

 Novologues KU-32 and KU-596 bind at the C-terminal domain of Hsp90 and exploit a long-range allosteric network to modulate the chaperone cycle.
Novologues KU-32 and KU-596 bind at the C-terminal domain of Hsp90 and exploit a long-range allosteric network to modulate the chaperone cycle.
Abstract
Unique to targeting the C-terminal domain of Hsp90 (C-Hsp90) is the ability to uncouple the cytotoxic and cytoprotective outcomes of Hsp90 modulation. After the identification of novobiocin as a C-Hsp90 interacting ligand a diverse gamut of novologues emerged, from which KU-32 and KU-596 exhibited strong neuroprotective activity. However, further development of these ligands is hampered by the difficulty to obtain structural information on their complexes with Hsp90. Using saturation transfer difference (STD) NMR spectroscopy, we found that the primary binding epitopes of KU-32 and KU596 map at the ring systems of the ligands and specifically the coumarin and biphenyl structures, respectively. Based on both relative and absolute STD effects, we identified KU-596 sites that can be explored to design novel third-generation novologues. In addition, chemical shift perturbations obtained by methyl-TROSY reveal that novologues bind at the cryptic, C-Hsp90 ATP-binding pocket and produce global, long-range structural rearrangements to dimeric Hsp90.
Introduction
Chaperones are ubiquitous molecular machineries assigned the daunting task of maintaining a balanced protein homeostasis (proteostasis) by continuously sampling and assessing the conformational state of polypeptides.1 They do so through a plethora of cellular processes from de novo, cotranslational and posttranslational folding of nascent polypeptides, to conformational maturation during later stages of protein folding, polypeptide disaggregation and protein degradation. Many diseases, including neurodegenerative diseases and several types of cancer are all characterised to some extent by misregulated proteostasis.2,3 Therefore, modulation of chaperones by small molecules has become a very attractive strategy for therapeutic intervention, particularly in cancer and neurodegenerative diseases. The heat shock protein family (Hsp), which includes Hsp100, Hsp90, Hsp70, chaperonins and small heat-shock proteins, together with their cognate cochaperones, comprise the major class of protein chaperone systems.
Among them, the ATP-dependent Hsp90 machinery is a key regulator of pathologies. In cancer, the role of Hsp90 is unique. First, its substrates (clients) are implicated in all hallmarks that lead to a neoplastic state, and although signalling proteins dominate its clientele (e.g. protein kinases and transcription factors) other proteins such as the telomerase, MMPs and survivin that assist in maintaining cell transformation are also regulated by Hsp90. Second, in contrast to normal tissues, where it predominantly exists in a free state, in tumors it is found as part of a multi-chaperone complex where it exhibits significantly enhanced affinity towards small molecules that modulate its essential ATPase activity.4,5 Therefore, Hsp90 inhibition presents a new model in cancer chemotherapy, where a combinatorial effect as opposed to individual target inhibition is achieved in a tumor-selective manner, leading to proteasomal degradation of clients and cytotoxicity.6,7 However, this is associated with an important mechanistic disadvantage. The heat shock factor 1 (Hsf1) is itself an Hsp90 client, and inhibition with “conventional” Hsp90 inhibitors leads to its dissociation from the chaperone and activation of the heat shock response, which is a very effective pro-survival cellular mechanism.8 Evidently, this becomes an advantage when considering suppression of neurodegenerative diseases, which are characterized by accumulation of misfolded proteins and protein aggregates in the cell.9 Indeed, Hsp chaperones can act as neuroprotective agents, as it was shown that their overexpression correlates with decreased aggregate formation in polyQ diseases,10 increased tau association with microtubules,11 and lower levels of misfolded or aggregated α-synuclein.12
At a molecular level, Hsp90 is a homodimer comprised of three domains. The N-terminal domain (N-Hsp90), which contains the ATP binding pocket, the middle domain (M-Hsp90) and the C-terminal domain (C-Hsp90), which mediates dimerization.13,14 During the chaperone cycle, all three domains interact with cochaperones and to some extent with substrates. Original studies with natural products, such as geldanamycin, which is the prototypical Hsp90 inhibitor,15 and radicicol,16 as well as semisynthetic analogues such as tanespimycin (17-AAG),17,18 have led to numerous structure activity relationship efforts that generated several second- and third-generation inhibitors that have been or are currently being evaluated in clinical trials.19,20 Common to all these ligands is the inhibition of Hsp90 by binding at N-Hsp90 in an ATP-competitive manner, resulting in chaperone cycle arrest, client degradation and the undesirable induction of the pro-survival heat shock response.
