

Abstract
TRF1 is a critical regulator of telomere length. As such, TRF1 levels are regulated by ubiquitin-dependent proteolysis via an SCF E3 ligase where Fbx4 contributes substrate specification. Here we report the crystal structure of the Fbx4-TRF1 complex at 2.4 Å resolution. Fbx4 contains a unusual substrate-binding domain that adopts a small GTPase fold. Strikingly, this atypical GTPase domain of Fbx4 binds to a globular domain of TRF1 through an intermolecular β sheet, instead of recognizing short peptides/degrons as often seen in other F-box protein-substrate complexes. Importantly, mutations in this interface abrogate Fbx4-dependent TRF1 binding and ubiquitination. Furthermore, the data demonstrate that recognition of TRF1 by SCFFbx4 is regulated by another telomere protein TIN2. Our results reveal an atypical small GTPase domain within Fbx4 as a substrate-binding motif for SCFFbx4 and uncover a mechanism for selective ubiquitination and degradation of TRF1 in telomere homeostasis control.

INTRODUCTION
Telomeres cap the ends of chromosomes and are essential for genome integrity in all eukaryotes (Blackburn, 2001). Telomeres consist of tandem repeat sequences that are synthesized by a special reverse transcriptase called telomerase (Cech, 2004). Telomerase activity is suppressed in somatic cells, where telomere tract shortening is associated with cellular senescence and acts as a molecular clock that regulates cellular and organismal lifespan (Wright and Shay, 1992). In contrast, telomerase activity is constantly maintained in stem cells and most tumor cells (Kim et al., 1994). Telomere length in these telomerase-containing cells does not increase without control, but instead it is tightly regulated within a narrow range of size distribution (Counter et al., 1992). A key regulator of human telomere length is the telomere repeat binding protein TRF1, which negatively regulates telomere elongation by telomerase (van Steensel and de Lange, 1997). Binding along the duplex region of telomeres, TRF1 functions as a measuring device to monitor telomere length and act in cis to regulate access of telomerase to chromosome termini (van Steensel and de Lange, 1997). The amount of TRF1 that is bound to telomeres directly correlates with telomere length; overexpression of TRF1 results in gradual telomere shortening, whereas inhibition of TRF1 binding to telomeres induces a progressive increase in telomere length (van Steensel and de Lange, 1997).
Recent genetic and molecular studies have revealed that telomere-TRF1 association is tightly regulated by a series of sequential post-translational modification events. TRF1 is initially poly-(ADP-ribosyl)ated by tankyrase 1, a multifunctional poly(ADP-ribose) polymerases (Cook et al., 2002; Smith and de Lange, 2000; Smith et al., 1998). ADP-ribosylation of TRF1 by tankyrase 1 releases TRF1 from telomeres (Chang et al., 2003). TRF1 is then polyubiquitinated and degraded through the proteasome pathway (Chang et al., 2003; Lee et al., 2006). Although poly-(ADPribosyl)ation is not required for TRF1 ubiquitination in vitro, the catalytic activity of tankyrase 1 is essential for TRF1 degradation in vivo; that is, TRF1 is ubiquitinated only upon release from telomeres (Chang et al., 2003).
Ubiquitination of TRF1 is mediated by the F-box protein, Fbx4 (Lee et al., 2006). F-box proteins typically function as substrate-specific adaptor subunits of the SCF (Skp1Cul1/Rbx1-F-box protein) ubiquitin E3 ligases, which regulate a broad range of cellular processes in eukaryotes (Petroski and Deshaies, 2005). Besides an F-box domain that binds to Skp1, the F-box proteins contain various substrate interaction domains, based on which, F-box proteins fell into three major classes (Jin et al., 2004). The two largest classes, named Fbw and Fbl, contain WD-40 repeat and leucine-rich repeat domains, respectively (Jin et al., 2004). Fbx4 belongs to the third class – Fbx, and lacks previously identified protein interaction domains outside of the F-box (Figure 1A). Therefore, the molecular basis of how Fbx4 recognizes TRF1 and mediates its degradation remains unknown.
Figure 1. Overview of the Fbx4G-TRF1TRFH Complex Structure.

(A) Domain organization of the Fbx4 and TRF1 polypeptide chains. In Fbx4, the F-box is colored in green and the C-terminal Fbx4G domain in cyan. In TRF1, the N-terminal acidic region is in red, the C-terminal Myb domain in slate, and the TRFH domain in yellow. The shaded area between Fbx4 and TRF1 indicates that the Fbx4-TRF1 interaction is mediated by Fbx4G and TRF1TRFH.
(B) Ribbon diagram of the dimeric Fbx4G-TRF1TRFH complex. Fbx4G and TRF1TRFH are colored in cyan and yellow, respectively, in one complex, and blue and orange in the other. The secondary structure elements are labeled.
(C) Surface representation indicates that the dimeric Fbx4G-TRF1TRFH complex adopts a saddle-shaped conformation. The orientation of complex is rotated by 90° about a horizontal axis relative to the complex in (B).
Here, we report the 2.4 Å resolution crystal structure of the Fbx4-TRF1 complex, which unexpectedly reveals that Fbx4 contains a small GTPase fold. This substrate-binding domain recognizes a globular domain of TRF1 instead of short degron peptides seen in all other available F-box protein-substrate complex structures (Petroski and Deshaies, 2005; Ravid and Hochstrasser, 2008). We show that the telomere bound TRF1 is protected from SCFFbx4 mediated ubiquitination by another telomere protein TIN2 (Chen et al., 2008; Kim et al., 1999). Combining structural, mutational and biochemical analyses, we propose a coherent model for TRF1 regulation by SCFFbx4.
RESULTS
Overview of the Fbx4-TRF1 Complex
A previous study reported that a fragment of Fbx4 (residues 171–308) C-terminal to the F-box motif interacts with the TRFH domain of TRF1 (TRF1TRFH) (Lee et al., 2006). However, attempts to express and purify Fbx4171–308 yielded insoluble proteins (data not shown). Examination of the primary sequence and predicted secondary structure of Fbx4 identified a putative globular domain (residue 162–387). Indeed, recombinant Fbx4162–387 formed a stable complex with TRF1TRFH (Figures S1A and S1B).
To gain insights into the molecular basis of TRF1 recognition by Fbx4, we obtained crystals of the Fbx4162–387-TRF1TRFH complex and determined its structure by multiple-wavelength anomalous dispersion (MAD) at a resolution of 2.4 Å (Figure 1B). The calculated electron density map allowed unambiguous tracing of most of the complex except a 38-residue disordered loop in Fbx4 (residues 240–277) (Figure S1C). The complex structure has been refined to an R-value of 23.7% (Rfree = 26.3%) with good geometry (Table 1).
Table 1.
Data collection, phasing and refinement statistics
| Se-Met | ||
|---|---|---|
| Data collection | ||
| Peak | Inflection | |
| Space group | P43212 | P43212 |
| Cell dimensions | ||
| a, b, c (Å) | 68.097, 68.097, 234.421 | 68.197, 68.197, 234.562 |
| α, β, γ (º) | 90, 90, 90 | 90, 90, 90 |
| Wavelength (Å) | 0.9792 | 0.9794 |
| Resolution (Å) (high res. shell) | 50–2.4(2.49–2.4) | 50–2.5 (2.59–2.5) |
| Rmerge (%) (high res. shell) | 0.081(0.279) | 0.082(0.502) |
| I/σ (high res. cell) | 54.3(4.4) | 35.1 (2.5) |
| Completeness (%) (high res. shell) | 96.2(76.5) | 92.3(41.8) |
| Redundancy (high res. shell) |
11.4 (5.1) |
12.3(6.2) |
| Phasing | Acentric Phasing Power | Centric Phasing Power |
| Se peak anomalous | 1.782 | |
| Se inflection isomorphous | 0.253 | 0.237 |
| Se inflection anomalous |
0.539 |
|
|
FOM |
||
| Acentric reflections | 0.387 | |
| Centric reflections |
0.110 |
|
| Refinement | ||
| Resolution (Å) | 50–2.4 | |
| No. reflections | 38,593 | |
| Rwork/Rfree (%) | 23.74/26.34 | |
| No. atoms | ||
| Protein | 3077 | |
| Water | 60 | |
| B-factors | ||
| Protein | 56.9 | |
| Water | 45.8 | |
| R.m.s deviations | ||
| Bond lengths (Å) | 0.008023 | |
| Bond angles (º) | 1.28514 | |
The Fbx4162–387-TRF1TRFH complex structure reveals a 2:2 stoichiometry between Fbx4162–387 and TRF1TRFH, consistent with the observed molecular weight of the complex (~ 97 kDa) by gel filtration chromatographic analysis (Figure S1A). The complex has a saddle-shaped structure measuring linear dimensions of approximately 100 Å × 85 Å × 75 Å (Figures 1B and 1C). Fbx4162–387 and TRF1TRFH interact through complementary hydrophobic surfaces (Figure S1D). Each Fbx4162–387 molecule contacts only one subunit of TRF1TRFH that does not overlap the TRF1TRFH dimeric interface (Figure 1B). The formation of the binary complex causes the burial of ~ 1,300 Å of surface area at the interface. The homodimeric TRF1TRFH forms the ‘pommel’ and ‘cantle’ of the ‘saddle’ with each TRF1TRFH comprising ten α helices (Figures 1B and 1C). Superposition analysis shows that the structure of TRF1TRFH remains largely unchanged with or without Fbx4162–387 bound except for loop L34 (Figure S1E). L34 is highly flexible and partially disordered in the unliganded TRF1TRFH (Fairall et al., 2001).
