increases risk of pancreatic cancer. Hamori et al42 reported that among pancreatitis patients who were followed longitudinally (1–40 years), those with CFTR-related chronic pancreatitis (all of whom were smokers) had a SIR of 26.5 (95% CI, 8.6–61.9) for pancreatic cancer. In contrast, among patients with pancreatic cancer, CFTR-variants are uncommon.26,43 McWilliams et al43 compared the frequency of 39 common CFTR variants in 949 white patients and 13,340 white controls and found a significant association between pancreatic cancer and CFTR variant carrier status (odds ratio (OR), 1.40; 95% CI, 1.04–1.89).43 Thus, CFTR appears to be linked to pancreatic cancer when it causes chronic pancreatitis (often early onset), but is not a major, direct cause of pancreatic cancer.
The field of pancreatic genetics is rapidly expanding with the discovery of multiple susceptibility genes and disease modifying factors. Most of the pathogenic variants are rare, have small independent effect sizes, or are part of complex risk signatures with other factors. However, the three extensively studied genes, PRSS1, SPINK1 and CFTR, provide insight into a more general mechanism of pancreatic cancer – generation of recurrent injury and/or inflammation in the gland.
Cell Growth and Cell Cycle Control –TP53, ATM, CHEK2, CDKN2A, and STK11
Proper functioning of proteins that regulate cell growth and the cell cycle provide a critical protection against oncogenesis. Loss of their regulation drives uncontrolled proliferation, and activation of the DNA damage checkpoint occurs in the early stages of intraductal papillary mucinous neoplasms (IPMNs) and PanIN lesions.44,45 This concept is further evidenced by the contribution of mutations in TP53 and KRAS – two critical early initiators of pancreatic oncogenesis.
KRAS (Gene ID: 3845) codes for the KRAS proto-oncogene GTPase, an important regulator of cellular response to cytokines, hormones and growth factors, cell proliferation,46,47 transcriptional silencing of tumor suppressor genes,48 and the inflammatory response.49,50 Activating mutations in KRAS are implicated in various malignancies, including colorectal carcinoma, lung cancer and pancreatic cancer. The most consistent feature of pancreatic cancer is the somatic KRAS activating mutation p.G12D (KRASG12D).51 Genetically engineered mouse models that include KRASG12D develop pancreatic cancer, especially after induction of acute pancreatitis.52 However, germline KRASG12D gain-of-function variants are rare in humans, and loss-of-function variants have not been linked to pancreatic cancer.
Tumor protein p53 (TP53, Gene ID: 7157) codes for a tumor suppressor that regulates cell proliferation, DNA repair and apoptosis in response to cellular stress. KRAS and TP53 are the two genes most frequently detected to contain somatic mutations in PDAC (93.7% and 56%, respectively),5 and particularly KRAS in PanIN1A lesions (92.3%).53,54 Mutations in TP53 can cause Li-Fraumeni syndrome (OMIM: 151623), which is characterized by a high risk for cancer (in one report, 60% by age 45 years and 95% by age 70),55 including a relative risk (RR) of 7.3 for pancreatic cancer.56 Other tumor suppressors in which mutations are associated with pancreatic cancer include ATM, CHEK2, CDKN2A, and STK11.
ATM (Gene ID: 472) codes for a protein kinase that regulates cell proliferation and detects DNA damage to coordinate DNA repair.57 Biallelic mutations in ATM cause Ataxia-telangiectasia (OMIM: 208900), a rare disorder characterized by progressive ataxia, telangiectasias, a weakened immune system, and an increased risk for cancer–especially leukemia and lymphoma. The prevalence of ATM mutations have been found to be significantly greater in familial pancreatic cancer cases than spouse controls (2.4% v. 0%),58 and have also been identified in sporadic cases.59 A whole genome sequencing study detected a relatively high number of rare deleterious ATM variants in familial pancreatic cancer cases, among other familial pancreatic cancer genes.60 Furthermore, somatic biallelic inactivation of ATM is found more frequently in tumors from familial pancreatic cancer cases than in sporadic controls.61 One study estimated a RR of 2.41 (95% CI, 0.34–17.1) for pancreas cancer in heterozygous ATM mutation carriers.62
CHEK2 (Gene ID: 11200) codes for a protein kinase that functions in response to DNA damage and interacts with several proteins including p53. It is also a contributing factor to Li-Fraumeni syndrome (OMIM: 609265). CHEK2 was initially reported as a multi-organ cancer susceptibility gene associated with breast, prostate, colon and pancreas cancer, with low-to-moderate penetrance.63 More evidence for significant associations between CHEK2 and pancreatic cancer have been identified, but larger studies are needed to fully elucidate interactions and risks associated with CHEK2 mutations.64,65 Furthermore, limitations in our understanding and its low-to-moderate penetrance make interpretation and risk counseling difficult when CHEK2 mutations are identified in isolation.
CDKN2A codes for several proteins that regulate cell growth and division, including p16 (INK4A) and p14 (ARF). The p14 protein protects p53 from degradation.66 Familial atypical multiple mole melanoma (FAMMM, OMIM: 155601 & 606719) has been associated with CDKN2A mutations, but with reduced penetrance and variable expressivity. FAMMM is typically characterized by multiple atypical nevi, melanoma and increased risk for internal malignancies, especially pancreatic cancer (13–22 fold in FAMMM, and 38-fold for CDKN2A FAMMM).67 A combined study spanning three continents reported a significant association between pancreatic cancer and CDKN2A mutations in North America (P = 0.02) and Europe (P < 0.001).68 A large cohort study also identified deleterious CDKN2A mutations in 13 pancreatic cancer cases with a positive familial history out of 727 unrelated probands.69 Still, screening for CDKN2A mutations is controversial due gaps in our knowledge of CDKN2A genotype-phenotype relationships.
STK11 (Gene ID: 6794) codes for a kinase that regulates AMP-activated protein kinase (AMPK) family members, which function in a variety of processes, including cell growth. Mutations in STK11 cause Peutz-Jeghers syndrome (PJS, OMIM: 175200), an autosomal dominant disorder characterized by gastrointestinal hamartomatous polyps, mucocutaneous pigmentation, and elevated cancer risks (gastrointestinal, breast, colon). Somatic loss of STK11 heterozygosity has been observed in carriers that develop pancreatic cancer, consistent with the ‘two-hit hypothesis’ for the role of tumor suppressors in cancer initiation.70 One report identified a RR of 139.7 (95% CI, 61.1–276.4) in Italian patients with PJS.71 Mutations in STK11 are rare, and over 340 mutations associated with PJS have been identified. Therefore, knowledge is limited by small sample sizes for individual variants.