Alternatively, Hsp90 inhibition can be achieved by targeting C-Hsp90. The natural product novobiocin (Fig. 1) was found to interact with a cryptic ATP-binding site on C-Hsp90 (ref. 21 and 22) and trigger the degradation of oncogenic clients such as Raf-1, mutated p53, v-Src and HER2.23 Since then, it was recognized that C-Hsp90 inhibitors provide an exceptional therapeutic opportunity to uncouple the cytotoxic and neuroprotective outcomes of Hsp90 inhibition. In this respect, it was shown that for the first- and second-generation of novobiocin analogues KU-32 and KU-596 (Fig. 1), the concentration required to induce the heat shock response was three orders of magnitude lower than the concentration required to trigger client degradation.24,25 On the other hand, the novobiocin analogue KU-174 triggered client degradation and exhibited broad cytotoxicity, without inducing the heat shock response.26
Fig. 1. Chemical structure of the C-Hsp90 binding inhibitor, novobiocin, and its analogues KU-174, KU-32 and KU-596. The three distinct fragments that are identified in KU-32 and KU-596, namely, the noviose analogue, the ring system and the amide, are highlighted in green, blue and green boxes, respectively.

The ability of C-Hsp90 inhibitors to tune the functional outcomes of the Hsp90 chaperone cycle and exert differential therapeutic outcomes27 makes them very attractive drug candidates against either different types of cancer or neurologic disorders. However, comprehensive mechanistic understanding of the mode of action of such ligands, as well as further rational SAR studies are hampered by the absence of structural or other experimental data characterizing their interaction with Hsp90. In the present study we used saturation transfer difference (STD) NMR spectroscopy as a tool to obtain molecular insights into the mode of KU-32 and KU-596 binding to Hsp90. We show that the primary binding epitope of the two ligands is localised in the ring systems, and we propose specific sites on KU-596 that can be explored for the design of novologues with optimized binding properties. In addition, the use of methyl-TROSY NMR data acquired with full-length Hsp90 allowed us to expand our knowledge on the molecular mechanism by which these ligands modulate the function of Hsp90. Similar to novobiocin and other C-Hsp90 ligands, KU-32 and KU-596 binding elicits long range allosteric structural rearrangements that are propagated to N-Hsp90.
Results and discussion
As compared to the structure of novobiocin (Fig. 1), the noviose moiety of both KU-32 and KU-596 is not functionalized with a carbamoyl group at the 3′ position, but instead it exists in a 2′,3′-diol form. In KU-596 the coumarin skeleton of novobiocin is substituted by a biphenyl system where the B-ring is fluorinated at the meta position, while both ligands contain an acetamide arm, which is essential in producing a neuroprotective activity.
Both ligands exhibit good solubility properties in buffer solutions and their 1H NMR spectra show little signal overlap and narrow lines (Fig. 2), and therefore the STD approach can be utilized as a tool to characterize their binding epitopes. The STD spectra of KU-32 and KU-596 acquired at a ligand : Hsp90 ratio of 164 show that to some extent all ligand signals are enhanced (Fig. 2), suggesting that the noviose, the ring system and the acetamide arm moieties are all interacting with the C-Hsp90 binding site. However, as the amount of saturation received by a given ligand-proton depends on its distance to the protein, STD intensities can be utilised to report proximity to the binding pocket and thus evaluate the relative contributions of each of the functional groups in ligand binding. The highest STD intensities for KU-32 and KU-596 was observed for HK and HL, respectively, and were set as 100%, relative to which the STD effect of other protons was normalized and classified in four bins (Fig. 3).
Fig. 2. STD-NMR experiments of KU-32 (A) and KU-596 (B). The regular 1H spectrum of each ligand in the presence of Hsp90α is shown in black, while the STD spectra acquired with saturation transfer periods of 0.3 s and 2.0 s are shown in orange and green, respectively. For KU-596 a “negative control” STD spectrum was acquired in the absence of Hsp90, with a saturation transfer period of 2 s (red). STD spectra are 10× amplified. Amplification factors (ASTD) discussed in the text were obtained using a saturation transfer of 2 s.

Fig. 3. Relative STD effects for KU-32 and KU-596. STD effects for KU-32 and KU596 were normalized relative to the STD effect measured for HK and HL, respectively. The values on the top report the STD effect measured for a 2 s saturation time (STD2s), while the values on the bottom report the STD effect at zero saturation time (STD0s), as determined by fitting the ASTDs of a series of experiments with saturation times ranging from 0.1 to 5 s. Four distinct bins (marked with different colours) were used to rank the STD effect observed for each proton.

Considering first the protons of the aromatic system, for KU-596, HM and HK of the B-ring show substantial overlap (at 7.03 ppm) and thus their individual contributions remain uncertain. However, when integrated as a single peak the resulting “overall” STD effect, is close to the maximum STD, observed for HL (98%). This is followed by the STD of HH, on the A-ring, which is 86%. All other protons of the ring system exhibit significant STD effects, and for HJ, HN and HI, these were found to be 73%, 60% and 53%, respectively. For KU-32, the only probe on the B-ring is HK of the lactone, which shows the maximum STD for this ligand, and therefore the relative proximity of the other positions to Hsp90 cannot be evaluated. A direct comparison between the STD effects of A-ring protons HH and HI of KU-596 and KU-32 is possible, as these are found at equivalent positions with the respect to the noviose moiety. The STD values of HH and HI of KU-32 are 55% and 85%, respectively, and fall in different bins as compared to the corresponding positions of KU-596, indicating distinct relative contributions of these positions in Hsp90 binding. On the other hand, HJ in KU-32 is substituted for a methyl group, which shows a high STD signal (44%), suggesting that this position may still provide contacts with Hsp90. Thus, derivatizing the equivalent position in ring-A of the biphenyl scaffold of KU-596 offers an opportunity of improving the interaction with Hsp90.