The structure of Fbx4162–387 adopts a compact, globular fold featuring a highly curved seven-stranded β sheet surrounded by five α helices (Figures 1B, 2B, and S2). Strikingly, although primary sequence analysis failed to identify any known protein motif in Fbx4162–387, an unbiased search of the database using Dali (Holm and Sander, 1991) revealed unequivocal structural resemblance of Fbx4162–387 with more than 400 Ras superfamily small GTPases (Colicelli, 2004; Wennerberg et al., 2005). Each of these GTPases can be superimposed onto Fbx4162–387 with a root-mean-square deviation (rmsd) of ~3Å in the positions of over 135 Cα atoms of equivalent residues. Thus, this analysis identified Fbx4162–250 as a domain belonging to the Ras superfamily of small GTPases. Hereafter we will refer to Fbx4162–250 as Fbx4G (Fbx4 GTPase domain) (Figure 1A).
Figure 2. Fbx4G is an Atypical Small GTPase Domain without GTP Binding Activity.
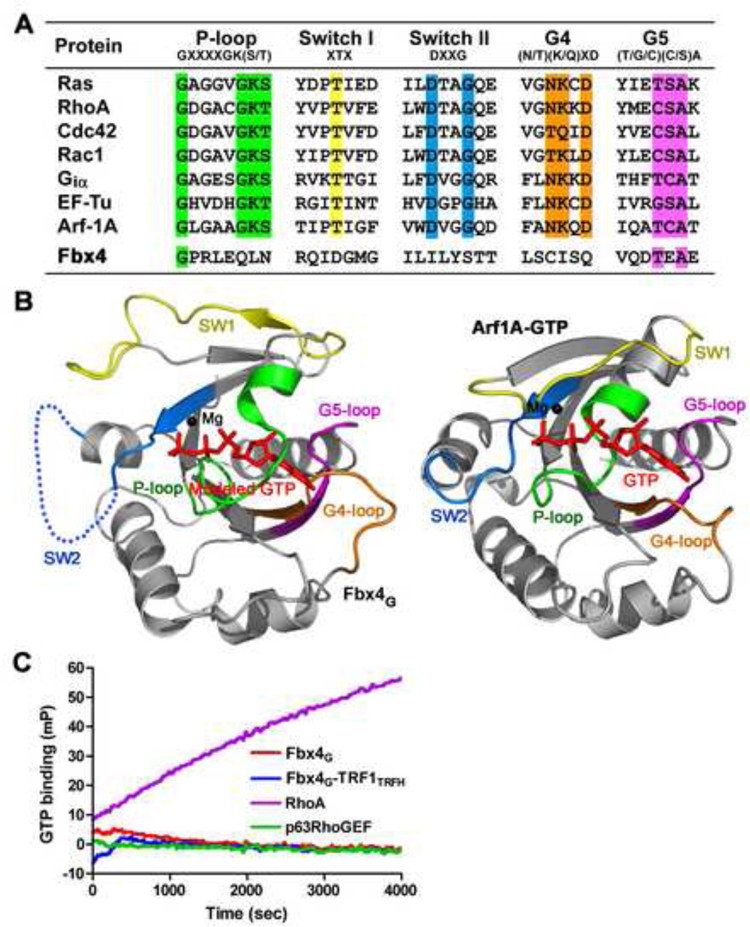
(A) Sequence alignment in the loop regions of Fbx4G and a group of small GTPases. The conserved residues important for nucleotide binding are highlighted in different colors;the P-loop is in green, Switch I in yellow, Switch II in cyan, G4 in orange and G5 in magenta.
(B) The conformation of Fbx4G observed in the crystal structure is incompatible with GTP binding. Left: ribbon representation of Fbx4G with modeled GTP and Mg2+. The Ploop, Switch II and the G4-loop of Fbx4G block the binding of GTP. Right: ribbon representation of the Arf1A-GTP complex (PDB: 2J59). The coloring scheme of the loop regions is the same as in (A). GTP is shown as a stick model and Mg2+ a black ball.
(C) Times courses of GTP binding for Fbx4G and the Fbx4G-TRF1TRFH complex. GTP binding was monitored by the increase in fluorescence millipolarization (mP) of a fluorescent GTP analog as it was bound. In this experiment, neither Fbx4G nor the Fbx4GTRF1TRFH complex bound to GTP. RhoA (a small GTPase) and p63RhoGEF (a non GTPase protein) were used as positive and negative controls, respectively.
Fbx4G Contains an Atypical Small GTPase Domain
The GTP hydrolyzing activity of classical Ras superfamily GTPases relies on five highly conserved loop regions: the P-loop is responsible for binding of the α- and β-phosphate groups; Switch I and Switch II provide residues for Mg2+ and γ -phosphate binding; and the G4 and G5 loops recognize the guanine base (Figure 2A) (Paduch et al., 2001). Despite the high degree of overall structural similarity, the loop regions of Fbx4G, relative to classical small GTPases, are markedly divergent. First, Fbx4G lacks most of the conserved residues that are important for GTP binding (Figure 2A). Second, the P- and G4 loops adopt different conformation than those observed in classical small GTPase proteins, and preclude Fbx4G from binding of GTP (Figure 2B). Third, Fbx4G has a very long Switch II loop (~ 40 residues as compared to ~8 residues in classical GTPases), which is disordered in the Fbx4G-TRF1TRFH complex structure (Figure 2B). Thus, these sequence and structural analyses suggested that Fbx4G would not bind GTP. Through use of a fluorescent polarization assay we found that neither Fbx4G nor the Fbx4G-TRF1TRFH complex detectably bound to GTPγS, whereas RhoA, a well characterized small GTPase, associated with the nucleotide (Figure 2C). Furthermore, we have also tested several other nucleotides, including ATP, ADP and TTP, and found out none of these nucleotides could associate with Fbx4 (data not shown). Collectively, we conclude that while Fbx4G contains an atypical small GTPase domain, it lacks any detectable potential to effectively bind or coordinate GTP.
TRF1 Recognition by Fbx4
The structure of the Fbx4G-TRF1TRFH complex reveals the molecular basis by which Fbx4 recognizes TRF1 (Figure 3). The primary feature of the Fbx4G-TRF1TRFH interaction is an anti-parallel intermolecular β sheet interaction involving β6 of Fbx4G and an induced β strand βA between helices α2 and α3 of TRF1TRFH, resulting in an eight-strand β sheet extending over both molecules (Figure 3A). βA corresponds to loop L23 in the unliganded TRF1TRFH structure (Fairall et al., 2001). There are three backbone hydrogen-bonding interactions between Fbx4G β6 and TRF1TRFH βA (Figure 3B). It is a common theme that small GTPase proteins interact with their effectors by forming an intermolecular β-sheet (Herrmann, 2003). However, Fbx4G uses β6 to bind to TRF1TRFH, whereas most GTPases employ their β2 on the GTP-binding edge of the β sheet to contact their effectors (Figure S3) (Herrmann, 2003).
Figure 3. Recognition of TRF1 by Fbx4.
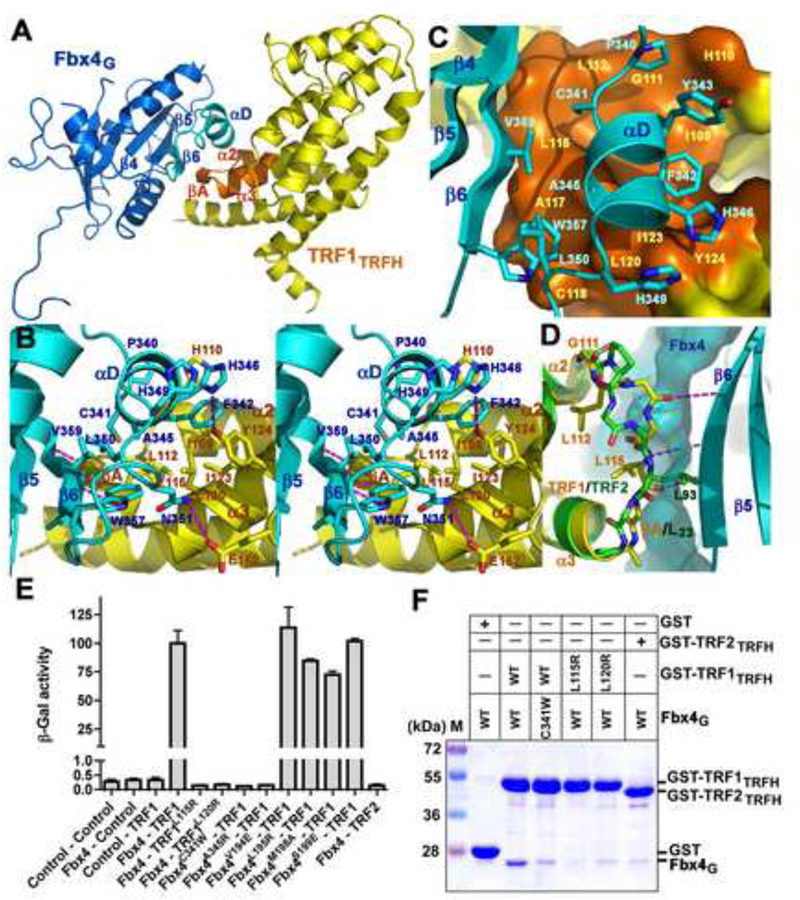
(A) An overall view of the Fbx4G-TRF1TRFH interaction. The interacting structural elements of Fbx4G (β6 and αD) and TRF1TRFH (βA, α2 and α3) are colored in cyan and orange, respectively, and the rest of Fbx4G and TRF1TRFH in blue and yellow.