DNA Repair – BRCA1, BRCA2, PALB2, FA genes, and MMR genes
DNA repair refers to a critical set of processes that recognize and repair DNA damage to maintain genome integrity against endogenous (replication errors, reactive oxygen species) and exogenous (radiation, mutagens, etc.) sources. Cells can acquire up to 1 million DNA lesions per day.72 Patients with inherited errors of DNA repair are particularly susceptible to acquired mutations and development of cancer. Hereditary Breast and Ovarian Cancer (HBOC, OMIM: 604370 & 612555) syndrome is the most well studied high-risk cancer syndrome caused by mutations that disrupt DNA. Other hereditary diseases of impaired DNA repair include Fanconi anemia (FA, see OMIM: 227650) and Lynch syndrome (OMIM: 120435).
HBOC is characterized by a familial pattern of multiple and early-onset breast and ovarian cancers, though patients also have increased risk for additional cancers such as pancreas, prostate, melanoma and brain. HBOC is caused by mutations in BRCA1 (Gene ID: 672) and BRCA2 (Gene ID: 675), which code for proteins that function in homologous recombination mediated double strand break (DSB) repair.73,74 BRCA1 also functions in DNA damage signaling, chromatin remodeling, and transcriptional regulation.73 Thompson et al observed a RR of 2.26 (95% CI, 1.26–4.06) in BRCA1 carriers.75 However, other studies failed to identify any significant association between BRCA1 mutations and pancreatic cancer, suggesting a low penetrance for pancreatic malignancy.76,77 In contrast, BRCA2 mutations have been established as the most common genetic cause of familial pancreatic cancer (RR, 3.51–4.1)76,78 and are estimated to account for up to 19% of families.69,79–81 Many truncating variants have been identified by sequencing, and several founder mutations such as BRCA1 185delAG, BRCA1 5382insC, and BRCA2 6174delT are found in familial pancreatic cancer families of Ashkenazi Jewish descent.82–84
Fanconi anemia is a genetically heterogeneous disease characterized by bone marrow failure (leading to aplastic anemia), physical abnormalities, and a high lifetime risk for cancer, particularly acute myeloid leukemia (AML). The cumulative risk has been estimated at 37% for leukemia at age of 29 and 76% for solid tumor by age 45.85 Currently, 21 genes have been identified in the FA pathway – a signaling pathway that responds to interstrand crosslinks.86 The gene product of one of these genes, PALB2 (Gene ID: 79728), affiliates with BRCA1 and BRCA2 during homologous recombination,87 and PALB2 variants have been identified in familial pancreatic cancer families, but with a relatively small prevalence (~3%).69,88 PALB2 carriers have a significantly earlier mean onset of PDAC than non-carriers (51 v. 63 years).89 Inherited variants in other FA genes have also been associated with pancreatic cancer, including FANCA, FANCC and FANCG (Gene ID: 2175, 2176, 2189).90–92
Lynch syndrome (hereditary non-polyposis colorectal cancer; HNPCC) is caused by mutations in the mismatch repair (MMR) genes (MLH1, MSH2, MSH6, PMS2, Gene ID: 4292, 4436, 2956, 5395). Deletions at the 3’ end of the non-MMR gene, EPCAM (Gene ID: 4072), silence downstream MSH2, disrupting mismatch repair to cause Lynch syndrome.93 Cancer arises following the acquisition of a second somatic mutation (two-hits), which results in defective DNA repair and microsatellite instability. Patients have a high risk for colorectal cancer (22–74% in MLH1/MSH2 carriers),94 as well as an increased risk for extra colorectal cancers including pancreatic cancer (9–11 fold) and endometrial cancer.95,96 One study found that the majority of pancreatic cancers are diagnosed <60 years in HNPCC families, however, this finding needs to be replicated.97
Nijmegen breakage syndrome (OMIM 251260), prevalently identified with Slavic founder mutation 657del5 (c.657_661delACAAA) in NBN (Gene ID: 4683), is a rare autosomal recessive disease predisposed to multiple malignancies, especially lymphomas.98 It is not yet a well-established pancreatic cancer risk gene, but a recent study in the Czech Republic reported a significant PDAC risk for this founder mutation (OR, 9.7; 95% CI, 1.9–50.2).99 No other risk variant has been reported.
Cell Mobility and Adhesion – PALLD, APC
Cell mobility and adhesion genes play a pivotal role in the late malignant transformation, influencing the adhesion, invasion and migration ability of cells, especially cancer stem cells. The APC gene (Gene ID: 324) codes for a tumor suppressor that associates with other proteins to control cell proliferation (Wnt pathway agonist), stabilize microtubules, and mediate cell migration and adhesion.100 Mutations in APC are a key contributor to colorectal cancer development and progression, and are found in >80% of sporadic colorectal tumors.101 Pathogenic variants in APC cause Familial adenomatous polyposis (FAP, OMIM: 175100), a colon cancer syndrome characterized by development of hundreds to thousands of colorectal adenomas, typically by late adolescence, and a 100% risk for colon cancer without intervention. These patients also have an elevated risk for pancreatic cancer (RR, 4.46; 95% CI, 1.2–11.4),102,103 but the risk appears to vary by mutation type and sex.104,105
PALLD (Gene ID: 23022) codes for Palladin, a crucial component of the actin cytoskeleton that mediates cell morphology, adhesion and contraction. Though PALLD has been found to be associated with familial pancreatic cancer in one large family,106 the association remains controversial and unreplicated over the past decade.107–111 Still, expression and functional studies support a role for palladin in tumor invasion and metastasis through its overexpression in the non-neoplastic stroma of pancreatic ductal adenocarcinoma.112–114
Overexpression of the epithelial cell adhesion molecule (EPCAM; mentioned above as a cause of lynch syndrome) has been identified in >50% of pancreas tumors and found to be correlated with poorer patient outcomes.115 However, variants in EPCAM have not emerged in association studies with pancreatic cancer, and it is likely to be a poor predictor of pancreatic cancer.
