

Abstract
Olefin-olefin metathesis has led to important advances in diverse fields of research, including synthetic chemistry, materials science and chemical biology. The corresponding carbonyl-olefin metathesis also enables direct carbon-carbon bond formation from readily available precursors, however, currently available synthetic procedures are significantly less advanced. This Synpacts article provides an overview of recent achievements in the field of Lewis acid-mediated and Lewis-acid catalyzed carbonyl-olefin metathesis reactions.
Keywords: Carbonyl-olefin metathesis, Lewis acid, iron, base metal
Graphical Abstract
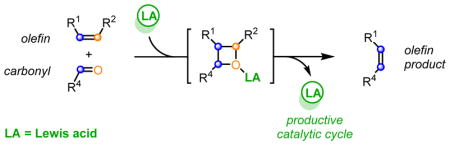
The formation of carbon-carbon bonds is of fundamental importance in the field of synthetic organic chemistry and is therefore invaluable for the synthesis of many important biologically active molecules, including current pharmaceuticals and complex natural products. The development of new, sustainable, efficient and selective catalytic procedures for carbon-carbon bond formation, therefore, represents a key research goal in synthetic chemistry. The metathesis reaction between two alkenes 1 and 2 (Scheme 1) to result in the formation of two distinct olefinic products 3 and 4 is among the most powerful catalytic carbon-carbon bond forming reactions known and has led to profound synthetic developments in the petroleum, materials, agricultural, and pharmaceutical industries.1 The corresponding carbonyl-olefin metathesis reaction between an olefin 5 and a carbonyl 6 (Scheme 1) similarly enables the direct construction of carbon-carbon bonds, resulting in the formation of olefin 7 and carbonyl 8,2,3,4 and has the potential to have an analogous impact on synthetic strategy (Scheme 1).5 However, compared to the olefin-olefin metathesis reaction, currently available procedures for carbonyl-olefin metathesis are significantly less advanced. This Synpacts article is focused on our work in the area of Lewis acid-mediated and Lewis acid-catalyzed carbonyl-olefin metathesis reactions.6
Scheme 1.
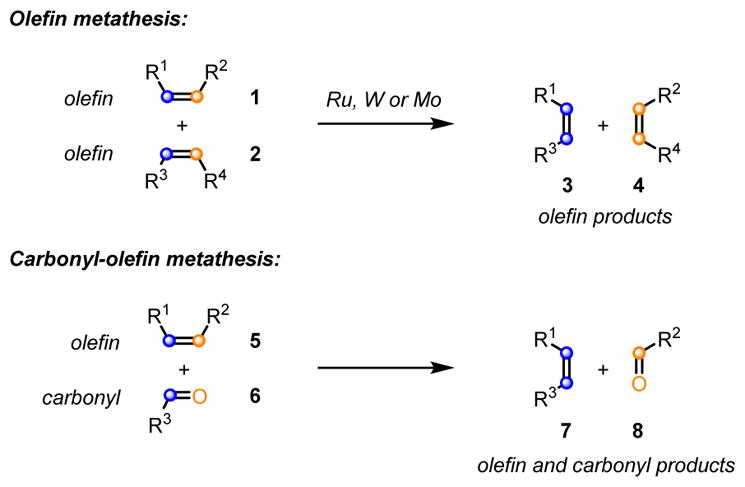
Olefin-olefin metathesis versus carbonyl-olefin metathesis.
1. Lewis Acid-Mediated Carbonyl-Olefin Metathesis
In their studies towards the synthesis of carotol sesquiterpenes, Demole, Enggist and Borer reported the exclusive formation of oxetane 10 in 58% yield upon treatment of cycloheptanone 9 with 20 mol% SnCl4.7 The authors propose a stepwise mechanistic pathway relying on an intramolecular [2+2]-cycloaddition to form the corresponding cis-oxetane 10 as the sole product (Scheme 2). In 1984, Snider and coworkers revisited the original reports by Demole in the course of their studies of intramolecular ene-reactions.8 When cycloheptanone 9 was treated with stoichiometric amounts of MeAlCl2/Me2AlCl (in a 2:1 ratio) in dichloromethane for 10 hours, the formation of diene 11 was observed in 30% yield. Snider and coworkers describe 11 as the product of a metathesis reaction which they presume to proceed via a stepwise cycloaddition to form intermediate oxetane 10 and a subsequent retro-cycloaddition to result in 11 along with the loss of acetone (Scheme 2). Notably, the authors report that no reaction occurred upon treatment of 9 with Me1.5AlCl1.5, however, a complex mixture resulted when cycloheptanone 9 was subjected to stoichiometric amounts of MeAlCl2, indicating the importance of Lewis acid strength in mediating the carbonyl-olefin metathesis reaction.
Scheme 2.
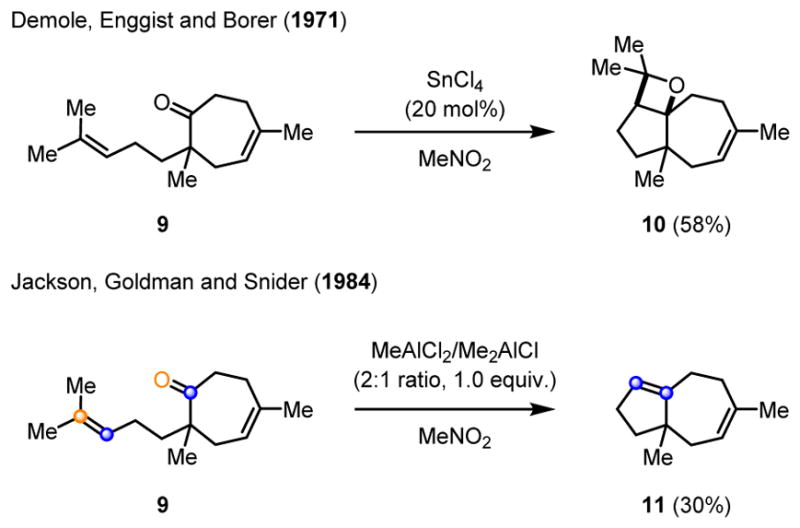
First example of a Lewis acid-mediated carbony-olefin metathesis.
