

Abstract
Intrahepatic cholangiocarcinoma (iCCA) is a primary liver cancer epidemiologically linked with liver injury which has poorly understood incipient stages and lacks early diagnostics and effective therapies. While iCCA is conventionally thought to arise from the biliary tract, studies have suggested that both hepatocytes and biliary cells (cholangiocytes) may give rise to iCCA. Consistent with the plasticity of these cell lineages, primary liver carcinomas exhibit a phenotypic range from hepatocellular carcinoma (HCC) to iCCA with intermediates along this spectrum. Here we generated mouse models to examine the consequence of targeting mutant Kras and Tp53, common alterations in human iCCA, to different adult liver cell types. Selective induction of these mutations in the SOX9+ population, predominantly of mature cholangiocytes, resulted in iCCA emerging from premalignant biliary intraepithelial neoplasia (BillN). By contrast, adult hepatocytes were relatively refractory to these mutations and formed rare hepatocellular carcinomas (HCC). In this context, injury accelerated hepatocyte-derived tumorigenesis and promoted a phenotypic switch to iCCA. BilIN precursor lesions were absent in the hepatocyte-derived iCCA models, pointing towards distinct and direct emergence of a malignant cholangiocytic phenotype from injured, oncogenically primed hepatocytes. Tp53 loss enhanced reprogramming of hepatocytes to biliary cells, which may represent a mechanism facilitating formation of hepatocyte-derived iCCA. Overall, our work shows iCCA driven by Kras and Tp53 may originate from both mature cholangiocytes and hepatocytes, and factors such as chronic liver injury and underlying genetic mutations determine the path of progression and resulting cancer phenotype.
Introduction
iCCA is a lethal primary liver cancer that exhibits histologic features of the biliary tract and commonly harbors mutations in IDH1/2, BAP1, KRAS, TP53, SMAD4, and ARID1A (1, 2). Epidemiologic associations include conditions affecting the biliary compartment (e.g. primary sclerosing cholangitis, liver fluke infection) as well as the hepatocellular compartment (e.g. Hepatitis B and C, cirrhosis) (3). In light of its histologic semblance, the mature cholangiocyte has traditionally been the assumed cell of origin in iCCA. However, the existence of mixed hepatocellular/cholangiocarcinoma (HCC-iCCA) tumors and the association of iCCA with chronic hepatocellular injury suggest the possibility of other cell types giving rise to iCCA including hepatocytes. Studies using transgenic mice have demonstrated that iCCA can develop from cholangiocytes; however, in the setting of severe carcinogenic injury or genetic activation of Notch signaling, a driver of biliary differentiation, iCCA can also arise from hepatocytes (4–7). This observation is consistent with models of acute liver injury in which hepatocytes can be reprogrammed to assume a biliary phenotype, a process that requires Notch activation (8, 9). The genetic and physiologic contexts in which this hepatocyte plasticity is relevant to iCCA remain to be fully understood.
In the present study, we tested the oncogenic impact of targeting Kras and Tp53 mutations to either adult liver compartment. In mature hepatocytes, these mutations only promoted iCCA in the context of liver injury. By contrast, the SOX9+ compartment consisting predominantly of cholangiocytes was highly sensitive to iCCA development driven by these mutations. Moreover, only the latter model was associated with the multi-stage progression of premalignant biliary iCCA precursors suggesting distinct premalignant lesions may underlie iCCA pathogenesis which are dictated by the cell of origin. Finally, our results suggest that hepatocyte-to-cholangiocyte reprogramming serves as a barrier to KRAS-driven iCCA development, and that TP53 is a critical regulator of this process.
Materials and Methods
Mice
Mice were of mixed genetic background. All mouse strains (Alb-Cre, Sox9-CreERT2, KrasLSL-G12D, and Tp53f) used are previously studied. Mice were fed 0.1% DDC (3,5-diethoxycarbonyl-1,4-dihydrocollidine) diet for 2 weeks to 2 months, depending on the experiment (Custom Animal Diets, AD5001). In survival studies, mice were monitored for signs of illness including abdominal bloating, diminished activity, and/or poor grooming.
Tamoxifen and AAV8 Injections
To generate hepatocyte-specific mutations for experiments relating to Figure 1 and Supplemental Figure 1, mice 10-25 weeks old were injected with 2.5 × 1011 viral AAV8-TBG-Cre particles via tail-vein injection or retro-orbital injection. To generate hepatocyte-specific mutations for experiments relating to Figure 4, mice 8-12 weeks old were injected with 2.5 × 1011 viral AAV8-TBG-Cre particles via retro-orbital injection. To generate mutations in the SOX9+ compartment, Sox9-CreERT2 mice were injected with tamoxifen for one week.