(B) Stereo view of the Fbx4G-TRF1TRFH interface. Fbx4G and TRF1TRFH interacting residues are presented as stick models. The Fbx4G-TRF1TRFH intermolecular hydrogen bonds are shown as dashed magenta lines.
(C) The αD helix of Fbx4G sits on a hydrophobic surface formed by μA and helices of α2 and α3 of TRF1TRFH.
(D) Fbx4G binding is TRF1TRFH specific. Loop L23 of TRF2TRFH (green) adopts a different conformation than that of TRF1TRFH (yellow) so that it cannot form an intermolecular β-sheet interaction with Fbx4G. The surface representation shows that the side chain of TRF2 Leu93 collides with μ6 of Fbx4G.
(E) Effects of the Fbx4 and TRF1 mutations on the Fbx4-TRF1 interaction in a yeast two-hybrid assay. Interaction of LexA-TRF1 with GAD-Fbx4 was measured as β-galactosidase activity. Data are average of three independent β-galactosidase measurements normalized to the wild-type Fbx4-TRF1 interaction, arbitrarily set to 100. Error bars in the graph represent standard deviation.
(F) GST pull-down assay of the wild-type and mutant Fbx4G-TRF1TRFH interactions.
In addition to the intermolecular β sheet, the formation of the Fbx4G-TRF1TRFH complex is reinforced by the αD helix of Fbx4G, which contacts TRF1 through extensive wan der Waals interactions. This short helix packs against a concaved-shaped hydrophobic surface that spans both molecules with αC, β4, β5 and β6 of Fbx4G on one side and α2, α3 and βA of TRF1TRFH on the other (Figures 3A, 3B, and 3C). Particularly, located at the center of the αD helix is Phe342, whose side chain sits snugly on a complementary surface formed by a cluster of hydrophobic residues of TRF1TRFH (Figures 3B and3C). These van der Waals contacts are further buttressed by two electrostatic interactions. The side chain amino group of Fbx4 Asn351 forms a hydrogen bond with the carboxyl group of TRF1 Glu162, while the imidazole ring of Fbx4 His346 donates another one to the phenol ring of TRF1 Tyr124 (Figure 3B).
To corroborate our structural analysis, we examined whether mutations of the interface residues of TRF1TRFH or Fbx4G could weaken or disrupt the Fbx4-TRF1 interaction. In support of the crystal structure, substitution of the conserved leucine residues of TRF1TRFH (Leu115 or Leu120) on the interface with a positively charged and bulkier arginine residue was sufficient to abolish the interaction with Fbx4G in both yeast-two-hybrid and in vitro GST-pull-down assays (Figures 3B, 3C, 3E, and 3F). Similarly, Fbx4G mutations C341W and A345R on the other side of the interface also impaired the interaction (Figures 3B, 3C, 3E, and 3F). By contrast, four control mutations of Fbx4 or TRF1, designed to eliminate the crystal packing contacts between Fbx4G and TRF1TRFH, had no detectable effect on complex formation (Figure 3E). Therefore, we conclude that the hydrophobic interface between Fbx4 and TRF1 is necessary for the binding of Fbx4 to TRF1.
Fbx4 Does Not Bind to TRF2
TRF1 and TRF2 are two closely related paralogous proteins (Broccoli et al., 1997). Despite the structural similarities of the TRFH domains of TRF1 and TRF2, there are some sequence variations in the TRFH domains between these two paralogs (Chen et al., 2008; Fairall et al., 2001). We asked whether Fbx4 also binds TRF2. However, we failed to detect Fbx4-TRF2 binding using both yeast two-hybrid and GST pull-down assays (Figures 3E and 3F). Comparison of the TRFH domains of TRF1 and TRF2 revealed that structural differences in loop L23 are likely responsible for the ability of Fbx4 to distinguish between these two paralogous proteins. In TRF1, a glycine residue (Gly111) is at the beginning of loop L23 (strand βA in the Fbx4G-TRF1TRFH complex) (Figure 3D). Its unusual dihedral angles allow L23 to adopt a distinct conformation that lines up the backbone of L23 for the optimal intermolecular β-sheet interaction with β6 of Fbx4G (Figure 3D). In addition, the side-chains of Leu112 and Leu115 of TRF1 rotate away from strand β6 of Fbx4 and form part of the hydrophobic surface that contacts helix αD of Fbx4G (Figure 3D). In contrast, the overall geometry of TRF2 loop L23 prevents the formation of the intermolecular β-sheet with Fbx4G (Figure 3D). Most notably, the sidechain of TRF2 Leu93 rotates almost 180° relative to the position of Leu115 in TRF1 resulting in a clash with β5 of Fbx4G (Figure 3D). This structural analysis provides rationale for the bind specificity of Fbx4 for TRF1. Substituting the corresponding residues from TRF2 for TRF1 abrogates TRF1 ubiquitination strongly supported this notion (data not shown).
SCFFbx4 Is an E3 Ubiquitin Ligase for TRF1
To examine the functional significance of the Fbx4-TRF1 interaction, we reconstituted Fbx4-dependent TRF1 ubiquitination in vitro using highly purified recombinant protein components. We initially incubated wild-type (wt) TRF1 with recombinant ubiquitin, E1, Ubc5a (E2), and the SCFFbx4 complex (E3) together with adenosine triphosphate (ATP). Polyubiquitination of wt TRF1 was readily detected in an F-box- and ubiquitin-dependent manner (Figure 4A, compare lanes 2 and 8 with 10). Omission of any component required in the ubiquitin transfer reaction abrogated polyubiquitination of TRF1 (Figure 4A, compare lanes 3, 4, 5, 6, and 7 with 10). In addition, replacement of Fbx4 with another F-box protein, Skp2, polyubiquitination of TRF1 was drastically reduced, confirming the substrate specificity of Fbx4 (Figure 4A, compare lanes 9 with 10). Consistent with the yeast two-hybrid and in vitro binding data (Figures 3E and 3F), the ubiquitination of a TRF1 mutant (TRF1L115R/L120R) was greatly reduced compared with that of wt TRF1 (Figure 4B, compare lanes 2 with 6). Similarly, addition of a mutant SCFFbx4 complex (SCFFbx4C341W/A345R) instead of the wt complex in the assay also impaired the TRF1 ubiquitination (Figure 4B, compare lanes 4 with 6). Furthermore, addition of the Fbx4G domain (Figure 4C, lanes 3–6) but not BSA (Figure 4C, lanes 7–10) inhibited the TRF1 ubiquitination, suggesting that the observed ubiquitination of TRF1 was not due to any co-purified contaminant activities. Collectively, these results indicated that the binding of Fbx4 to TRF1 is essential for SCFFbx4-mediated ubiquitination of TRF1.
Figure 4. The Fbx4-TRF1 Interface Is Essential for Fbx4-dependent TRF1 Ubiquitination.
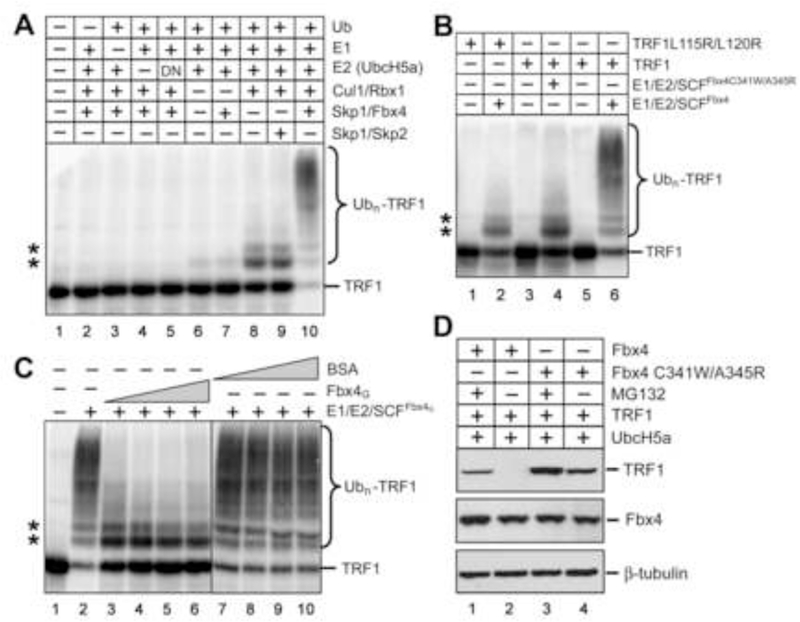
(A) Fbx4-dependent of ubiquitination of TRF1. Purified recombinant TRF1 was 33Plabeled by protein kinase GST-cyclin B/Cdk1 and 33P-TRF1 was separated from GSTcyclin B/Cdk1 by depletion with GSH beads. 33P-TRF1 was then incubated with purified recombinant E1, E2(UbcH5a), E3(SCFFbx4) enzymes and ubiquitin, methylated ubiquitin, ubiquitin aldehyde and MG 132. Reaction mixtures were separated by SDS-PAGE, followed by PhosphorImaging analysis. In panels (A), (B) and (C), asterisks indicate the non-specific ubiquitination products of TRF1 due to the presence of Cul1/Rbx1.