Considering the acetamide arm of the two ligands it is evident that they show moderate STD effects. In particular, protons HO, HP and HR of KU-596 exhibit STD effects of 30%, 26% and 35%, respectively, while that of HM, of KU-32, is 46%. Nevertheless, comparison of the absolute STD values (Fig. S1†) of the methyl group, which is possible since the two STD series were acquired under identical ligand concentrations and ligand : Hsp90 ratios, reveals that HM of KU-32 receives a significantly higher saturation than the equivalent HR of KU-596. Therefore, the position of the methyl group of KU-32 in the binding pocket as specifically imposed when the acetamide is attached on the lactone ring of the coumarin optimizes the interaction with C-Hsp90.
Finally, as compared to the protons of the ring systems of both KU-596 and KU-32, the protons of the noviose moiety show only a moderate-to-low STD effect, with the highest values obtained for HG and HF (Fig. 3). This is in agreement with previous STD studies performed on novobiocin,28 where the prenylated phenol ring and the coumarin were identified as the primary interaction epitope with Hsp90 and to a lesser extent, the noviose ring. Thus, although noviose was originally identified as a critical structure in providing specificity for the C-Hsp90 binding site, the ring system still makes more intimate contacts with the protein. We note that the lowest STD effect observed for KU-32 is for HD, which is therefore identified as yet another site that potentially can be derivatized to further optimize contacts with Hsp90.
In addition to the proximity of a ligand to the protein, several other factors affect the magnitude of the measured STD effect and thus epitope mapping. These include the saturation time, the residence time of the ligand in the bound state, the longitudinal relaxation rates (R1) (particularly for ligands that exhibit large variations in the R1 values of their proton sites), as well as spin diffusion. The most appropriate approach to minimize biases from these factors is to determine initial STD effects or STD effects at zero saturation time (STD0).29,30 Therefore we compared the STD effects described above, obtained with a saturation time of 2 s (STD2s), to STD0. Saturation built-up was followed by a series of twelve experiments where the saturation times ranged between 0.1–5 s, and STD0 was obtained as the initial slope of the curve (Fig. 3 and S2†). Although the R1 rates of free KU-32 and KU-596 protons were not evaluated in the present study, the results show that overall there is a very good agreement between STD2s and STD0 values (Fig. S2†). However, notable exceptions are protons from the noviose ring, where STD deviations significantly greater than 10% are observed. In particular, STD0 for protons HA1 and HA2 show a ∼50% increase relative to the STD effect measured using a 2 s saturation time for both ligands. In addition, for KU-596, protons HO and HP of the acetamide arm also experience enhanced STD0. Still, only HA1 and HA2 of KU-32, and HA1 of KU-596 are classified in different bins when STD2s and STD0 are considered, suggesting that the two approaches yield the same relative contributions to Hsp90 binding, that is the ring system is involved in closer contacts with the binding site, as compared to the noviose and acetamide moieties.
Although local perturbations, such as the interference with C-Hsp90 dimerization31 are attributed to novobiocin binding, the signature molecular effect observed with C-Hsp90 interacting ligands is the allosterically induced conformational changes occurring throughout Hsp90 that regulate ligand and co-chaperone binding at N-Hsp90.21–23,31,32 This long range crosstalk between N- and C-Hsp90 is also observed for activating allosteric modulators of Hsp90, which bind to C-Hsp90 and drastically improve the inherently low basal ATPase activity of N-Hsp90 by accelerating the rate-limiting N-Hsp90 dimerization.33,34 Therefore, we next sought to investigate whether the KU-32 and KU-596 analogues discussed in here modulate the function of Hsp90 in a similar fashion. Towards this end we used a perdeuterated sample of Hsp90 at which 1H/13C was site-specifically incorporated at the methyl group of Ile residues (δ position) and monitored the interaction with KU-596, using methyl-TROSY.35 This approach has been successfully applied to NMR studies of Hsp90 complexes36–40 and other high molecular weight systems.41–43
The overlay of the 13C-HMQC spectrum of Hsp90 acquired in the absence and presence of KU32 or KU-596 (Fig. 4 and S3†), reveals prominent chemical shift changes for a common set of signals that belong to Ile residues of C-Hsp90 (referred to as “C1” and “C2”). Hence, KU-32 and KU-596 share a common mode of binding, and similarly to novobiocin, they interact with C-Hsp90. When the shifts caused by the two ligands are compared, C1 shifts towards a different direction for KU-596 than for KU-32, while C2 shifts towards the same direction for both ligands. Since the noviose moiety is common between KU-596 and KU-32, the chemical shift change of C2 is attributed to the interaction with the noviose moiety, and the chemical shift change of C1 to the interaction with the ring system and/or the acetamide arm. In addition to these two signals from C-Hsp90, another set of signals from N- and M-Hsp90 experience small (<20 Hz), but measurable chemical shift changes (Fig. S3†). Notably, some of these signals belong to Ile residues that are found: (i) in the vicinity of domain interfaces within a protomer (I214, I361 and I494) or (ii) in the vicinity of the N-/N-Hsp90 