Targeted Treatment of Pancreatic Cancer
Taking this pathway-level view of genetic and other risk factors facilitates a comprehensive understanding of both single and multifactorial risk for pancreatic cancer. Such a perspective is already utilized in the selection of targeted therapies for pathway-specific perturbations in pancreatic cancer. For example, PARPi (poly ADP-ribose polymerase inhibitors) and cytotoxic agents such as cisplatin, gemcitabine and mitomycin C have been implemented as first-line therapies for patients that harbor pathogenic mutations in DNA damage repair pathways, including the ATM, BRCA1, BRCA2, PALB2 and Fanconi anemia genes.116–118 Multiple clinical trials for pancreatic cancer with germline mutations have investigated PARP inhibitors, including Olaparib (ClinicalTrials.gov Identifier: NCT01078662, NCT02184195)119, Veliparib (NCT01585805, NCT02890355)120 and Rucaparib (NCT02042378, NCT03140670)121. Olaparib has reported a promising overall survival for patients with BRCA1/2 mutations.119 Immunotherapy, including anti-PD-1 (anti-Programmed cell Death protein 1) and anti-PD-L1 (PD ligand 1) alone or in combination with anti-CTLA4 (cytotoxic T-lymphocyte Antigen 4), have been administered in trials for patients with different mismatch repair deficiency (MLH1, MSH2, MSH6, PMS2, EPCAM) cancers (including pancreas).122 This approach is based on a higher immune burden of mutation-associated neoantigens.123 Wee-1 inhibitors and APR-246, targeting mutant TP53, have also shown positive results in cancer trials.124,125 Since loss of STK11 results in activated mTOR (mammalian target of rapamycin), mTOR inhibitors may be effective against pancreatic cancer in Peutz-Jeghers syndrome patients.126 CDK4/6 inhibitors targeting loss of CDKN2A,127 and Wnt/β-catenin signaling inhibitors for APC proved to be effective in pancreatic cancer cell lines or mouse models,128–130 both of which are being evaluated in clinical trials (NCT03065062; NCT01351103). The importance of germline variants is that every cell in the body contains the variants, not just a limited population of tumor subclones. As predicted by this perspective, the effectiveness of targeted cancer treatments appears to be more effective when targeting germline variants than tumor variants.
CONCLUSION
The complex genetic basis of pancreatic cancer is evident - many genes with suspected variants have been identified and reported across years of meticulous research. However, no single gene has emerged as a primary contributor to pancreatic cancer. Gene panels are well recognized as a useful tool to improve risk assessment, but larger panels are complicated by trade-offs including variants of uncertain significance (VUS) and result ambiguity.131,132 Still, the interpretation of germline genetic data within known cancer risk genes is important now and of clear benefit for many patients with and at high-risk of pancreatic cancer.
The well-curated information presented here can help providers with the clinical interpretation of variants on genetic testing and provides a framework for examining the effects of multiple perturbations on a pathway-specific level. Continued recognition and definition of additional genes and their functional placement in models of PDAC oncogenesis will facilitate rapid application of research data to refined patient management.
In the area of complex disorders such as pancreatic cancer, two themes are emerging. First, large amounts of data must be analyzed and sorted within defined systems and models.133–135 Second, this information must be integrated into easy to understand decision support tools at the point of care, and with much more patient partnership and feedback than ever before.136 New tools that link health records with clinical and research databases to provide continually updated information are critical because physicians cannot comprehend or calculate the meaning of all data that is important for patient management.136 These are the requirements for precision medicine, and they are coming into reach.
Supplementary Material
Acknowledgments
Funding:
Financial support: China Scholarship Council (WZ), NIH DK063922 (CAS), Consortium for the Study of Pancreatitis: Pittsburgh Clinical Center, 1U01DK108306 (REB, DCW) and Wayne Fusaro Pancreatic Cancer Research Fund (DCW).
Footnotes
Disclosure:
The authors declare no conflicts of interest related to the manuscript.
References
- 1.Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975–2013. Available at: https://seer.cancer.gov/statfacts/html/pancreas.html. Accessed December 3, 2016.
- 2.Andersen DK, Andren-Sandberg Å, Duell EJ, et al. Pancreatitis-diabetes-pancreatic cancer: summary of an NIDDK-NCI workshop. Pancreas. 2013;42:1227–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Whitcomb DC, Shelton CA, Brand RE. Genetics and Genetic Testing in Pancreatic Cancer. Gastroenterology. 2015;149:1252–1264.e4. [DOI] [PubMed] [Google Scholar]
- 4.Solomon S, Das S, Brand R, et al. Inherited pancreatic cancer syndromes. Cancer J. 2012;18(6):485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Di Marco M, Astolfi A, Grassi E, et al. Characterization of pancreatic ductal adenocarcinoma using whole transcriptome sequencing and copy number analysis by single-nucleotide polymorphism array. Mol Med Report. 2015;12:7479–7484. [DOI] [PubMed] [Google Scholar]
- 6.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McWilliams RR, Rabe KG, Olswold C, et al. Risk of malignancy in first-degree relatives of patients with pancreatic carcinoma. Cancer. 2005;104:388–394. [DOI] [PubMed] [Google Scholar]
- 10.Molina-Montes E, Gomez-Rubio P, Márquez M, et al. Risk of pancreatic cancer associated with family history of cancer and other medical conditions by accounting for smoking among relatives. Int J Epidemiol. 2018;47:473–483 [DOI] [PubMed] [Google Scholar]
- 11.Lowenfels AB, Maisonneuve P, DiMagno EP, et al. Hereditary pancreatitis and the risk of pancreatic cancer. International Hereditary Pancreatitis Study Group. J Natl Cancer Inst. 1997;89:442–446. [DOI] [PubMed] [Google Scholar]
- 12.Lowenfels AB, Maisonneuve P, Whitcomb DC, et al. Cigarette smoking as a risk factor for pancreatic cancer in patients with hereditary pancreatitis. JAMA. 2001;286:169–170. [DOI] [PubMed] [Google Scholar]
- 13.Shelton CA, Umapathy C, Stello K, et al. Hereditary Pancreatitis in the United States: Survival and Rates of Pancreatic Cancer. (in review). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hingorani SR, Petricoin EF, Maitra A, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. [DOI] [PubMed] [Google Scholar]