Figure 1. Targeting Kras and Tp53 mutations to the hepatocyte compartment promotes iCCA in the setting of injury.
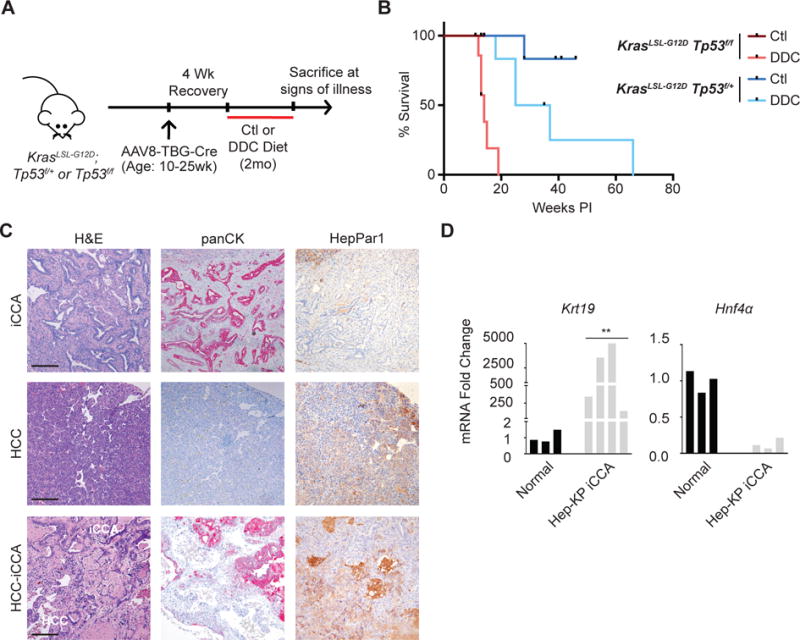
A. Hepatocyte-specific Cre-mediated recombination was induced in KrasLSL-G12D;Tp53f/f and KrasLSL-G12D;Tp53f/+ mice ages 10-25 weeks by injection with AAV8-TBG-Cre. Mice were provided control (Ctl) or DDC-diet for 2 months and followed until signs of illness. B. Liver tumor-specific survival of Hep-KP mice. C. Representative images of H&E and immunohistochemistry (IHC) for panCK and HepPar1 on liver tumors from DDC treated Hep-KP mice. Scale bars, 200μm. D. qPCR analyses for biliary marker Krt19 and hepatocyte marker Hnf4a in normal liver and Hep-KP derived iCCA.
Figure 4. Tp53 restricts hepatocyte-to-cholangiocyte reprogramming.
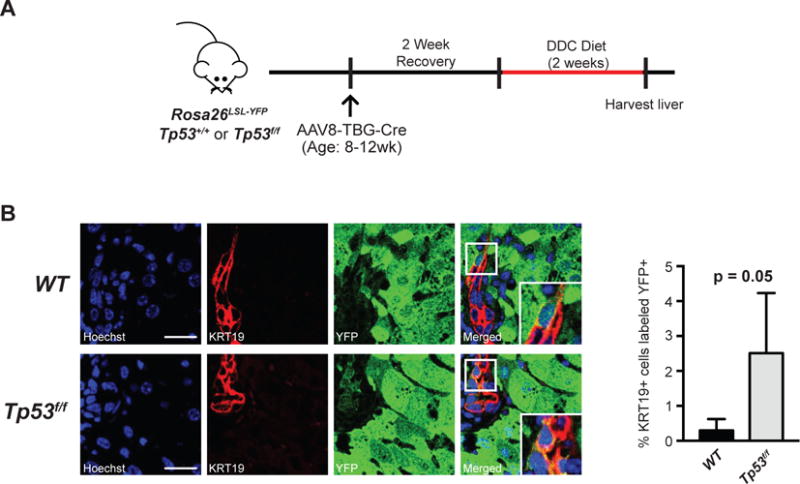
A. Tp53f/f or Tp53+/+ mice with the Rosa26LSL-YFP reporter were injected with AAV8-TBG-Cre to induce hepatocyte-specific Cre-mediated recombination. After recovery, mice were put on a 2-week DDC diet to induce hepatocyte-to-cholangiocyte reprogramming. B. Quantification of YFP+/KRT19+ cells. Representative images are shown of YFP+/KRT19+ cells and adjacent YFP−/KRT19+ cells (n=4 mice/cohort, 15 portal fields/mouse p = 0.05 by unpaired t-test). Scale bars, 25μm. Insets show 3× magnification view.