(B) The Fbx4-TRF1 interface is essential for Fbx4-dependent ubiquitination of TRF1. Reactions were performed as in (A), but TRF1L115R/L120R was used as substrate in one reaction and SCFFbx4C341W/A345R as E3 ligase in another.
(C) Addition of Fbx4G suppresses Fbx4-dependent ubiquitination of TRF1. Ubiquitination assays were performed as in (A), but increasing amount of Fbx4G (3, 6, 12, 24 μM) or BSA (3, 6, 12, 24 μM) was added in the reactions.
(D) HEK 293T cells were transfected with TRF1, UbcH5a, Fbx4 WT or Fbx4 C341W/A345R. Four hours after transfection, cells were split into two plates and grown for 24 hours prior to being treated with 2 µM MG 132 or DMSO for 16 hours. Cells were lysed and cell extracts were immunoblotted with indicated antibodies.
To facilitate quantitative detection of the ubiquitinated substrate we labeled TRF1 by phosphorylation using cyclin B/Cdk1 in the presence of 33P-γ-ATP before the in vitro ubiquitination assay. Although phosphorylation of TRF1 is dispensable for its association with Fbx4 as indicated by the crystal structure (Figures 1B, 3B, and 3C), it remains plausible that TRF1 phosphorylation by cyclin B/Cdk1 may facilitate TRF1 ubiquitination due to an allosteric effect. To rule out this possibility, we carried out the in vitro ubiquitination assay using TRF1 substrate in the presence of cold ATP with or without phosphorylation by cyclin B/Cdk1, and then detected the ubiquitinated product by western blot. As shown in Figure S4, both phosphorylated and unphosphorylated TRF1 were efficiently ubiquitinated by SCFFbx4. In fact, TRF1 phosphorylation by cyclin B/Cdk1 slightly inhibited the SCFFbx4 mediated TRF1 unbiquitination (Figure S4, compare lanes 4 and 9). Therefore, we concluded that, unlike most other F-box proteins that recognize the phosphorylated degron peptide sequences of their substrates, phosphorylation is unlikely to be a prerequisite for TRF1 ubiquitination by SCFFbx4.
Fbx4 Promotes TRF1 Degradation in Cells
To examine whether the observed Fbx4-TRF1 interaction is important in Fbx4-dependent degradation of TRF1 in vivo, we co-transfected TRF1 and UbcH5a together with Fbx4 or its TRF1-binding deficient mutant (Fbx4C341W/A345R) in human embryonic kidney 293T cells and then monitored the steady state levels of TRF1. Overexpression of wt Fbx4 but not Fbx4C341W/A345R greatly reduced the levels of TRF1 (Figure 4D, compares lanes 2 and 4). Addition of proteasome inhibitor MG132 significantly elevates TRF1 in Fbx4transfected cells suggesting that reduction of TRF1 reflects proteasome mediated degradation (Figure 4D, lane 1). We conclude that Fbx4-TRF1 association is essential for SCFFbx4 mediated-TRF1 degradation.
Inhibition of the SCFFbx4-mediated TRF1 Ubiquitination by TIN2
Telomere localization of TRF1 is crucial for its stability. It has been proposed that the DNA-binding Myb domain of TRF1 might contain the ubiquitin acceptor lysine and that the telomere binding through this domain protects TRF1 from ubiquitination (Chang et al., 2003). However, a TRF1 mutant lacking the Myb domain, TRF1ΔMyb, can be efficiently modified in our ubiquitination assay, demonstrating the existence of lysines outside of the Myb domain that can be ubiquitinated (Figure 5A). Furthermore, addition of either single-stranded or double-stranded telomeric DNAs had no effect on TRF1 ubiquitination by SCFFbx4 (Figure 5B). Therefore, we concluded that telomere binding itself is unlikely the mechanism of why telomere-bound TRF1 is protected from ubiquitination and degradation.
Figure 5. TIN2 Blocks the Fbx4-TRF1 Interaction and Inhibits the Ubiquitination of TRF1 Mediated by SCFFbx4.
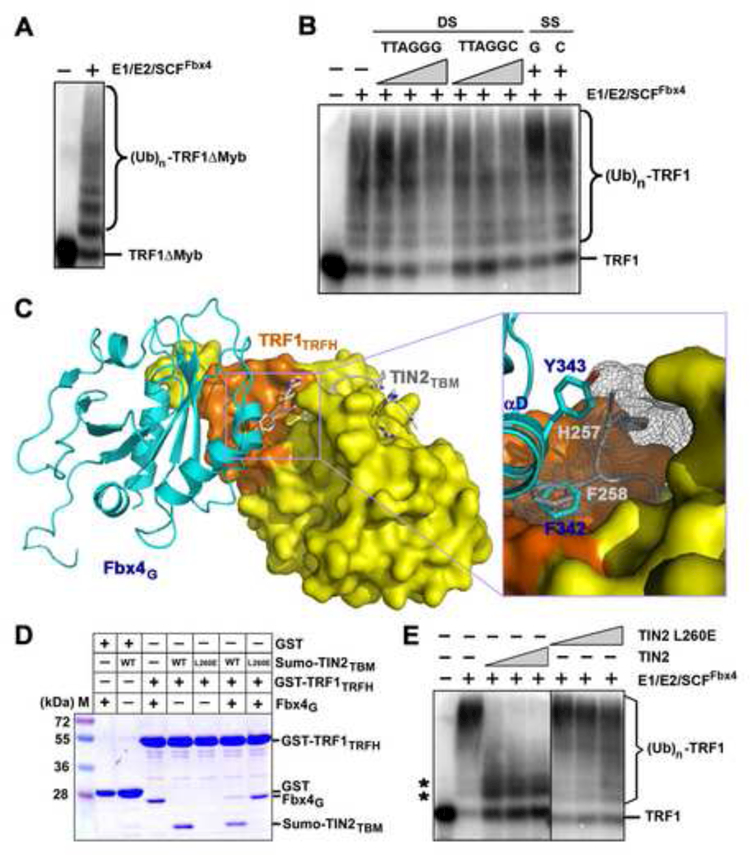
(A) TRF1 deletion mutant lacking the DNA-binding Myb domain, TRF1ΔMyb, can be efficiently ubiquitinated by SCFFbx4.
(B) Addition of either wt or mutant telomere DNAs has no effect on TRF1 ubiquitination. In vitro ubiquitination reactions containing 33P-labeled TRF1 were carried out in the absence (−) or presence of double-stranded (DS; 1, 12 or 36 μM TTAGGG or TTAGGC) repeat DNA or single-stranded [SS; 36 μM TTAGGG (G) or AATCCC (C)] repeat DNA.
(C) Superposition of the Fbx4G-TRF1TRFH structure with the crystal structure of the TRF1TRFH-TIN2TBM complex. Fbx4G (cyan) and TIN2TBM (gray) share a common hydrophobic interacting surface on TRF1TRFH (orange). An enlarged view of the collision between Fbx4G αD helix and the TIN2TBM peptide is highlighted in the box.
(D) GST pull-down competition assay of the Fbx4G-TRF1TRFH interaction in the presence of wt or the L260E mutant TIN2TBM peptides. For clarity, Sumo-fused TIN2TBM peptides were used in this assay. Sumo itself does not bind to TRF1TRFH (data not shown).
(E) TIN2 inhibits Fbx4 mediated ubiquitination of TRF1. TRF1 ubiquitination assays were performed as in (A), but increasing amount of TIN2 (3, 9, 27, 81 μM) or mutant TIN2L260E (3, 9, 27, 81 μM) was added in the reactions. Asterisks indicate the non-specific ubiquitination products of TRF1 due to the presence of Cul1/Rbx1.
Given TRF1 interacts with many proteins at telomeres, it is likely that a TRF1 associated protein would inhibit TRF1 recognition by SCFFbx4 and thus protect TRF1 from ubiquitination and degradation. A possible candidate is TIN2, a component of the telomeric shelterin complex (de Lange, 2005), which binds to both TRF1 and TRF2 and is essential for their stability at telomeres (Kim et al., 2008; Kim et al., 1999; Ye and de Lange, 2004). In a previous study, we determined the crystal structure of TRF1TRFH complexed with a short peptide of TIN2, TIN2TBM (TRF1 binding motif) (Figure S5)(Chen et al., 2008). When the TRF1TRFH-TIN2TBM and the Fbx4G-TRF1TRFH crystal structures are superimposed, it is clear that the TRF1TRFH-TIN2TBM interaction interferes with Fbx4G binding to TRF1TRFH by introducing significant spatial hindrance; the Nterminus of the TIN2TBM peptide collides with helix αD of Fbx4G (Figure 5C). Notably, the two phenol rings from Phe342 of Fbx4G and Phe258 of TIN2TBM occupy exactly the same binding site on TRF1TRFH (Figure 5C). Phe258 of TIN2 is a critical amino acid for the TRF1TRFH-TINTBM interaction; the F258A mutation greatly reduced the TRF1 binding affinity of TIN2TBM by more than 30 folds (Chen et al., 2008). Thus, this observation suggested that TIN2 and Fbx4 cannot bind to TRF1 simultaneously.