In 1994, Bickelhaupt, van Schaik and Vijn reported the reaction of benzaldehyde 12 and isobutylene 13 or 2-methyl-2-butene 15 to form styrene (14) and β-methylstyrene (16) in 15% and 30%, respectively as the carbonyl-olefin metathesis products (Scheme 3).9 The reaction is promoted by EPZ-10, a solid Lewis acid catalyst consisting of clay supported ZnCl2 (ZnCl2 content 12%).9 Importantly, higher conversion was observed at elevated temperatures and longer reaction times, however, it was accompanied by increased byproduct formation.
Scheme 3.

Carbonyl-olefin metathesis of benzaldehyde using the solid Lewis acid EPZ-10.
The authors propose a stepwise mechanism proceeding via initial Lewis acid activation of the carbonyl oxygen in 12 and subsequent nucleophilic addition of olefin 15 to form carbocation 18. Oxetane 19 results from an intramolecular addition, followed by a Lewis acid-assisted fragmentation to yield the carbonyl-olefin metathesis product β-methylstyrene 16. Importantly, Bickelhaupt and coworkers report the reaction to be limited to carbonyl compounds lacking α-hydrogen substituents which result in competing aldol condensation reactions with the acetone byproduct formed in the carbonyl-olefin metathesis reaction.
Efforts undertaken by Khripach and coworkers to protect the carbonyl moiety in seco steroid 21 as a dithioketal upon treatment with 3 equivalents of BF3·Et2O resulted in the formation of the carbonyl-olefin metathesis product 23 in 60% yield (Scheme 4).11 Subsequent investigations by Khripach and coworkers showed that the corresponding Z-olefin analog of 21 failed to undergo carbonyl-olefin metathesis under otherwise identical reaction conditions. An intramolecular, Lewis acid-promoted [2+2]-cycloaddition and immediate cycloreversion of the resulting oxetane 22 was suggested as a mechanistic hypothesis for the formation of cyclopentene 23.
Scheme 4.
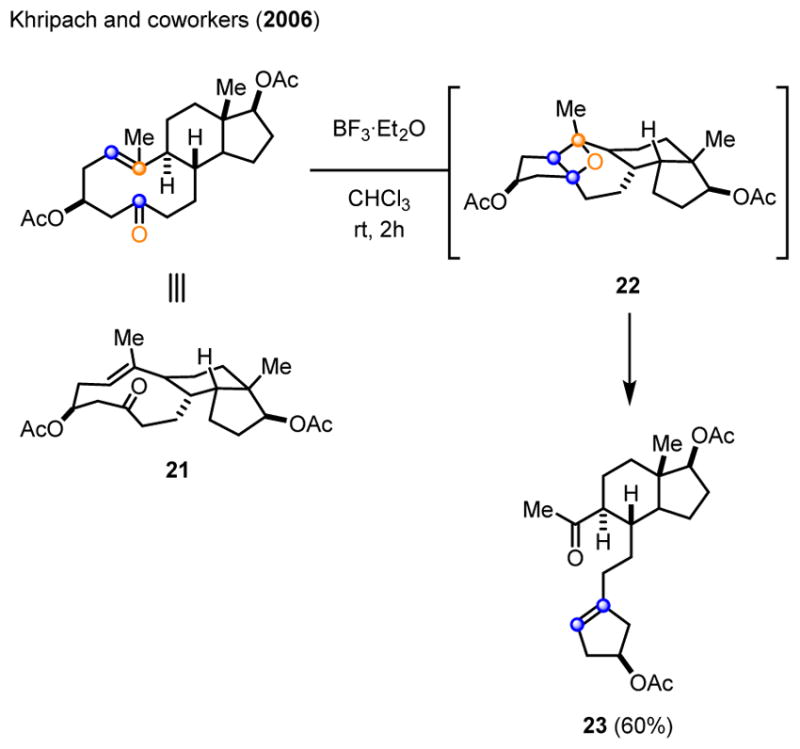
Carbonyl-olefin metathesis of 3β,17β-diacetoxy-5,10-secoandrost-1(10)-en-5-one 21 upon treatment with BF3·Et2O.
During their studies towards the marine natural product pestalone, Schmalz and coworkers observed the formation of indene 25 as a byproduct in 20% yield, upon deprotection attempts of the methylether subunits in ortho-prenylated benzophenone 24 (Scheme 5).12 Subsequent efforts resulted in improved reaction conditions which formed the carbonyl-olefin metathesis product 25 in 87% yield upon treatment with 1.5 equivalents of BF3·Et2O. The generality of this protocol for carbonyl-olefin metathesis was later investigated for a series of acetophenones, bearing prenyl (26), geranyl (28) and homoprenyl (29) sidechains in the ortho position. When these substrates were subjected to the optimized reaction conditions relying on 1.5 equivalents of BF3·Et2O in dichloromethane at −40°C for one hour, the corresponding indene 27 was formed in 87% and 38% yield, respectively, from acetophenones 26 and 28. Importantly, the dihydronaphthalene 30 was obtained in 75% yield in a carbonyl-olefin metathesis reaction to form a 6-membered ring system. Schmalz and coworkers also favor a stepwise mechanistic pathway, proceeding via initial exo-trig cyclization of 31 upon Lewis acid activation of acetophenone 26 with BF3·Et2O (Scheme 6). The resulting tertiary carbocation 32 is presumed to isomerize to the more stable benzylic carbocation 34 via intermediate oxetane 33. Fragmentation of carbocation 34 upon the loss of acetone as byproduct results in the formation of indene 27 as the desired carbonyl-olefin metathesis product.
Scheme 5.
