

Abstract
Tpt1 catalyzes the transfer of an internal 2′-monophosphate moiety (2′-PO4) from a “branched” 2′-PO4 RNA splice junction to NAD+ to form a “clean” 2′-OH, 3′-5′ phosphodiester junction, ADP-ribose 1″-2″ cyclic phosphate, and nicotinamide. First discovered as an essential component of the Saccharomyces cerevisiae tRNA splicing machinery, Tpt1 is widely distributed in nature, including in taxa that have no yeast-like RNA splicing system. Here we characterize the RslTpt1 protein from the bacterium Runella slithyformis, in which Tpt1 is encoded within a putative RNA repair gene cluster. We find that (i) expression of RslTpt1 in yeast complements a lethal tpt1Δ knockout, and (ii) purified recombinant RslTpt1 is a bona fide NAD+-dependent 2′-phosphotransferase capable of completely removing an internal 2′-phosphate from synthetic RNAs. The in vivo activity of RslTpt1 is abolished by alanine substitutions for conserved amino acids Arg16, His17, Arg64, and Arg119. The R64A, R119A, and H17A mutants accumulate high levels of a 2′-phospho-ADP-ribosylated RNA reaction intermediate (2′-P-ADPR, evanescent in the wild-type RslTpt1 reaction), which is converted slowly to a 2′-OH RNA product. The R16A mutant is 300-fold slower than wild-type RslTpt1 in forming the 2′-P-ADPR intermediate. Whereas wild-type RsTpt1 rapidly converts the isolated 2′-P-ADPR intermediate to 2′-OH product in the absence of NAD+, the H17A, R119A, R64A, and R16A mutant are slower by factors of 3, 33, 210, and 710, respectively. Our results identify active site constituents involved in the catalysis of step 1 and step 2 of the Tpt1 reaction pathway.
Keywords: 2′-phosphate splice junction, RNA repair, tRNA splicing
INTRODUCTION
Cells and viruses exploit a variety of RNA repair pathways to maintain or manipulate RNA structure in response to purposeful RNA breakage inflicted by transesterifying endoribonucleases, e.g., during physiological RNA processing (tRNA splicing) and under conditions of cellular stress (virus infection, unfolded protein response) (Popow et al. 2012). RNA cleavage by transesterification yields 2′,3′-cyclic-PO4 and 5′-OH ends. RNA repair enzymes capable of sealing 2′,3′-cyclic-PO4/5′-OH breaks are present in diverse taxa in all phylogenetic domains of life.
There are two styles of repair pathways that differ with respect to whether the RNA ligation event results either in a “clean” 3′-5′ phosphodiester at the repair junction or a junction in which there is a covalent modification of the 2′-OH adjacent to the newly formed 3′-5′ phosphodiester. The clean junction pathways are exemplified by: (i) the two-component bacteriophage T4 RNA repair system in which an end-healing enzyme polynucleotide kinase-phosphatase (Pnkp) converts 2′,3′-cyclic-PO4 and 5′-OH ends to 2′-OH,3′-OH and 5′-PO4 ends that are then sealed by an ATP-dependent 3′-OH/5′-PO4 RNA ligase (Rnl1) (Wang et al. 2002, 2003; Das and Shuman 2013); and (ii) the Escherichia coli GTP-dependent RNA ligase RtcB that joins 2′,3′-cyclic-PO4 and 5′-OH ends via RNA-3′-PO4 and RNA-(3′)pp(5′)G intermediates (Tanaka et al. 2011; Chakravarty et al. 2012).
There are two types of 2′-OH junction modifications that can occur during, or be coupled to, enzymatic RNA ligation. In the case of the Hen1-Pnkp RNA repair system found in diverse bacteria, the Hen1 methyltransferase installs a ribose 2′-OCH3 mark at the 3′ terminal nucleoside of the RNA break, such that subsequent ATP-dependent 3′-OH/5′-PO4 RNA ligation embeds the 2′-OCH3 mark at the splice junction (Chan et al. 2009a,b; Jain and Shuman 2010, 2011). The advantage of this system is that the 2′-OCH3 modification immunizes the repaired RNA against iterative cleavage by a transesterifying endoribonuclease.
The second type of junction modification pertains to the tRNA splicing system of fungi and plants, wherein a multidomain tRNA ligase catalyzes three reactions that heal and seal the 2′,3′-cyclic-PO4 and 5′-OH ends of the tRNA exons (Greer et al. 1983; Sawaya et al. 2003). In the healing phase of fungal tRNA splicing, the 5′-OH end is converted by a polynucleotide kinase to a 5′-PO4, and the 2′,3′-cyclic-PO4 is hydrolyzed to a 3′-OH,2′-PO4 end by a cyclic phosphodiesterase (CPDase). An ATP-dependent RNA ligase then joins these ends to form a spliced tRNA containing a 2′-PO4, 3′-5′ phosphodiester at the splice junction. The fungal/plant RNA ligase is unique among polynucleotide sealing enzymes in that ligation is strictly dependent on the 2′-PO4 moiety of the 3′-OH strand (Remus and Shuman 2013). Discrimination of 2′-PO4 versus 2′-OH ends provides a quality control checkpoint to ensure that only the broken RNAs that have undergone healing by the CPDase can be sealed. A potential disadvantage of this requirement is that the 2′-PO4 at the tRNA splice junction in the anticodon loop precludes proper tRNA function in codon recognition during translation (Spinelli et al. 1997). Thus, it is necessary to remove the junction 2′-PO4 as the final step in fungal tRNA splicing, a reaction performed by a dedicated tRNA 2′-phosphotransferase enzyme named Tpt1 that was discovered and characterized by the laboratory of Eric Phizicky (McCraith and Phizicky 1990, 1991).
Tpt1, which is essential for growth of the budding yeast Saccharomyces cerevisiae (Culver et al. 1997), catalyzes the transfer of the tRNA 2′-PO4 to NAD+ to form ADP-ribose 1″-2″ cyclic phosphate and nicotinamide (Culver et al. 1993). The Tpt1 mechanism entails two chemical steps (Spinelli et al. 1999; Steiger et al. 2005). First, NAD+ reacts with the tRNA 2′-phosphate to expel nicotinamide and generate a 2′-phospho-ADP-ribosylated RNA intermediate. Then, transesterification of the ADP-ribose 2′-OH to the tRNA 2′-phosphate displaces the tRNA product and generates ADP-ribose 1″-2″ cyclic phosphate. Tpt1 exemplifies a family of structurally homologous proteins distributed widely in eukaryal, archaeal, and bacterial proteomes (Spinelli et al. 1998). Because Tpt1 homologs are found in bacterial species (e.g., E. coli) that have no known intron-containing tRNAs and no known pathways to generate RNAs with internal 2′-PO4 modifications, it is possible that members of the Tpt1 family from such taxa may dephosphorylate other (non-RNA) substrates in vivo via a shared mechanism of phosphoryl transfer to NAD+ to form ADP-ribose 1″-2″ cyclic phosphate. However, it is certainly not the case that such Tpt1 homologs are specific for non-RNA substrates, insofar as the E. coli homolog (named KptA) can perform the same NAD+-dependent RNA 2′-phosphoryl transfer reactions in vitro as yeast Tpt1 (Spinelli et al. 1999; Steiger et al. 2001) and can complement an otherwise lethal S. cerevisiae tpt1Δ mutation in vivo (Spinelli et al. 1998; Sawaya et al. 2005).
An alternative scenario is that bacteria may indeed have a capacity to install internal RNA 2′-PO4 groups, conceivably during RNA repair pathways that are as yet uncharacterized, and that such RNAs are potential substrates for Tpt1. Our laboratory seeks to discover new RNA repair enzymes and pathways from diverse taxa by integrating biochemical and structural insights into known repair enzymes with phylogenetic analyses of cellular and viral proteomes. Strong clues to the existence of a novel repair pathway in microbial taxa comes from the genetic clustering of putative repair enzymes (e.g., prompting the discovery of the bacterial Hen1-Pnkp repair cassette) (Martins and Shuman 2005; Chan et al. 2009a,b; Jain and Shuman 2010, 2011). We recently uncovered and began characterizing a candidate four-gene repair cluster in the bacterium Runella slithyformis, comprising: (i) HD-Pnk, a bifunctional end-healing enzyme composed of an N-terminal 2′,3′-phosphoesterase HD domain and a C-terminal 5′-OH polynucleotide kinase domain (Munir and Shuman 2017); (ii) a putative ATP-dependent polynucleotide ligase, encoded by an ORF immediately upstream of HD-Pnk, such that the 3′ end of the ligase ORF overlaps the 5′ end of the HD-Pnk ORF; and (iii) a two-gene cassette, separated from the ligase ORF by a 152-nucleotide spacer, that encodes a homolog of Tpt1 and a predicted bifunctional AdoMet-dependent methyltransferase (MTase)–histidine-triad (HIT) phosphotransferase (Fig. 1). The HIT family includes repair enzymes that resolve abortive nucleic acid reaction intermediates generated by polynucleotide ligases (Tumbale et al. 2011).
