

Abstract
The quality of RNA sequencing data relies on specific priming by the primer used for reverse transcription (RT-primer). Nonspecific annealing of the RT-primer to the RNA template can generate reads with incorrect cDNA ends and can cause misinterpretation of data (RT mispriming). This kind of artifact in RNA-seq based technologies is underappreciated and currently no adequate tools exist to computationally remove them from published data sets. We show that mispriming can occur with as little as two bases of complementarity at the 3′ end of the primer followed by intermittent regions of complementarity. We also provide a computational pipeline that identifies cDNA reads produced from RT mispriming, allowing users to filter them out from any aligned data set. Using this analysis pipeline, we identify thousands of mispriming events in a dozen published data sets from diverse technologies including short RNA-seq, total/mRNA-seq, HITS-CLIP, and GRO-seq. We further show how RT mispriming can lead to misinterpretation of data. In addition to providing a solution to computationally remove RT-misprimed reads, we also propose an experimental solution to completely avoid RT-mispriming by performing RNA-seq using thermostable group II intron derived reverse transcriptase (TGIRT-seq).
Keywords: RNA sequencing (RNA-seq), reverse transcriptase, reverse transcription, mispriming, artifacts, GRO-seq, HITS-CLIP, short RNA-seq, TGIRT, RNA-binding, EZH2, polycomb repressive complex (PRC2)
INTRODUCTION
RNA-seq technologies are widely used to address biological questions relevant to transcriptional, cotranscriptional, and post-transcriptional regulation of gene expression. Some methods involve measurement of read coverage across an entire gene or exon while others utilize the specific positions of read pile-ups. A key step in all these RNA-seq technologies involves reverse transcription followed by library construction and sequencing. In some experiments, RNA adapters are first ligated to RNA 3′ ends followed by reverse transcription (RT) using a primer complementary to the ligated adapter (RT-primer). Alternatively, RT is first performed using random primers and then adapters are ligated to the cDNA molecules (Fig. 1A). The latter approach is utilized by standard Illumina TruSeq RNA-seq kits. The former is a cost-efficient approach to retain strand information and is thus used in many technologies (Levin et al. 2010; Hafner et al. 2012b; Podnar et al. 2014; Hrdlickova et al. 2017). Accurate cDNA synthesis relies on binding of the RT-primer specifically to the 3′ adapter ligated to RNA or the unbiased pairing of random primer to RNA. Nonspecific binding of the RT-primer can produce artefactual reads due to RT mispriming and lead to misinterpretations of read counts and cDNA lengths in RNA-seq experiments (Fig. 1A; van Gurp et al. 2013).
FIGURE 1.

Strategies for cDNA library preparation. (A) One approach involves reverse transcription with random primers first, followed by adapter ligations and sequencing (left). The other approach is to first sequentially ligate 3′ and 5′ adapters, then perform cDNA synthesis using a primer complementary to the adapter (RT-primer) followed by sequencing (right). On using RT-primer with a specific sequence, mispriming could occur due to annealing of the RT-primer to transcript sequences with some complementarity (RT mispriming). (B) Genome browser view showing enrichment of sequencing reads at a specific exon in EZH2 short RNA RIP compared to IgG IP and input controls across different cell lines and independent replicates (Rep). The inset shows sense-strand reads (red) mapping to the peak region.
Aside from a couple of publications, RT mispriming has not been recognized as a potential problem in transcriptome-wide studies. Experimental methods to avoid RT mispriming artifacts were recently proposed for NET-seq (native elongating transcript sequencing) and HITS-CLIP (high-throughput sequencing of RNA isolated by crosslinking and immunoprecipitation) (Ule et al. 2003; Chi et al. 2009; Mayer et al. 2015; Gillen et al. 2016). Although these methods will be useful for future experiments, there is still a need to identify and remove misprimed reads from existing data sets. Failure to account for or remove reads produced from mispriming during analysis of published data sets can lead to misinterpretation of data.
The current approach to identifying RT mispriming events involves looking for genomic regions close to cDNA peaks that are complementary to the first 6–7 bases of the RT-primer (matching the 3′ adapter) (Mayer et al. 2015; Gillen et al. 2016). We however find that mispriming can occur with just two bases followed by scattered complementarity to the RT-primer. Thus, existing approaches underestimate the extent of mispriming in the data. Here, we provide an analysis pipeline to remove RT-misprimed reads and apply this to several published data sets. Using this approach we identify RT mispriming events in data from multiple RNA-seq technologies including HITS-CLIP, short RNA-seq, total/mRNA-seq, and GRO-seq (global run-on followed by sequencing) and further show how RT mispriming could lead to misinterpretation of data (Danko et al. 2015). As an alternative to existing solutions, we propose cDNA library construction using the template-switching activity of novel thermostable group II intron-encoded reverse transcriptases (TGIRT-seq) as a reliable approach to avoid RT mispriming (Nottingham et al. 2016; Qin et al. 2016).