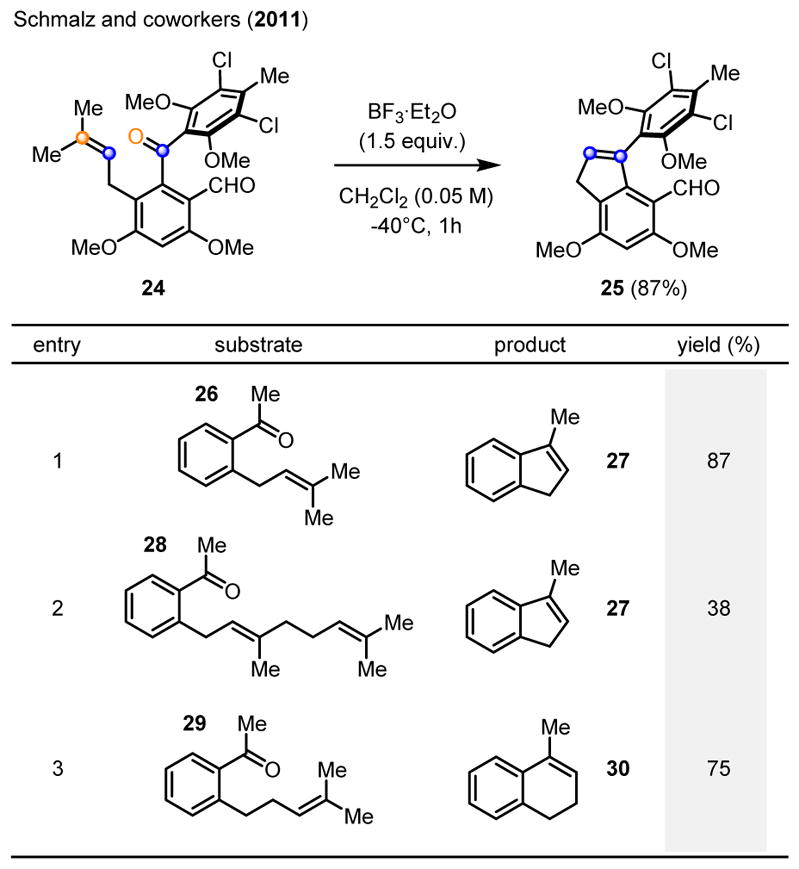
BF3-promoted intramolecular carbonyl-olefin metathesis of ortho-prenylaryl ketones.
Scheme 6.
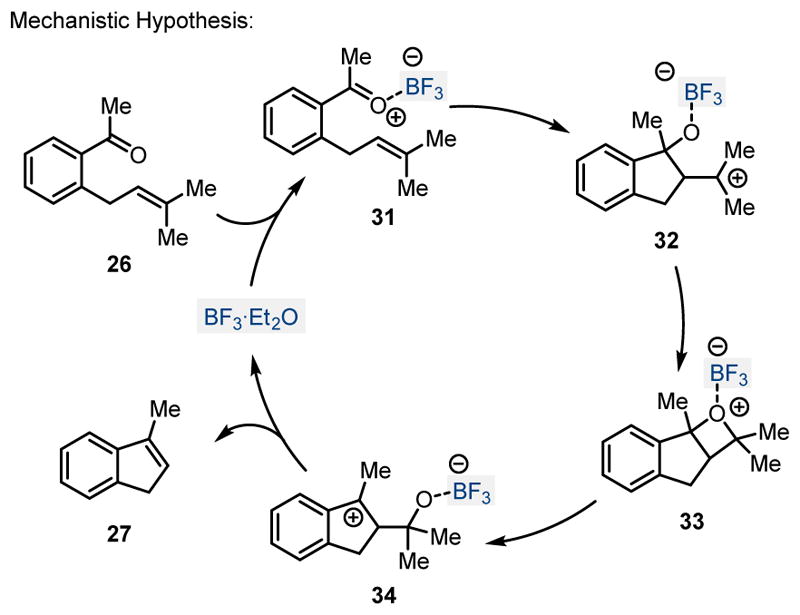
Mechanistic hypothesis for the BF3·Et2O promoted formation of indene 27 from acetophenone derivative 26.
2. Lewis Acid-Catalyzed Carbonyl-Olefin Metathesis
Franzén and coworkers were able to build on the results obtained previously by Bickelhaupt and developed an organocatalytic carbonyl-olefin metathesis approach to trans β-alkylstyrenes relying on trityl tetrafluoroborate (TrBF4) as a catalyst.13 The authors reasoned that the carbocation-based Lewis acid catalyst TrBF4 37 has the potential to exhibit distinct reactivity compared to metal- or metalloid-based Lewis acid catalysts. Further efforts led to optimized reaction conditions for the carbonyl-olefin metathesis of aromatic aldehydes 35 and trisubstituted alkenes 36 relying on 20 mol% TrBF4 37 as catalyst to result in the formation of trans β-alkylstyrenes in up to 85% yield (Scheme 7).
Scheme 7.
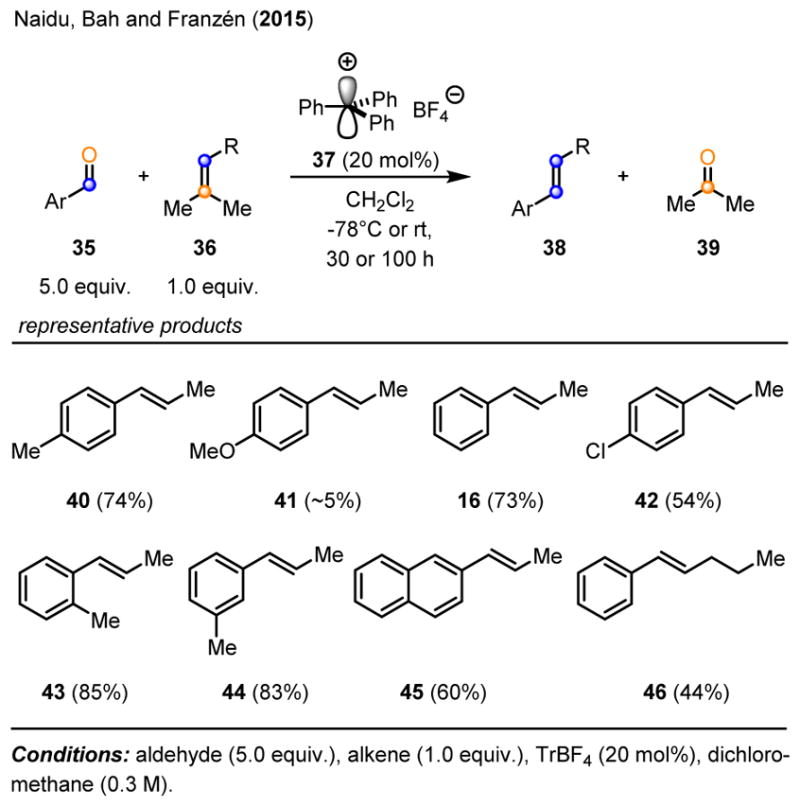
Organocatalytic carbonyl-olefin metathesis resulting in trans β-alkylstyrenes.