- 15.Habbe N, Shi G, Meguid RA, et al. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc Natl Acad Sci U S A. 2008;105:18913–18918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wagner M, Greten FR, Weber CK, et al. A murine tumor progression model for pancreatic cancer recapitulating the genetic alterations of the human disease. Genes Dev. 2001;15:286–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aguirre AJ, Bardeesy N, Sinha M, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17:3112–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosenfeldt MT, O’Prey J, Morton JP, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504:296–300. [DOI] [PubMed] [Google Scholar]
- 19.Murtaugh LC. Pathogenesis of pancreatic cancer: lessons from animal models. Toxicol Pathol. 2014;42:217–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Waddell N, Pajic M, Patch A-M, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lowenfels AB, Maisonneuve P, Cavallini G, et al. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N Engl J Med. 1993;328:1433–1437. [DOI] [PubMed] [Google Scholar]
- 22.Mantovani A, Allavena P, Sica A, et al. Cancer-related inflammation. Nature. 2008;454:436–444. [DOI] [PubMed] [Google Scholar]
- 23.Medzhitov R Origin and physiological roles of inflammation. Nature. 2008;454:428–435. [DOI] [PubMed] [Google Scholar]
- 24.Löhr M, Klöppel G, Maisonneuve P, et al. Frequency of K-ras mutations in pancreatic intraductal neoplasias associated with pancreatic ductal adenocarcinoma and chronic pancreatitis: a meta-analysis. Neoplasia. 2005;7:17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kirkegård J, Cronin-Fenton D, Heide-Jørgensen U, et al. Acute Pancreatitis and Pancreatic Cancer Risk: A Nationwide Matched-cohort Study in Denmark. Gastroenterology. 2018;154:1729–1736. [DOI] [PubMed] [Google Scholar]
- 26.Schubert S, Traub F, Brakensiek K, et al. CFTR, SPINK1, PRSS1, and CTRC mutations are not associated with pancreatic cancer in German patients. Pancreas. 2014;43:1078–1082. [DOI] [PubMed] [Google Scholar]
- 27.Howes N, Lerch MM, Greenhalf W, et al. Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clin Gastroenterol Hepatol. 2004;2:252–261. [DOI] [PubMed] [Google Scholar]
- 28.Rebours V, Boutron-Ruault M-C, Schnee M, et al. Risk of pancreatic adenocarcinoma in patients with hereditary pancreatitis: a national exhaustive series. Am J Gastroenterol. 2008;103:111–119. [DOI] [PubMed] [Google Scholar]
- 29.Raimondi S, Lowenfels AB, Morselli-Labate AM, et al. Pancreatic cancer in chronic pancreatitis; aetiology, incidence, and early detection. Best Pract Res Clin Gastroenterol. 2010;24:349–358. [DOI] [PubMed] [Google Scholar]
- 30.Witt H, Luck W, Hennies HC, et al. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat Genet. 2000;25:213–216. [DOI] [PubMed] [Google Scholar]
- 31.Pfützer RH, Barmada MM, Brunskill AP, et al. SPINK1/PSTI polymorphisms act as disease modifiers in familial and idiopathic chronic pancreatitis. Gastroenterology. 2000;119:615–623. [DOI] [PubMed] [Google Scholar]
- 32.Masamune A, Mizutamari H, Kume K, et al. Hereditary pancreatitis as the premalignant disease: a Japanese case of pancreatic cancer involving the SPINK1 gene mutation N34S. Pancreas. 2004;28:305–310. [DOI] [PubMed] [Google Scholar]
- 33.Lempinen M, Paju A, Kemppainen E, et al. Mutations N34S and P55S of the SPINK1 gene in patients with chronic pancreatitis or pancreatic cancer and in healthy subjects: a report from Finland. Scand J Gastroenterol. 2005;40:225–230. [DOI] [PubMed] [Google Scholar]
- 34.Midha S, Sreenivas V, Kabra M, et al. Genetically determined chronic pancreatitis but not alcoholic pancreatitis is a strong risk factor for pancreatic cancer. Pancreas. 2016;45:1478–1484. [DOI] [PubMed] [Google Scholar]
- 35.LaRusch J, Jung J, General IJ, et al. Mechanisms of CFTR functional variants that impair regulated bicarbonate permeation and increase risk for pancreatitis but not for cystic fibrosis. PLoS Genet. 2014;10:e1004376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weiss FU, Simon P, Bogdanova N, et al. Complete cystic fibrosis transmembrane conductance regulator gene sequencing in patients with idiopathic chronic pancreatitis and controls. Gut. 2005;54:1456–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cohn JA, Neoptolemos JP, Feng J, et al. Increased risk of idiopathic chronic pancreatitis in cystic fibrosis carriers. Hum Mutat. 2005;26:303–307. [DOI] [PubMed] [Google Scholar]
- 38.Schneider A, Larusch J, Sun X, et al. Combined bicarbonate conductance-impairing variants in CFTR and SPINK1 variants are associated with chronic pancreatitis in patients without cystic fibrosis. Gastroenterology. 2011;140:162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosendahl J, Landt O, Bernadova J, et al. CFTR, SPINK1, CTRC and PRSS1 variants in chronic pancreatitis: is the role of mutated CFTR overestimated? Gut. 2013;62:582–592. [DOI] [PubMed] [Google Scholar]
- 40.Gelrud A, Sheth S, Banerjee S, et al. Analysis of cystic fibrosis gener product (CFTR) function in patients with pancreas divisum and recurrent acute pancreatitis. Am J Gastroenterol. 2004;99:1557–1562. [DOI] [PubMed] [Google Scholar]
- 41.Bertin C, Pelletier A-L, Vullierme MP, et al. Pancreas divisum is not a cause of pancreatitis by itself but acts as a partner of genetic mutations. Am J Gastroenterol. 2012;107:311–317. [DOI] [PubMed] [Google Scholar]
- 42.Hamoir C, Pepermans X, Piessevaux H, et al. Clinical and morphological characteristics of sporadic genetically determined pancreatitis as compared to idiopathic pancreatitis: higher risk of pancreatic cancer in CFTR variants. Digestion. 2013;87:229–239. [DOI] [PubMed] [Google Scholar]
- 43.McWilliams RR, Petersen GM, Rabe KG, et al. Cystic fibrosis transmembrane conductance regulator (CFTR) gene mutations and risk for pancreatic adenocarcinoma. Cancer. 2010;116:203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miyasaka Y, Nagai E, Yamaguchi H, et al. The role of the DNA damage checkpoint pathway in intraductal papillary mucinous neoplasms of the pancreas. Clin Cancer Res. 2007;13:4371–4377. [DOI] [PubMed] [Google Scholar]
- 45.Koorstra J-BM, Hong S-M, Shi C, et al. Widespread activation of the DNA damage response in human pancreatic intraepithelial neoplasia. Mod Pathol. 2009;22:1439–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zimmermann G, Papke B, Ismail S, et al. Small molecule inhibition of the KRAS-PDEδ interaction impairs oncogenic KRAS signalling. Nature. 2013;497:638–642. [DOI] [PubMed] [Google Scholar]