FIGURE 1.

RslTpt1 genetic organization and tRNA 2′-phosphotransferase activity in yeast. (A) (Top panel) The ORF encoding Runella slithyformis Tpt1 is followed by three co-oriented ORFs encoding putative enzymes with likely roles in nucleic acid repair. (Bottom panel) The amino acid sequence of R. slithyformis (Rsl) Tpt1 (NCBI accession AEI48611) is aligned to that of Saccharomyces cerevisiae (Sce) Tpt1 (NCBI accession ABE08504). Positions of side chain identity/similarity are denoted by dots. Gaps in the alignments are denoted by dashes. Four conserved amino acids identified by alanine scanning as essential for SceTpt1 function in vivo are shown in white font on black background. The RslTpt1 residues Arg16, His17, Arg64, and Arg119 that were mutated to alanine in the present study are denoted by [ | ]. (B) Genetic complementation of S. cerevisiae tpt1Δ by expression of RslTpt1. Serial dilutions of tpt1Δ p[CEN SceTPT1], tpt1Δ p[CEN RslTPT1], and tpt1Δ p[2µ RslTPT1] cells recovered after plasmid shuffle were spotted to YPD agar plates, which were incubated at 20°C, 30°C, and 37°C.
We previously reported a purification and biochemical analysis of the HD-Pnk end-healing enzyme (Munir and Shuman 2017). Preliminary study of the ligase-like protein indicated that it reacts with ATP to form a covalent enzyme–AMP intermediate characteristic of classic polynucleotide ligases, but does not ligate any of the DNA and RNA substrates with 3′-OH and 5′-PO4 termini that we tested. Because the natural substrates for repair by the Runella HD-Pnk and ligase enzymes are not known, we tentatively hypopthesize that genetic clustering with a Tpt1 homolog might signal the participation of an RNA 2′-phosphotransferase in a novel bacterial repair pathway. To provide a foundation for this idea, we undertook a biochemical and genetic analysis of the Runella slithyformis Tpt1 enzyme. We show here that RslTpt1 is biologically active as an RNA 2′-phosphotransferase in vivo, as gauged by yeast tpt1Δ complementation, and biochemically active as an NAD+-dependent RNA 2′-phosphotransferase in vitro, by using a series of chemically synthesized RNAs with an internal 2′-PO4 modification. We define by alanine scanning four conserved amino acids that are essential for RslTpt1 function and illuminate their distinct roles during the two chemical steps of the Tpt1 reaction.
RESULTS
Runella slithyformis Tpt1 is biologically active as a tRNA 2′-phosphotransferase in yeast
An alignment of the amino acid sequences of S. cerevisiaie Tpt1 (SceTpt1, a 230-aa polypeptide) and Runella slithyformis Tpt1 (RslTpt1, a 183-aa polypeptide) highlights 89 positions of side chain identity/similarity (Fig. 1A). Prior studies had shown that Tpt1 is essential for growth of budding yeast by virtue of its requirement for removal of the 2′-PO4 at the tRNA splice junctions formed by yeast Trl1 (Spinelli et al. 1997; Schwer et al. 2004). To gauge if RslTpt1 can perform this function, we cloned the RslTPT1 ORF into yeast CEN and 2µ plasmids wherein its expression is driven by the S. cerevisiae TPI1 promoter. We tested complementation by plasmid shuffle in a S. cerevisiae tpt1Δ p[CEN URA3 TPT1] strain. RslTPT1 complemented tpt1Δ when expressed from either a single copy or multicopy plasmid. As shown in Figure 1B, the RslTPT1 strains grew as well as the SceTPT1 control when spot-tested on YPD agar medium at 20°C–37°C.
RslTpt1 has RNA 2′-phosphotransferase activity in vitro
Having affirmed that the RslTpt1 is biologically active in RNA processing, we produced RslTpt1 in E. coli and purified the protein as described in Materials and Methods. Our aim was to elucidate the biochemical activities of RslTpt1 and its reaction mechanism. The principal stumbling blocks to early mechanistic studies of Tpt1 were the difficulty in preparing a defined 2′-PO4 RNA substrate in sufficient quantity and the need for post-reaction processing steps for separation/quantification of substrates and products. Later, the Phizicky laboratory deployed chemically synthesized 2′-PO4 RNAs (Kierzek et al. 2000) as substrates for SceTpt1 and E. coli KptA, which facilitated dissection of the two-step reaction pathway shared by SceTpt1 and EcoKptA (Spinelli et al. 1999; Steiger et al. 2001, 2005).
To assay RslTpt1 activity in the present study, we synthesized a 15-mer RNA oligonucleotide containing an internal 2′-PO4 that corresponds to the splice junction of the anticodon stem-loop of yeast tRNATrp (Fig. 2A). The procedures for the chemical synthesis and analysis of RNA purity are described in Materials and Methods. The 15-mer 2′-PO4 RNA was 5′ 32P-labeled and gel-purified prior to use in Tpt1 activity assays. We expected to resolve the 2′-PO4 RNA substrate from the 2′-OH product via denaturing PAGE, but were frustrated to find that this could not be achieved for a 15-mer RNA. Thus, we had to improvise a new approach to detect removal of the 2′-PO4 from the 15-mer RNA.
FIGURE 2.

Recombinant RslTpt1 has RNA 2′-phosphotransferase activity in vitro. (A, left side) An internal 2′-PO4 is an obstacle to 3′ resection by polynucleotide phosphorylase. PNPase reaction mixtures (10 µL) containing 20 mM Tris-HCl (pH 7.5) 5 mM MgCl2, 30 µM (NH4)2HPO4, 20 nM (0.2 pmol) 5′ 32P-labeled 15-mer 2′-PO4 RNA (sequence shown at bottom), and increasing amounts of M. smegmatis PNPase as specified were incubated at 37°C for 15 min. The PNPase reactions were quenched by adding an equal volume of 90% formamide, 50 mM EDTA. (Right side of panel A) Removal of the 2′-PO4 by RslTpt1 eliminates the block to PNPase digestion. Tpt1 reaction mixtures (10 µL) containing 100 mM Tris-HCl (pH 7.5), 20 nM 5′ 32P-labeled 15-mer 2′-PO4 RNA, 1 mM NAD+ (where indicated by +), 5 mM MgCl2 (where indicated by +), and 0 or 1 pmol RslTpt1 as specified were incubated at 37°C for 30 min, then adjusted to contain 30 µM (NH4)2HPO4 and 5 pmol PNPase and incubated at 37°C for 15 min. The PNPase digestions were quenched by adding an equal volume of 90% formamide, 50 mM EDTA. The products were analyzed by electrophoresis (at 55 W constant power) through a 40-cm 20% polyacrylamide gel containing 7 M urea in 45 mM Tris-borate, 1 mM EDTA and visualized by autoradiography. A partial alkaline hydrolysis ladder of the 5′ 32P-labeled 15-mer 2′-PO4 RNA was analyzed in parallel in the rightmost lane; the alkaline ladder, when overexposed, provided markers to assign the size of the tetranucleotide PNPase reaction end product. The site in the RNA sequence at which PNPase halts 3′ digestion of the 15-mer 2′-PO4 RNA substrate to yield an 11-mer 2′-PO4 RNA is indicated at bottom by (|–––). The tetranucleotide generated by PNPase digestion after removal of the 2′-PO4 by RslTpt1 is indicated at bottom by the leftward arrow. (B) NAD+ titration. Tpt1 reaction mixtures (10 µL) containing 100 mM Tris-HCl (pH 7.5), 20 nM 5′ 32P-labeled 15-mer 2′-PO4 RNA, 5 mM MgCl2, and 0 or 1 pmol RslTpt1 as specified, and 0, 0.01, 0.1, 1, or 10 µM NAD+ were incubated at 37°C for 30 min, then adjusted to contain 30 µM (NH4)2HPO4 and 2.5 pmol PNPase and incubated for at 37°C for 15 min. The PNPase digestions were quenched by adding an equal volume of 90% formamide, 50 mM EDTA. The products were analyzed by urea-PAGE and visualized by autroradiography.
The solution to the problem of discriminating a 2′-PO4 from a 2′-OH was to treat the 5′ 32P-labeled RNA with purified recombinant Mycobacterium smegmatis polynucleotide phosphorylase (MsmPNPase) prior to RNA analysis by denaturing PAGE. MsmPNPase catalyzes iterative Mg2+- dependent phosphorolysis of RNA 3′ ends to liberate an NDP at each step (Unciuleac and Shuman 2013a). Processive phosphorolysis of 5′ 32P-labeled RNA by MsmPNPase generates a short radiolabeled oligonucleotide product (e.g., a trimer) derived from the 5′ end of the substrate RNA (Unciuleac and Shuman 2013b). PNPase spearheads a major pathway of 3′ RNA decay in bacteria. It was shown previously that internal tracts of deoxynucleotides within an RNA substrate elicit a kinetic obstacle to phosphorolysis by MsmPNPase (Unciuleac and Shuman 2013b). We suspected that MsmPNPase might also be affected by an internal 2′-PO4 group. Indeed, we found that 3′ resection of the 15-mer 2′-PO4 RNA was arrested after the removal of only four 3′-terminal nucleotides to yield an 11-mer 2′-PO4 RNA, i.e., MsmPNPase was unable to cleave the 3′–O–P bond vicinal to the 2′-PO4 (Fig. 2A).