Though a remarkable advance, the reaction does require the addition of the carbonyl substrate in large access (5:1 ratio) compared to the olefin starting material and long reaction times of up to 4 days. Weakly electron-withdrawing groups (F, Cl, Br) as well as weakly electron-donating groups (Me, Ar) were tolerated well under the reaction conditions, however, stronger electron-donating aromatic substituents (e.g. MeO) resulted in low yields of the trans β-alkylstyrene products. Additionally, aliphatic aldehydes and tetrasubstituted alkenes were found unreactive under the optimized reaction conditions. Importantly, Franzén and coworkers also evaluated metal-based Lewis acids for their ability to catalyze the carbonyl-olefin metathesis of aromatic aldehydes 25 and amylene analogs 36. Zn(II) salts and SnCl4 were found inactive under the reaction conditions, while low yields of the desired products were observed with InCl3 and AlCl3. Notably, BF3·Et2O and HBF4·Et2O promoted the formation of trans β-alkylstyrenes 38 in 41% and 44% yield, respectively.
The authors propose a mechanism relying on a stepwise [2+2]-cycloaddition pathway to form an intermediate oxetane 49 which subsequently fragments to yield the desired carbonyl-olefin metathesis product (Scheme 8). Upon activation of aldehyde 12 with the Lewis acid 37, oxonium ion 47 is formed, which can be subsequently attacked by alkene 36 to form the tertiary carbocation 48. Oxetane 49 results upon intramolecular nucleophilic addition as the product of a formal [2+2]-cycloaddition reaction. This reactive intermediate can undergo final Lewis acid-promoted fragmentation to yield trans β-alkylstyrene 16 and acetone as the corresponding carbonyl-olefin metathesis products.
Scheme 8.
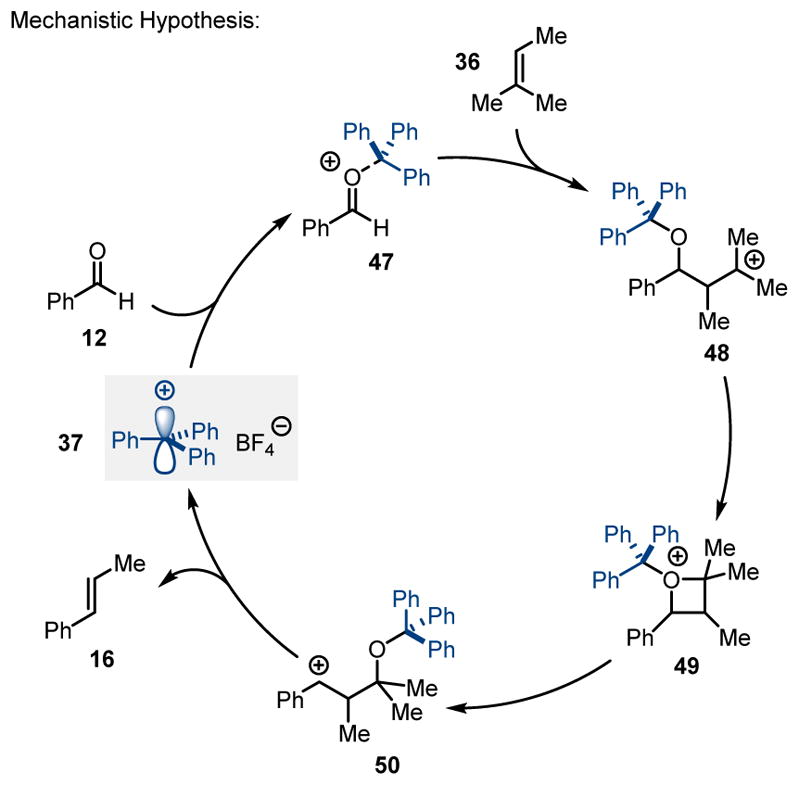
Trityl cation-catalyzed carbonyl-olefin metathesis.
In 2015, our laboratory was able to contribute to this growing area of research by developing a catalytic, carbonyl-olefin ring-closing metathesis reaction for a variety of aryl ketones. Building on the earlier reports of Bickelhaupt, Khripach and Schmalz, relying on stoichiometric amounts of Lewis acids, we envisioned an efficient reaction design for carbonyl-olefin metathesis. Upon activation with a suitable Lewis acid, carbonyl-derivatives 6 and alkenes 5 are able to undergo in situ formation of oxetanes 51 as reactive intermediates, which subsequently fragment to the corresponding carbonyl-olefin metathesis products 7 and 8, thus allowing for catalyst turnover (Scheme 9).14 Many challenges are associated with implementing this type of design principle for catalytic carbonyl-olefin metathesis. Specifically, a catalyst capable of promoting both the [2+2]-cycloaddition, as well as the subsequent [2+2]-cycloreversion, needs to be identified. Additionally, minimizing potential side reactions after [2+2]-cycloreversion, such as polymerization and regeneration of starting materials, is important for an efficient reaction protocol. In addition to side reactions, control of chemoselectivity is vital because unsaturated ketone substrates are known to undergo many different Lewis acid-mediated reactions, including carbonyl-ene, Prins, and enolate alkylations. We envisioned that the judicious choice of a metal-derived Lewis acid could address these challenges and function as an efficient activator for both oxetane formation and cycloreversion to provide carbonyl-olefin metathesis products. To identify a single Lewis acid capable of catalyzing the desired transformation, we initially investigated the reaction of β-ketoester 52 with various Lewis acids (Scheme 10). Overall, we found FeCl3 (Scheme 10, entry 1) to be the best catalyst for this reaction. Lowering the oxidation state of iron from iron(III) to iron(II) results in no conversion (Scheme 10, entry 2), which could be a result of FeCl2 having low solubility in dichloroethane. FeBr3, GaCl3, and BF3·Et2O (Scheme 10, entries 5, 6, and 8) all provide the desired metathesis product, albeit in lower yield than FeCl3. Considering the demonstrated potential for metal salts to contain catalytically relevant impurities, it was important to determine whether or not FeCl3 was functioning as the active catalytic species. When commercially available, sublimed grade FeCl3 (>99% trace metal basis) was used under otherwise identical reaction conditions, the quantitative formation of 53 was observed (Scheme 10, entry 11), matching the result obtained with reagent grade FeCl3 and validating FeCl3 as the active catalyst in this transformation. Finally, Brønsted acids (Scheme 10, entries 12 and 13) did not provide any conversion to the metathesis product, showing that metal-based Lewis acids are uniquely suited to serve as catalysts for this transformation.