Histology
Tissue was fixed in 10% formalin, paraffin embedded, cut into 4μm sections and put on slides. Deparaffinized sections underwent antigen retrieval in citrate buffer (Dako) for 30 minutes, blocked for 4 minutes in Background Sniper (Biocare Medical). Sections were incubated with primary antibodies overnight at 4°C and secondary antibodies for 2 hours at room temperature, then incubated with ABC complex (Vectastain) and VECTOR Red (Vector Laboratories) or HSS-HRP and DAB (Vector Laboratories), then stained with hematoxylin. Immunofluorescence sections were incubated with Hoechst for 45 minutes at room temperature. Antibodies: anti-PanCK (Dako #Z0622), anti-hepatocyte (Biocare Medical #166), anti-JAG1 (Santa Cruz #8303), anti-KRT19 (DSHB), anti-WNT7B (Novus Biologicals #NBP1-59564), anti-GFP (Cell Signaling #2956), biotinylated anti-rabbit (Vector BA-1000), Alexa Fluor 488 α-rabbit (Thermo A-11008), Alexa Fluor 555α-rat (Thermo A-21434).
qPCR
RNA was extracted from frozen liver tissue using the RNeasy Mini Kit (Qiagen). cDNA was created using a high capacity cDNA reverse transcriptase kit (Applied Biosystems). All qPCR reactions were performed in triplicate, using SYBR green master mix (Bio-Rad). Expression level was normalized to RhoA. The following primer sets were used. Axin2: 5′-GCCAATGGCCAAGTGTCTCT-3′ and 5′-GCGTCATCTCCTTGGGCA-3′; Dll1: 5′-GATGGATCTCTGCGGCTCTTC-3′ and 5′-GCACCGGCACAGGTAAGAGT-3′; Hes1: 5′-AAAGCCTATCATGGAGAAGAGGCG-3′ and 5′-GGAATGCCGGGAGCTATCTTTCTT-3′; Hey1: 5′-ACACTGCAGGAGGGAAAGGTT-3′ and 5′-CAAACTCCGATAGTCCATAGCCA-3′; Hnf4a: 5′-GGTAGGGGAGAATGCGACTC-3′ and 5′-AAACTCCAGGGTGGTGTAGG-3′; Jag1: 5′-CCCACGTGTTCCACAAACATC-3′ and 5′-CCATGGGAACAGTTATTTGGAGA-3′; Krt19: 5′-TGCTGGATGAGCTGACTCTG-3′ and 5′-AATCCACCTCCACACTGACC-3′; Lef1: 5′-CCCACACGGACAGTGACCTA-3′ and 5′-TGGGCTCCTGCTCCTTTCT-3′; Wnt10a: 5′-TGGAGACTCGGAACAAAGTC-3′ and 5′-AGCTTCCGACGGAAAGCTTC-3′; Wnt7b: 5′-GGTGTGGCAGTGTACCTGCAA-3′ and 5′-GTGAAGACCTCGGTGCGCT-3′.
Statistics
Statistics for qPCR were calculated on the raw, log-scale data using a two-tailed t-test. For visualization, data was represented as fold change and normalized to control liver samples. Statistics for Kaplan-Meier analysis were calculated according to the log-rank (Mantel-Cox) test. To calculate the statistical significance of mice with exclusively HCC tumors, the Fisher’s exact test was employed. To analyze Figure 4, 15 random portal fields per mouse were independently quantified by three blinded observers for the proportion of KRT19+ cells that were YFP+; the average of these three values was used for statistical analysis.
Study Approval
All animal studies were conducted in accordance with the AAALAC accredited University Committee on Animal Resources (UCAR).
Results and Discussion
We previously established that targeting Kras and Tp53 mutations to the mouse liver using the Alb-Cre transgene (Alb-Cre;KrasLSL-G12D;Tp53f/f) alleles (AKP model) results in development of tumors with a histologic spectrum ranging from exclusively iCCA to exclusively HCC (10). The Alb-Cre allele recombines floxed alleles in both hepatocytes and cholangiocytes, beginning in late embryogenesis (11). To evaluate the response of mature hepatocytes to these genetic alterations, we employed intravenous injection of an adeno-associated-virus with hepatocyte-specific tropism to deliver Cre recombinase expressed under the hepatocyte-specific thyroid-binding globulin promoter (AAV8-TBG-Cre) (8, 9). Lineage tracing using a RosaLSL-YFP reporter allele, revealed efficient recombination in hepatocytes, and the absence of YFP+/KRT19+ cells confirmed that this strategy does not target cholangiocytes (Supplemental Figure 1). Adult KrasLSL-G12D;Tp53f/f (n=4) or KrasLSL-G12D;Tp53f/+ (n=6) mice were injected with virus and monitored until signs of illness (Hep-KP mice) (Figure 1A,B). While each animal had visible tumors at necropsy, all were extra-hepatic (lung and soft tissue). Moreover, histologic analysis revealed only a single microscopic focus of early HCC among the first ten mice surveyed (Supplemental Table 1). Thus, mature hepatocytes are relatively refractory to Kras-Tp53 mutation as compared to other tissues where AAV8-TBG-Cre exhibits ectopic activity.