The salient finding was that PNPase treatment of the RNA after it had been reacted in vitro with RslTpt1 in the presence of 1 mM NAD+ and 5 mM Mg2+ yielded a more rapidly migrating 5′ 32P-labeled tetranucleotide as the limit product of PNPase digestion under these conditions (Fig. 2A). This result indicates that Tpt1 effected quantitative removal of the 2′-PO4 obstacle to PNPase digestion. By systematic omission of components from the Tpt1 reaction mixture, we found that removal of the PNPase-blocking 2′-PO4 was strictly dependent on Tpt1 and NAD+ (Fig. 2A). To roughly gauge the dependence of Tpt1 activity on NAD+ concentration, we reacted 100 nM RslTpt1 with 20 nM 5′ 32P-labeled 15-mer 2′-PO4 RNA substrate in the absence of added NAD+ or in the presence of 0.01, 0.1, 1, or 10 µM NAD+, prior to treating the reaction mixtures with PNPase (Fig. 2B). The RNA was dephosphorylated completely in the presence of 10 or 1 µM NAD+. That fact that the Tpt1 reaction was 89% complete at 0.1 µM NAD+ (i.e., fivefold molar excess over the input RNA substrate and equimolar to input enzyme) attests to the efficiency with which NAD+ is used as the phosphate acceptor by RslTpt1. Even at 0.01 µM NAD+ (when NAD+, at half the concentration of the input RNA substrate, was limiting), 16% of the substrate was dephosphorylated, i.e., one-third of the input NAD+ was consumed by Tpt1.
To provide additional proof that Tpt1 removed the 2′-PO4 group, we subjected the substrate and the reaction product to partial alkaline hydrolysis, which generates a ladder of 5′ 32P-labeled RNAs by virtue of chemical transesterification at every phosphodiester with a vicinal ribose 2′-OH (Fig. 3A). The akaline ladder derived from the 15-mer 2′-PO4 RNA substrate has a “skipped” step corresponding to the position of the internal 2′-PO4 group. Reaction of the substrate with Tpt1 and NAD+ resulted in a gain of signal at this step in the alkaline ladder, to a level of hydrolysis commensurate with the flanking phosphodiesters (i.e., consistent with complete removal of the 2′-PO4). Restoration of the skipped step by Tpt1 depended on the inclusion of NAD+ in the reaction mixture, but did not require an added divalent cation (Fig. 3A). Collectively, the results of these experiments establish that RslTpt1 is a bona fide NAD+-dependent RNA 2′-phosphotransferase.
FIGURE 3.

RslTpt1 product analysis and assay of binding to 2′-PO4 RNA. (A) Reaction mixtures (10 µL) containing 100 mM Tris acetate (pH 6.0), 20 nM (0.2 pmol) 5′ 32P-labeled 15-mer 2′-PO4 RNA (shown at bottom), 1 mM NAD+ (where indicated by +), 5 mM MgCl2 (where indicated by +), and RslTpt1 as specified were incubated at 37°C for 30 min. Reaction components were omitted where indicated by –. For partial alkaline hydrolysis, the mixtures were adjusted to 40 mM NaHCO3 (pH 8.0) and 60 mM Na2CO3 (pH 10.5) and heated at 95°C for 5 min. The samples were mixed with an equal volume of 90% formamide, 50 mM EDTA and analyzed by urea-PAGE. A scan of the gel is shown. The arrowhead at right denotes the position of the alkaline-sensitive internucleotide phosphodiester resulting from removal of the internal 2′-PO4 by RslTpt1. (B) Reaction mixtures (10 µL) containing 100 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 0.1 µM (1 pmol) 5′ 32P-labeled 15-mer 2′-PO4 RNA, 5% (v/v) glycerol, and RslTpt1 as specified were incubated at 22°C for 10 min. The samples were supplemented with glycerol to 15% final concentration and then analyzed by electrophoresis (for 2 h at 110 V) through a 15-cm native 6% polyacrylamide gel in 22.5 mM Tris-borate buffer. An autoradiograph of the dried gel is shown. The positions of the free 15-mer RNA and a Tpt1•RNA complex of retarded mobility are indicated on the right.
To query whether RslTpt1 binds to the 15-mer 2′-PO4 RNA substrate in the absence of NAD+, we used a native gel electrophoretic mobility shift assay. Incubation of RslTpt1 with 0.1 µM labeled RNA resulted in the formation of a discrete protein–RNA complex that migrated more slowly than the free RNA during native PAGE (Fig. 3B). The extent of complex formation increased with input protein and was nearly complete at 1 and 2 µM RslTpt1. In separate gel-shift experiment comparing 5′ 32P-labeled 15-mer synthetic RNAs that had either a 2′-PO4 or a 2′-OH at the position corresponding to the anticodon-loop splice junction, we observed that formation of a gel-shifted RslTpt1•RNA complex depended on the 2′-PO4 group (Supplemental Fig. S1).
Direct assay of RslTpt1 activity with 6-mer and 3-mer 2′-PO4 RNA substrates
In order to simplify the Tpt1 assay format, we synthesized shorter RNA substrates with an internal 2′-PO4 (6-mer and 3-mer; see Fig. 4) that we predicted could be resolved by denaturing PAGE from their respective 2′-dephosphorylated derivatives. As shown in Figure 4, reaction of 100 nM Tpt1 with 100 nM 5′ 32P-labeled 6-mer and 3-mer 2′-PO4 RNA substrates resulted in their quantitative conversion to more slowly migrating species corresponding to the 2′-OH product. Removal of the 2′-PO4 from the 6-mer and 3-mer substrates required added NAD+ but not divalent cation (Fig. 4). Activity of RslTpt1 on a “minimal” trinucleotide RNA substrate with an internal 2′-PO4 is consistent with previous studies showing that E. coli KptA and yeast Tpt1 can efficiently dephosphorylate this trinucleotide (Spinelli et al. 1999; Steiger et al. 2001).
FIGURE 4.

Direct assay of RslTpt1 activity with 6-mer and 3-mer 2′-PO4 RNA substrates. (A) Reaction mixtures (10 µL) containing 100 mM Tris-HCl (pH 7.5), 0.1 µM (1 pmol) 5′ 32P-labeled 6-mer 2′-PO4 RNA or 3-mer 2′-PO4 RNA (shown at bottom), 1 mM NAD+ (where indicated by +), 5 mM MgCl2 (where indicated by +), and 0 or 1 pmol RslTpt1 as specified were incubated at 37°C for 30 min, then mixed with an equal volume of 90% formamide, 50 mM EDTA. The reaction products were analyzed by urea-PAGE and visualized by autoradiography. (B) Reaction mixtures (10 µL) containing 100 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 1 mM NAD+, 0.2 µM (2 pmol) 5′ 32P-labeled 6-mer 2′-PO4 RNA, and RslTpt1 as specified were incubated at 37°C for 30 min, then mixed with three volumes of cold 90% formamide, 50 mM EDTA. The reaction products were analyzed by urea-PAGE and visualized by autoradiography (top panel). The extent of 2′-OH RNA product formation was quantified by scanning the gel with a Fujifilm FLA-7000 imaging device and is plotted as a function of input RslTpt1 (bottom panel). Each datum in the graph is the average of three separate enzyme titration experiments ±SEM.
The extent of conversion of the 5′ 32P-labeled 6-mer 2′-PO4 RNA substrate into a 2′-OH product was proportional to the amount of RslTpt1 added (Fig. 4B). From the slope of the enzyme titration curve we calculated a specific activity of 470 fmol of 2′-OH RNA formed per fmol of RslTpt1 in a 30 min reaction, which translates into a turnover number of 15.7 min−1 (Fig. 4B).
Identification of conserved amino acids essential for RslTpt1 activity in vivo
Prior alanine-scanning mutational analysis of 14 amino acids of yeast Tpt1 that are conserved in homologous proteins from fungi, metazoa, protozoa, bacteria, and archaea identified four residues–Arg23, His24, Arg71, and Arg138–as essential for Tpt1 function in vivo (Sawaya et al. 2005). These amino acids are highlighted in white font on black background in Figure 1A. The activity of E. coli KptA in complementing tpt1Δ was abolished by alanine substitutions at the equivalent side chains: Arg21, His22, Arg69, and Arg125 (Sawaya et al. 2005). To gauge whether RslTpt1 relies on the same constellation of putative active site residues, we introduced alanines in lieu of RslTpt1 Arg16, His17, Arg64, and Arg119 and tested them by plasmid shuffle for tpt1Δ complementation. All four mutant alleles were lethal in vivo in yeast, i.e., they failed to give rise to FOA-resistant colonies at 37°C, 30°C, or 20°C (Fig. 5A and data not shown).