Scheme 9.
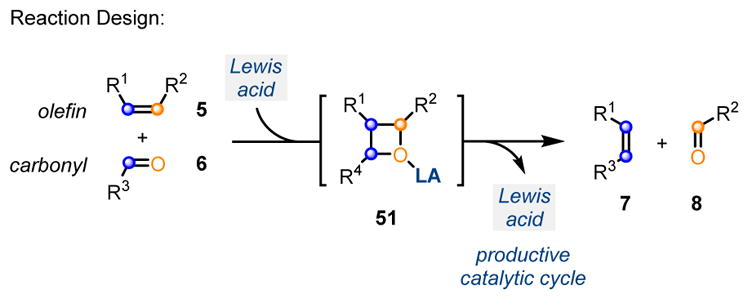
Design principle for Lewis acid-catalyzed carbonyl-olefin ring-closing metathesis.
Scheme 10.
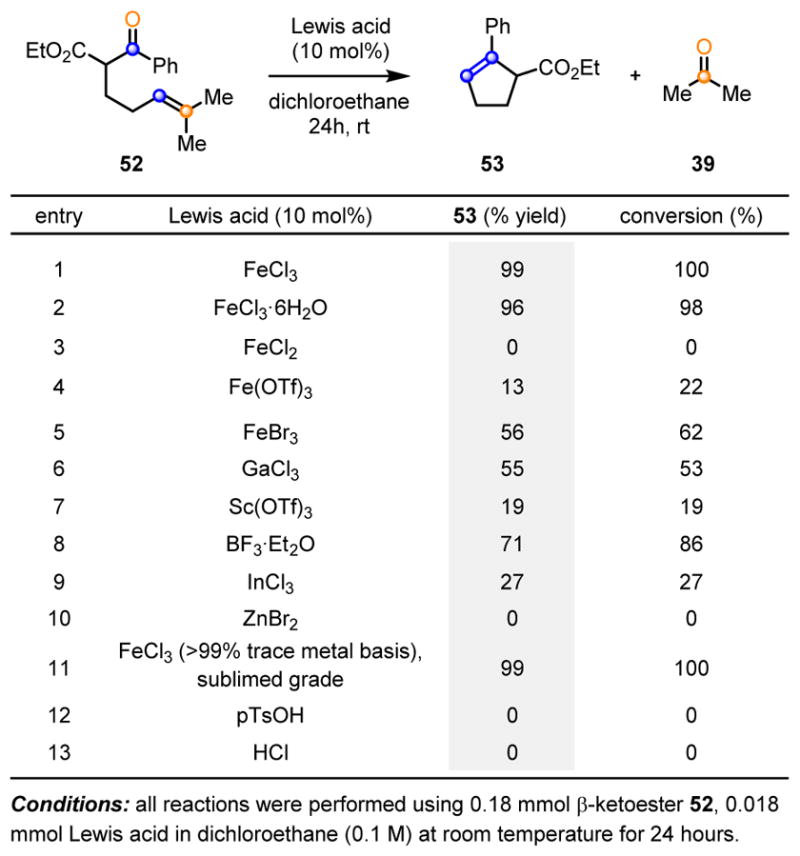
Evaluation of various Lewis acids and Brønsted acids in the catalytic carbonyl-olefin ring-closing metathesis of 52.
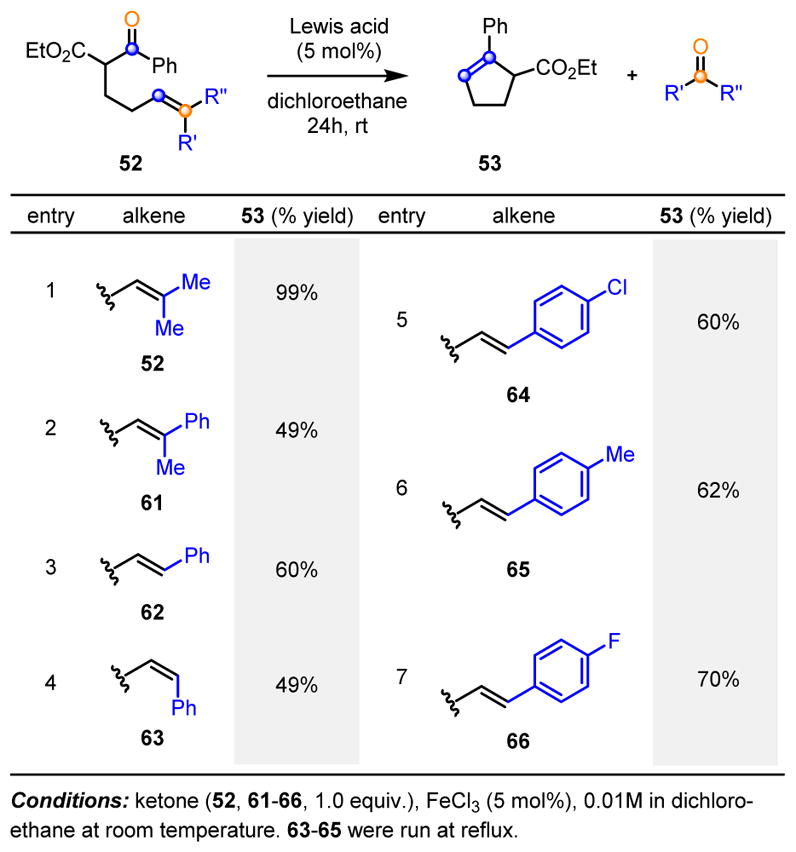
Additional efforts focused on mechanistic experiments to trap potential carbocation intermediates. Substrate 59 bearing a pendant alcohol with the potential to trap a benzylic carbocation intermediate 58 resulted in the isolation of the corresponding metathesis product 60 in 87% yield as a single product (Scheme 11). The iron(III)-catalyzed carbonyl-olefin metathesis reaction of 52 has also been conducted under the optimized reaction conditions with the addition of equimolar amounts of various nucleophiles (methanol, isopropanol, benzoic acid, acetic acid, nitromethane and acetonitrile, Scheme 11, entries 1–6) which have the potential to trap carbocation intermediates as the corresponding nucleophilic addition or subsequent elimination products. The desired carbonyl-olefin metathesis product 53 was isolated as the sole product in all cases in up to 98% yield. No formation of products resulting from intermediate carbocations was observed. The mechanism of this reaction was further studied computationally, using the ZStruct method.15 This method identified one highly favorable route from starting material to product, involving a concerted, asynchronous [2+2]-cycloaddition between the alkene and the carbonyl to give an oxetane and a concerted, asynchronous oxetane cycloreversion to give the desired metathesis product together with the carbonyl byproduct. We initially considered two possible mechanistic scenarios that both utilize oxetane 55 as a key intermediate. The pathway relying on carbocation intermediates (Scheme 12, right) starts with coordination of ketone 52 to FeCl3 resulting in intermediate 54 that is activated for nucleophilic addition by the nearby alkene to result in diastereomeric carbocations 56 and 57. Ring closure to oxetane 55 is feasible for carbocation 57 but geometrically difficult for 56, and it is therefore plausible that the two diastereomers interconvert through activated ketone 54, an explanation that is validated by the high yields observed in many cases. Ring opening of 55 provides benzylic carbocation 58, which can fragment to produce metathesis product 53 and carbonyl byproduct 39. Alternatively, a concerted reaction pathway (Scheme 12, left) relies on an iron-mediated [2+2]-cycloaddition to provide oxetane 55 that then endures a [2+2]-cycloreversion to form metathesis product 53 and carbonyl byproduct 39. Taken together, the carbocation trapping experiments and computational investigations point towards a concerted yet asynchronous reaction pathway where carbocation intermediates are not essential for catalysis. Subsequent investigation of the substrate scope revealed that a variety of aryl substituted alkene substrates 52 afford the expected metathesis product 53 and the corresponding benzaldehyde