- 47.Yang MH, Nickerson S, Kim ET, et al. Regulation of RAS oncogenicity by acetylation. Proc Natl Acad Sci U S A. 2012;109:10843–10848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Serra RW, Fang M, Park SM, et al. A KRAS-directed transcriptional silencing pathway that mediates the CpG island methylator phenotype. elife. 2014;3:e02313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.di Magliano MP, Logsdon CD. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology. 2013;144:1220–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Logsdon CD, Lu W. The significance of ras activity in pancreatic cancer initiation. Int J Biol Sci. 2016;12:338–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bournet B, Muscari F, Buscail C, et al. KRAS G12D mutation subtype is A prognostic factor for advanced pancreatic adenocarcinoma. Clin Transl Gastroenterol. 2016;7:e157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Daniluk J, Liu Y, Deng D, et al. An NF-κB pathway-mediated positive feedback loop amplifies Ras activity to pathological levels in mice. J Clin Invest. 2012;122:1519–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morris JP, Wang SC, Hebrok M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat Rev Cancer. 2010;10:683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kanda M, Matthaei H, Wu J, et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology. 2012;142:730–733.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lustbader ED, Williams WR, Bondy ML, et al. Segregation analysis of cancer in families of childhood soft-tissue-sarcoma patients. Am J Hum Genet. 1992;51:344–356. [PMC free article] [PubMed] [Google Scholar]
- 56.Ruijs MWG, Verhoef S, Rookus MA, et al. TP53 germline mutation testing in 180 families suspected of Li-Fraumeni syndrome: mutation detection rate and relative frequency of cancers in different familial phenotypes. J Med Genet. 2010;47:421–428. [DOI] [PubMed] [Google Scholar]
- 57.Awasthi P, Foiani M, Kumar A. ATM and ATR signaling at a glance. J Cell Sci. 2015;128:4255–4262. [DOI] [PubMed] [Google Scholar]
- 58.Roberts NJ, Jiao Y, Yu J, et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov. 2012;2:41–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shindo K, Yu J, Suenaga M, et al. Deleterious germline mutations in patients with apparently sporadic pancreatic adenocarcinoma. J Clin Oncol. 2017;35:3382–3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Roberts NJ, Norris AL, Petersen GM, et al. Whole genome sequencing defines the genetic heterogeneity of familial pancreatic cancer. Cancer Discov. 2016;6:166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim H, Saka B, Knight S, et al. Having pancreatic cancer with tumoral loss of ATM and normal TP53 protein expression is associated with a poorer prognosis. Clin Cancer Res. 2014;20:1865–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thompson D, Duedal S, Kirner J, et al. Cancer risks and mortality in heterozygous ATM mutation carriers. J Natl Cancer Inst. 2005;97:813–822. [DOI] [PubMed] [Google Scholar]
- 63.Cybulski C, Górski B, Huzarski T, et al. CHEK2 is a multiorgan cancer susceptibility gene. Am J Hum Genet. 2004;75:1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mohelnikova-Duchonova B, Havranek O, Hlavata I, et al. CHEK2 gene alterations in the forkhead-associated domain, 1100delC and del5395 do not modify the risk of sporadic pancreatic cancer. Cancer Epidemiol. 2010;34:656–658. [DOI] [PubMed] [Google Scholar]
- 65.Lener MR, Scott RJ, Kluźniak W, et al. Do founder mutations characteristic of some cancer sites also predispose to pancreatic cancer? Int J Cancer. 2016;139:601–606. [DOI] [PubMed] [Google Scholar]
- 66.Saiki Y, Horii A. Molecular pathology of pancreatic cancer. Pathol Int. 2014;64:10–19. [DOI] [PubMed] [Google Scholar]
- 67.Soura E, Eliades PJ, Shannon K, et al. Hereditary melanoma: Update on syndromes and management: Genetics of familial atypical multiple mole melanoma syndrome. J Am Acad Dermatol. 2016;74:395–407; quiz 408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Goldstein AM, Chan M, Harland M, et al. Features associated with germline CDKN2A mutations: a GenoMEL study of melanoma-prone families from three continents. J Med Genet. 2007;44:99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhen DB, Rabe KG, Gallinger S, et al. BRCA1, BRCA2, PALB2, and CDKN2A mutations in familial pancreatic cancer: a PACGENE study. Genet Med. 2015;17:569–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Su GH, Hruban RH, Bansal RK, et al. Germline and somatic mutations of the STK11/LKB1 Peutz-Jeghers gene in pancreatic and biliary cancers. Am J Pathol. 1999;154:1835–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Resta N, Pierannunzio D, Lenato GM, et al. Cancer risk associated with STK11/LKB1 germline mutations in Peutz-Jeghers syndrome patients: results of an Italian multicenter study. Dig Liver Dis. 2013;45:606–611. [DOI] [PubMed] [Google Scholar]
- 72.Lodish H, ed. Molecular Biology of the Cell. 5th ed. New York: Freeman; 2004. [Google Scholar]
- 73.Savage KI, Harkin DP. BRCA1, a “complex” protein involved in the maintenance of genomic stability. FEBS J. 2015;282:630–646. [DOI] [PubMed] [Google Scholar]
- 74.Fradet-Turcotte A, Sitz J, Grapton D, et al. BRCA2 functions: from DNA repair to replication fork stabilization. Endocr Relat Cancer. 2016;23:T1–T17. [DOI] [PubMed] [Google Scholar]
- 75.Thompson D, Easton DF, Breast Cancer Linkage Consortium. Cancer Incidence in BRCA1 mutation carriers. J Natl Cancer Inst. 2002;94:1358–1365. [DOI] [PubMed] [Google Scholar]
- 76.Moran A, O’Hara C, Khan S, et al. Risk of cancer other than breast or ovarian in individuals with BRCA1 and BRCA2 mutations. Fam Cancer. 2012;11:235–242. [DOI] [PubMed] [Google Scholar]
- 77.Mersch J, Jackson MA, Park M, et al. Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer. 2015;121:269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Breast Cancer Linkage Consortium. Cancer risks in BRCA2 mutation carriers. J Natl Cancer Inst. 1999;91:1310–1316. [DOI] [PubMed] [Google Scholar]
- 79.Couch FJ, Johnson MR, Rabe KG, et al. The prevalence of BRCA2 mutations in familial pancreatic cancer. Cancer Epidemiol Biomarkers Prev. 2007;16:342–346. [DOI] [PubMed] [Google Scholar]
- 80.Hahn SA, Greenhalf B, Ellis I, et al. BRCA2 germline mutations in familial pancreatic carcinoma. J Natl Cancer Inst. 2003;95:214–221. [DOI] [PubMed] [Google Scholar]