RESULTS
Short RNA sequencing experiments show spurious peaks from coding exons
To identify short RNAs that could potentially be interacting with EZH2, a chromatin modifier protein that also binds RNAs, we used a modified RIP-seq approach where we omitted all RNA digestion steps but instead size selected for 20–50 nt long RNAs (Zhao et al. 2010). We performed replicate short RNA RIP-seq experiments for EZH2 and analyzed them by comparing to two negative controls—an immunoprecipitation (IP) with nonspecific IgG and input RNA that was not subject to IP but otherwise processed in parallel—in two glioblastoma multiforme (GBM) cell lines. We found RNA sense-strand reads piling up as peaks localized to a short region within specific exons from several genes (Fig. 1B). Although we found several hundreds of these exonic cDNA peaks to be highly enriched in the EZH2 IP compared to the IgG control IP (fold change >2 and FDR-corrected P-value <0.05 using DESeq2), these peaks were detectable in both controls (Supplemental Fig. S1A).
Strikingly, the exonic cDNA peaks showed flush 3′ ends suggestive of mis-alignment or spliced reads (Figs. 1B, 2A; Supplemental Fig. S1A–C). The raw reads from these cDNA peaks however showed no evidence of a novel splice junction or misalignment. To further understand their identity we checked for sequence biases at genomic regions flanking the short RNA peaks. We found sequences similar to the first few bases of the 3′ adapter adjacent to the short RNA peaks (Supplemental Fig. S1B,C). The nucleotide composition at genomic regions adjacent to the 3′ ends of all EZH2-enriched short RNA peaks showed a clear bias for a sequence similar to the 3′ adapter (Fig. 2B). Recently, similar peaks enriched for the 3′ adapter sequence were identified in HITS-CLIP data that were tagged as false positive peaks produced by mispriming during reverse transcription (Gillen et al. 2016).
FIGURE 2.

Characteristics of EZH2 enriched cDNA peaks from short RNA-seq. (A) Heatmap showing EZH2-enriched cDNA peaks with flush ends. The heatmap shows the distribution of reads spanning 200 bases around EZH2-enriched peaks. The intensity of red color represents mapped read counts. Each row represents an EZH2-enriched cDNA peak. (B) Sequence logo showing sequence enrichment spanning 40 bp around an EZH2-enriched cDNA peak with its 3′ end at position 0. (C) A cDNA peak adjacent to a sequence with its first two bases matching the 3′ adapter followed by scattered matches. (D) Sequence matches between bases adjacent to the 3′ end of all EZH2-enriched cDNA peaks and the sequence of the 3′ adapter (showing up to 19 bases). (Top) Overall proportion of peaks showing a match at each position. (Bottom) Heatmap of individual matches. Filled cells represent a match and empty cells correspond to positions that do not match the 3′ adapter. Rows are ordered by the number of matches starting from the left, which corresponds to the 3′ end of the adapter.
Mispriming sites were previously identified by looking for regions matching the first 6–7 bases of the 3′ adapter proximal to cDNA peaks. We however found artefactual exonic cDNA peaks produced from genomic regions with only partial matches to the first 6–7 bases of the 3′ adapter. For 80% of all EZH2-enriched exonic cDNA peaks, only the first two bases matched the 3′ adapter, and for 48%, the first seven bases matched the 3′ adapter with two mismatches allowed (Fig. 2C,D; Supplemental Fig. S1C). This suggests that based on the criteria previously used (seven bases complementary to the RT-primer with two mismatches allowed), only 48% of the exonic cDNA peaks would be identified as a false positive mispriming artifact.
Pipeline to identify sites of mispriming from RNA sequencing data sets
The short RNA library preparation protocol involves ligation of the 3′ adapter followed by reverse transcription with an RT-primer complementary to the adapter (Hafner et al. 2012b; Luo 2012). False cDNA peaks are produced when the RT-primer binds to regions of complementarity on the RNA molecule and synthesizes cDNA (Fig. 3A). Based on the properties we observed for cDNA peaks in short RNA-seq experiments, we defined the following criteria to identify mispriming artifact peaks as distinct from true cDNA peaks (Fig. 3B). (i) Sites of mispriming should have at least two bases matching the 3′ end of the 3′ adapter, (ii) cDNA peaks produced from mispriming should have flush 3′ ends with at least 10 reads high pile-up and, (iii) There should be no other cDNA peak with 3′ flush ends resembling the misprimed peak but not matching the RT-primer within 20 bases flanking the misprimed peak. This is critical to avoid false mispriming calls at regions of high expression that likely contain many reads with flush ends as a result of high read density. We implemented these criteria in a computational pipeline to identify mispriming sites. The first step in the pipeline is alignment of sequencing reads using a global aligner (BWA). Since miRNAs and other small RNAs have defined ends, we then filter reads that do not map to non-protein-coding genes. From the filtered alignment file, we identify genomic positions where cDNA peaks with flush ends (>10 reads) are adjacent to (i) dinucleotides matching the 3′ adapter (k-mer sites) and, (ii) dinucleotides that do not match the 3′ adapter (non-k-mer sites). Finally, mispriming sites are identified as k-mer sites that do not contain a non-k-mer site within 20 bases. For data sets containing misprimed reads, a significant fraction of mispriming sites identified by our pipeline is expected to match more than the first two bases of the 3′ adapter.