Liver injury is an important risk factor for both iCCA and HCC and can precipitate hepatic cellular plasticity (3, 8, 12). Furthermore, the signaling pathways required to facilitate such transdifferentiation events are implicated in iCCA pathogenesis (5, 8, 13, 14). Based on these considerations, we evaluated the impact of liver injury on mature Kras-Tp53 mutant hepatocytes using the AAV8-TBG-Cre model. The DDC diet is a non-carcinogenic liver injury model that causes ductular reaction, fibrosis, and inflammation (4, 15). Following administration of AAV8-TBG-Cre and a recovery period, adult KrasLSL-G12D;Tp53f/f (n=7) or KrasLSL-G12D;Tp53f/+ (n=6) mice were placed on DDC diet for 2 months and then monitored until signs of illness (Figure 1A). In this cohort, 12 of 13 mice were sacrificed due to significant liver tumor burden (range, 12-66 weeks post-injection), with several mice exhibiting multiple discrete liver tumors (Figure 1B). Histologic examination of the 18 liver tumors revealed iCCA (n=7), HCC (n=7), and mixed iCCA-HCC with discernible zones of histologic transition (n=4) (Figure 1C, Supplementary Table 1). We verified that iCCA and HCC had the expected immunohistochemical (IHC) staining profiles for panCK and HepPar1 (Figure 1C). Moreover, qRT-PCR confirmed expression of the ductal marker, Krt19 and loss of the hepatocyte marker Hnf4a in Hep-KP-derived iCCAs (Figure 1D). Similar results were observed in the AKP model, where the DDC diet markedly accelerated tumor development (median survival, 54 vs. 34 weeks) and shifted the tumor spectrum from one of equal proportions of HCC, iCCA, and mixed HCC-iCCA to one with all tumors consisting of iCCA or mixed HCC-iCCA phenotypes (Supplemental Figure 2A-D). Thus, DDC treatment both sensitizes Kras-Tp53 mutant hepatocytes to malignant transformation, but confers a phenotypic switch specifically favoring an iCCA histopathogenesis.
Next, we assessed the sensitivity of the mature ductal compartment to Kras-Tp53 mutations by generating a cohort of Sox9-CreERT2;KrasLSL-G12D;Tp53f/f mice (SOX9-KP model) (16). The Sox9-CreERT2 transgene enables recombination of floxed alleles in the liver bile ducts, while largely sparing the hepatocytes (Supplemental Figure 3A,B), as well as several extrahepatic cell types. All SOX9-KP mice developed liver masses following tamoxifen injection (average latency, 30 weeks; Figure 2A,B). Histologic analyses revealed that 7/9 mice had iCCA with characteristic ductal and glandular features and positive panCK staining, whereas a single animal developed HCC (Figure 2C, Supplemental Table 2). Notably, adjacent liver to iCCA exhibited extensive ductular reactions and stromal proliferation, as well as dysplastic, panCK-positive biliary lesions resembling biliary intraepithelial neoplasia (BilIN), an established precursor of human iCCA (Figure 2C). Thus, the biliary compartment is readily transformed by Kras and Tp53 mutations, which generate a phenotype consisting of staged ductal precursor lesions and iCCA development.
Figure 2. Targeting Kras and Tp53 mutations to the SOX9+ compartment promotes BilIN and iCCA.