interprotomer interface (I110) observed in the “closed” conformation of AMP-PNP bound yeast Hsp90 (ref. 44) (Fig. 4). These chemical shift changes are not a result of ligand binding at the “canonical” ATP binding site, as addition of KU-596 to 15N-labeled N-Hsp90α produces no changes (shift or broadening) to the HSQC spectrum of the isolated domain (Fig. S3†). Therefore, similarly to novobiocin or the activating ligands, KU-32 and KU-596 binding to Hsp90 elicit global conformational changes to the Hsp90 dimer. Since apo-Hsp90α was utilised in the present study, this ligand-induced structural rearrangement may represent a shift to a novel N-Hsp90 dimerized state, which is distinct from the AMP-PNP stabilized state,45 or a redistribution of conformations in the reorientation space sampled by the NM-Hsp90 domains of the two protomers, or may simply reflect an intra-protomer N-/C-Hsp90 communication relayed through M-Hsp90.
Fig. 4. The interaction of Hsp90α with KU-32, KU-596 and AMP-PNP monitored by methyl-TROSY NMR. (A) Isoleucine residues of Hsp90 that experience changes in chemical shift are mapped (grey spheres) on the structure of dimeric yeast Hsp90 (“closed state”). The assignment of C-Hsp90 fragment is not currently available and the two C-Hsp90 methyl groups that shift are referred as C1 and C2. (B) Selected regions of the 1H–13C HMQC spectrum of Hsp90 in the absence (black) and presence of KU-596 (orange), KU-32 (green) or AMP-PNP (red), showing isoleucine signals that experience changes in chemical shift.

To gain further insights to the mode of novologue recognition by Hsp90 we also compared the changes observed by KU-32 and KU-596 to the spectral signature of AMP-PNP binding. Upon addition of AMP-PNP to full-length Hsp90 many N-Hsp90 signals experience large changes in chemical shift (Fig. S3†), which is evidently caused by binding at the “canonical” ATP-binding site of N-Hsp90. Furthermore, in agreement with previous findings that indicated the presence of a “cryptic” ATP-binding site at C-Hsp90,21,22,46 addition of AMP-PNP caused significant shifts to signals C1 and C2 of C-Hsp90, without affecting other C-Hsp90 isoleucine signals. Therefore, as expected on the basis of the proposed competitive binding between novobiocin and ATP,21 we find that the novologues KU-32 and KU-596 interact with the cryptic ATP-binding pocket of C-Hsp90. Intriguingly, the same set of signals from Ile residues located at the vicinity of domain interfaces that were perturbed by KU-32 and KU-596 (I214, I361 and I494) were also shifted by AMP-PNP, suggesting that the two ligands may exploit a common pathway to propagate long-range conformational changes to dimeric Hsp90.
Conclusions
Since the discovery of novobiocin as an Hsp90 inhibitor, several SAR studies have been performed aiming to design potent analogues that produce antiproliferative or prosurvival effects. Through a series of extensive modifications, KU-32 and its analogue KU-596, were developed as novologues with neuroprotective activity. Both ligands are able to improve psychosensory, electrophysiologic, morphologic, and bioenergetic deficits associated with diabetic sensory neuropathy and reverse its pathological consequences.47,48
Despite this success, further development of these ligands is hampered by the difficulty to obtain high-resolution structural information of their complexes with C-Hsp90 or the full-length protein. In the present NMR study, substantial STD effects were measured for all three distinct fragments of KU-32 and KU-596, suggesting that to some extend all interact with Hsp90 and thus both ligands are embraced within the binding cleft. Nevertheless, it is the coumarin moiety of KU-32 and the biphenyl moiety of KU-596 that make the most intimate contacts with the protein and therefore define the major binding epitopes of these ligands. In addition, the STD effects provide key information as which parts of these fragments can be utilized as starting points for further derivatization of KU-596 to create third generation analogues. First, considering that the methyl group attached on the A-ring of the coumarin fragment of KU-32 exhibits a large STD effect and assuming a similar positioning of the two ligands in the complex, substitution of this position may to improve the binding properties of KU-596. Second, although the improved properties of KU-596 as compared to KU-32 are attributed to the substitution of the coumarin for the biphenyl moiety, the contribution of the acetamide arm in providing contacts with Hsp90 is compromised. This is evident from the relative STD effects observed for each of the two ligands but also from the direct comparison of the absolute STD values measured for the methyl group where a 36% drop is observed. Thus, the ethyl derivative does indeed positions the acetamide group two atoms away from ring-A, which is essential in conferring neuroprotective activity to the ligand, but derivatization of positions O or P is expected to decrease mobility and provide new interactions with Hsp90. For both ligands, position D of the noviose ring exhibits the lowest STD effect and thus provides the smallest contribution to the interface. We expect that derivatization of this position will further improve the binding properties of KU-596. Finally, analysis of the CSP data obtained through the use of methyl-TROSY NMR shows that KU-32 and KU-596 binding at the cryptic ATP-binding site of C-Hsp90 brings global structural rearrangements to Hsp90, suggesting that these ligands act as allosteric modulators of the chaperone cycle through either intra- or inter-protomer pathways.