FIGURE 3.
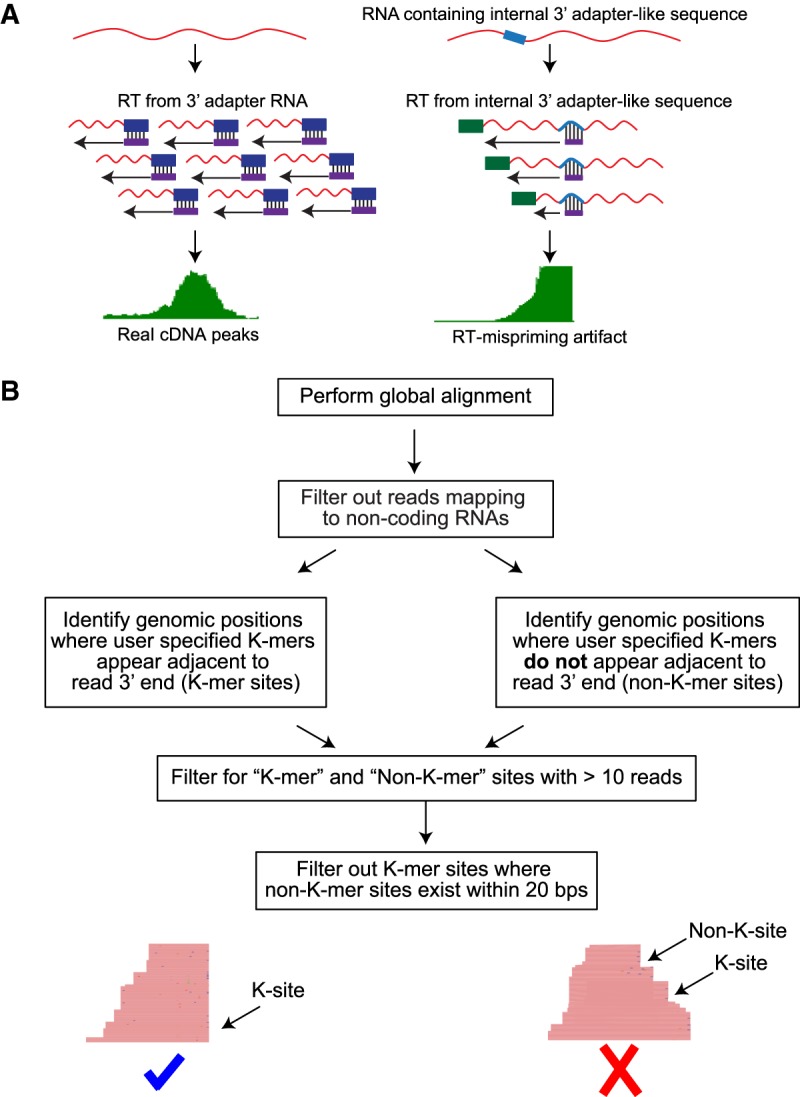
Identification of mispriming events in RNA-seq data sets. (A) Schematic comparing bonafide cDNA peaks with peaks from mispriming events. RNA molecules that are properly ligated and reverse transcribed from specific RT primer-3′ adapter interaction produce a pile-up of cDNA reads that have staggered ends (left). On the other hand, when RT-primer pairs with a sequence similar to the 3′ adapter present within an RNA molecule, cDNA peaks with flush ends next to the priming site are produced. (B) Pipeline to identify sites of mispriming.
With this approach we were able to identify mispriming sites in several short RNA-seq data sets with ∼95% success rate (∼1700 out of ∼1800 short RNA peaks containing two bases complementary to the RT-primer). As expected, we found enrichment for the 3′ adapter sequence downstream from mispriming sites identified by our pipeline in all short RNA-seq data sets (Supplemental Fig. S2A). By identifying precisely the sites of mispriming, we were also able to filter out reads that were produced as a result of RT mispriming (Supplemental Fig. S2B). In addition to short RNA peaks enriched in EZH2 IP samples, we were able to identify more than 10,000 mispriming sites per data set (Supplemental Table 1). We found that the number of mispriming sites decreased as the complexity of the library increased, with the input library showing the least amount of mispriming. We also checked for mispriming in an independent short RNA-seq data set that we downloaded from GEO (GSE68254) (Cass et al. 2016). Similar to our input short RNA-seq samples, we also observed thousands of mispriming sites in this published data set (Supplemental Table 2; Supplemental Fig. S2C). Since we were able to detect mispriming events in short RNA-seq input libraries, we suspected that mRNA-seq libraries might also be contaminated with misprimed reads. Using our pipeline, we identified several misprimed reads in total/mRNA-seq data sets we generated (T98G and U87MG cells) that coincided with the position of misprimed peaks from short RNA-seq experiments. Similarly, we also found several mispriming events in published mRNA-seq data sets that we downloaded from GEO (Supplemental Fig. S3).