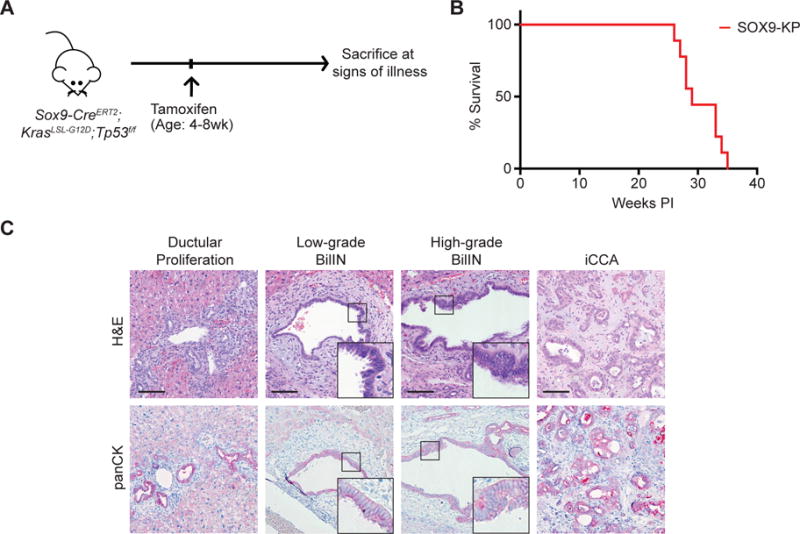
A. Cre-mediated recombination was induced in Sox9-CreERT2;KrasLSL-G12D;Tp53f/f mice via tamoxifen injections between ages 4-8 weeks, and mice were followed for signs of illness (n=9 mice). B. Kaplan-Meier analysis displaying time until illness of SOX9-KP mice. C. H&E and panCK IHC staining of liver sections revealing progressive spectrum of biliary lesions in the SOX9-KP model. Scale bars, 100μm. Insets show 3× magnification view.
This expansion of premalignant biliary precursors was specific to the SOX9-KP model. Despite the prominent formation of iCCA in the Hep-KP and AKP-DDC models, there was a virtual absence of associated BilIN lesions. Serial analysis of DDC-treated AKP-mice prior to signs of illness further corroborated these findings. In particular, among the cohort analyzed (4 mice), there was only a single case where an isolated focus of iCCA was found in zone three of the liver, distant from the bile ducts without evidence of any ductal metaplasia (Supplemental Figure 2D). Thus, evident biliary precursor lesions do not precede development of iCCA from hepatocytes in this model, suggesting that the observed phenotypic shift may represent the direct transformation and transdifferentiation of hepatocytes. Overall, these data reveal that the tumor suppressive barriers and steps of histopathologic progression are distinct in iCCA arising from either liver cell lineage.
To understand the signaling events contributing to hepatocyte (Hep-KP) and biliary (SOX9-KP)-derived iCCA, we examined the Notch and Wnt pathways, which are implicated in liver cell fate decisions and in the sustained growth of iCCA (8, 17–20). Hep-KP-derived iCCAs show high expression of the Notch ligand Jag1 (p=0.005) and target genes Hey1 (p=0.009) and Hes1 (p=0.026) compared to non-malignant liver (Figure 3A). Furthermore, in contrast to hepatocytes in a DDC-injured liver, which show minimal JAG1 staining compared to adjacent biliary cells, and mirroring the SOX9-KP iCCA model, Hep-KP iCCA shows co-staining of JAG1 with the biliary marker KRT19 in the malignant epithelium, indicating expression of JAG1 is acquired during hepatocyte-to-iCCA transformation (Figure 3B). Among most HCC-iCCA transitional zones JAG1 staining correlates with KRT19 staining, suggesting Notch signaling may drive differentiation towards a biliary-like fate (Supplemental Figure 4A). Evaluation of the Wnt pathway revealed Wnt ligands Wnt7b (p=0.001) and Wnt10a (p=0.001) and the target gene Lef1 (p=0.096) are also expressed at high levels in Hep-KP-derived iCCAs, reflecting observations in biliary-derived iCCA (Supplemental Figure 4B). Furthermore, immunofluorescence reveals strong WNT7B staining in the KRT19 compartment, as well as in occasional stromal cells, consistent with previous observations (Supplemental Figure 4C) (19). Together these data indicate that Notch signaling is activated during hepatocyte-to-iCCA transformation, which may promote the biliary differentiation characteristic of iCCA. Furthermore, Hep-KP and SOX9-KP-derived iCCAs exhibit activity in Notch and Wnt signaling, suggesting they may share similar biologic programming regarding these important targetable pathways.
Figure 3. Hep-KP-derived iCCA exhibits activity in Notch signaling.