or acetophenone byproducts 39. All alkenes bearing aryl substituents resulted in diminished yields as compared to the dimethyl-substituted alkene, a result that can be explained as an increased stabilization of positive charge leading to less efficient reactivity. Importantly, both 62 (E)- and 63 (Z)-styrenes convert to cyclization product 53 showing that either alkene stereoisomer is a viable substrate. Additional investigation of the substrate scope focused on arene substituents, β-substitution, and chain length. Aryl and heteroaryl ketones bearing distinct substituents resulted in good yields of the desired metathesis products (Scheme 14, 67–72). Substrates incorporating a-quaternary carbon centers cyclize to the corresponding metathesis products (73–78) with remarkable efficiency. Additionally, a plethora of β-substituents are compatible with the optimized reaction conditions for carbonyl-olefin ring-closing metathesis to result in the formation of products containing ester (79, 80), amide (81), aryl ketone (82) and sulfone (83) fragments. Metathesis products 84–87 stem from substrates lacking a 1,3-dicarbonyl functionality, ultimately establishing that this moiety is not necessary for the carbonyl-olefin metathesis reaction to proceed. Finally, homologation of the alkyl chain enables access to cyclohexenes 89–92. Collectively, these examples demonstrate that the iron-catalyzed carbonyl-olefin metathesis reaction is general and highly tolerant of a variety of functional groups.
Scheme 11.
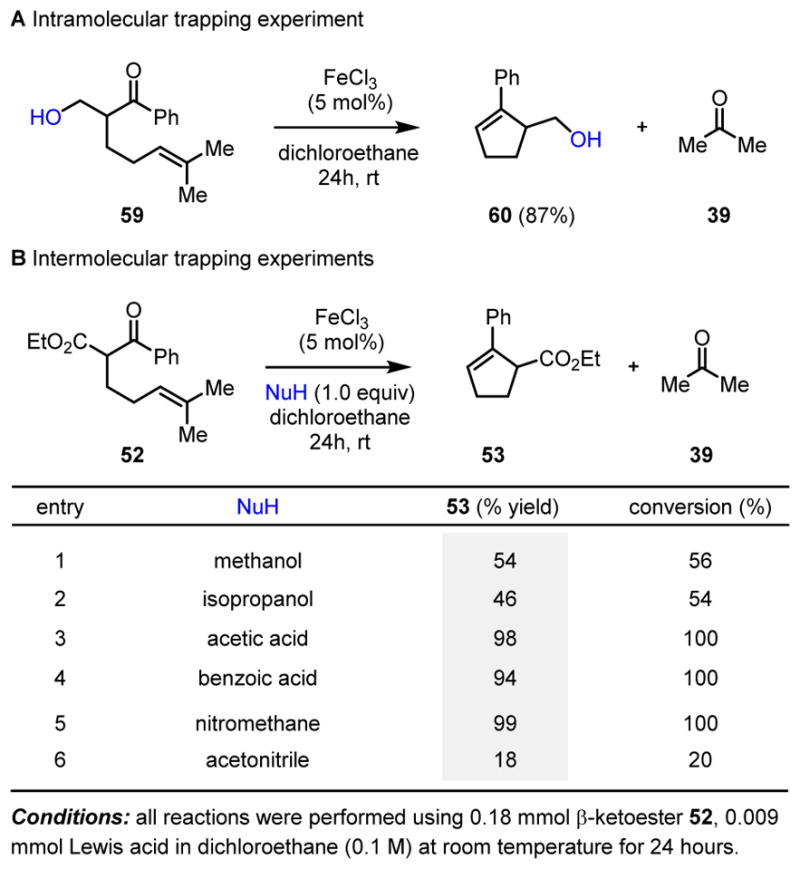
Intramolecular and intermolecular trapping experiments of the iron(III)-catalyzed carbonyl-olefin metathesis reaction.
Scheme 12.

Possible mechanistic pathways for the iron(III)-catalyzed carbonyl-olefin metathesis reaction.
Scheme 14.

Representative product scope of the iron(III)-catalyzed carbonyl-olefin ring-closing metathesis reaction.
After developing the carbonyl-olefin metathesis reaction for the synthesis of functionalized cyclopentenes and cyclohexenes, we became interested in applying this process to the synthesis of polycyclic aromatic compounds (PACs).16 PACs represent important structural motifs in materials science, natural product synthesis, and asymmetric catalysis. The optimized conditions for iron(III)-catalyzed carbonyl-olefin metathesis proved efficient for a wide range of functionalized biaryl ketones 93 to result in the formation of the desired metathesis products 94 in high yields (Scheme 15). Interestingly, while dimethyl-substituted olefins had previously proven superior for the carbonyl-olefin metathesis reaction described above, the styrenyl olefin 93 provided the most efficient conversion to PACs while avoiding carbonyl-ene side reactions. A variety of higher order polycyclic aromatics is accessible based on this carbonyl-olefin metathesis approach, including benzo[c]chrysene 98, pyrene 99, and picene 101. Furthermore, substrate 103, containing two olefins and two carbonyls, efficiently undergoes a double carbonyl-olefin metathesis reaction to give 102 as the single product in 95% yield (Scheme 16).