RT mispriming occurs in multiple RNA-seq based technologies and leads to misinterpretation of data
In addition to short RNA-seq experiments, several technologies utilize 3′ adapter ligation followed by reverse transcription for library preparation (Fig. 1A) including HITS-CLIP, NET-seq, GRO-seq, total/mRNA-seq, RIP-seq, and RIBO-seq. Many of these technologies rely on specific positions of cDNA peaks to identify the binding sites of RNA binding proteins (CLIP based approaches), or RNA polymerase (GRO-seq and NET-seq), or ribosome footprinting sites (RIBO-seq). Since RT mispriming produces false cDNA peaks, we hypothesized that mispriming could lead to misinterpretation of data from sequencing technologies that rely on the specific position of cDNA peaks (Ule et al. 2003; Aspden et al. 2014; Danko et al. 2015; Mayer et al. 2015; Nojima et al. 2015). In HITS-CLIP, RT mispriming was recently shown to produce false peaks identified by looking for sequences matching the first 6–7 bases of the 3′ adapter in a 200 bp region spanning the cDNA peaks (Gillen et al. 2016). This approach would fail to identify mispriming sites that have scattered matches to the 3′ adapter beyond the first two bases. Using our pipeline, we were able to detect and filter out several mispriming sites in two independent published CLIP data sets (Fig. 4A–C; Supplemental Table 2; Haecker et al. 2012; Xue et al. 2013). In addition to mispriming sites with 6–7 base matches, we identified mispriming sites with scattered matches to the 3′ adapter that would be missed by the existing pipelines (Supplemental Fig. S4A,B). In order to check if RT mispriming could lead to false positive peak calls, we compared the number of peaks identified using the peak caller pyicoclip in one of the downloaded CLIP data sets before and after filtering for misprimed reads (Althammer et al. 2011). We detected ∼2.5% peaks as false positive peak calls that could be misinterpreted as binding sites.
FIGURE 4.

Mispriming can cause misinterpretation of binding sites identified from HITS-CLIP data sets. (A) Sequence logos showing sequence enrichment spanning 20 bp around mispriming sites identified by our pipeline for two published HITS-CLIP data sets. (B,C) View of a HITS-CLIP peak adjacent to a sequence matching the 3′ adapter.
We next applied our pipeline to identify mispriming events in GRO-seq data sets, another technology that relies on the specific position of cDNA peaks. GRO-seq is primarily used to identify sites of elongating RNA polymerase based on the position of cDNA peaks. High density of cDNA reads close to a gene′s transcription start site relative to the gene body indicates RNA polymerase promoter-proximal pausing. The extent of pausing is measured in terms of the pausing index (PI), calculated as the ratio of the number of reads per kilobase mapping close to transcription start sites (within 300 bp spanning the TSS) to reads mapping to the gene body (TSS + 250 bp to TSS + 2250 bp) (Min et al. 2011; Adelman and Lis 2012; Williams et al. 2015). In several GRO-seq data sets we were able to identify 10,000–50,000 mispriming sites accounting for millions of reads in some data sets (Fig. 5A,B; Supplemental Table 2; Supplemental Fig. S5; Andersson et al. 2014; Salony et al. 2016). Since GRO-seq utilizes the position-specific count of cDNA reads to detect RNA polymerase pausing, we suspected that artefactual fluctuations in read densities as a result of mispriming could lead to mis-identification of pause sites. To test for this, we identified and filtered out misprimed reads from a published GRO-seq data set and analyzed it for differences in pausing index before and after filtering. Differences in PI between unfiltered and filtered data would highlight cases of erroneous measurements of RNA polymerase pausing. For one of the GRO-seq data sets (GSE71898: SRR2153508), we found 230 protein-coding genes where the unfiltered data set showed at least twofold difference in PI values compared to the filtered data set (Fig. 5C,D; Salony et al. 2016). These protein-coding genes include cases where mispriming leads to higher PI (indicating promoter-proximal pausing) and some that show lower PI (indicating higher elongation rates).
FIGURE 5.

Mispriming can cause misinterpretation of RNA polymerase pausing. (A) Sequence logos showing sequence enrichment spanning 20 bp around mispriming sites identified by our pipeline for two published GRO-seq data sets. (B) View of GRO-seq peaks adjacent to sequence matching the 3′ adapter sequence. (C) Heatmap showing pausing index (PI) for 230 genes that show at least twofold difference in PI before and after filtering for mispriming reads. Rows are ordered by decreasing ratio of PI in filtered to PI in unfiltered data set. (D) Heatmap showing distribution of reads proximal to transcription start site and gene body in unfiltered (left) and filtered data set (right) for 230 genes shown in C.