We next determined whether the TRF1-TIN2 interaction sequesters TRF1 from recognition by Fbx4. In the GST pull-down assay, when both Fbx4G and TIN2TBM were incubated with GST-tagged TRF1TRFH, only TIN2TBM associated with GST-TRF1TRFH, while both proteins could be pulled down individuals by GST-TRF1TRFH (Figure 5D). In contrast, a TRF1-binding deficient mutant of TIN2TBM (TIN2TBML260E) failed to compete with Fbx4G for GST-TRF1TRFH binding (Figure 5D), indicating that TIN2 has a higher binding affinity to TRF1 than Fbx4 does. Furthermore, when recombinant TIN2 was added in the in vitro ubiquitination assay, ubiquitination of TRF1 was clearly inhibited (Figure 5E). This inhibition was not due to some non-specific effects of TIN2 since the TIN2 L260E mutant had no effect on TRF1 ubiquitination (Figure 5E). Collectively, these structural and biochemical data showed that TIN2 can inhibit TRF1 recognition by Fbx4 and prevent SCFFbx4-mediated TRF1 ubiquitination in vitro.
TIN2 Suppresses Fbx4-dependent TRF1 Degradation in Cells
If TIN2 inhibits Fbx4-mediated ubiquitination and degradation of TRF1 in cells, overexpression of TIN2 should protect TRF1 from degradation and cause it to accumulate. The protection effect should be more pronounced in wt cells than Fbx4 knockdown cells. To test this hypothesis, we first knocked down the endogenous Fbx4 in HeLa cells using a lentiviral short hairpin RNA (shRNA) that targets Fbx4. In agreement with the previous observations, stable expression of Fbx4 shRNA but not the control shRNA causes a reduction in Fbx4 expression (Figure 6A) (Chang et al., 2003; Lee et al., 2006). Subsequently, Flag-tagged TRF1 and TIN2 (or empty vector) were transiently transfected into Fbx4 and control shRNA treated cells, and the amount of Flag-TRF1 was assayed by immunoblotting. Notably, whereas no significant change in the amount of Flag-TRF1 was observed in Fbx4 knockdown cells with or without TIN2 (Figure 6A, compare lanes 1 and 2), there is a significant increase in Flag-TRF1 levels when TIN2 was cotransfected in control shRNA cells (Figure 6A, compare lanes 3 and 4). Since the expression levels of TIN2 were comparable in control or Fbx4 shRNA treated cells (Figure 6A, compare lanes 1 and 3), these results suggested that TIN2 induced stabilization of TRF1 requires Fbx4 and TIN2 does not afford additional stabilization of TRF1 without Fbx4.
Figure 6. TRF1 stabilization by TIN2 depends on Fbx4.
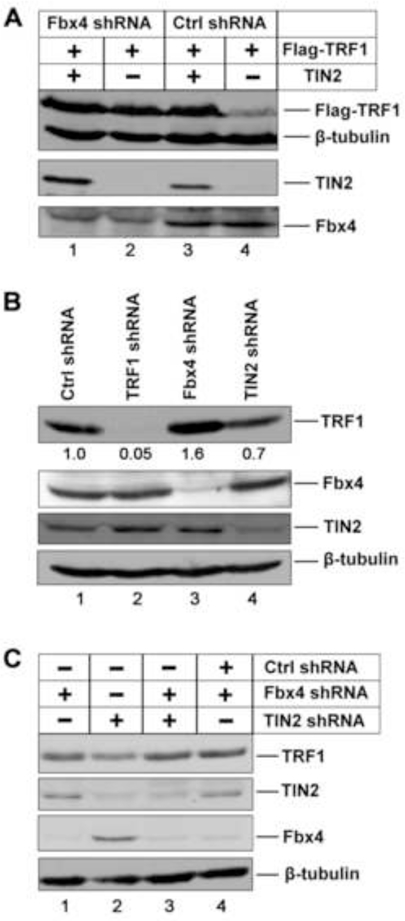
(A) HeLa cells expressing control or Fbx4 shRNA were transfected with Flag-TRF1 along with either TIN2 or an empty expression vector. The expression levels of Flag-TRF1, TIN2 and Fbx4 were analyzed Immunoblotting with the respective antibodies. β-tubulin was blotted as the loading control.
(B) Immunoblotting analysis of the endogenous levels of TRF1 in HeLa cells stably expressing control, TRF1, Fbx4 or TIN2 shRNA vectors with the indicated antibodies.
(C) Endogenous levels of TRF1 in Fbx4 or TIN2 single or double knockdown cells as determined by immunoblotting with the indicated antibodies.
To examine the roles of Fbx4 and TIN2 in regulating the endogenous levels of TRF1, we determined the effect of depletion of Fbx4 and TIN2 on accumulation of TRF1 in cells. Consistent with previous studies, knockdown of Fbx4 and TIN2 reproducibly resulted in increased and decreased levels of TRF1, respectively (Figure 6B) (Lee et al., 2006; Ye and de Lange, 2004), suggesting that Fbx4 promotes endogenous TRF1 degradation whereas TIN2 stabilizes the TRF1 protein level. To establish the direct connection between TIN2 and Fbx4 in TRF1 regulation, we next asked whether the reduction of TRF1 protein level in TIN2 shRNA cells depends upon Fbx4. As shown in Figure 6C, reduction of TRF1 caused by TIN2 depletion can be abrogated by depletion of Fbx4 (compare lanes 2 and 3). The level of TRF1 in TIN2 and Fbx4 double knockdown cells is virtually identical to that in Fbx4 knockdown cells (Figure 6C, compare lanes 3 and 4), indicating that the TIN2 induced stabilization of endogenous TRF1 depends on Fbx4. Taken together, our in vitro and in vivo studies provide a mechanism by which interaction between TIN2 and TRF1 inhibits the ubiquitination of TRF1 by SCFFbx4, and thus prevents TRF1 from degradation.
DISCUSSION
An Unusual Substrate Recognition Domain in Fbx4
SCF ubiquitin ligases regulate a myriad of cellular and development processes and the molecular basis of how they selectively recruit substrates is a key issue in ubiquitin biology. The substrate receptor F-box proteins invariably contain an F-box domain at the N-terminal region and substrate-recognition domain at the C-terminal regions. Unlike the well-characterized Fbw and Fbl families of F-box proteins, the molecular mechanisms of how Fbx family proteins recognize their substrates are largely unknown. One of the few known examples is the Fbs subfamily, which uses a β-sandwich-containing sugar binding domain (SBD) to recognize N-glycan (Yoshida et al., 2002). In this work, our structural analysis revealed that the substrate receptor Fbx4 contains an unsuspected atypical small GTPase domain. Functional analysis confirmed that key residues within this domain directly mediate binding to TRF1. Atypical small GTPases represent a class of signaling molecules different from the classical small GTPases by virtue of their deficiency in nucleotide binding and distinct biological processes they regulate (Aspenstrom et al., 2007). The Fbx4-TRF1 complex structure and subsequent biochemical analysis validated the hypothesis that the atypical small GTPase domain functions as a protein-protein interaction module rather than a GTP hydrolyzing enzymatic domain (Aspenstrom et al., 2007; Chang et al., 2006). Since the atypical small GTPase cannot be readily predicted from the primary sequence, it will be important to determine whether other Fbx proteins may also harbor this functional domain. Utilization of an atypical small GTPase fold for substrate recruitment may not be limited to SCF. RhoBTBs (1, 2 and 3), which contain both an atypical small GTPase domain and at least one Broad Complex/Tramtrack/Brica-brac (BTB) domain, have recently been shown to be the adapters and substrate receptors of CUL3 ubiquitin ligases (Wilkins et al., 2004). The substrates for RhoBTB1–3 have not yet been identified. Based on our studies it is tempting to speculate that the atypical GTPase domains may be the substrate interaction motif for CUL3 ligases.
Specificity of Substrate Recognition by Fbx4
Unlike many well-characterized SCF substrates, which invariably undergo posttranslational modifications (e.g. phosphorylation, sugar modification, hydroxylation of proline) or denaturation for recognition by the respective F-box adaptors (Petroski and Deshaies, 2005; Ravid and Hochstrasser, 2008), SCFFbx4 recognizes an unmodified structural motif of TRF1 (Figure 1B). The remarkable specificity of Fbx4-substrate interaction is further underscored by stringent substrate discrimination between TRF1 and TRF2. Hence, structural insights into the Fbx4-TRF1 complex revealed that there is much greater specificity for a subset of SCF-substrate binding than was previously thought. There are many examples of E3-short peptide substrate complex structures (Hao et al., 2007; Hao et al., 2005; Min et al., 2002; Wu et al., 2003; Zhuang et al., 2009). While short peptide do exhibit specific binding to the F-box proteins, it is possible that these short peptides do not represent all of the binding interfaces of the full-length substrates. The Fbx4-TRF1 structure, which contains an extended substrate, raises a possibility that F-box protein may recognize a highly structured substrate in addition to the short degron peptide signal.
In addition to TRF1, recent work has also identified cyclin D1 as another substrate for Fbx4-dependent ubiquitination (Lin et al., 2006). Ubiquitination of cyclin D1 requires phosphorylation at Thr286 and the presence of an adapter protein αB-crystallin (Lin et al., 2006). In contrast, our data showed that TRF1 ubiquitination by SCFFbx4 is αB-crystallin independent and phosphorylation of TRF1 is dispensable for its association with Fbx4. Thus, Fbx4 is capable of mediating both phosphorylated and unphosphorylated substrates ubiquitination and degradation in the context of two different SCF ligases, SCFFbx4 and SCFFbx4-αB Crystallin, respectively. Definitive clarification of different modes of substrate recognition by Fbx4 has to wait until the crystal structure of the Fbx4-cyclin D1-αB-crystallin complex becomes available.