FIGURE 5.

Four conserved amino acids are essential for RslTpt1 activity in vivo. (A) Test of RslTPT1-Ala mutants for S. cerevsiae tpt1Δ complementation by plasmid shuffle. Yeast tpt1Δ p[CEN URA3 TPT1] cells were transformed with CEN plasmids bearing wild-type SceTPT1 (positive control), empty CEN vector (negative control), and wild-type RslTPT1 or the indicated RslTPT1-Ala mutant alelles. Single transformants were selected and streaked on agar medium containing 5-FOA. The plate was photographed after incubation for 7 d at 37°C. (B) Aliquots (5 µg) of recombinant RslTpt1 were analyzed by SDS-PAGE. The Coomassie blue-stained gel is shown. The positions and sizes (kDa) of marker polypeptides are indicated on the left. (C) Reaction mixtures (10 µL) containing 100 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 1 mM NAD+, 0.1 µM (1 pmol) 5′ 32P-labeled 6-mer 2′-PO4 RNA, and 0.05 µM (0.5 pmol) RslTpt1 (wild-type or mutant) were incubated at 37°C for 30 min, then mixed with an equal volume of 90% formamide, 50 mM EDTA. The products were resolved by urea-PAGE and visualized by autoradiography.
Mutational effects on RslTpt1 activity in vitro
To gauge the biochemical impact of the lethal alanine mutations, we produced and purified RslTpt1 proteins R16A, H17A, R64A, and R119A in parallel with the wild-type protein. Each of the Tpt1 proteins was subjected to a final Superdex-200 gel filtration step during which they eluted as monomers. SDS-PAGE analysis of the peak Superdex fractions verified that similar purity was attained for each preparation (Fig. 5B). An initial assay of the reaction of 0.5 pmol of RslTpt1 with 1 pmol of 5′ 32P-labeled 6-mer 2′-PO4 RNA revealed distinctive effects of each mutation on reaction outcome. Whereas wild-type RslTpt1 converted all of the 2′-PO4 substrate into a single 2′-OH product of slower mobility, the R64A and R119A mutants generated a novel species, migrating above the 2′-OH product (Fig. 5C), that we will show corresponds to the 2′-phospho-ADP-ribosylated RNA intermediate in the Tpt1 reaction pathway (Spinelli et al. 1999). The 2′-P-ADPR species was more abundant than the 2′-OH product in the R64A and R119A reactions (Fig. 5C). In contrast, the R16A mutant was virtually unreactive with the 6-mer 2′-PO4 RNA at this level of input enzyme. These results suggest that the three essential arginines might act at different steps in the Tpt1 reaction pathway. Although the H17A mutant converted most of the input 2′-PO4 substrate to 2′-OH product, residual 2′-P-ADPR RNA was detectable in this single-point assay.
Mutational effects on the kinetics of 2′-PO4 removal under conditions of enzyme excess
We tracked the time course of the reaction of 0.5 µM RslTpt1 (wild-type and mutants) with 0.2 µM 5′ 32P-labeled 6-mer 2′-PO4 RNA. The product analyses by PAGE are shown in Figure 6. The distribution of 2′-OH product and 2′-P-ADPR RNAs (expressed as the percentage of total labeled RNA) as a function of time are plotted in Figure 7, with each datum being the average of three independent experiments. Wild-type RslTpt1 converted 75% of the 2′-PO4 substrate to 2′-OH product in 5 sec (the earliest time sampled in the experiment shown in Fig. 6) and conversion was complete in 10 sec. In reactions sampled at 3 sec, wild-type RslTpt1 generated 65% 2′-OH RNA and 4.6% 2′-P-ADPR RNA (Fig. 7). Thus, it seems that step 2 of the 2′-phosphotransferase pathway (Fig. 6) catalyzed by wild-type RslTpt1 is faster than step 1. We derived an apparent step 1 rate constant of 21.7 min−1 for wild-type RslTpt1 (Fig. 7, bottom right panel) by plotting the sum of the 2′-OH and 2′-P-ADPR RNAs as a function of time and fitting the data by nonlinear regression to a one-phase association.
FIGURE 6.

Mutational effects on the kinetics of 2′-PO4 removal. Reaction mixtures containing 100 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 1 mM NAD+, 0.2 µM 5′ 32P-labeled 6-mer 2′-PO4 RNA, and 0.5 µM RslTpt1 (wild-type or mutant as specified) were incubated at 37°C and quenched at the times specified (above the lanes) by addition of 3 volumes of cold 90% formamide, 50 mM EDTA. The reaction products were analyzed urea-PAGE and visualized by autoradiography. The inset box depicts the 6-mer 2′-PO4 RNA substrate, 2′-P-ADPR RNA intermediate, and 2′-OH RNA product of the two-step Tpt1 reaction.
FIGURE 7.

Step-specific effects of mutations on the kinetics of 2′-PO4 removal. Assays were performed as described in the legend to Figure 6. The reaction products were analyzed by urea-PAGE and quantified by scanning the gels. The distributions of the 2′-P-ADPR RNA intermediate and 2′-OH RNA product (expressed as percentage of total RNA) are plotted as a function of reaction time. Each datum in the graphs is the average of three independent time course experiments ±SEM. The apparent rate constants for step1 shown in the table at bottom right were derived by plotting the sum of the 2′-P-ADPR RNA intermediate and 2′-OH RNA product as a function of time and fitting the data by nonlinear regression to a one-phase association in Prism.
The kinetic profile of the R64A enzyme was profoundly different. R64A converted 78% of the 2′-PO4 substrate to 2′-P-ADPR RNA in 15 sec, after which the 2′-P-ADPR RNA was transformed slowly into 2′-OH RNA over the remainder of a 30 min reaction (Figs. 6 and 7). These data establish a clear precursor-product relationship between the 2′-P-ADPR RNA reaction intermediate and the 2′-OH RNA end product (as depicted in the two-step reaction pathway in Fig. 6) and that replacing Arg64 with alanine selectively slows step 2 of the pathway. The apparent step 1 rate constant of 7.68 min−1 for the R64A mutant (Fig. 7, bottom right panel) was threefold slower than that of the wild-type RslTpt1.
The H17A mutant was faster at end-product formation than R64A, insofar as virtually all of the input 2′-PO4 RNA was converted to 2′-OH RNA in 5 min (Fig. 6). H17A accumulated the 2′-P-ADPR intermediate, which comprised 31% of the total RNA at 30 and 45 sec (Figs. 6 and 7). The H17A step 1 rate constant of 3.49 min−1 was sixfold slower than the wild-type rate.
The R119A mutant also displayed a step 2 kinetic defect, seen as the early accumulation of the 2′-P-ADPR intermediate (peaking at 48%–49% of total RNA at the 2 and 3 min sampling times) and its subsequent slow conversion to 2′-OH product over 30 min, with the additional feature that the rate of consumption of the substrate was slowed compared to wild-type, R64A, and H17A (Figs. 6 and 7). The R119A step 1 rate constant of 0.81 min−1 was 27-fold slower than the wild-type rate.
The R16A enzyme exhibited a much different defect, whereby it effected a very slow and incomplete consumption of the 2′-PO4 substrate over a 30 min reaction that yielded a mixture of 2′-P-ADPR RNA and 2′-OH product (Figs. 6 and 7). Thus, the R16A mutation drastically affects the first step of the Tpt1 reaction pathway. The R16A step 1 rate constant of 0.073 min−1 was 300-fold slower than the wild-type rate.
Isolation of the 2′-P-ADPR RNA intermediate allows study of step 2 per se
We scaled up the reaction of RslTpt1-R64A with the 5′ 32P-labeled 6-mer 2′-PO4 RNA substrate and then gel-purified the 2′-P-ADPR RNA intermediate. The experiment in Figure 8A shows that a 1-min incubation of 500 nM wild-type RslTpt1 with 50 nM isolated 2′-P-ADPR RNA in the absence of NAD+ elicited its quantitative conversion to a 2′-OH RNA product. The H17A mutant also effected complete formation of 2′-OH product in 1 min (Fig. 8A). In contrast, the R119A and R64A proteins achieved 56% and 22% conversion of 2′-P-ADPR RNA to 2′-OH RNA in 1 min, which increased to 97% and 75% conversion after 10 min. The R16A protein converted one-fourth of the input 2′-P-ADPR RNA to 2′-OH RNA in 10 min (Fig. 8A).
FIGURE 8.