RT mispriming can be avoided by using TGIRT-seq
One way to address sequencing artifacts arising from RT mispriming, as proposed for NET-seq and HITS-CLIP, is to include a degenerate barcode on the 5′ end of the 3′ adapter sequence (Mayer et al. 2015; Gillen et al. 2016). With this approach, misprimed reads can be removed by collapsing reads with identical 3′ adapter barcode sequences. Although this approach helps with filtering out mispriming-induced artifacts from downstream analysis, there would be a substantial loss of sequencing reads. As an alternative, we propose RNA-seq using thermostable group II intron-encoded reverse transcriptase (TGIRT-seq) to avoid RT mispriming. TGIRT-seq is a relatively new RNA-seq workflow that utilizes template switching to link 3′ adapter sequences to the synthesized cDNA. In contrast to other small RNA library preparation methods (Fig. 1A, right), TGIRT-seq synthesizes cDNA by template switching from a preannealed 3′ adapter RNA/DNA heteroduplex, skipping the priming step (Fig. 6A; Nottingham et al. 2016; Qin et al. 2016). Since this approach does not involve cDNA synthesis dependent on specific priming by RT-primer, we hypothesized that artifacts from RT mispriming would not occur in data generated using TGIRT-seq. To test this, we performed short RNA sequencing of immunoprecipitated RNA from nonspecific control IgG using TGIRT-seq. By performing TGIRT-seq on the control IP sample, we were able to compare short RNA peaks from coding exons with background peaks from other classes of short RNAs (miRNAs, snoRNAs, etc.) as an internal control. Although we found several thousand reads mapping to multiple classes of short RNAs, we were unable to detect short RNA peaks from coding exons we previously observed with the NEB small RNA library preparation kit (Fig. 6B). This shows that TGIRT-seq can help avoid RT mispriming without compromising on read coverage at transcripts that exist in the cell.
FIGURE 6.

Mispriming can be experimentally addressed with TGIRT-seq. (A) Overview of TGIRT-seq protocol. 3′ adapter RNA–DNA hybrid is annealed and used in a template switching reaction with TGIRT to synthesize cDNA molecules linked to the 3′ adapter. This is followed by ligation of 5′ adapter, PCR amplification, and sequencing. (B) Genome browser views of misprimed exonic cDNA peaks (left) and miRNA peaks (right) in a short RNA-seq library prepared with NEB small RNA library kit and TGIRT-seq.
DISCUSSION
RNA-seq based technologies are widely used to answer questions relevant to gene expression changes, protein–RNA interactions, RNA–RNA interactions, identifying RNA secondary structure and RNA polymerase dynamics during transcription (Ule et al. 2003; König et al. 2010; Hafner et al. 2012a; Ding et al. 2014; Rouskin et al. 2014; Mayer et al. 2015). The quality and accuracy of sequencing data relies on efficient priming by the RT-primer during cDNA synthesis, ligation of adapters and priming by primers during PCR amplification. Although sequencing bias during PCR amplification and adapter ligations have been previously addressed and solutions proposed, bias during reverse transcription is underappreciated (Schwartz et al. 2011; van Gurp et al. 2013). Data from RNA-seq technologies can be contaminated with incorrect cDNA ends as a result of mispriming by the primer used for reverse transcription (RT primer). If unaccounted for during analysis, misprimed reads can lead to misinterpretation of data. Here we developed an analysis pipeline aimed at identifying and eliminating misprimed reads from aligned RNA-seq data sets. An earlier approach to identifying mispriming events was based on looking for cDNA pile-ups adjacent to sequences matching the first 6–7 bases of the 3′ adapter (complementary to the RT primer). Based on detailed analysis of short RNA-seq data sets, we found that the RT-primer does not necessarily require base-pairing over a stretch of 6–7 bases to cause mispriming but instead can occur at sites with a match to only the first two bases followed by scattered complementarity. Thus, the earlier approach relying on a 6–7 base match is insufficient to identify all mispriming sites. We applied our mispriming identification pipeline to filter out mispriming artifacts in several published data sets from multiple technologies including short RNA-seq, total RNA-seq, HITS-CLIP, and GRO-seq. We further show how failure to remove misprimed reads could lead to misinterpretation of data.