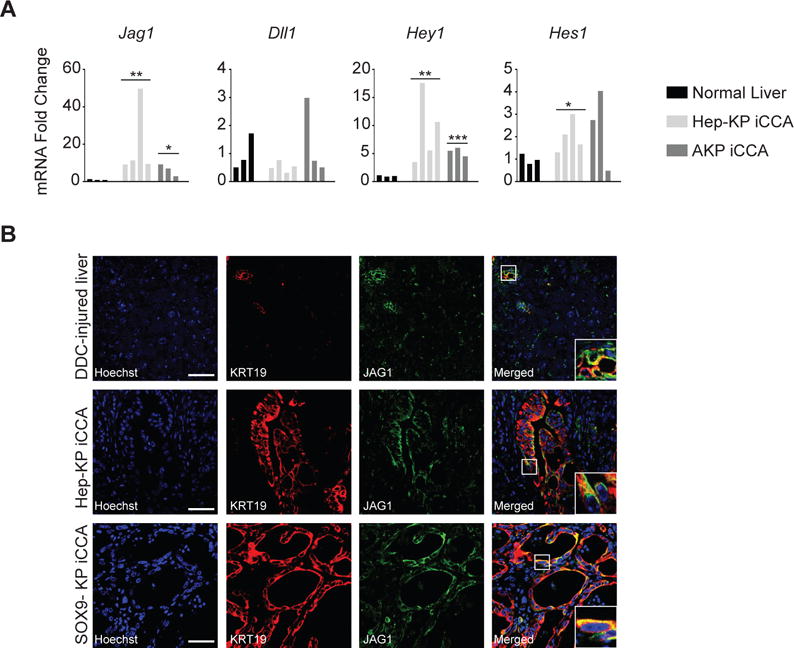
A. qPCR analyses for Notch ligands Jag1 and Dll1 and Notch target genes Hes1 and Hey1. B. Immunofluorescence (IF) for the Notch ligand JAG1 and the biliary marker KRT19 in DDC injured liver demonstrating expression in the CK-19+ biliary compartment and in iCCA in both SOX9 and Hep-KP models. Scale bars, 50μm. Insets show 3× magnification view.
Having established that hepatocyte-derived cancer can acquire biliary identity in the setting of Kras and Tp53 mutations, we sought to determine if the capacity to transdifferentiate is determined by the underlying mutational context. It was previously demonstrated that deletion of Tp53 in the liver promotes undifferentiated tumors that express the stem cell marker Nestin (21). Furthermore, Kras mutations in the absence of Tp53 mutations can drive HCC (7, 10, 22). Taken together, these observations suggest that inactivation of Tp53 in hepatocytes may be critical in enabling biliary differentiation associated with iCCA; therefore, we sought to specifically evaluate the role of Tp53 in the process of hepatocyte-to-cholangiocyte reprogramming in states of injury when a small proportion of resulting KRT19+ biliary-like cells may be derived from hepatocytes (8). We hypothesized that deletion of Tp53 in hepatocytes could lead to increased transdifferentiation events, and therefore more hepatocyte-derived KRT19+ cells. To test this, mice (n = 4/cohort) carrying Tp53f/f or Tp53+/+ and the Rosa26LSL-YFP reporter were injected with AAV8-TBG-Cre to generate hepatocyte-specific mutations and lineage trace this compartment. Following two weeks of recovery, mice were provided a 2-week DDC diet to induce injury and provoke transdifferentiation (Figure 4A). Quantifications of randomly imaged portal fields for the percentage of hepatocyte-derived cholangiocytes (YFP+/KRT19+) relative to the total number cholangiocytes (KRT19+) revealed over a 5-fold increase, from 0.6% in Tp53+/+ to 3.4% in the Tp53f/f cohort (p = 0.05, Welch’s t-test) (Figure 4B). This substantial increase in hepatocyte-derived cholangiocytes is most likely due to an increased rate of transdifferentiation rather than preferential expansion of Tp53-null cholangiocytes since targeting deletion of Tp53 to cholangiocytes as well as hepatocytes via Alb-Cre did not increase cholangiocyte expansion following DDC-injury (Supplemental Figure 5A,B). Furthermore, we observe only a relatively small increase in Ki67 labeling in Tp53f/f vs. Tp53+/+ AAV8-TBG-Cre targeted hepatocytes post-DDC injury (Supplemental Figure 6A,B), suggesting the 5-fold change we observe in co-staining cells is unlikely to be caused by this modest proliferative advantage. Thus, we find that Tp53 restricts hepatocyte-to-cholangiocyte reprogramming and offers a potential mechanism by which Tp53 mutation enables hepatocyte-derived iCCA.
In summary, we show that the mature cholangiocytes and hepatocytes can both give rise to iCCA in the setting of Kras and Tp53 mutations, and liver injury plays a key role in both priming hepatocytes for malignant transformation and facilitating iCCA development. Importantly, we show that the cell of origin may dictate the histopathologic route to iCCA, with cholangiocyte-derived iCCA uniquely being associated with BilIN precursor lesions. Finally, our data indicate that hepatocyte-to-cholangiocyte reprogramming is an important barrier to tumorigenesis and is restricted by Tp53 function.