NAD+-independent conversion of 2′-P-ADPR RNA intermediate to 2′-OH product. (A) Reaction mixtures containing 100 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 0.05 µM isolated 5′ 32P-labeled 6-mer 2′-P-ADPR RNA (lanes 2′-P-ADPR), and 0.5 µM RslTpt1 (wild-type or mutant as specified) were incubated at 37°C and quenched at the times specified (1 min or 10 min) by addition of 3 volumes of cold 90% formamide, 50 mM EDTA. RslTpt1 was omitted from the reaction in lane –. The reaction products were analyzed urea-PAGE and visualized by autoradiography. Control reactions containing 0.05 µM 5′ 32P-labeled 6-mer 2′-PO4 RNA substrate, 1 mM NAD+, and either 0.5 µM RslTpt1 (2′-P, lane WT) or no added enzyme (2′P, lane [–]) were analyzed in parallel in the leftmost lanes. (B) Reaction mixtures containing 100 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 0.05 µM 5′ 32P-labeled 6-mer 2′-P-ADPR RNA, and 0.5 µM RslTpt1 (wild-type or mutant as specified) were incubated at 37°C and quenched at the times specified by addition of 3 volumes of cold 90% formamide, 50 mM EDTA. The reaction products were analyzed by urea-PAGE and quantified by scanning the gels. The extents of 2′-OH RNA product formation (expressed as percentage of total RNA) are plotted as a function of reaction time with x-axis units in seconds (left panel) or minutes (right panel). Each datum in the graphs is the average of three independent time course experiments ±SEM. The curves and apparent step 2 rate constants were obtained by fitting the data by nonlinear regression to a one-phase association in Prism.
A finer kinetic analysis of the isolated step 2 reaction of the wild-type and mutant enzymes is shown in Figure 8B. The wild-type, H17A, R119A, and R64A reactions all proceeded to completion on a time-scale ranging from 7 sec (wild-type) to 30 min (R64A). The R16A reaction reached an apparent endpoint of 58% conversion of 2′-P-ADPR RNA to 2′-OH RNA after 80 min. The kinetic profiles were fit to a one-phase association to derive apparent step 2 rate constants as follows: WT (34.9 min−1); H17A (11.8 min−1); R119A (1.05 min−1); R64A (0.163 min−1); R16A (0.049 min−1). Thus, the H17A, R119A, R64A, and R16A mutations slowed the rate of the isolated step 2 reaction by factors of 3, 33, 210, and 710, respectively.
Mutational effects on 2′-phophotransferase specific activity
The studies of mutational effects on RslTpt1 activity in the preceding sections were performed under conditions in which RslTpt1 was present in molar excess over the RNA substrate, a state unlikely to apply in vivo when the RslTpt1 mutants were tested for tpt1Δ complementation in yeast and found to be lethal. In the experiments shown in Figure 9A–D, the impact of the four lethal alanine changes on 2′-phosphotransferase specific activity was gauged by enzyme titration across a broad range spanning substoichiometeric to super-stoichiometeric RslTpt1:RNA conditions. The H17A, R119A, R64A, and R16A proteins generated substantial amounts of 2′-P-ADPR RNA intermediate at the lower range of input enzyme tested. The intermediate declined and the 2′-OH RNA product accumulated steadily as the H17A, R119A, and R64A protein concentrations were increased, with nearly complete conversion to 2′-OH product being achieved at the highest RslTpt1 levels tested. The specific activities of the mutants in forming the 2′-OH RNA product were calculated from the slopes of the titration curves in the linear range of enzyme dependence, as determined by linear regression in Prism. The specific activity values, expressed as pmol of 2′-OH RNA generated per pmol of input RslTpt1 in a 30 min reaction, were as follows: 3.59 ± 0.22 for H17A; 0.751 ± 0.056 for R119A; 0.90 ± 0.035 for R64A; and 0.098 ± 0.0059 for R16A. Compared to the wild-type RslTpt1 specific activity of 470 (Fig. 4B), the H17A, R119A, R64A, and R16A mutants exhibited specific activity decrements of 130-fold, 625-fold, 520-fold, and 4800-fold, respectively.
FIGURE 9.

Mutational effects on 2′-phophotransferase specific activity. Reaction mixtures (10 µL) containing 100 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 1 mM NAD+, 0.2 µM (2 pmol) 5′ 32P-labeled 6-mer 2′-PO4 RNA, and RslTpt1 mutants H17A (panel A), R119A (panel B), R64A (panel C), and R16A (panel D) as specified were incubated at 37°C for 30 min, then mixed with 3 volumes of cold 90% formamide, 50 mM EDTA. The reaction products were analyzed by urea-PAGE. The extents of 2′-P-ADPR RNA intermediate and 2′-OH RNA product formation were quantified by scanning the gels and are plotted as a function of input RslTpt1. Each datum in the graphs is the average of three separate enzyme titration experiments ±SEM.
DISCUSSION
The present study enhances our understanding of the two-step pathway of NAD+-dependent RNA 2′-phosphate removal by Tpt1 enzymes, whereby attack of the 2′-phosphate on the nicotinamide ribose of NAD+ leads to the formation of a 2′-phospho-ADP-ribosylated RNA intermediate that is subsequently converted to an RNA2′OH product via transesterification of the phosphate to the ribose O2″ (Spinelli et al. 1999; Steiger et al. 2005). We characterize the Tpt1 enzyme from the bacterium Runella slithyformis as a bona fide NAD+-dependent RNA 2′-phosphotransferase that can perform this essential reaction in vivo in budding yeast and in vitro using as substrates short synthetic RNAs with an internal 2′-phosphate. The biological activity of RslTpt1 relies on a conserved histidine and three conserved arginines that were shown to be essential for the in vivo activity of S. cerevisiae Tpt1 and E. coli KptA (Sawaya et al. 2005). However, the earlier structure-function studies lacked a biochemical correlate, because an idoneous RNA substrate for SceTpt1 and EcoKptA was not in hand. Here we find that the biologically defective RslTpt1 mutants R16A, H17A, R64A, and R119A display distinctive biochemical defects that are mechanistically informative, as discussed below.
The properties of wild-type RslTpt1 highlight its efficiency. Under conditions of RNA substrate excess, RslTpt1 performed 470 rounds of 6-mer RNA 2′-dephosphorylation per enzyme during a 30 min reaction at 37°C, without detectable accumulation of a 2′-phospho-ADP-ribosylated RNA intermediate in the linear range of the enzyme titration. Under conditions of enzyme excess, RslTpt1 converted the 6-mer substrate to product in just a few seconds. After 3 sec (the earliest time we could sample manually), the 2′-OH product and 2′-P-ADPR intermediate comprised 65% and 4.6% of the total RNA, respectively, suggesting that step 1 is likely to be rate-limiting. Indeed, the kinetic data for wild-type RslTpt1 (in Fig. 7) fit well by linear regression to a unidirectional two-step mechanism with step 1 and step 2 rate constants of 21.0 ± 0.62 min−1 and 221 ± 51 min−1, respectively (i.e., step 2 is 10-fold faster than step 1). Consistent with formation of the 2′-P-ADPR RNA intermediate being rate-limiting, the single-turnover step 1 rate constant of 21 min−1 was similar to the turnover number of 15.7 min−1 observed by enzyme titration under steady-state conditions. Note that the turnover number of RslTpt1 with the 6-mer substrate pCCAA2′pAU was similar to the kcat value of 19 min−1 reported for SceTpt1 with the 8-mer substrate pGUAA2′pAUCU (Steiger et al. 2001).
The intermediacy of the 2′-P-ADPR species was affirmed by the ability of wild-type RslTpt1 to effect rapid and complete conversion of the gel-purified 6-mer 2′-P-ADPR RNA to the 2′-OH RNA product in the absence of NAD+. However, the apparent single-turnover rate constant of 34.9 min−1 for step 2 in isolation was sixfold slower than the step 2 rate derived for the reaction of RslTpt1 with 2′-PO4 RNA substrate at the same concentration of input enzyme (0.5 µM). We suspect that the isolated step 2 reaction with 2′-P-ADPR RNA in solution may be subject to a rate-limiting RNA binding (or precatalytic conformational) step that does not apply when the 2′-P-ADPR intermediate is formed in situ on the enzyme.
The four RslTpt1-Ala mutants studied here had grossly reduced specific activities as 2′-phosphotransferases in vitro, as gauged by enzyme titrations. Given the likelihood that Tpt1 must act iteratively in vivo to handle the flux of 2′-phosphate-containing splice junctions formed by yeast tRNA ligase, we would attribute the lethality of the RslTpt1-Ala mutants in yeast to their reduced specific activities, which could reflect a defect in substrate recognition, chemical catalysis, or a combination thereof. To focus on mutational effects on the chemical steps, we tracked the kinetics of the reaction of the RslTpt1-Ala proteins with the 6-mer 2′-PO4 RNA substrate under conditions of enzyme excess. We thereby found that, unlike wild-type RslTpt1, the H17A, R119A, and R64A mutants accumulated substantial levels of the 2′-P-ADPR intermediate, which comprised 31%, 50%, and 78% of the total RNA at their peak times, before slowly converting the intermediate to 2′-OH product. Deriving apparent step 1 rate constants by fitting the sum of the intermediate and product RNAs versus time to a one phase association showed that the rates of step 1 for H17A, R64A, and R119A were slower than wild-type RslTpt1 by factors of 6, 3, and 27, respectively. A more severe defect was seen for the R16A mutant, which slowly generated intermediate and product in roughly equal amounts over a 30 min period and for which the apparent step 1 rate constant was 300-fold slower than wild-type.