In our short RNA-seq data sets, we found reads mapping to coding exons in addition to known classes of short RNAs (miRNAs, snoRNA, etc.). These cDNA read pile-ups at coding exons showed flush 3′ ends that ended exactly before sequence matching the 3′ adapter sequence that we had used to generate cDNA libraries. This is a key characteristic of a mispriming event that was previously shown for HITS-CLIP and NET-seq (Mayer et al. 2015; Gillen et al. 2016). In the case of both HITS-CLIP and NET-seq, mispriming artifacts were previously detected by finding sequences spanning a region around cDNA peaks that matched the first 6–7 bases of the 3′ adapter (complementary to the RT primer). We however were able to detect several cDNA peaks with only scattered complementarity beyond the first two bases of RT-primer. This suggests that the mispriming sites previously identified for HITS-CLIP and NET-seq represent only a subset of all mispriming sites.
Using our pipeline we were able to identify thousands of mispriming sites in multiple short RNA-seq data sets. We found that the number of mispriming sites were significantly lower for input libraries (short RNAs from total RNA pool) compared to immunoprecipitated samples. This is likely attributable to a relatively smaller pool of distinct RNA molecules in the immunoprecipitated sample. We next applied this pipeline to several published data sets from several technologies including short RNA-seq, total/mRNA-seq, HITSCLIP, and GRO-seq. We were able to detect hundreds to thousands of mispriming sites from multiple data sets. The extent of RT mispriming in different technologies can vary depending on the library complexity. RNA-seq is primarily performed to detect differential expression of genes and splicing changes across transcripts between two conditions. In both kinds of analysis, results are based on read coverage across a large region of a gene or transcript. Even though we were able to detect hundreds of mispriming events in multiple mRNA-seq data sets, they had negligible impact on gene expression and splicing (data not shown) (Lubas et al. 2015; Polioudakis et al. 2015). On the contrary, with technologies that rely on specific positions of cDNA peaks (HITS-CLIP and GRO-seq), the mispriming events that we identified showed a much greater impact on data interpretation. In the case of HITS-CLIP, we identified thousands of peaks that would be misidentified as binding sites, and for GRO-seq we identified several genes where mispriming could lead to misinterpretation of RNA polymerase elongation dynamics. This shows that RT mispriming affects multiple RNA-seq data sets and can lead to widespread misinterpretation of data.
One proposed experimental approach to avoid RT mispriming is to alter the 5′ end of the 3′ adapter sequence to contain degenerate barcodes that can later be used to collapse reads with identical 3′ adapter sequence. Although this approach helps remove misprimed reads from downstream analysis, this can lead to loss of sequencing data. As an alternative, we propose the use of TGIRT-seq that lacks the RT-priming step during library preparation. This approach completely eliminates mispriming artifacts from the library (Fig. 6B).
In this manuscript, we provide evidence for how RT mispriming contaminates and leads to misinterpretation of data for several RNA-seq libraries across multiple technologies. As a solution we provide an analysis pipeline to filter out misprimed reads from sequencing data that will be useful for future analysis utilizing published data sets. We also provide an alternative experimental approach to avoid RT mispriming during RNA-seq library preparation.
MATERIALS AND METHODS
Cell lines and reagents
T98G and U87MG (ATCC-CRL-1690 and ATCC-HTB14) were grown in EMEM with 10% FBS. All cell lines were maintained at 37°C and 5% CO2. Antibodies used were: EZH2 (Active Motif 39875) and IgG2a (Sigma Aldrich M5409).
Nuclear lysis
All RIP-seq experiments were performed using nuclear lysates. Cells were incubated in hypotonic solution (10 mM KCl, 10 mM HEPES pH 7.5, 1.5 mM MgCl2, and 2 mM DTT) for 5 min and spun down by centrifugation. A total of 1 mg/mL digitonin (Sigma Aldrich D141) was then added to the lysate resuspension and further incubated for 10 min with constant mixing. Cells were then mechanically lysed using a Dounce homogenizer (15 times) and spun down. Pelleted cells were then resuspended in NP40 lysis buffer (150 mM KCl, 50 mM HEPES pH 7.5, 5 mM EDTA, 0.5 % IGEPAL, and 2 mM DTT) and subjected to mild sonication (6 cycles, 10 sec on and 30 sec off). All the steps in this procedure were performed at 4°C.
RNA-seq and RIP-seq
RNA-seq experiments were performed on poly-A selected mRNA (Bioo 512980) as previously described (Polioudakis et al. 2015). RNA immunoprecipitations were performed on nuclear lysates from 4 × 150 cm2 plates of cells using 10 µg of specific antibody per experiment. Magnetic bead preparation and immunoprecipitations (IP) were performed as previously described (Polioudakis et al. 2015). 10% of the nuclear lysate was separated as input for total RNA-seq. Following IP, beads were resuspended in TRIzol (Thermo Fisher Scientific 15596026) and RNA was extracted as per manufacturer′s instructions. Small RNA RIP-seq libraries were prepared using NEBNext small RNA library preparation kit (NEB E7330) for IP and input. Following library preparation, 20–50 bases long products were size-selected from 6% TBE gel as per manufacturer′s instructions. TGIRT-seq libraries from IP and input RNA samples were prepared as previously described (Nottingham et al. 2016; Qin et al. 2016).