- 81.Murphy KM, Brune KA, Griffin C, et al. Evaluation of candidate genes MAP2K4, MADH4, ACVR1B, and BRCA2 in familial pancreatic cancer: deleterious BRCA2 mutations in 17%. Cancer Res. 2002;62:3789–3793. [PubMed] [Google Scholar]
- 82.Stadler ZK, Salo-Mullen E, Patil SM, et al. Prevalence of BRCA1 and BRCA2 mutations in Ashkenazi Jewish families with breast and pancreatic cancer. Cancer. 2012;118:493–499. [DOI] [PubMed] [Google Scholar]
- 83.Ferrone CR, Levine DA, Tang LH, et al. BRCA germline mutations in Jewish patients with pancreatic adenocarcinoma. J Clin Oncol. 2009;27:433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Holter S, Borgida A, Dodd A, et al. Germline BRCA Mutations in a Large Clinic-Based Cohort of Patients With Pancreatic Adenocarcinoma. J Clin Oncol. 2015;33:3124–3129. [DOI] [PubMed] [Google Scholar]
- 85.Alter BP. Cancer in Fanconi anemia, 1927–2001. Cancer. 2003;97:425–440. [DOI] [PubMed] [Google Scholar]
- 86.Mamrak NE, Shimamura A, Howlett NG. Recent discoveries in the molecular pathogenesis of the inherited bone marrow failure syndrome Fanconi anemia. Blood Rev. 2017;31:93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Park J-Y, Zhang F, Andreassen PR. PALB2: the hub of a network of tumor suppressors involved in DNA damage responses. Biochim Biophys Acta. 2014;1846:263–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Slater EP, Langer P, Niemczyk E, et al. PALB2 mutations in European familial pancreatic cancer families. Clin Genet. 2010;78:490–494. [DOI] [PubMed] [Google Scholar]
- 89.Borecka M, Zemankova P, Vocka M, et al. Mutation analysis of the PALB2 gene in unselected pancreatic cancer patients in the Czech Republic. Cancer Genet. 2016;209:199–204. [DOI] [PubMed] [Google Scholar]
- 90.van der Heijden MS, Yeo CJ, Hruban RH, et al. Fanconi anemia gene mutations in young-onset pancreatic cancer. Cancer Res. 2003;63:2585–2588. [PubMed] [Google Scholar]
- 91.Rogers CD, van der Heijden MS, Brune K, et al. The genetics of FANCC and FANCG in familial pancreatic cancer. Cancer Biol Ther. 2004;3:167–169. [DOI] [PubMed] [Google Scholar]
- 92.Rogers CD, Couch FJ, Brune K, et al. Genetics of the FANCA gene in familial pancreatic cancer. J Med Genet. 2004;41:e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kempers MJE, Kuiper RP, Ockeloen CW, et al. Risk of colorectal and endometrial cancers in EPCAM deletion-positive Lynch syndrome: a cohort study. Lancet Oncol. 2011;12:49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Syngal S, Brand RE, Church JM, et al. ACG clinical guideline: Genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol. 2015;110:223–62; quiz 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kastrinos F, Mukherjee B, Tayob N, et al. Risk of pancreatic cancer in families with Lynch syndrome. JAMA. 2009;302:1790–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Win AK, Young JP, Lindor NM, et al. Colorectal and other cancer risks for carriers and noncarriers from families with a DNA mismatch repair gene mutation: a prospective cohort study. J Clin Oncol. 2012;30:958–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Geary J, Sasieni P, Houlston R, et al. Gene-related cancer spectrum in families with hereditary non-polyposis colorectal cancer (HNPCC). Fam Cancer. 2008;7:163–172. [DOI] [PubMed] [Google Scholar]
- 98.Chrzanowska KH, Gregorek H, Dembowska-Bagińska B, et al. Nijmegen breakage syndrome (NBS). Orphanet J Rare Dis. 2012;7:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Borecka M, Zemankova P, Lhota F, et al. The c.657del5 variant in the NBN gene predisposes to pancreatic cancer. Gene. 2016;587:169–172. [DOI] [PubMed] [Google Scholar]
- 100.Zhang L, Shay JW. Multiple roles of APC and its therapeutic implications in colorectal cancer. J Natl Cancer Inst. 2017;109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rowan AJ, Lamlum H, Ilyas M, et al. APC mutations in sporadic colorectal tumors: A mutational “hotspot” and interdependence of the “two hits”. Proc Natl Acad Sci U S A. 2000;97:3352–3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Grady WM. Genetic testing for high-risk colon cancer patients. Gastroenterology. 2003;124:1574–1594. [DOI] [PubMed] [Google Scholar]
- 103.Giardiello FM, Offerhaus GJ, Lee DH, et al. Increased risk of thyroid and pancreatic carcinoma in familial adenomatous polyposis. Gut. 1993;34:1394–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Leshno A, Shapira S, Liberman E, et al. The APC I1307K allele conveys a significant increased risk for cancer. Int J Cancer. 2016;138:1361–1367. [DOI] [PubMed] [Google Scholar]
- 105.Liberman E, Kraus S, Sagiv E, et al. The APC E1317Q and I1307K polymorphisms in non-colorectal cancers. Biomed Pharmacother. 2007;61:566–569. [DOI] [PubMed] [Google Scholar]
- 106.Pogue-Geile KL, Chen R, Bronner MP, et al. Palladin mutation causes familial pancreatic cancer and suggests a new cancer mechanism. PLoS Med. 2006;3:e516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zogopoulos G, Rothenmund H, Eppel A, et al. The P239S palladin variant does not account for a significant fraction of hereditary or early onset pancreas cancer. Hum Genet. 2007;121:635–637. [DOI] [PubMed] [Google Scholar]
- 108.Slater E, Amrillaeva V, Fendrich V, et al. Palladin mutation causes familial pancreatic cancer: absence in European families. PLoS Med. 2007;4:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Klein AP, Borges M, Griffith M, et al. Absence of deleterious palladin mutations in patients with familial pancreatic cancer. Cancer Epidemiol Biomarkers Prev. 2009;18:1328–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schneider R, Slater EP, Sina M, et al. German national case collection for familial pancreatic cancer (FaPaCa): ten years experience. Fam Cancer. 2011;10:323–330. [DOI] [PubMed] [Google Scholar]
- 111.Ghiorzo P, Fornarini G, Sciallero S, et al. CDKN2A is the main susceptibility gene in Italian pancreatic cancer families. J Med Genet. 2012;49:164–170. [DOI] [PubMed] [Google Scholar]
- 112.Goicoechea SM, García-Mata R, Staub J, et al. Palladin promotes invasion of pancreatic cancer cells by enhancing invadopodia formation in cancer-associated fibroblasts. Oncogene. 2014;33:1265–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cannon AR, Owen MK, Guerrero MS, et al. Palladin expression is a conserved characteristic of the desmoplastic tumor microenvironment and contributes to altered gene expression. Cytoskeleton (Hoboken). 2015;72:402–411. [DOI] [PubMed] [Google Scholar]