Methods
Synthesis of KU-32 and KU-596
KU-32 was synthesized as previously described.27 Briefly, acetic anhydride (0.02 mL, 0.21 mmol) was added to a solution of aniline (0.05 g, 0.13 mmol) in CH2Cl2 (1 mL) at room temperature. After reaction completion as indicated by thin-layer chromatography, the solution was concentrated and purified via column chromatography (SiO2, 0–5% MeOH in CH2Cl2) to afford the acetylated product as a white amorphous solid (0.05 g, 85% yield). This product was then added to a 10% v/v solution of Et3N in MeOH (1 mL) at room temperature. After reaction completion as indicated by thin-layer chromatography, the solution was concentrated and purified via column chromatography (SiO2, 0–5% MeOH in CH2Cl2) to afford KU-32 as a white amorphous solid (0.03 g, 70% yield). 1H NMR (400 MHz, CDCl3) δ 8.62 (s, 1H), 7.98 (s, 1H), 7.31 (d, J = 8.7 Hz, 1H), 7.18 (d, J = 8.8 Hz, 1H), 5.61 (d, J = 1.7 Hz, 1H), 4.29–4.20 (m, 2H), 3.61 (s, 3H), 3.41–3.34 (m, 1H), 2.58 (d, J = 2.3 Hz, 1H), 2.56 (d, J = 3.2 Hz, 1H), 2.27 (s, 3H), 2.23 (s, 3H), 1.38 (s, 3H), 1.14 (s, 3H). KU-596 was obtained following the procedure described in reference was synthesized as previously described,24 as a colorless amorphous solid (0.03 g, 78% yield over 2 steps). 1H NMR (500 MHz, CDCl3) δ 7.37 (td, J = 8.0, 6.0 Hz, 1H), 7.20 (d, J = 8.5 Hz, 1H), 7.10–7.04 (m, 2H), 7.03 (dd, J = 8.5, 2.7 Hz, 1H), 6.99 (ddd, J = 9.7, 2.4, 1.6 Hz, 1H), 6.88 (d, J = 2.7 Hz, 1H), 5.54 (d, J = 2.3 Hz, 1H), 5.27 (s, 1H), 4.20 (d, J = 8.1 Hz, 1H), 4.16 (s, 1H), 3.59 (s, 1H), 3.33 (d, J = 9.1 Hz, 1H), 3.27 (q, J = 7.1 Hz, 2H), 2.74 (t, J = 7.2 Hz, 2H), 2.61 (s, 1H), 2.57 (s, 1H), 1.87 (s, 3H), 1.36 (s, 3H), 1.21 (s, 3H).
Expression and purification of Hsp90α and N-Hsp90α
Hsp90α was overexpressed from a pET28 vector encoding for a N-terminal His-tag, using BL21(DE3) cells in minimal media. Cells were incubated at 37 °C until an OD600 ∼ 0.6 and were subsequently chilled for 10 minutes in a water/ice bath. Overexpression was induced by the addition of 0.5 mM IPTG at 18 °C for 20 hours. Perdeuterated/CH3-labelled Hsp90 (Ileδ) was overexpressed using the same protocol, using glucose-d7 as the sole carbon source and adding 50 mg L–1 α-ketobutyric acid to the media 40 minutes before induction. Cells were resuspended in 20 mM Tris, pH = 8.0, 500 mM NaCl, 10 mM imidazole, 3 mM DTT, 1 mM PMSF, protease cocktail and 0.1 mg ml–1 lysozyme. After sonication the lysate was clarified by centrifugation and loaded to a Ni2+-affinity column. Unbound proteins were washed with 20 column volumes of lysis buffer and Hsp90 was eluted in the same buffer containing 400 mM imidazole. Fractions were concentrated and loaded to a Superdex 200 26/600 equilibrated with 50 mM Tris, pH = 8.0, 1 M NaCl, 0.5 mM EDTA and 3 mM DTT. Dimeric Hsp90 was further purified through a 10 ml HiTrap Q Sepharose FF using a 50 mM–1 M NaCl gradient in 25 mM Tris, pH = 7.5, 4 mM EDTA and 3 mM DTT.