While phosphorylation of TRF1 by Cyclin B/Cdk1 is dispensable for TRF1 ubiquitination in vitro, we cannot rule out the possibility that TRF1 phosphorylation by cyclin B/Cdk1 may promote its ubiquitination by SCFFbx4 in vivo. Cdk1 activity is required for telomere addition in yeast (Frank et al., 2006) and Cdc13 appears to be a target of Cdk1 (Li et al., 2009). In this vein, phosphorylation of TRF1 by cyclin B/Cdk1 could affect its ubiquitination by SCFFbx4. Further studies are required to address the physiological significance of TRF1 phosphorylation by cyclin B/Cdk1.
Posttranslational Modifications of TRF1 and Their Roles in TRF1 Dynamic Regulation
TRF1 stability and its dynamic localization to telomeres are regulated by multiple posttranslational modifications. In human cells, TRF1 is poly(ADP-ribosyl)ated by tankyrase 1 and this leads to TRF1 eviction from telomeres, and subsequently ubiquitination and degradation by the ubiquitin/proteasome pathway (Smith et al., 1998). Previous studies by others indicated that TIN2 stabilizes TRF1 on telomeres by protecting TRF1 from poly(ADP-ribosyl)ation by tankyrase 1 (Ye and de Lange, 2004). Here, our structural and biochemical studies reveal another link between TIN2 and posttranslational modification of TRF1. TIN2 suppresses TRF1 polyubiquitination by sequestering its degradation motif from recognition by Fbx4 when both are assembled to form the shelterin complex to cap the chromosome ends. Thus, TIN2 serves as a double safe to prevent TRF1 release from telomeres in human cells. Our data also suggest that the telomere-unbound form of TRF1 is relatively unstable, and subjected to Fbx4-mediated ubiquitination and degradation (Chang et al., 2003; Lee et al., 2006). However, we do not yet know whether TIN2 disassociates with TRF1 after TRF1 release from telomeres. A recent in vivo quantitative analysis of shleterin proteins demonstrated that most of TRF1 molecules are associated with telomeres with no detectable telomere-unbound TRF1 in cells (Takai et al., 2009), indicating that telomere-unbound TRF1 is unstable and subjected to degradation. Taken together, it is very likely that TRF1 is no longer associated with TIN2 after evicted from telomeres. Further work is needed to test this hypothesis.
Degradation by the proteasome is not the only fate of polyubiquitinated TRF1. A recent study showed that polyubiquitinated TRF1 can also be deubiquitinated and stabilized by ubiquitin-specific protease 22 (Usp22) in a Gcn5-containing SAGA complex (Atanassov et al., 2009). Notably, our studies reveal that depletion of TIN2 only yielded modest reduction in TRF1 level (Figure 6B). This is consistent with the new finding that a portion of ubiquitinated TRF1 is deubiquitinated, stabilized and possibly recycled back to telomeres.
The dynamic regulation of TRF1 on and off telomeres by posttranslational modifications and their controllers (for example TIN2 and tankyrase 1) appears to play an important role in telomere homeostasis maintenance (Smith, 2009). TRF1 serves as a negative regulator of telomere length maintenance (van Steensel and de Lange, 1997). Therefore, release of TRF1 from telomeres may be necessary to allow telomerase to access the chromosome ends for telomere extension. Furthermore, it has been shown that functional human telomeres adopt a partially open conformation and are recognized as damaged DNAs in the G2 phase of the cell cycle (Deng et al., 2009; Verdun et al., 2005; Verdun and Karlseder, 2006). It is likely that after replication, TRF1 also needs to be reorganized on telomeres in order to have telomeres properly processed to form a functional protective structure. On the other hand, protecting TRF1 from ubiquitination by TIN2 on telomeres and recycling TRF1 back to telomeres through deubiquitination may be important in S phase, during which TRF1 is required for efficient telomere DNA replication (Sfeir et al., 2009). In addition, TIN2 is the central component of shelterin and interacts with TRF1, TRF2 and TPP1 simultaneously, providing a potential mechanism through which the regulation of TRF1 posttranslational modification is linked to other shelterin proteins. Working out how and when all these complicated and interdigitated mechanisms cooperate to control TRF1 dynamics and telomere homeostasis remains a major challenge for the future. Nevertheless, given the specificity of the TRF1-Fbx4 binding interface, it is possible to develop selective small molecule inhibitors that specifically perturb interaction between Fbx4 and TRF1. Such inhibitors may be novel anti-tumor therapeutic agents as they are expected to cause shortening of telomeres and apoptosis of cancer cells.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
Human TRF1TRFH (residues 58 – 268) was cloned into a GST fusion protein expression vector, pGEX6p-1 (GE healthcare) and Fbx4G (residues 162–387) into a modified pET28b vector with a Sumo protein fused at the N-terminus after the His6 tag (Wang et al., 2007). The Fbx4G-TRF1TRFH complex was coexpressed in E. coli BL21(DE3). After induction for 16 hours with 0.1 mM IPTG at 25°C, the cells were harvested by centrifugation and the pellets were resuspended in lysis buffer (50 mM Tris-HCl pH 8.0, 50 mM NaH2PO4, 400 mM NaCl, 3 mM imidazole, 10% glycerol, 1 mM PMSF, 0.1 mg/ml lysozyme, 2 mM 2-mercaptoethanol, and home-made protease inhibitor cocktail). The cells were then lysed by sonication and the cell debris was removed by ultracentrifugation. The supernatant was mixed with Ni-NTA agarose beads (Qiagen) and rocked for 6 hours at 4˚C before elution with 250 mM imidazole. Then Ulp1 protease was added to remove the His6-Sumo tag. The complex was then mixed with glutathione sepharose beads (GE Healthcare) and rocked for 8 hours at 4˚C before elution with 15 mM glutathione. Protease 3C was added to remove the GST-tag. Finally, the Fbx4G-TRF1TRFH complex was further purified by passage through Mono-Q ion-exchange column and by gel-filtration chromatography on Hiload Superdex200 equilibrated with 25 mM Tris-HCl pH 8.0, 150 mM NaCl and 5 mM dithiothreitol (DTT). The purified Fbx4G-TRF1TRFH complex was concentrated to 7 mg/ml and stored at −80˚C.
For the GST-pull-down assay, wt and mutant GST-TRF1TRFH, GST-TRF2TRFH, His6-Sumo-Fbx4G and His6-Sumo-TIN2TBM were expressed in E. coli and purified following the same procedure as described above except for only one affinity chromatography step was used according to the tags of the proteins. The His6-Sumo tag of Fbx4G was cleaved by Ulp1 protease.
For the in vitro ubiquitination assay, TRF1 deletion mutant TRF1ΔMyb and full-length wt and mutant TIN2 and TRF1 proteins were expressed in E. coli as N-terminal His6-Sumo tag fusions, and they were purified by Ni-NTA affinity chromatography followed by Ulp1 digestion to cleave the His6-Sumo tag and finally by gel filtration chromatography. GST-tagged Fbx4 with two deletions (Residues 1–54 and 150–170) was coexpressed with truncated Skp1 (Schulman et al., 2000) as a dicistronic message in E. coli and the complex was purified by glutathione-affinity chromatography followed by thrombin cleavage of the GST tag and by gel filtration chromatography. Deletions of Fbx4 and Skp1 were made to increase the solubility and stability of the complex. The human E1, UbcH5 (E2), Cul1-Rbx1, and Skp1-Skp2 were produced as described (Chen et al., 2008; Hao et al., 2005).
Crystallization, Data Collection and Structure Determination of Fbx4G-TRF1TRFH
The Se-Met substituted Fbx4G-TRF1TRFH complex crystals were grown by hanging drop vapor diffusion at 16ºC. The precipitant/well solution contained 300 mM NH4H2PO4, 100 mM sodium citrate pH 6.2, 10% n-propanol and 10 mM DTT. Crystals were gradually transferred into a harvesting solution containing 200 mM NH4H2PO4, 100 mM sodium citrate pH 6.2, 10mM DTT, and 5 M sodium formate. To improve the diffraction resolution, the crystals were briefly soaked in the harvesting solution in the presence of 100 mM NaBr for ~ 10 minutes before flash-frozen in liquid nitrogen for storage and data collection under cryogenic conditions (100K). Se-Met-MAD data sets at the Se peak and inflection wavelengths were collected using one crystal at beam line 21ID-D at APS. Crystals belong to space group P43212 and with unit cell parameters refined to a = b = 68.097 Å and c = 234.421 Å for the peak wavelength data set. The two data sets were processed using HKL2000 (Otwinowski and Minor, 1997). 12 selenium atoms were located and refined, and the MAD phases calculated using SHARP (La Fortelle and Bricogne, 1997). The initial MAD map was significantly improved by solvent flattening. A model was automatically built into the modified experimental electron density using ARP/WARP (Lamzin et al., 2001); the model was then further refined using simulated-annealing and positional refinement in CNS (Brunger et al., 1998) with manual rebuilding (O) (Jones et al., 1991).
Fluorescence polarization (FP) Assay
We monitored the polarization of light emitted after the addition of 1 μM BODIPY FL GTPγS (excitation/emission maxima ~ 503/512 nm; Invitrogen) to a reaction mixture containing 2 μM protein sample (Fbx4G, Fbx4G-TRF1TRFH, RhoA or p63RhoGEF) in 20 mM HEPES pH 8.0, 150 mM NaCl, 10% glycerol, 10 mM MgCl2, 1 mM DTT. Samples were excited with plane-polarized light using a BMG LABTECH PHERAstar with 485/520 FP Module. Millipolarization (mP) of the emitted light was plotted versus time. The signal from the fluorophore alone is first subtracted from each curve.