- 114.Salaria SN, Illei P, Sharma R, et al. Palladin is overexpressed in the non-neoplastic stroma of infiltrating ductal adenocarcinomas of the pancreas, but is only rarely overexpressed in neoplastic cells. Cancer Biol Ther. 2007;6:324–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fong D, Steurer M, Obrist P, et al. Ep-CAM expression in pancreatic and ampullary carcinomas: frequency and prognostic relevance. J Clin Pathol. 2008;61:31–35. [DOI] [PubMed] [Google Scholar]
- 116.Chiorean EG, Coveler AL. Pancreatic cancer: optimizing treatment options, new, and emerging targeted therapies. Drug Des Devel Ther. 2015;9:3529–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sahin IH, Lowery MA, Stadler ZK, et al. Genomic instability in pancreatic adenocarcinoma: a new step towards precision medicine and novel therapeutic approaches. Expert Rev Gastroenterol Hepatol. 2016;10:893–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhang Y, Yang C, Cheng H, et al. Novel agents for pancreatic ductal adenocarcinoma: emerging therapeutics and future directions. J Hematol Oncol. 2018;11:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kaufman B, Shapira-Frommer R, Schmutzler RK, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33:244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lowery MA, Kelsen DP, Capanu M, et al. Phase II trial of veliparib in patients with previously treated BRCA-mutated pancreas ductal adenocarcinoma. Eur J Cancer. 2018;89:19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Domchek SM, Hendifar AE, McWilliams RR, et al. RUCAPANC: An open-label, phase 2 trial of the PARP inhibitor rucaparib in patients (pts) with pancreatic cancer (PC) and a known deleterious germline or somatic BRCA mutation. J Clin Oncol. 2016;34:4110.27863191 [Google Scholar]
- 122.Diaz LA, Marabelle A, Delord J-P, et al. Pembrolizumab therapy for microsatellite instability high (MSI-H) colorectal cancer (CRC) and non-CRC. J Clin Oncol. 2017;35:3071. [Google Scholar]
- 123.Gubin MM, Zhang X, Schuster H, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Oza AM, Weberpals JI, Provencher DM, et al. An international, biomarker-directed, randomized, phase II trial of AZD1775 plus paclitaxel and carboplatin (P/C) for the treatment of women with platinum-sensitive, TP53-mutant ovarian cancer. J Clin Oncol. 2015;33:5506. [DOI] [PubMed] [Google Scholar]
- 125.Gourley C, Green J, Gabra H, et al. PISARRO: A EUTROC phase Ib study of APR-246 in combination with carboplatin (C) and pegylated liposomal doxorubicin (PLD) in platinum sensitive relapsed high grade serous ovarian cancer (HGSOC). J Clin Oncol. 2016;34:5571. [Google Scholar]
- 126.Klümpen H-J, Queiroz KCS, Spek CA, et al. mTOR inhibitor treatment of pancreatic cancer in a patient With Peutz-Jeghers syndrome. J Clin Oncol. 2011;29:e150–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Franco J, Witkiewicz AK, Knudsen ES. CDK4/6 inhibitors have potent activity in combination with pathway selective therapeutic agents in models of pancreatic cancer. Oncotarget. 2014;5:6512–6525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Pramanik KC, Fofaria NM, Gupta P, et al. Inhibition of β-catenin signaling suppresses pancreatic tumor growth by disrupting nuclear β-catenin/TCF-1 complex: critical role of STAT-3. Oncotarget. 2015;6:11561–11574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chartier C, Raval J, Axelrod F, et al. Therapeutic Targeting of Tumor-Derived R-Spondin Attenuates β-Catenin Signaling and Tumorigenesis in Multiple Cancer Types. Cancer Res. 2016;76:713–723. [DOI] [PubMed] [Google Scholar]
- 130.Chou A, Froio D, Nagrial AM, et al. Tailored first-line and second-line CDK4-targeting treatment combinations in mouse models of pancreatic cancer. Gut. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Slavin TP, Niell-Swiller M, Solomon I, et al. Clinical Application of Multigene Panels: Challenges of Next-Generation Counseling and Cancer Risk Management. Front Oncol. 2015;5:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mets S, Tryon R, Veach PM, et al. Genetic Counselors’ Experiences Regarding Communication of Reproductive Risks with Autosomal Recessive Conditions found on Cancer Panels. J Genet Couns. 2016;25:359–372. [DOI] [PubMed] [Google Scholar]
- 133.Whitcomb DC. What is personalized medicine and what should it replace? Nat Rev Gastroenterol Hepatol. 2012;9:418–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gligorijević V, Malod-Dognin N, Pržulj N. Integrative methods for analyzing big data in precision medicine. Proteomics. 2016;16:741–758. [DOI] [PubMed] [Google Scholar]
- 135.Alyass A, Turcotte M, Meyre D. From big data analysis to personalized medicine for all: challenges and opportunities. BMC Med Genomics. 2015;8:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kohane IS. HEALTH CARE POLICY. Ten things we have to do to achieve precision medicine. Science. 2015;349:37–38. [DOI] [PubMed] [Google Scholar]
- 137.Petersen GM. Familial pancreatic cancer. Semin Oncol. 2016;43:548–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Goral V Pancreatic Cancer: Pathogenesis and Diagnosis. Asian Pac J Cancer Prev. 2015;16:5619–5624. [DOI] [PubMed] [Google Scholar]
- 139.Stoffel EM. Screening in GI cancers: the role of genetics. J Clin Oncol. 2015;33:1721–1728. [DOI] [PubMed] [Google Scholar]
- 140.Ghiorzo P Genetic predisposition to pancreatic cancer. World J Gastroenterol. 2014;20:10778–10789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Reznik R, Hendifar AE, Tuli R. Genetic determinants and potential therapeutic targets for pancreatic adenocarcinoma. Front Physiol. 2014;5:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rustgi AK. Familial pancreatic cancer: genetic advances. Genes Dev. 2014;28:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Fendrich V, Langer P, Bartsch DK. Familial pancreatic cancer--status quo. Int J Colorectal Dis. 2014;29:139–145. [DOI] [PubMed] [Google Scholar]
- 144.Cassidy LD, Liau S-S, Venkitaraman AR. Chromosome instability and carcinogenesis: insights from murine models of human pancreatic cancer associated with BRCA2 inactivation. Mol Oncol. 2014;8:161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bartsch DK, Gress TM, Langer P. Familial pancreatic cancer--current knowledge. Nat Rev Gastroenterol Hepatol. 2012;9:445–453. [DOI] [PubMed] [Google Scholar]