Identification of EZH2-enriched exonic cDNA peaks
Small RNA RIP-seq reads were mapped to the human genome (UCSC version hg19) using BWA, and reads mapping to exons were counted using bedtools (Quinlan and Hall 2010; Quinlan 2014). For GBM cells where RIP-seq experiments were performed in replicates, reads mapping to exons were compared to negative controls (IgG and input) using DESeq2 to identify exons significantly enriched in the EZH2 short RNA RIP (Love et al. 2014). To be identified as an EZH2-enriched coding exon, the number of normalized reads mapping to exons were required to be significantly higher (FDR-corrected P < 0.05) by at least twofold in the EZH2 RIP in comparison to both negative controls. In addition, exonic cDNA peaks were called using pyicoclip, and only peaks containing EZH2-enriched coding exon were included (Althammer et al. 2011).
Analysis of published data sets
All external data sets listed in Supplemental Table 2 were downloaded from GEO. Adapters from Fastq files were removed using cutadapt and mapped to the human genome (UCSC version hg19) using BWA (Li and Durbin 2009; Martin 2011). Mispriming sites were identified from aligned files using custom Python scripts, and misprimed reads were removed using bedtools. Scripts used for this analysis are available on Github (https://github.com/haridh/RT-mispriming). For HITS-CLIP, peaks were called using pyicoclip before and after filtering out misprimed reads. For GRO-seq, the pausing index at genes containing misprimed reads was compared before and after filtering out misprimed reads. The pausing index was calculated as the ratio of the number of reads per kilobase mapping close to transcription start sites (within 300 bp spanning TSS) and gene body (TSS + 250 bp to TSS + 2250 bp).
DATA DEPOSITION
Primary sequencing data generated in this study are available at NCBI's GEO database (www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE85163). Python scripts used to identify RT mispriming events are available on Github (github.com/haridh/RT-mispriming).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Nathan Abell for optimizing the nuclear lysis protocol, Anna Battenhouse for assistance with aligning sequencing data, Alan Lambowitz, Ryan Nottingham, and Douglas Wu for providing TGIRT-seq reagents, helpful discussion regarding the protocol and comments on the manuscript, Robert Darnell and Christopher Park for helpful discussions about RT mispriming, the Genomic Sequencing and Analysis Facility at UT Austin and the MD Anderson Cancer Center-Science Park NGS Facility for Illumina sequencing. We also thank the Texas Advanced Computing Center (TACC) at UT Austin for the use of computational facilities. This work was funded in part by grants from the Cancer Prevention and Research Institute of Texas (CPRIT RP120194) and the National Institutes of Health (NIH) (CA198648) to V.R.I. The Science Park NGS Facility was supported by CPRIT Core Facility support grant RP120348.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.066217.118.
REFERENCES
- Adelman K, Lis JT. 2012. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet 13: 720–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Althammer S, González-Vallinas J, Ballaré C, Beato M, Eyras E. 2011. Pyicos: a versatile toolkit for the analysis of high-throughput sequencing data. Bioinformatics 27: 3333–3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson R, Refsing Andersen P, Valen E, Core LJ, Bornholdt J, Boyd M, Heick Jensen T, Sandelin A. 2014. Nuclear stability and transcriptional directionality separate functionally distinct RNA species. Nat Commun 5: 5336. [DOI] [PubMed] [Google Scholar]
- Aspden JL, Eyre-Walker YC, Phillips RJ, Amin U, Mumtaz MAS, Brocard M, Couso J-P. 2014. Extensive translation of small Open Reading Frames revealed by Poly-Ribo-Seq. Elife 3: e03528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cass AA, Bahn JH, Lee J-H, Greer C, Lin X, Kim Y, Hsiao Y-HE, Xiao X. 2016. Global analyses of endonucleolytic cleavage in mammals reveal expanded repertoires of cleavage-inducing small RNAs and their targets. Nucleic Acids Res 44: 3253–3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi SW, Zang JB, Mele A, Darnell RB. 2009. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 460: 479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danko CG, Hyland SL, Core LJ, Martins AL, Waters CT, Lee HW, Cheung VG, Kraus WL, Lis JT, Siepel A. 2015. Identification of active transcriptional regulatory elements from GRO-seq data. Nat Methods 12: 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Tang Y, Kwok CK, Zhang Y, Bevilacqua PC, Assmann SM. 2014. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 505: 696–700. [DOI] [PubMed] [Google Scholar]