N-Hsp90α was expressed from a pGEX-4 T3 vector49 in minimal media supplemented with 15N NH4Cl. Cell cultures were allowed to reach an OD600 of 0.5 before inducing with 0.25 mM IPTG, for 5 hours at 37 °C. Cells were collected, resuspended in 50 mM Tris, pH = 8.0, 200 mM NaCl, 1 mM EDTA, 2 mM DTT, and lysed by sonication. The lysate was subsequently clarified by centrifugation and the fusion N-Hsp90 was purified over a GST column and digested overnight at 4 °C by thrombin. N-Hsp90 was purified through a second GST and finally through a Superdex 200 column.
NMR spectroscopy
STD experiments for both KU-32 and KU-596 were run under identical conditions, in 50 mM potassium phosphate buffer, pH = 7.5, 100 mM KCl and 2 mM DTT in 100% D2O. The ligand concentration was 770 μM and the Hsp90α concentration was 4.7 μM. The Agilent dpfgse_satxfer2 pulse sequence was used, with on-resonance irradiation frequencies set at 0.08 ppm and 0.004 ppm respectively for KU-32 and KU-596, respectively, while for both ligands the off-resonance frequency was set at 30 ppm. The same regions of both the on- and off-resonance experiments were integrated to quantify the STD effect, and the results are presented as the amplification factor (ASTD), according to (1):
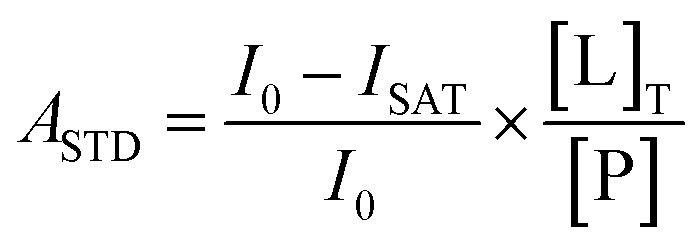 |
1 |
where, I0 is the integral of a signal in the off-resonance spectrum, ISAT is the integral of the same signal in the on-resonance spectrum, [L]T is the ligand concentration, and [P] is the Hsp90α concentration. Reported ASTD values were obtained using a saturation time of 2 s. The amplification factor at 0 s saturation time (A0) was quantified by the method of initial slopes.30 Specifically, saturation build-up curves were obtained by performing a set of 12 experiments with increasing saturation times (0.1, 0.3, 0.5, 0.75, 1.0, 1.25, 1.5, 2.0, 2.5, 3.0, 4.0, and 5.0 s). The rate constant ksat and the calculated STDmax were derived by fitting the curves to (2):
| STD(tsat) = STDmax × (1 – e–ksat×tsat) | 2 |
from which A0 was obtained as STDmax × ksat.
Methyl-TROSY titrations of Hsp90 were acquired in 20 mM deuterated Tris, pH = 7.5, 100 mM NaCl, 0.5 mM EDTA, 1.5 mM DTT in D2O, at a concentration of 100 μM. The same buffer was used for the titration of N-Hsp90, but in 7.5% D2O. Ligand concentration was 0.9, 1.4 and 2.5 mM for KU-32, KU-596 and AMP-PNP. For KU-32 and KU-596, the spectra of free and ligand bound Hsp90 were acquired in the presence of the same amount of deuterated DMSO (1.15%). For AMP-PNP the spectra of free and ligand bound Hsp90 contained the same MgSO4 concentration (5 mM).
The assignment of Hsp90α methyl groups has been reported previously by others.40
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was supported by the US National Institutes of Health grants GM115854 (I. G.) and CA120458 (B. S. J. B.).