The appreciation of cellular plasticity in liver regeneration has led to reconsideration of cellular origins of HCC and iCCA (5, 8). Here we demonstrate how the interplay between the cellular compartment, oncogenic mutations, and presence or absence of tissue injury determines the histopathologic paths to cancer and the resulting tumor phenotype (4, 10, 22). In the case of Kras and Tp53 mutations, co-existing mutations in human tumors, there are distinct barriers and premalignant precursor lesions depending on whether the cholangiocytes or hepatocytes are targeted. In ductal cells, such mutations drive multi-stage progression of ductal lesions (BilIN) to iCCA. Kras-Tp53 mutant hepatocytes, however, can be pushed towards this same malignant fate in the setting of injury but without evidence of ductal precursor lesions. Tp53 may play a key role in enabling hepatocyte-derived iCCA in this context. TP53 has been shown to control plasticity in a number of different cellular contexts and is known to control stemness qualities among primary liver cancers via regulation of Nestin (21, 23). We now identify a Tp53-dependent inhibition of hepatocyte-to-cholangiocyte reprogramming as an important mechanism suppressing the development of hepatocyte-derived iCCA.
Overall, iCCAs deriving from distinct cells of origin may have differences in prognosis and therapeutic susceptibility, warranting investigation into associated biomarkers. Our models point to the potential impact of differing cells-of-origin on disease biology; in particular, the prevalence of precursor lesions in each model could be relevant for screening and prevention strategies. Furthermore, although each has a unique route of histologic progression to iCCA, cholangiocyte-derived and hepatocyte-derived iCCA both exhibit activity in Notch and Wnt signaling, suggesting similar biologic programming in these targetable pathways, regardless of the cell of origin.
Supplementary Material
Significance.
The histopathogenesis of the biliary tract cancer, driven by Tp53 and Kras mutations, can be differentially impacted by the cell of origin within the mature liver as well by major epidemiological risk factors.
Acknowledgments
A.H. is supported by the NCI (1R01CA172302) and For Pete’s Sake Golf Tournament in memory of Pete Osterling and N.B. by the NIH (P01 CA117969-07, R01 CA133557-05, P50CA1270003), Gallagher Endowed Chair in Gastrointestinal Cancer Research, and TargetCancer Foundation.
Abbreviations
- iCCA
Intrahepatic cholangiocarcinoma
- HCC
hepatocellular carcinoma
- BilIN
biliary intraepithelial neoplasia
- DDC
3,5-diethyoxycarbonyl-1,4-dihydrocollidine
Footnotes
Conflict of Interest: The authors declare no competing financial interests.
References
- 1.Nakamura H, Arai Y, Totoki Y, Shirota T, Elzawahry A, Kato M, et al. Genomic spectra of biliary tract cancer. Nat Genet. 2015;47(9):1003–10. doi: 10.1038/ng.3375. [DOI] [PubMed] [Google Scholar]
- 2.Jusakul A, Cutcutache I, Yong CH, Lim JQ, Huang MN, Padmanabhan N, et al. Whole-Genome and Epigenomic Landscapes of Etiologically Distinct Subtypes of Cholangiocarcinoma. Cancer Discov. 2017 doi: 10.1158/2159-8290.CD-17-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Banales JM, Cardinale V, Carpino G, Marzioni M, Andersen JB, Invernizzi P, et al. Expert consensus document: Cholangiocarcinoma: current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA) Nat Rev Gastroenterol Hepatol. 2016;13(5):261–80. doi: 10.1038/nrgastro.2016.51. [DOI] [PubMed] [Google Scholar]
- 4.Guest RV, Boulter L, Kendall TJ, Minnis-Lyons SE, Walker R, Wigmore SJ, et al. Cell lineage tracing reveals a biliary origin of intrahepatic cholangiocarcinoma. Cancer Res. 2014;74(4):1005–10. doi: 10.1158/0008-5472.CAN-13-1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fan B, Malato Y, Calvisi DF, Naqvi S, Razumilava N, Ribback S, et al. Cholangiocarcinomas can originate from hepatocytes in mice. J Clin Invest. 2012;122(8):2911–5. doi: 10.1172/JCI63212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sekiya S, Suzuki A. Intrahepatic cholangiocarcinoma can arise from Notch-mediated conversion of hepatocytes. J Clin Invest. 