It is noteworthy that none of the temporal reaction profiles of the RslTpt1-Ala mutants fit well to a simple unidirectional two-step kinetic mechanism, the problem being that the observed decay of the the 2′-P-ADPR species and conversion to product at later reaction times was too slow for a good fit. The “least poor” of the fits were seen for the R64A (k1 of 5.4 ± 0.9 min−1 and k2 of 0.089 ± 0.005 min−1) and R119A (k1 of 0.62 ± 0.07 min−1 and k2 of 0.15 ± 0.01 min−1). The step 2 rates for R64A and R119A calculated from fitting to the unidirectional two-step scheme are twofold and sixfold slower than the observed rates of the isolated step 2 reaction with the purified 2′-P-ADPR RNA intermediate (0.163 min−1 and 1.05 min−1, respectively). The results are more in keeping with a branched kinetic mechanism for the mutant enzymes, in which a fraction of the 2′-P-ADPR RNA formed during step 1 reversibly enters an off-pathway state that is most simply postulated to entail dissociation of 2′-P-ADPR RNA from the mutant enzyme and rebinding from solution in order to proceed through step 2 (Supplemental Fig. S2). A key point is that the single turnover reaction with the 2′-PO4 RNA substrate (0.2 µM) is performed in the presence of 1 mM NAD+, a sensible condition given that the intracellular concentration of NAD+ in E. coli is 2.6 mM (Bennett et al. 2009). Consequently, any 2′-P-ADPR RNA that dissociates from Tpt1 after step 1 is competing with a ≥5000-fold excess of NAD+ for binding to the enzyme active site (Supplemental Fig. S2), wherein it is likely that the ADP-ribose moieties of the 2′-P-ADPR RNA and NAD+ occupy the same or closely overlapping positions, such that their binding is mutually exclusive.
By focusing on mutational effects on the step 1 rate and the rate of the isolated step 2 reaction, we can conclude that Arg64 is selectively required for the step 2 transesterification reaction: viz., the R64A mutation slows the step 1 rate by only threefold while slowing the step 2 rate by a factor of 214 vis à vis wild-type RslTpt1. In contrast, the other mutants studied here have relatively less skew in their impacts on step 1 versus step 2. To wit, H17A is slowed sixfold and threefold; R119A is slowed 27-fold and 33-fold; and R16A is slowed 300-fold and 710-fold in steps 1 and 2, respectively. We hypothesize that Arg64 makes atomic contacts in the active site that are uniquely pertinent to the chemistry of step 2, e.g., contacts to the RNA 2′-phosphate moiety of the 2′-phospho-ADP-ribosylated RNA intermediate. A bidentate interaction of Arg64 with the 2′-phosphate oxygens would stabilize the extra negative charge in the putative penta-coordinated phosphorane transition-state of the step 2 transesterification reaction. (This model is appealing given that we find that RslTpt1 does not require a divalent cation for activity in vitro.) In contrast, the chemistry of step1 occurs at the ribose C1 carbon of NAD+ and, although Arg64 might promote the binding of the 2′-PO4 RNA to the enzyme via interactions with the 2′-phosphate, there is no implicit need for such contacts in order for the 2′-phosphate to act as a nucleophile in step 1. Previously, Steiger et al (2005) identified a double-mutant K69A-R71S of SceTpt1 that was lethal in vivo and had ∼1000-fold lower specific activity in dephosphorylating a trinucleotide 2′-PO4 RNA substrate in vitro, yet had the distinctive property of accumulating substantial amounts of the 2′-P-ADPR intermediate. Arg71 of SceTpt1 (which results in lethality in vivo when mutated singly to alanine) is the equivalent of Arg64 in RslTpt1.
The observation that the R16A, H17A, and R119A mutations affect both steps of the Tpt1 pathway with varying but balanced severity suggests that Arg16, His17, and Arg119 either make particular contacts to the RNA or NAD+ substrates that are important for both steps or make different atomic contacts during steps 1 and 2, both of which are important. A priori, in a metal-independent mechanism, there is a likely need for a general base catalyst to deprotonate the ADP-ribose O2″ position so it can act as the nucleophile during step 2 transesterification and a histidine side chain is a good candidate for this role (as well as potentially contributing to NAD+ binding during step 1). Similarly, there is a presumptive need for a general acid catalyst to donate a proton to the RNA ribose O2′ leaving group during step 2 transesterification. Clarification of the basis for substrate recognition and reaction chemistry by Tpt1 obviously hinges on obtaining an ensemble of atomic structures of an exemplary Tpt1 enzyme in complexes with substrates, reaction intermediates, and products. At present, there is available a crystal structure of the apoenzyme form of a biochemically uncharacterized Tpt1 homolog from the archeaon Aeropyrum pernix (Kato-Murayama et al. 2005). In the ApeTpt1 enzyme, shown in Figure 10, the conserved side chains corresponding to RslTpt1 Arg16, His17, Arg64, and Arg119 are clustered together in a groove between the two lobes of the enzyme in the vicinity of two chloride anions modeled in the refined structure and rendered in Figure 10 as yellow spheres. The chlorides, which are separated by 6.4 Å, are potential mimetics of RNA phosphates. The ApeTpt1 structure is consistent with our conclusion that Arg16, His17, Arg64, and Arg119 are indeed constituents of the Tpt1 active site.
FIGURE 10.
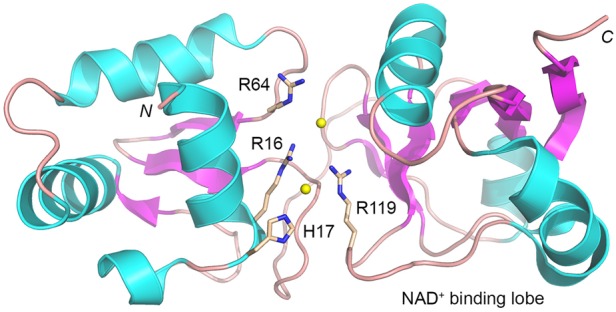
Conserved essential Tpt1 amino acids colocalize at the putative active site. The tertiary structure of Aeropyrum pernix Tpt1 (pdb 1WFX) is depicted as a cartoon trace with cyan α helices and magenta β strands. The amino (N) and carboxl (C) termini of the protein are indicated. The C-terminal lobe is implicated in NAD+ binding on the basis of its homolog to the NAD+-binding module of Diphtheria toxin (pdb 1TOX). The four conserved amino acid side chains that are essential for the activity of SceTpt1, EcoKptA, and RslTpt1 are rendered as stick models with beige carbons and numbered according to their position in RslTpt1. Two chloride anions modeled in the ApeTpt1 structure are depicted as yellow spheres.
Finally, it is worth highlighting our observation that an internal 2′-PO4 modification arrests 3′ RNA resection by PNPase, which is a biologically relevant agent of RNA 3′ decay in bacteria. Kierzek et al. (2000) reported that an internal 2′-PO4 group did not prevent cleavage of the vicinal RNA 3′-5′ phosphodiester by snake venom phosphodiesterase. If our findings anent PNPase extend to the 3′ exonuclease activity of the RNA exosome (which has structural homology with PNPase), then the question is raised as to whether Tpt1 enzymes might facilitate the turnover of cellular RNAs that are inappropriately spliced or repaired by a yeast-like ligase. A related question is whether and how yeast tRNAs that retain a 2′-PO4 post-splicing (e.g., when Tpt1 is conditionally inactivated) are handled in vivo.
MATERIALS AND METHODS
Plasmids for expression of RslTPT1 in yeast
The RslTPT1 ORF was amplified by PCR from Runella slithyformis (ATCC 49304) genomic DNA with primers that introduced a BamHI site at the translation start codon and an XhoI site flanking the stop codon. The resulting PCR product was digested with BamHI and XhoI and then inserted into yeast expression vectors p413-TPIB (CEN HIS3) and p423-TPIB (2μ HIS3), wherein expression of RslTPT1 is driven by the constitutive yeast TPI1 promoter. Alanine mutations (R16A, H17A, R64A, and R119A) were introduced into the RslTPT1 gene via a two-stage overlap extension polymerase chain reaction using Phusion polymerase. The mutated PCR products were digested with BamHI and XhoI and inserted into BamHI/XhoI-cut p423-TPIB plasmids. The RslTPT1 ORF was sequenced completely in the p413-RslTPT1 and p423-RslTPT1 plasmids to confirm that no unwanted coding changes were introduced during PCR amplification and cloning.