- Gillen AE, Yamamoto TM, Kline E, Hesselberth JR, Kabos P. 2016. Improvements to the HITS-CLIP protocol eliminate widespread mispriming artifacts. BMC Genomics 17: 338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haecker I, Gay LA, Yang Y, Hu J, Morse AM, McIntyre LM, Renne R. 2012. Ago HITS-CLIP expands understanding of Kaposi′s sarcoma-associated herpesvirus miRNA function in primary effusion lymphomas. PLoS Pathog 8: e1002884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Lianoglou S, Tuschl T, Betel D. 2012a. Genome-wide identification of miRNA targets by PAR-CLIP. Methods 58: 94–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Renwick N, Farazi TA, Mihailović A, Pena JTG, Tuschl T. 2012b. Barcoded cDNA library preparation for small RNA profiling by next-generation sequencing. Methods 58: 164–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrdlickova R, Toloue M, Tian B. 2017. RNA-seq methods for transcriptome analysis. Wiley Interdiscip Rev RNA 8: e1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, Turner DJ, Luscombe NM, Ule J. 2010. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol 17: 909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin JZ, Yassour M, Adiconis X, Nusbaum C, Thompson DA, Friedman N, Gnirke A, Regev A. 2010. Comprehensive comparative analysis of strand-specific RNA sequencing methods. Nat Methods 7: 709–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubas M, Andersen PR, Schein A, Dziembowski A, Kudla G, Jensen TH. 2015. The human nuclear exosome targeting complex is loaded onto newly synthesized RNA to direct early ribonucleolysis. Cell Rep 10: 178–192. [DOI] [PubMed] [Google Scholar]
- Luo S. 2012. MicroRNA expression analysis using the Illumina microRNA-seq platform. Methods Mol Biol 822: 183–188. [DOI] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal 17: 10. [Google Scholar]
- Mayer A, di Iulio J, Maleri S, Eser U, Vierstra J, Reynolds A, Sandstrom R, Stamatoyannopoulos JA, Churchman LS. 2015. Native elongating transcript sequencing reveals human transcriptional activity at nucleotide resolution. Cell 161: 541–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min IM, Waterfall JJ, Core LJ, Munroe RJ, Schimenti J, Lis JT. 2011. Regulating RNA polymerase pausing and transcription elongation in embryonic stem cells. Genes Dev 25: 742–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nojima T, Gomes T, Grosso ARF, Kimura H, Dye MJ, Dhir S, Carmo-Fonseca M, Proudfoot NJ. 2015. Mammalian NET-seq reveals genome-wide nascent transcription coupled to RNA processing. Cell 161: 526–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nottingham RM, Wu DC, Qin Y, Yao J, Hunicke-Smith S, Lambowitz AM. 2016. RNA-seq of human reference RNA samples using a thermostable group II intron reverse transcriptase. RNA 22: 597–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podnar J, Deiderick H, Huerta G, Hunicke-Smith S. 2014. Next-generation sequencing RNA-seq library construction. Curr Protoc Mol Biol 106: 4.21.1–4.21.19. [DOI] [PubMed] [Google Scholar]
- Polioudakis D, Abell NS, Iyer VR. 2015. MiR-191 regulates primary human fibroblast proliferation and directly targets multiple oncogenes. PLoS One 10: e0126535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y, Yao J, Wu DC, Nottingham RM, Mohr S, Hunicke-Smith S, Lambowitz AM. 2016. High-throughput sequencing of human plasma RNA by using thermostable group II intron reverse transcriptases. RNA 22: 111–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR. 2014. BEDTools: the Swiss-army tool for genome feature analysis. Curr Protoc Bioinformatics 47: 11.12.1–11.12.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouskin S, Zubradt M, Washietl S, Kellis M, Weissman JS. 2014. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature 505: 701–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salony, Solé X, Alves CP, Dey-Guha I, Ritsma L, Boukhali M, Lee JH, Chowdhury J, Ross KN, Haas W, et al. 2016. AKT inhibition promotes nonautonomous cancer cell survival. Mol Cancer Ther 15: 142–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Oren R, Ast G. 2011. Detection and removal of biases in the analysis of next-generation sequencing reads. PLoS One 6: e16685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, Darnell RB. 2003. CLIP identifies Nova-regulated RNA networks in the brain. Science 302: 1212–1215. [DOI] [PubMed] [Google Scholar]
- van Gurp TP, McIntyre LM, Verhoeven KJF. 2013. Consistent errors in first strand cDNA due to random hexamer mispriming. PLoS One 8: e85583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams LH, Fromm G, Gokey NG, Henriques T, Muse GW, Burkholder A, Fargo DC, Hu G, Adelman K. 2015. Pausing of RNA polymerase II regulates mammalian developmental potential through control of signaling networks. Mol Cell 58: 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Ouyang K, Huang J, Zhou Y, Ouyang H, Li H, Wang G, Wu Q, Wei C, Bi Y, et al. 2013. Direct conversion of fibroblasts to neurons by reprogramming PTB-regulated microRNA circuits. Cell 152: 82–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Ohsumi TK, Kung JT, Ogawa Y, Grau DJ, Sarma K, Song JJ, Kingston RE, Borowsky M, Lee JT. 2010. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol Cell 40: 939–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.