Yeast Two-hybrid Assay
The yeast two-hybrid assays were performed using L40 strain harboring pBTM116 and PACT2 (Clontech) fusion plasmids. The colonies containing both plasmids were selected on –Leu –Trp plates. β-galactosidase activities were measured by liquid assay (Moretti et al., 1994).
In Vitro GST Pull-down Assay
5 μg of purified GST or GST-fusion proteins (GST-TRF2TRFH, GST-TRF1TRFH or its mutants (GST-TRF1TRFHL115R and GST-TRF1TRFHL120R)) were incubated with 10 μg wild-type or mutant (C341W) Fbx4G at 4°C for 16 hours in 40 μl binding buffer (25 mM Tris-HCl pH 8.0, 150 mM NaCl and 5 mM DTT) containing 5 μl glutathione sepharose beads. For the competition assay, 5 μg of GST-TRF1TRFH were incubated with 10 μg Fbx4G in the presence of 10 μg of His6-Sumo-TIN2TBM or its L260E mutant proteins. The glutathione sepharose beads were then washed three times by 100 μl binding buffer and finally resuspended in 20 μl 2× SDS protein sample buffer. The samples were analyzed by SDS-PAGE.
In Vitro Ubiquitination Assay
Wild-type or mutant TRF1 proteins were labeled with [γ−33P]ATP by in vitro kinase assay. TRF1 proteins (4 μM) were phosphorylated by incubating with the GST-cyclin B/Cdk1 complex (0.1 μM) in 1 × kinase reaction buffer containing 50 mM Tris-HCl, pH 8.0, 10 mM MgCl2, 10 µM ATP, and 2 µCi of [γ−33P]ATP for 1 hour. The phosphorylated TRF1 was separated from GST-cyclin B/Cdk1 by glutathione affinity chromatography. The ubiquitination assays were performed by incubating phosphorylated TRF1 with 0.5 μM E1, 5 μM UbcH5a (E2), 1 μM SCFFbx4 E3 complex, 5 μM ubiquitin (Sigma), 100 μM methylated ubiquitin (BostonBiochem), 1 μM ubiquitin aldehyde (BostonBiochem), 50 μM MG 132(Calbiochem) and 1 μl 20 × energy regeneration system (10 mM ATP, 20mM HEPES pH 7.4, 10 mM MgOAc, 300 mM creatine phosphate, 0.5 mg/ml creatine phosphokinase) in a final volume of 20 μl. The reactions were incubated at 30°C for 2 hours, terminated by boiling in SDS protein sample buffer and analyzed by SDS-PAGE. Gels were dried prior to phosphoImaging analysis.
For ubiquitination of TRF1 without phosphorylation, we incubated recombinant Flag-TRF1 protein purified from E. coli with the recombinant SCFFbx4 complex. AntiFlag M2 (Stratagene) was used in immunoblotting experiment to detect TRF1 ubiquitin conjugates.
To investigate whether telomeric DNAs inhibit TRF1 ubiquitination by SCFFbx4, oligonucleotides containing GATCTAGCT(TTAGGG)6 and (CCCTAA)6AGCTAGATC for DS (TTAGGG) or GATCTAGCT(TTAGGC)6 and (GCCTAA)6AGCTAGATC for DS (TTAGGC) and GATCTAGCT(TTAGGG)6 for SS(G) or GATCTAGCT(TTAGGC)6 for SS(C) were used. Oligonucleotides were purchased from Invitrogen. 33P-labeled TRF1 was incubated with DS or SS oligonucleotides at 30°C for 30 min prior to addition to the ubiquitination reactions.
Cell Culture, Transfection and Immunoblotting Analysis
293T cells were purchased from ATCC and maintained at 37°C in a 5% CO2 atmosphere in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS; Invitrogen), penicillin (100 IU/ml), streptomycin (100 mg/ml), and L-glutamine (2 mM, Invitrogen). The cDNAs encoding TRF1, Fbx4, Fbx4 C341W/A345R, UbcH5a, or TIN2 were subcloned into mammalian expression vector pEXL and used for the transfection of 293T cells by PEI. Four hours after transfection, one plate of cells were split into two plates. 24 hours after transfection, cells were treated with 2 µM MG132 or DMSO for 16 hours and harvested.
Cell lysates were prepared in the lysis buffer (50 mM Tris-HCl, pH 7.4, 200 mM NaCl, 1 mM EDTA, 1% NP40, 15% glycerol). The protein concentrations were measured by Bradford assay (Biorad). Samples were resolved on 12% SDS–PAGE and electrophoretically transferred to nitrocellulose. Western blots were performed using the anti-Flag M2 (Stratagene), anti-TRF1 (Abcam), anti-Fbx4 (Lin et al. 2006) and anti-Tubulin (ICN) antibodies. The proteins were detected using HRP conjugated goat-anti-mouse or rabbit secondary antibody (GEhealthcare) with an ECL WesternDura detection kit (Pierce).
Generation of Stable shRNA Knockdown Cell Lines
Stable cell lines expressing shRNA constructs targeting Fbx4, TIN2 or TRF1 were created by lentiviral-mediated gene transfer. Briefly, shRNA lentiviral constructs targeting Fbx4 (CCGATTGATGTACAGCTATAT), TIN2 (CGACGAGGAGCAGTTCGAA) and TRF1 (CGACGAGGAGCAGTTCGAA) were made in a modified lentiviral vector FG12-Puro (Qin et al., 2003). Lentiviruses were produced by transient transfection packing using 293T cells as described previously (Riquelme et al., 2006). Fresh viruses were used to infect HeLa S3 cells and stable cell lines were established by selecting with 2 µg/ml of puromycin for two weeks.
Supplementary Material
ACKNOWLEDGEMENTS
We thank P. Lu for the Fbx4 cDNA, H. Pinca-Worm for Cdk1 and cyclin B baculovirus stocks, B. Hao for Cul1-Rbx1, Skp1-Fbx4, and Sk1-Skp2 expression vectors, J.J. Tesmer for recombinant RhoA and p63RhoGEF proteins, T. de Lange for anti-TRF1 antibody, and S. Smith and S.H. Kim for anti-TIN2 antibodies and F. Wang and K. Wan for help at various stages of the project. M.L. is a Howard Hughes Medical Institute Early Career Scientist. Work was supported by NIH grants (GM 083015–01 to M.L. and CA 107098 to X.L.), an American Cancer Society Research Scholar grant and a Sidney Kimmel Scholar Award (to M.L.). The General Medicine and Cancer Institutes Collaborative Access Team has been funded in whole or in part with federal funds from the National Cancer Institute (grant Y1-CO-1020) and the National Institute of General Medical Science (grant Y1-GM-1104). Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract no. DE-AC02–06CH11357.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession number
The coordinates and structure factors of the Fbx4G-TRF1TRFH complex have been deposited in the RCSB Protein Data Bank under accession code 3L82.