Test of RslTPT1 function by plasmid shuffle
The S. cerevisiae tpt1Δ haploid strain YBS501 (MATa ura3–1 ade2–1 trp1–1 his3–11,15 leu2–3,11–2 can1–100 tpt1::LEU2 p360-TPT1), in which the TPT1 ORF was deleted and replaced by LEU2, is dependent for viability on the p360-TPT1 plasmid (CEN URA3 SceTPT1) (Schwer et al. 2004). YBS501 was transformed with: (i) the p413-RslTPT1 and p423-RslTPT1 plasmids carrying either a WT or Ala-mutant allele of RslTPT1; (ii) a p413-SceTPT1 plasmid (CEN HIS3 SceTPT1) plasmid as a positive control; and (iii) the empty CEN HIS3 vector as negative control. Transformants were selected at 30°C on His– agar medium. Three individual His+ colonies were patched to His– agar medium and cells from each isolate were then streaked on agar medium containing 0.75 mg/mL 5-FOA (5-fluoroorotic acid). The plates were incubated at 20°C, 30°C, and 37°C. The p413-SceTPT1 and the p413-RslTPT1 and p423-RslTPT1 plasmids with wild-type RslTPT1 alleles all supported the formation of FOA-resistant colonies. In contrast, the vector and the p423-RslTPT1-Ala plasmids did not allow formation of FOA-resistant colonies after 7 d at any of the temperatures tested. Thus, the R16A, H17A, R64A, and R119A mutations were deemed lethal in vivo. Viable FOA-resistant tpt1Δ p413-SceTPT1, tpt1Δ p413-RslTPT1, and tpt1Δ p423-RslTPT1 colonies were grown in YPD-Ad (yeast, peptone, 2% dextose, 0.1 mg/mL adenine) liquid medium at 30°C to mid-log phase (A600 0.6), then diluted to attain A600 of 0.1, and aliquots (3 µL) of serial 10-fold dilutions were spotted on YPD agar plates and incubated at 20°C, 30°C, and 37°C.
Recombinant RslTpt1 proteins
The RslTPT1 ORF was amplified by PCR from R. slithyformis genomic DNA with primers designed to introduce an NdeI site immediately 5′ of the translation initiation codon and XhoI site immediately 3′ of the stop codon. The resulting PCR product was digested with NdeI and XhoI and then inserted into pET28a expression vector so as to fuse RslTpt1 to an N-terminal His6 tag. Alanine mutations (R16A, H17A, R64A, and R119A) were introduced into the expression vector either by a two-step overlap extension PCR with mutagenic primers and insertion of the mutated genes into the pET28a plasmid or by quick-change PCR. The RslTPT1 ORF was sequenced completely in the pET plasmids to confirm that no unwanted coding changes were introduced during PCR amplification and cloning. The pET28a-His6RslTpt1 and pET28a-His6RslTpt1-Ala plasmids were transformed into Escherichia coli BL21(DE3)-CodonPlus. Cultures (1 liter) derived from single kanamycin and chloramphenicol resistant transformants were grown at 37°C in Luria–Bertani medium containing 50 µg/mL kanamycin and 35 µg/mL chloramphenicol until the A600 reached 0.5 to 0.6, at which time the cultures were adjusted to 0.5 mM isopropyl-β-D-thiogalactoside and 2% (v/v) ethanol and then incubated for 16 h at 17°C with continuous shaking. Cells were harvested by centrifugation, and the pellets were stored at −80°C. All subsequent procedures were performed at 4°C. Thawed bacteria were suspended in 50 mL buffer A (50 mM Tris-HCl, pH 7.5, 1 M NaCl, 20 mM imidazole, 0.05% Triton X-100, 10% glycerol) and lysozyme was added to a final concentration of 1 mg/mL along with one protease inhibitor tablet (Roche). After mixing for 50 min, the resulting lysates were sonicated to reduce viscosity and insoluble material was removed by centrifugation for 45 min at 14,000 rpm. The soluble extracts were mixed for 1 h with 5 mL of His60 Ni Superflow resin (Clontech) that had been equilibrated in buffer A. The resin was washed twice with 50 mL of buffer A, then serially with 30 mL of buffer B (50 mM Tris-HCl, pH 7.5, 3 M KCl), and 50 mL of 40 mM imidazole in buffer C (50 mM Tris-HCl, pH 7.5, 500 mM NaCl, 10% glycerol). The resin was then poured into a column and the bound material was serially step-eluted with 100 mM, 200 mM, 300 mM, and 500 mM imidazole in buffer C. The elution profiles were monitored by SDS-PAGE. The peak His6RslTpt1-containing fractions were pooled and adjusted to 10 mM ethylenediaminetetraacetic acid (EDTA) and 10 mM dithiothreitol (DDT). The His6RslTpt1 protein preparations were then concentrated by centrifugal ultrafiltration (to ∼15–20 mg/mL in 10 mL) and gel-filtered through a 120-ml Superdex 200 column equilibrated with buffer D (50 mM Tris-HCl, pH 7.0, 300 mM NaCl, 1 mM EDTA, 1 mM DTT, 10% glycerol) at a flow rate of 1 mL/min, while collecting 2-mL fractions. Peak His6RslTpt1 fractions were concentrated by centrifugal ultrafiltration and stored at −80°C. Protein concentrations were determined by using the BioRad dye reagent with bovine serum albumin as the standard. The yields of His6RslTpt1 proteins were: WT, 50 mg; R16A mutant, 80 mg; H17A mutant, 65 mg; R64A mutant, 60 mg; and R119A mutant, 65 mg.
General methods for solid phase synthesis of oligonucleotides
Oligoribonucleotide syntheses were carried out using an ABI 3400 DNA synthesizer (Applied Biosystems) on a Unylinker (ChemGenes) solid support at a 1 µmol scale. Conventional 2′-tert-butyl-dimethylsilyl (TBDMS) ribonucleoside and 2′-acetyl levulinyl (ALE) riboadenosine 3′-phosphoramidites (0.15 M in MeCN) (ChemGenes) were used. For 2′-phosphitylation at the internal rA position, bis-cyanoethyl,-N,N-diisopropyl-phosphoramidite (0.20 in MeCN) was used. Phosphoramidites were dissolved in MeCN and activated with 5-ethylthio-1H-tetrazole (0.25 M in MeCN). Capping was carried out by the simultaneous delivery of acetic anhydride in pyridine/tetrahydrofuran (THF) and N-methylimidazole (16% in THF) and contacting the solid support for 6 sec. Oxidation of the phosphite triester intermediates was effected with 0.1 M iodine in pyridine/H2O/THF (20 sec); a solution of 3% trichloroacetic acid in THF, delivered over 1.8 min, was used to deprotect DMTr groups. For 2′-phosphate-containing substrates, a solution of anhydrous TEA/MeCN (2:3 v/v) was used to remove cyanoethyl phosphate protecting groups, while a 0.5 M solution of hydrazine hydrate in pyridine/AcOH (3:2 v/v) was used to remove ALE protecting groups. All oligonucleotides were deprotected and cleaved from the solid support using an ammonium hydroxide and ethanol solution. TBDMS groups were removed using TREAT-HF. Crude oligonucleotides were purified via HPLC and characterized by LC-MS.
Synthesis of 2′-PO4 branchpoint-containing oligonucleotides
Synthesis of RNAs containing internal 2′-PO4 moieties was carried out in two parts, as per the synthesis of branched RNAs (Katolik et al. 2014). For the sequence: 5′-UCGACUCCAA(2′-PO4)AUCGA, the first section of the oligonucleotide, 5′-A(2′-ALE)AUCGA, was synthesized on the solid support in the conventional 3′ to 5′ solid phase synthesis. The A(2′-ALE) unit in this sequence was introduced by coupling a 5′-DMTr-2′-ALE-3′-OCE phosphoramidite (0.15 M in MeCN) for 15 min (all other nucleotides were introduced using the 2′-TBDMS monomers). Subsequently, to remove cyanoethyl phosphodiester protecting groups, anhydrous TEA/MeCN (2:3 v/v) solution was passed through the solid support (20 min, repeated four times). This step ensures that all phosphate 3′-5′ linkages including that vicinal to the 2′-ALE group is in the diester form, thus preventing chain cleavage in the ensuing removal of the ALE group. After washing with MeCN (20 min) and drying the solid support over an argon stream (10 min), the synthesis columns were temporarily removed from the synthesizer and dried in vacuo for 30 min. To remove the 2′-ALE groups, the columns were returned to the synthesizer and a freshly prepared solution of 0.5 M hydrazine hydrate in pyridine/AcOH (3:2 v/v) was flowed through the columns (20 sec flow + 3.75 min sleep, repeated four times). After washing (MeCN, 10 min) and drying (Ar gas, 10 min), the solid supports were dried again in vacuo (30 min). To phosphitylate at the newly exposed 2′-OH, bis-cyanoethyl-N,N-diisopropyl CED phosphoramidite (0.20 M in MeCN) was coupled for 30 min, and then further oxidized using 0.1 M iodine in pyridine/H2O/THF (20 sec). To complete the oligonucleotide, standard 3′ to 5′ synthesis was continued on the 5′ terminus of the growing oligonucleotide using the sequence 5′-UCGACUCCA to yield the desired 15mer oligonucleotide substrate. This synthetic route was repeated to provide the 5′-CCAA(2′-PO4)AU hexamer and 5′-AA(2′-PO4)A trimer substrates.