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c8md00151k
References
- Hartl F. U., Bracher A., Hayer-Hartl M. Nature. 2011;475:324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- Balch W. E., Morimoto R. I., Dillin A., Kelly J. W. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- Labbadia J., Morimoto R. I. Annu. Rev. Biochem. 2015;84:435–464. doi: 10.1146/annurev-biochem-060614-033955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal A., Thao L., Sensintaffar J., Zhang L., Boehm M. F., Fritz L. C., Burrows F. J. Nature. 2003;425:407–410. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- Moulick K., Ahn J. H., Zong H., Rodina A., Cerchietti L., Gomes DaGama E. M., Caldas-Lopes E., Beebe K., Perna F., Hatzi K., Vu L. P., Zhao X., Zatorska D., Taldone T., Smith-Jones P., Alpaugh M., Gross S. S., Pillarsetty N., Ku T., Lewis J. S., Larson S. M., Levine R., Erdjument-Bromage H., Guzman M. L., Nimer S. D., Melnick A., Neckers L., Chiosis G. Nat. Chem. Biol. 2011;7:818–826. doi: 10.1038/nchembio.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neckers L., Workman P. Clin. Cancer Res. 2012;18:64–76. doi: 10.1158/1078-0432.CCR-11-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman P., Burrows F., Neckers L., Rosen N. Ann. N. Y. Acad. Sci. 2007;1113:202–216. doi: 10.1196/annals.1391.012. [DOI] [PubMed] [Google Scholar]
- Zou J., Guo Y., Guettouche T., Smith D. F., Voellmy R. Cell. 1998;94:471–480. doi: 10.1016/s0092-8674(00)81588-3. [DOI] [PubMed] [Google Scholar]
- Pratt W. B., Gestwicki J. E., Osawa Y., Lieberman A. P. Annu. Rev. Pharmacol. Toxicol. 2015;55:353–371. doi: 10.1146/annurev-pharmtox-010814-124332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonini N. M. Proc. Natl. Acad. Sci. U. S. A. 2002;99(Suppl 4):16407–16411. doi: 10.1073/pnas.152330499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou F., Netzer W. J., Tanemura K., Li F., Hartl F. U., Takashima A., Gouras G. K., Greengard P., Xu H. Proc. Natl. Acad. Sci. U. S. A. 2003;100:721–726. doi: 10.1073/pnas.242720499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klucken J., Shin Y., Masliah E., Hyman B. T., McLean P. J. J. Biol. Chem. 2004;279:25497–25502. doi: 10.1074/jbc.M400255200. [DOI] [PubMed] [Google Scholar]
- Pearl L. H. Biopolymers. 2016;105:594–607. doi: 10.1002/bip.22835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schopf F. H., Biebl M. M., Buchner J. Nat. Rev. Mol. Cell Biol. 2017;18:345–360. doi: 10.1038/nrm.2017.20. [DOI] [PubMed] [Google Scholar]
- Whitesell L., Mimnaugh E. G., De Costa B., Myers C. E., Neckers L. M. Proc. Natl. Acad. Sci. U. S. A. 1994;91:8324–8328. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte T. W., Akinaga S., Soga S., Sullivan W., Stensgard B., Toft D., Neckers L. M. Cell Stress Chaperones. 1998;3:100–108. doi: 10.1379/1466-1268(1998)003<0100:arbttn>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnur R. C., Corman M. L., Gallaschun R. J., Cooper B. A., Dee M. F., Doty J. L., Muzzi M. L., DiOrio C. I., Barbacci E. G., Miller P. E. J. Med. Chem. 1995;38:3813–3820. doi: 10.1021/jm00019a011. [DOI] [PubMed] [Google Scholar]
- Schnur R. C., Corman M. L., Gallaschun R. J., Cooper B. A., Dee M. F., Doty J. L., Muzzi M. L., Moyer J. D., DiOrio C. I., Barbacci E. G. J. Med. Chem. 1995;38:3806–3812. doi: 10.1021/jm00019a010. [DOI] [PubMed] [Google Scholar]
- Chatterjee S., Burns T. F. Int. J. Mol. Sci. 2017;18:1978. doi: 10.3390/ijms18091978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuehlke A. D., Moses M. A., Neckers L. Philos. Trans. R. Soc., B. 2018;373:20160527. doi: 10.1098/rstb.2016.0527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcu M. G., Chadli A., Bouhouche I., Catelli M., Neckers L. M. J. Biol. Chem. 2000;275:37181–37186. doi: 10.1074/jbc.M003701200. [DOI] [PubMed] [Google Scholar]
- Soti C., Racz A., Csermely P. J. Biol. Chem. 2002;277:7066–7075. doi: 10.1074/jbc.M105568200. [DOI] [PubMed] [Google Scholar]
- Marcu M. G., Schulte T. W., Neckers L. J. Natl. Cancer Inst. 2000;92:242–248. doi: 10.1093/jnci/92.3.242. [DOI] [PubMed] [Google Scholar]