REFERENCES
- Aspenstrom P, Ruusala A, and Pacholsky D (2007). Taking Rho GTPases to the next level: the cellular functions of atypical Rho GTPases. Experimental Cell Research 313, 3673–3679. [DOI] [PubMed] [Google Scholar]
- Atanassov BS, Evrard YA, Multani AS, Zhang Z, Tora L, Devys D, Chang S, and Dent SY (2009). Gcn5 and SAGA regulate shelterin protein turnover and telomere maintenance. Molecular Cell 35, 352–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn EH (2001). Switching and signaling at the telomere. Cell 106, 661–673. [DOI] [PubMed] [Google Scholar]
- Broccoli D, Smogorzewska A, Chong L, and de Lange T (1997). Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2. Nature Genetics 17, 231–235. [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. (1998). Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr 54 (Pt 5), 905–921. [DOI] [PubMed] [Google Scholar]
- Cech TR (2004). Beginning to understand the end of the chromosome. Cell 116, 273–279. [DOI] [PubMed] [Google Scholar]
- Chang FK, Sato N, Kobayashi-Simorowski N, Yoshihara T, Meth JL, and Hamaguchi M (2006). DBC2 is essential for transporting vesicular stomatitis virus glycoprotein. Journal of Molecular Biology 364, 302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang W, Dynek JN, and Smith S (2003). TRF1 is degraded by ubiquitin-mediated proteolysis after release from telomeres. Genes Dev 17, 1328–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Yang Y, van Overbeek M, Donigian JR, Baciu P, de Lange T, and Lei M (2008). A shared docking motif in TRF1 and TRF2 used for differential recruitment of telomeric proteins. Science, 319, 1092–1096. [DOI] [PubMed] [Google Scholar]
- Colicelli J (2004). Human RAS superfamily proteins and related GTPases. Sci STKE 2004, RE13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook BD, Dynek JN, Chang W, Shostak G, and Smith S (2002). Role for the related poly(ADP-Ribose) polymerases tankyrase 1 and 2 at human telomeres. Molecular and Cellular Biology 22, 332–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counter CM, Avilion AA, LeFeuvre CE, Stewart NG, Greider CW, Harley CB, and Bacchetti S (1992). Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. The EMBO Journal 11, 1921–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange T (2005). Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev 19, 2100–2110. [DOI] [PubMed] [Google Scholar]
- Deng Y, Guo X, Ferguson DO, and Chang S (2009). Multiple roles for MRE11 at uncapped telomeres. Nature 460, 914–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairall L, Chapman L, Moss H, de Lange T, and Rhodes D (2001). Structure of the TRFH dimerization domain of the human telomeric proteins TRF1 and TRF2. Molecular Cell 8, 351–361. [DOI] [PubMed] [Google Scholar]
- Frank CJ, Hyde M, and Greider CW (2006). Regulation of telomere elongation by the cyclin-dependent kinase CDK1. Molecular Cell 24, 423–432. [DOI] [PubMed] [Google Scholar]
- Hao B, Oehlmann S, Sowa ME, Harper JW, and Pavletich NP (2007). Structure of a Fbw7-Skp1-cyclin E complex: multisite-phosphorylated substrate recognition by SCF ubiquitin ligases. Molecular Cell 26, 131–143. [DOI] [PubMed] [Google Scholar]
- Hao B, Zheng N, Schulman BA, Wu G, Miller JJ, Pagano M, and Pavletich NP (2005). Structural basis of the Cks1-dependent recognition of p27(Kip1) by the SCF(Skp2) ubiquitin ligase. Molecular Cell 20, 9–19. [DOI] [PubMed] [Google Scholar]
- Herrmann C (2003). Ras-effector interactions: after one decade. Current Opinion in Structural Biology 13, 122–129. [DOI] [PubMed] [Google Scholar]
- Holm L, and Sander C (1991). Database algorithm for generating protein backbone and side-chain co-ordinates from a C alpha trace application to model building and detection of co-ordinate errors. Journal of Molecular Biology 218, 183–194. [DOI] [PubMed] [Google Scholar]
- Jin J, Cardozo T, Lovering RC, Elledge SJ, Pagano M, and Harper JW (2004). Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev 18, 2573–2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, and Kjeldgaaed M (1991). Improved methods for building protein models in electron density maps and the location of errors in these methods. Acta Crystallogr A 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, and Shay JW (1994). Specific association of human telomerase activity with immortal cells and cancer. Science 266, 2011–2015. [DOI] [PubMed] [Google Scholar]
- Kim SH, Davalos AR, Heo SJ, Rodier F, Zou Y, Beausejour C, Kaminker P, Yannone SM, and Campisi J (2008). Telomere dysfunction and cell survival: roles for distinct TIN2-containing complexes. The Journal of Cell Biology 181, 447–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Kaminker P, and Campisi J (1999). TIN2, a new regulator of telomere length in human cells. Nature Genetics 23, 405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Fortelle E.d., and Bricogne G (1997). Maximum-likelihood Heavy-Atom Parameter Refinement for Multiple Isomorphous Replacement and Multiwavelength Anomalous Diffraction Methods. In Method in Enzymology (Academic Press; ), pp. 472–494. [DOI] [PubMed] [Google Scholar]
- Lamzin VS, Perrakis A, and Wilson KS (2001). The ARP/WARP suite for automated construction and refinement of protein models. In Int Tables for Crystallography Vol F: Crystallography of biological macromolecules (Rossmann MG & Arnold E eds) Dordrecht, Kluwer Academic Publishers, The Netherlands, pp. 720–722. [Google Scholar]
- Lee TH, Perrem K, Harper JW, Lu KP, and Zhou XZ (2006). The F-box protein FBX4 targets PIN2/TRF1 for ubiquitin-mediated degradation and regulates telomere maintenance. The Journal of Biological Chemistry 281, 759–768. [DOI] [PubMed] [Google Scholar]
- Li S, Makovets S, Matsuguchi T, Blethrow JD, Shokat KM, and Blackburn EH (2009). Cdk1-dependent phosphorylation of Cdc13 coordinates telomere elongation during cell-cycle progression. Cell 136, 50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DI, Barbash O, Kumar KG, Weber JD, Harper JW, Klein-Szanto AJ, Rustgi A, Fuchs SY, and Diehl JA (2006). Phosphorylation-dependent ubiquitination of cyclin D1 by the SCF(FBX4-alphaB crystallin) complex. Molecular Cell 24, 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min JH, Yang H, Ivan M, Gertler F, Kaelin WG Jr., and Pavletich NP (2002). Structure of an HIF-1alpha -pVHL complex: hydroxyproline recognition in signaling. Science 296, 1886–1889. [DOI] [PubMed] [Google Scholar]
- Moretti P, Freeman K, Coodly L, and Shore D (1994). Evidence that a complex of SIR proteins interacts with the silencer and telomere-binding protein RAP1. Genes Dev 8, 2257–2269. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, and Minor W (1997). Processing of X-ray Diffraction Data Collected in Oscillation Mode. In Method in Enzymology (Academic Press; ), pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Paduch M, Jelen F, and Otlewski J (2001). Structure of small G proteins and their regulators. Acta Biochimica Polonica 48, 829–850. [PubMed] [Google Scholar]
- Petroski MD, and Deshaies RJ (2005). Function and regulation of cullin-RING ubiquitin ligases. Nature Reviews 6, 9–20. [DOI] [PubMed] [Google Scholar]
- Qin XF, An DS, Chen IS, and Baltimore D (2003). Inhibiting HIV-1 infection in human T cells by lentiviral-mediated delivery of small interfering RNA against CCR5. Proceedings of the National Academy of Sciences of the United States of America 100, 183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravid T, and Hochstrasser M (2008). Diversity of degradation signals in the ubiquitin-proteasome system. Nature Reviews 9, 679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riquelme C, Barthel KK, Qin XF, and Liu X (2006). Ubc9 expression is essential for myotube formation in C2C12. Experimental Cell Research 312, 2132–2141. [DOI] [PubMed] [Google Scholar]
- Schulman BA, Carrano AC, Jeffrey PD, Bowen Z, Kinnucan ER, Finnin MS, Elledge SJ, Harper JW, Pagano M, and Pavletich NP (2000). Insights into SCF ubiquitin ligases from the structure of the Skp1-Skp2 complex. Nature 408, 381–386. [DOI] [PubMed] [Google Scholar]
- Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, and de Lange T (2009). Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138, 90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S (2009). The SAGA continues.to the end. Molecular Cell 35, 256–258. [DOI] [PubMed] [Google Scholar]
- Smith S, and de Lange T (2000). Tankyrase promotes telomere elongation in human cells. Curr Biol 10, 1299–1302. [DOI] [PubMed] [Google Scholar]
- Smith S, Giriat I, Schmitt A, and de Lange T (1998). Tankyrase, a poly(ADP-ribose) polymerase at human telomeres. Science 282, 1484–1487. [DOI] [PubMed] [Google Scholar]
- Takai KK, Hooper SM, Blackwood SL, Gandhi R, and de Lange T (2010). In vivo stoichiometry of shelterin components. The Journal of Biological Chemistry 285, 1457–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, and de Lange T (1997). Control of telomere length by the human telomeric protein TRF1. Nature 385, 740–743. [DOI] [PubMed] [Google Scholar]
- Verdun RE, Crabbe L, Haggblom C, and Karlseder J (2005). Functional human telomeres are recognized as DNA damage in G2 of the cell cycle. Molecular Cell 20, 551–561. [DOI] [PubMed] [Google Scholar]
- Verdun RE, and Karlseder J (2006). The DNA damage machinery and homologous recombination pathway act consecutively to protect human telomeres. Cell 127, 709–720. [DOI] [PubMed] [Google Scholar]
- Wang F, Podell ER, Zaug AJ, Yang Y, Baciu P, Cech TR, and Lei M (2007). The POT1-TPP1 telomere complex is a telomerase processivity factor. Nature 445, 506–510. [DOI] [PubMed] [Google Scholar]
- Wennerberg K, Rossman KL, and Der CJ (2005). The Ras superfamily at a glance. Journal of Cell Science 118, 843–846. [DOI] [PubMed] [Google Scholar]
- Wilkins A, Ping Q, and Carpenter CL (2004). RhoBTB2 is a substrate of the mammalian Cul3 ubiquitin ligase complex. Genes Dev 18, 856–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright WE, and Shay JW (1992). The two-stage mechanism controlling cellular senescence and immortalization. Experimental Gerontology 27, 383–389. [DOI] [PubMed] [Google Scholar]
- Wu G, Xu G, Schulman BA, Jeffrey PD, Harper JW, and Pavletich NP (2003). Structure of a beta-TrCP1-Skp1-beta-catenin complex: destruction motif binding and lysine specificity of the SCF(beta-TrCP1) ubiquitin ligase. Molecular Cell 11, 1445–1456. [DOI] [PubMed] [Google Scholar]
- Ye JZ, and de Lange T (2004). TIN2 is a tankyrase 1 PARP modulator in the TRF1 telomere length control complex. Nature Genetics 36, 618–623. [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Chiba T, Tokunaga F, Kawasaki H, Iwai K, Suzuki T, Ito Y, Matsuoka K, Yoshida M, Tanaka K, et al. (2002). E3 ubiquitin ligase that recognizes sugar chains. Nature 418, 438–442. [DOI] [PubMed] [Google Scholar]
- Zhuang M, Calabrese MF, Liu J, Waddell MB, Nourse A, Hammel M, Miller DJ, Walden H, Duda DM, Seyedin SN, et al. (2009). Structures of SPOP-substrate complexes: insights into molecular architectures of BTB-Cul3 ubiquitin ligases. Molecular Cell 36, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.