- 146.Pihlak R, Valle JW, McNamara MG. Germline mutations in pancreatic cancer and potential new therapeutic options. Oncotarget. 2017;8:73240–73257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Teo MY, O’Reilly EM. Is it time to split strategies to treat homologous recombinant deficiency in pancreas cancer? J Gastrointest Oncol 2016;7:738–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Brennan GT, Relias V, Saif MW. BRCA and pancreatic cancer. JOP. 2013;14:325–328. [DOI] [PubMed] [Google Scholar]
- 149.Mocci E, Milne RL, Méndez-Villamil EY, et al. Risk of pancreatic cancer in breast cancer families from the breast cancer family registry. Cancer Epidemiol Biomarkers Prev. 2013;22:803–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kim R, Byer J, Saif MW. BRCA and pancreatic cancer: selection of chemotherapy. JOP. 2012;13:180–181. [PubMed] [Google Scholar]
- 151.Brennan GT, Relias V, Saif MW. BRCA and pancreatic cancer. JOP. 2013;14:325–328. [DOI] [PubMed] [Google Scholar]
- 152.Kim R, Byer J, Saif MW. BRCA and pancreatic cancer: selection of chemotherapy. JOP. 2012;13:180–181. [PubMed] [Google Scholar]
- 153.Hu C, Hart SN, Bamlet WR, et al. Prevalence of Pathogenic Mutations in Cancer Predisposition Genes among Pancreatic Cancer Patients. Cancer Epidemiol Biomarkers Prev. 2016;25:207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Yang XR, Rotunno M, Xiao Y, et al. Multiple rare variants in high-risk pancreatic cancer-related genes may increase risk for pancreatic cancer in a subset of patients with and without germline CDKN2A mutations. Hum Genet. 2016;135:1241–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Al-Sukhni W, Rothenmund H, Borgida AE, et al. Germline BRCA1 mutations predispose to pancreatic adenocarcinoma. Hum Genet. 2008;124:271–278. [DOI] [PubMed] [Google Scholar]
- 156.Lal G, Liu G, Schmocker B, et al. Inherited predisposition to pancreatic adenocarcinoma: role of family history and germ-line p16, BRCA1, and BRCA2 mutations. Cancer Res. 2000;60:409–416. [PubMed] [Google Scholar]
- 157.Lener MR, Kashyap A, Kluźniak W, et al. The Prevalence of Founder Mutations Among Individuals from Families with Familial Pancreatic Cancer Syndrome. Cancer Res Treat. 2016;49:430–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lucas AL, Shakya R, Lipsyc MD, et al. High prevalence of BRCA1 and BRCA2 germline mutations with loss of heterozygosity in a series of resected pancreatic adenocarcinoma and other neoplastic lesions. Clin Cancer Res. 2013;19:3396–3403. doi: 10.1158/1078-0432.CCR-12-3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Golan T, Kanji ZS, Epelbaum R, et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br J Cancer. 2014;111:1132–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lowery M, Shah MA, Smyth E, et al. A 67-year-old woman with BRCA 1 mutation associated with pancreatic adenocarcinoma. J Gastrointest Cancer. 2011;42:160–164. [DOI] [PubMed] [Google Scholar]
- 161.Lucas AL, Frado LE, Hwang C, et al. BRCA1 and BRCA2 germline mutations are frequently demonstrated in both high-risk pancreatic cancer screening and pancreatic cancer cohorts. Cancer. 2014;120:1960–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Dagan E Predominant Ashkenazi BRCA1/2 mutations in families with pancreatic cancer. Genet Test. 2008;12:267–271. [DOI] [PubMed] [Google Scholar]
- 163.Ozçelik H, Schmocker B, Di Nicola N, et al. Germline BRCA2 6174delT mutations in Ashkenazi Jewish pancreatic cancer patients. Nat Genet. 1997;16:17–18. [DOI] [PubMed] [Google Scholar]
- 164.Goggins M, Schutte M, Lu J, et al. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res. 1996;56:5360–5364. [PubMed] [Google Scholar]
- 165.Schubert EL, Lee MK, Mefford HC, et al. BRCA2 in American families with four or more cases of breast or ovarian cancer: recurrent and novel mutations, variable expression, penetrance, and the possibility of families whose cancer is not attributable to BRCA1 or BRCA2. Am J Hum Genet. 1997;60:1031–1040. [PMC free article] [PubMed] [Google Scholar]
- 166.Figer A, Irmin L, Geva R, et al. The rate of the 6174delT founder Jewish mutation in BRCA2 in patients with non-colonic gastrointestinal tract tumours in Israel. Br J Cancer. 2001;84(4):478–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.White K, Held KR, Weber BH. A BRCA2 germ-line mutation in familial pancreatic carcinoma. Int J Cancer. 2001;91:742–744. [DOI] [PubMed] [Google Scholar]
- 168.Slater EP, Langer P, Fendrich V, et al. Prevalence of BRCA2 and CDKN2a mutations in German familial pancreatic cancer families. Fam Cancer. 2010;9:335–343. [DOI] [PubMed] [Google Scholar]
- 169.Goldstein AM. Familial melanoma, pancreatic cancer and germline CDKN2A mutations. Hum Mutat. 2004;23:630. [DOI] [PubMed] [Google Scholar]
- 170.McWilliams RR, Wieben ED, Rabe KG, et al. Prevalence of CDKN2A mutations in pancreatic cancer patients: implications for genetic counseling. Eur J Hum Genet. 2011;19:472–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Goldstein AM, Struewing JP, Fraser MC, et al. Prospective risk of cancer in CDKN2A germline mutation carriers. J Med Genet. 2004;41:421–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Vasen HF, Gruis NA, Frants RR, et al. Risk of developing pancreatic cancer in families with familial atypical multiple mole melanoma associated with a specific 19 deletion of p16 (p16-Leiden). Int J Cancer. 2000;87:809–811. [PubMed] [Google Scholar]
- 173.Potjer TP, Kranenburg HE, Bergman W, et al. Prospective risk of cancer and the influence of tobacco use in carriers of the p16-Leiden germline variant. Eur J Hum Genet. 2015;23:711–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Vasen H, Ibrahim I, Ponce CG, et al. Benefit of Surveillance for Pancreatic Cancer in High-Risk Individuals: Outcome of Long-Term Prospective Follow-Up Studies From Three European Expert Centers. J Clin Oncol. 2016;34:2010–2019. [DOI] [PubMed] [Google Scholar]
- 175.Bartsch DK, Sina-Frey M, Lang S, et al. CDKN2A germline mutations in familial pancreatic cancer. Ann Surg. 2002;236:730–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Bartsch DK, Krysewski K, Sina-Frey M, et al. Low frequency of CHEK2 mutations in familial pancreatic cancer. Fam Cancer. 2006;5:305–308. [DOI] [PubMed] [Google Scholar]
- 177.Jones S, Hruban RH, Kamiyama M, et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009;324:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Shimosegawa T, Kume K, Satoh K. Chronic pancreatitis and pancreatic cancer: prediction and mechanism. Clin Gastroenterol Hepatol. 2009;7:S23–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.