- Kusuma B. R., Zhang L., Sundstrom T., Peterson L. B., Dobrowsky R. T., Blagg B. S. J. Med. Chem. 2012;55:5797–5812. doi: 10.1021/jm300544c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y., Ansar S., Michaelis M. L., Blagg B. S. Bioorg. Med. Chem. 2009;17:1709–1715. doi: 10.1016/j.bmc.2008.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskew J. D., Sadikot T., Morales P., Duren A., Dunwiddie I., Swink M., Zhang X., Hembruff S., Donnelly A., Rajewski R. A., Blagg B. S., Manjarrez J. R., Matts R. L., Holzbeierlein J. M., Vielhauer G. A. BMC Cancer. 2011;11:468. doi: 10.1186/1471-2407-11-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S., Liu Y., Garg G., Anyika M., McPherson N. T., Ma J., Dobrowsky R. T., Blagg B. S. ACS Med. Chem. Lett. 2016;7:813–818. doi: 10.1021/acsmedchemlett.6b00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattin S., Panza M., Vasile F., Berni F., Goti G., Tao J. H., Moroni E., Agard D., Colombo G., Bernardi A. Eur. J. Org. Chem. 2016:3349–3364. doi: 10.1002/ejoc.201600420. [DOI] [Google Scholar]
- Angulo J., Nieto P. M. Eur. Biophys. J. 2011;40:1357–1369. doi: 10.1007/s00249-011-0749-5. [DOI] [PubMed] [Google Scholar]
- Mayer M., James T. L. J. Am. Chem. Soc. 2004;126:4453–4460. doi: 10.1021/ja0398870. [DOI] [PubMed] [Google Scholar]
- Allan R. K., Mok D., Ward B. K., Ratajczak T. J. Biol. Chem. 2006;281:7161–7171. doi: 10.1074/jbc.M512406200. [DOI] [PubMed] [Google Scholar]
- Yun B. G., Huang W., Leach N., Hartson S. D., Matts R. L. Biochemistry. 2004;43:8217–8229. doi: 10.1021/bi0497998. [DOI] [PubMed] [Google Scholar]
- D'Annessa I., Sattin S., Tao J., Pennati M., Sanchez-Martin C., Moroni E., Rasola A., Zaffaroni N., Agard D. A., Bernardi A., Colombo G. Chemistry. 2017;23:5188–5192. doi: 10.1002/chem.201700169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattin S., Tao J., Vettoretti G., Moroni E., Pennati M., Lopergolo A., Morelli L., Bugatti A., Zuehlke A., Moses M., Prince T., Kijima T., Beebe K., Rusnati M., Neckers L., Zaffaroni N., Agard D. A., Bernardi A., Colombo G. Chemistry. 2015;21:13598–13608. doi: 10.1002/chem.201502211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollerenshaw J. E., Tugarinov V., Kay L. E. Magn. Reson. Chem. 2003;41:843–852. doi: 10.1002/mrc.1819. [DOI] [PubMed] [Google Scholar]
- Bachman A. B., Keramisanou D., Xu W., Beebe K., Moses M. A., Vasantha Kumar M. V., Gray G., Noor R. E., van der Vaart A., Neckers L., Gelis I. Nat. Commun. 2018;9:265. doi: 10.1038/s41467-017-02711-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagoz G. E., Duarte A. M., Akoury E., Ippel H., Biernat J., Moran Luengo T., Radli M., Didenko T., Nordhues B. A., Veprintsev D. B., Dickey C. A., Mandelkow E., Zweckstetter M., Boelens R., Madl T., Rudiger S. G. Cell. 2014;156:963–974. doi: 10.1016/j.cell.2014.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karagoz G. E., Duarte A. M., Ippel H., Uetrecht C., Sinnige T., van Rosmalen M., Hausmann J., Heck A. J., Boelens R., Rudiger S. G. Proc. Natl. Acad. Sci. U. S. A. 2011;108:580–585. doi: 10.1073/pnas.1011867108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oroz J., Kim J. H., Chang B. J., Zweckstetter M. Nat. Struct. Mol. Biol. 2017;24:407–413. doi: 10.1038/nsmb.3380. [DOI] [PubMed] [Google Scholar]
- Park S. J., Kostic M., Dyson H. J. J. Mol. Biol. 2011;411:158–173. doi: 10.1016/j.jmb.2011.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelis I., Bonvin A. M., Keramisanou D., Koukaki M., Gouridis G., Karamanou S., Economou A., Kalodimos C. G. Cell. 2007;131:756–769. doi: 10.1016/j.cell.2007.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Religa T. L., Sprangers R., Kay L. E. Science. 2010;328:98–102. doi: 10.1126/science.1184991. [DOI] [PubMed] [Google Scholar]
- Saio T., Guan X., Rossi P., Economou A., Kalodimos C. G. Science. 2014;344:1250494. doi: 10.1126/science.1250494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali M. M., Roe S. M., Vaughan C. K., Meyer P., Panaretou B., Piper P. W., Prodromou C., Pearl L. H. Nature. 2006;440:1013–1017. doi: 10.1038/nature04716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid S., Gotz M., Hugel T. ChemPhysChem. 2018;19:1–7. doi: 10.1002/cphc.201800342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnier C., Lafitte D., Tsvetkov P. O., Barbier P., Leclerc-Devin J., Millot J. M., Briand C., Makarov A. A., Catelli M. G., Peyrot V. J. Biol. Chem. 2002;277:12208–12214. doi: 10.1074/jbc.M111874200. [DOI] [PubMed] [Google Scholar]
- Ma J., Farmer K. L., Pan P., Urban M. J., Zhao H., Blagg B. S., Dobrowsky R. T. J. Pharmacol. Exp. Ther. 2014;348:281–292. doi: 10.1124/jpet.113.210435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J., Pan P., Anyika M., Blagg B. S., Dobrowsky R. T. ACS Chem. Neurosci. 2015;6:1637–1648. doi: 10.1021/acschemneuro.5b00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana J., Fulton D., Chen Y., Fairchild T. A., McCabe T. J., Fujita N., Tsuruo T., Sessa W. C. Circ. Res. 2002;90:866–873. doi: 10.1161/01.res.0000016837.26733.be. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.