Scheme 15.
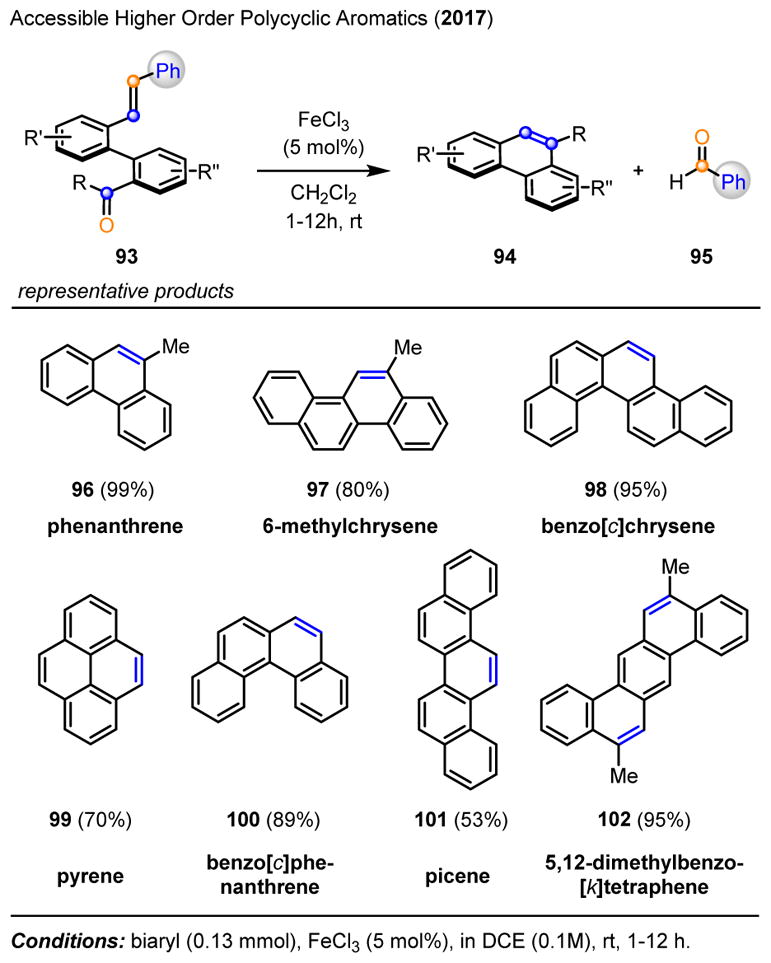
Representative examples of polycyclic aromatics accessible via the iron(III)-catalyzed carbonyl-olefin metathesis.
Scheme 16.
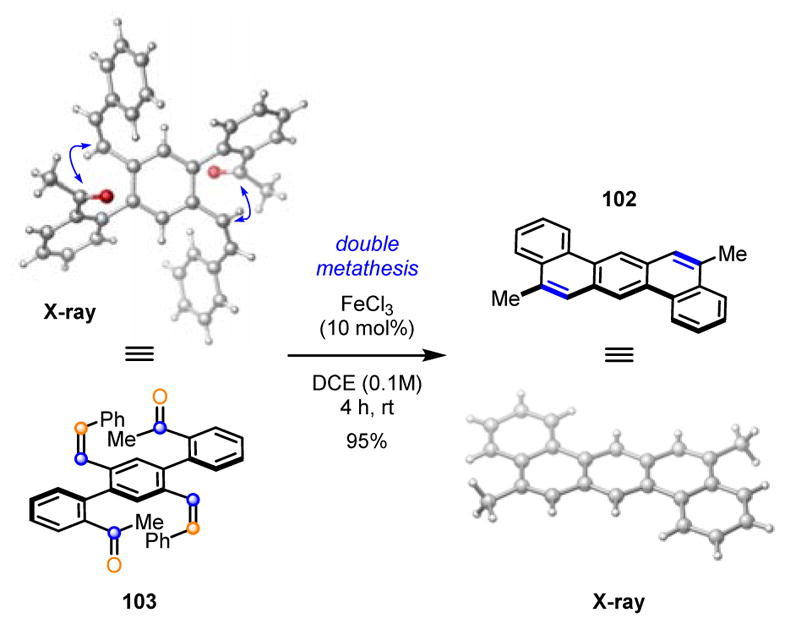
Double carbonyl-olefin metathesis approach for the synthesis of 5,12-dimethylbenzo[κ]tetraphene (102).
Catalytic carbonyl-olefin metathesis reactions have been a long sought after class of transformations in synthetic organic chemistry. Lewis acid-mediated approaches to carbonyl-olefin metathesis have seen a progression with early examples ranging from stoichiometric amounts of Lewis acids and harsh reaction conditions, to intermolecular examples relying on clay-supported Lewis acids resulting in moderate yields of the desired product. The first examples for Lewis acid-catalyzed carbonyl-olefin metathesis have recently been developed. Specifically, carbocation-based Lewis acids catalyze the formation of trans β-alkylstyrene products in carbonyl-olefin cross-metathesis reactions. The catalytic carbonyl-olefin ring-closing metathesis based on FeCl3 enables efficient access to a variety of 5- and 6-membered ring systems. The reaction relies on iron as an environmentally benign metal and provides a distinct advance towards the development of a sustainable and economical approach for carbonyl-olefin metathesis. This design principle for iron(III)-catalyzed carbonyl-olefin ring-closing metathesis has already enabled efficient access to a variety of complex molecules, including polycyclic aromatic hydrocarbons which are found as ubiquitous structural motifs in organic and pharmaceutical chemistry as well as materials science.
Scheme 13.
Reactivity of distinct alkene substituents in the iron(III)-catalyzed carbonyl-olefin metathesis reaction.