2012;122(11):3914–8. doi: 10.1172/JCI63065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ikenoue T, Terakado Y, Nakagawa H, Hikiba Y, Fujii T, Matsubara D, et al. A novel mouse model of intrahepatic cholangiocarcinoma induced by liver-specific Kras activation and Pten deletion. Sci Rep. 2016;6:23899. doi: 10.1038/srep23899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yanger K, Zong Y, Maggs LR, Shapira SN, Maddipati R, Aiello NM, et al. Robust cellular reprogramming occurs spontaneously during liver regeneration. Genes Dev. 2013;27(7):719–24. doi: 10.1101/gad.207803.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Font-Burgada J, Shalapour S, Ramaswamy S, Hsueh B, Rossell D, Umemura A, et al. Hybrid Periportal Hepatocytes Regenerate the Injured Liver without Giving Rise to Cancer. Cell. 2015;162(4):766–79. doi: 10.1016/j.cell.2015.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Dell MR, Huang JL, Whitney-Miller CL, Deshpande V, Rothberg P, Grose V, et al. Kras(G12D) and p53 mutation cause primary intrahepatic cholangiocarcinoma. Cancer Res. 2012;72(6):1557–67. doi: 10.1158/0008-5472.CAN-11-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu X, Kobayashi S, Qiao W, Li C, Xiao C, Radaeva S, et al. Induction of intrahepatic cholangiocellular carcinoma by liver-specific disruption of Smad4 and Pten in mice. J Clin Invest. 2006;116(7):1843–52. doi: 10.1172/JCI27282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Llovet JM, Zucman-Rossi J, Pikarsky E, Sangro B, Schwartz M, Sherman M, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2016;2:16018. doi: 10.1038/nrdp.2016.18. [DOI] [PubMed] [Google Scholar]
- 13.Kim W, Khan SK, Gvozdenovic-Jeremic J, Kim Y, Dahlman J, Kim H, et al. Hippo signaling interactions with Wnt/beta-catenin and Notch signaling repress liver tumorigenesis. J Clin Invest. 2017;127(1):137–52. doi: 10.1172/JCI88486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yimlamai D, Christodoulou C, Galli GG, Yanger K, Pepe-Mooney B, Gurung B, et al. Hippo pathway activity influences liver cell fate. Cell. 2014;157(6):1324–38. doi: 10.1016/j.cell.2014.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fickert P, Stoger U, Fuchsbichler A, Moustafa T, Marschall HU, Weiglein AH, et al. A new xenobiotic-induced mouse model of sclerosing cholangitis and biliary fibrosis. Am J Pathol. 2007;171(2):525–36. doi: 10.2353/ajpath.2007.061133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kopp JL, Dubois CL, Schaffer AE, Hao E, Shih HP, Seymour PA, et al. Sox9+ ductal cells are multipotent progenitors throughout development but do not produce new endocrine cells in the normal or injured adult pancreas. Development. 2011;138(4):653–65. doi: 10.1242/dev.056499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zong Y, Panikkar A, Xu J, Antoniou A, Raynaud P, Lemaigre F, et al. Notch signaling controls liver development by regulating biliary differentiation. Development. 2009;136(10):1727–39. doi: 10.1242/dev.029140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boulter L, Govaere O, Bird TG, Radulescu S, Ramachandran P, Pellicoro A, et al. Macrophage-derived Wnt opposes Notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nat Med. 2012;18(4):572–9. doi: 10.1038/nm.2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boulter L, Guest RV, Kendall TJ, Wilson DH, Wojtacha D, Robson AJ, et al. WNT signaling drives cholangiocarcinoma growth and can be pharmacologically inhibited. J Clin Invest. 2015;125(3):1269–85. doi: 10.1172/JCI76452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guest RV, Boulter L, Dwyer BJ, Kendall TJ, Man T-Y, Minnis-Lyons SE, et al. Notch3 drives development and progression of cholangiocarcinoma. Pro Natl Acad Sci U S A. 2016;113(43):12250–5. doi: 10.1073/pnas.1600067113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tschaharganeh DF, Xue W, Calvisi DF, Evert M, Michurina TV, Dow LE, et al. p53-dependent Nestin regulation links tumor suppression to cellular plasticity in liver cancer. Cell. 2014;158(3):579–92. doi: 10.1016/j.cell.2014.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saha SK, Parachoniak CA, Ghanta KS, Fitamant J, Ross KN, Najem MS, et al. Mutant IDH inhibits HNF-4a to block hepatocyte differentiation and promote biliary cancer. Nature. 2014:1–18. doi: 10.1038/nature13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang H, Xu Z, Zhong P, Ren Y, Liang G, Schilling HA, et al. Cell cycle and p53 gate the direct conversion of human fibroblasts to dopaminergic neurons. Nat Commun. 2015;6:10100. doi: 10.1038/ncomms10100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.