Acknowledgments
We thank the Petroleum Research Fund (PRF#54688-DNI1), the University of Michigan Office of Research and the NIH/National Institute of General Medical Sciences (GM118644) for financial support. C.S.S. thanks the David and Lucile Packard Foundation. J.R.L. thanks the National Science Foundation for a predoctoral fellowship.
Biographies
Jacob R. Ludwig grew up in Lake Orion Michigan and received his BSc in chemistry from Michigan State University in 2014 where he performed organic chemistry research in the laboratory of Prof. Jetze J. Tepe. After graduation, he joined the Schindler lab at the University of Michigan to pursue his PhD degree in organic chemistry.
Corinna S. Schindler grew up in Schwäbisch Hall, Germany and received her diploma in chemistry from the Technical University of Munich. After a research stay with Prof. K.C. Nicolaou at the Scripps Research Institute in La Jolla, CA, she joined the group of Prof. Erick M. Carreira at the ETH Zürich, Switzerland for her graduate studies. She then returned to the US to conduct postdoctoral studies with Prof. Eric N. Jacobsen at Harvard University before starting her independent career at the University of Michigan in 2013.
References
- 1.Grubbs RH, Wenzel AG. Handbook of Metathesis. 2. Wiley-VCH; 2015. pp. 1–3. [Google Scholar]
- 2.For metal-mediated carbonyl-olefin metathesis reactions, see: Schopov I, Jossifov C. Makromol Chem, Rapid Commun. 1983;4:659.Fu GC, Grubbs RH. J Am Chem Soc. 1993;115:3800.
- 3.For carbonyl-olefin metathesis reactions proceeding via oxetane photoadducts, see: Jones G, II, Schwartz SB, Marton MT. J Chem Soc, Chem Comm. 1973;11:374.Jones G, II, Acquadro MA, Carmody MA. J Chem Soc, Chem Comm. 1975;6:206.Carless HAJ, Trivedi HS. J Chem Soc, Chem Commun. 1979;8:382.D’Auria M, Racioppi R, Viggiani L. Photochem Photobiol Sci. 2010;9:1134. doi: 10.1039/c0pp00076k.Pérez-Ruiz R, Gil S, Miranda MA. J Org Chem. 2005;70:1376. doi: 10.1021/jo048708+.Pérez-Ruiz R, Miranda MA, Alle R, Meerholz K, Griesbeck AG. Photochem Photobiol Sci. 2006;5:51. doi: 10.1039/b513875b.Valiulin RA, Arisco TM, Kutateladze AG. J Org Chem. 2011;76:1319. doi: 10.1021/jo102221q.Valiulin RA, Arisco TM, Kutateladze AG. J Org Chem. 2013;78:2012. doi: 10.1021/jo301909j.
- 4.For catalytic carbonyl-olefin metathesis reactions proceeding via (3+2)/retro-(3+2)-cycloaddition, see: Griffith AK, Vanos CM, Lambert TH. J Am Chem Soc. 2012;134:18581. doi: 10.1021/ja309650u.Hong X, Liang Y, Griffith AK, Lambert TH, Houk KN. Chem Sci. 2014;5:471.
- 5.For the application of carbonyl-olefin metathesis in complex molecule synthesis, see: Stille JR, Grubbs RH. J Am Chem Soc. 1986;108:855.Stille JR, Santarsiero BD, Grubbs RH. J Org Chem. 1990;55:843.Nicolaou KC, Postema MHD, Claiborne CF. J Am Chem Soc. 1996;118:1565.Rainier JD, Allwein SP. J Org Chem. 1998;63:5310. doi: 10.1021/jo001514j.Rainier JD, Allwein SP, Cox JM. J Org Chem. 2001;66:1380. doi: 10.1021/jo001514j.Majumder U, Rainier JD. Tet Lett. 2005;46:7209.Iyer K, Rainier JD. J Am Chem Soc. 2007;129:12604. doi: 10.1021/ja073880r.Heller ST, Kiho T, Narayan ARH, Sarpong R. Angew Chem Int Ed. 2013;52:11129. doi: 10.1002/anie.201304687.Hong B, Li H, Wu J, Zhang J, Lei X. Angew Chem Int Ed. 2015;54:1011. doi: 10.1002/anie.201409503.
- 6.For a related approach, see: Ma L, Li W, Xi H, Bai X, Ma E, Yan X, Li Z. Angew Chem Int Ed. 2016;55:10410. doi: 10.1002/anie.201604349.
- 7.Demole E, Enggist P, Borer MC. Helv Chim Acta. 1971;54:1845. doi: 10.1002/hlca.19750580614. [DOI] [PubMed] [Google Scholar]
- 8.Jackson AC, Goldman BE, Snider BB. J Org Chem. 1984;49:3988. [Google Scholar]
- 9.van Schaik HP, Vijn RJ, Bickelhaupt F. Angew Chem Int Ed. 1994;33:1611. [Google Scholar]
- 10.Lee KY, Ko KY. Bull Korean Chem Soc. 2004;25:1929. [Google Scholar]
- 11.Khripach VA, Zhabinskii VN, Kuchto AI, Zhiburtovich YY, Gromak VV, Groen MB, van der Louw J, de Groot A. Tet Lett. 2006;47:6715. [Google Scholar]
- 12.Soicke A, Slavov N, Neudörfl JM, Schmalz HG. Synlett. 2011;17:2487. [Google Scholar]
- 13.Naidu VR, Bah J, Franzén J. Eur J Org Chem. 2015;8:1834. [Google Scholar]
- 14.This work was first reported as Ludwig JR, Gianino JB, Schindler C. Abstracts of Papers, 250th ACS National Meeting & Exposition; Boston, MA, United States. August 16th–20th, 2015; ORGN-388.; Ludwig JR, Zimmerman PM, Gianino JB, Schindler CS. Nature. 2016;533:374. doi: 10.1038/nature17432.
- 15.(a) Zimmerman PM. J Comput Chem. 2013;34:1385. doi: 10.1002/jcc.23271. [DOI] [PubMed] [Google Scholar]; (b) Zimmerman PM. J Chem Theory Comput. 2013;9:3043. doi: 10.1021/ct400319w. [DOI] [PubMed] [Google Scholar]
- 16.McAtee CC, Riehl PS, Schindler CS. J Am Chem Soc. 2017 doi: 10.1021/jacs.7b01114. [DOI] [PMC free article] [PubMed] [Google Scholar]