Deprotection
Deprotection and cleavage of oligonucleotides from the solid support was achieved by treatment with 1 mL of cold 29% aqueous ammonia/ethanol (3:1, v/v) for 48 h at room temperature. Samples were centrifuged and the supernatant was transferred to a clean 1.5 mL eppendorf tube and vented for 30 min, chilled on dry ice, and evaporated to dryness. Removal of the 2′-TBDMS protecting groups was achieved by treatment with a 300 µL solution of NMP/Et3N/TREAT-HF (3:4:6, v/v) for 90 min at 65°C, followed by quenching with 3 M NaOAc buffer (50 µL; pH 5.5) and precipitation of the crude oligonucleotide from cold butanol (1 mL, −20°C). Samples were chilled on dry ice for 30 min and then centrifuged. After removing the supernatant, the remaining pellet (containing RNA) was evaporated to dryness, taken up in autoclaved milli-Q water (1 mL), filtered, and quantitated by UV spectroscopy.
Oligonucleotide purification
Crude oligonucleotides were purified by ion exchange (IE) or reverse phase (RP) HPLC. RNAs six nucleotides and longer were purified by IE-HPLC using a Waters Protein-Pak DEAE 5PW anion exchange column (21.5 × 150 mm). A mobile phase of 1 M aqueous LiClO4 in milli-Q water was used for analysis and purification (for 15mers: 0%–24% LiClO4 over 35 min, 4 m/min, 60°C; for hexamers: 0%–20% LiClO4 over 30 min, 4 mL/min, 60°C). Following collection of the desired peaks, fractions were combined and excess LiClO4 salts were removed using Gel Pak 2.5 size exclusion columns (Glen Research). Trimers were purified by RP-HPLC using a Hamilton PRP-1 column. A mobile phase of ACN in 0.1 M triethylammonium acetate (TEAA) at a flow rate of 4 mL/min was used (0%–11% over 15 min, 60°C). Purified oligonucleotides were characterized by electrospray ionization-mass spectrometry (the HPLC elution profiles and MS analyses are shown in Supplemental Fig. S3) and quantitated by UV spectroscopy. Extinction coefficients were determined using the IDT OligoAnalyzer tool (www.idtdna.com/analyzer/Applications/OligoAnalyzer).
5′ 32P-labeled 2′-PO4 RNA substrates
The 15-mer, 6-mer, and 3-mer RNA oligonucleotides containing an internal 2′-phosphate were 5′ 32P-labeled by reaction with phosphatase-dead T4 polynucleotide kinase (Pnkp-D167N) in the presence of [γ32P]ATP. The reaction was quenched with 90% formamide, 50 mM EDTA, 0.01% xylene cyanol and the radiolabeled RNAs were purified by electrophoresis through a 40-cm 20% polyacrylamide gel containing 7 M urea in 45 mM Tris-borate, 1 mM EDTA. The radiolabeled RNAs were eluted from excised gel slices, recovered by ethanol precipitation, and resuspended in 10 mM Tris-HCl, pH 6.8, 1 mM EDTA (TE) and stored at −20°C.
Isolation of 5′ 32P-labeled 2′-P-ADPR RNA intermediate
Reaction mixtures (4 × 20 µl) containing 100 mM Tris-HCl, pH 7.5, 1 mM NAD+ (Sigma), 5 mM MgCl2, 100 pmol 5′ 32P-labeled 6-mer 2′-PO4 RNA, and 100 pmol RslTpt1-R64A were incubated for 20 sec at 37°C. The reactions were quenched with 3 volumes of cold 90% formamide, 50 mM EDTA, 0.01% xylene cyanol. The radiolabeled 2′-P-ADPR RNA species was separated from substrate and product RNAs by urea-PAGE (as described above), eluted from excised gel slices, ethanol precipitated, resuspended in TE buffer, and stored at −20°C.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the U.S. National Institutes of Health (R35GM122575 to S.S.), the Geoffrey Beene Cancer Research Center (to S.S.), and the National Science and Engineering Council of Canada (to M.D.).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.067165.118.
REFERENCES
- Bennett BD, Kimball EH, Gao M, Osterhout R, Van Dien SJ, Rabinowitz JD. 2009. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat Chem Biol 5: 593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarty AK, Subbotin R, Chait BT, Shuman S. 2012. RNA ligase RtcB splices 3′-phosphate and 5′-OH ends via covalent RtcB-(histidinyl)-GMP and polynucleotide-(3′)pp(5′)G intermediates. Proc Natl Acad Sci USA 109: 6072–6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CM, Zhou C, Huang RH. 2009a. Reconstituting bacterial RNA repair and modification in vitro. Science 326: 247. [DOI] [PubMed] [Google Scholar]
- Chan CM, Zhou C, Brunzelle JS, Huang RH. 2009b. Structural and biochemical insights into 2′-O-methylation at the 3′-terminal nucleotide of RNA by Hen1. Proc Natl Acad Sci USA 106: 17699–17704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culver GM, McCraith SM, Zillman M, Kierzek R, Michaud N, LaReau RD, Turner DH, Phizicky EM. 1993. An NAD derivative produced during transfer RNA splicing: ADP-ribose 1″–2″ cyclic phosphate. Science 261: 206–208. [DOI] [PubMed] [Google Scholar]
- Culver GM, McCraith SM, Consaul SA, Stanford DR, Phizicky EM. 1997. A 2′-phosphotransferase implicated in tRNA splicing is essential in Saccharomyces cerevisiae. J Biol Chem 272: 13203–13210. [DOI] [PubMed] [Google Scholar]
- Das U, Shuman S. 2013. Mechanism of RNA 2′,3′-cyclic phosphate end-healing by T4 polynucleotide kinase-phosphatase. Nucleic Acids Res 41: 355–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer CL, Peebles CL, Gegenheimer P, Abelson J. 1983. Mechanism of action of a yeast RNA ligase in tRNA splicing. Cell 32: 537–546. [DOI] [PubMed] [Google Scholar]
- Jain R, Shuman S. 2010. Bacterial Hen1 is a 3′ terminal RNA ribose 2′O-methyltransferase component of a bacterial RNA repair cassette. RNA 16: 316–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain R, Shuman S. 2011. Active site mapping and substrate specificity of bacterial Hen1, a manganese-dependent 3′ terminal RNA ribose 2′O-methyltransferase. RNA 17: 429–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katolik A, Johnsson R, Montemayor EJ, Hart PJ, Damha MJ. 2014. Regiospecific solid-phase synthesis of branched oligoribonucleotides that mimic intronic lariat RNA intermediates. J Org Chem 79: 963–975. [DOI] [PubMed] [Google Scholar]
- Kato-Murayama M, Bessho Y, Shirouzu M, Yokoyama S. 2005. Crystal structure of the RNA 2′-phosphotransferase from Aeropyrum pernix K1. J Mol Biol 348: 295–305. [DOI] [PubMed] [Google Scholar]
- Kierzek R, Steiger MS, Spinelli SL, Turner DH, Phizicky EM. 2000. The chemical synthesis of oligoribonucleotides with selectively placed 2′-O-phosphates. Nucleosides Nucleotides Nucleic Acids 19: 917–933. [DOI] [PubMed] [Google Scholar]
- Martins A, Shuman S. 2005. An end-healing enzyme from Clostridium thermocellum with 5′ kinase, 2′,3′ phosphatase, and adenylyltransferase activities. RNA 11: 1271–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCraith SM, Phizicky EM. 1990. A highly specific phosphatase from Saccharomyces cerevisiae implicated in tRNA splicing. Mol Cell Biol 10: 1049–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCraith SM, Phizicky EM. 1991. An enzyme from Saccharomyces cerevisiae uses NAD+ to transfer the splice junction 2′-phosphate from ligated tRNA to an acceptor molecule. J Biol Chem 266: 11986–11992. [PubMed] [Google Scholar]
- Munir A, Shuman S. 2017. Characterization of Runella slithyformis HD-Pnk, a bifunctional DNA/RNA end-healing enzyme composed of an N-terminal 2′,3′ -phosphoesterase HD domain and a C-terminal 5′-OH polynucleotide kinase domain. J Bacteriol 199: e00739-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popow J, Schlieffer A, Martinez J. 2012. Diversity and roles of (t)RNA ligases. Cell Mol Life Sci 69: 2657–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remus BS, Shuman S. 2013. A kinetic framework for tRNA ligase and enforcement of a 2′-phosphate requirement for ligation highlights the design logic of an RNA repair machine. RNA 19: 659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya R, Schwer B, Shuman S. 2003. Genetic and biochemical analysis of the functional domains of yeast tRNA ligase. J Biol Chem 278: 43298–43398. [DOI] [PubMed] [Google Scholar]
- Sawaya R, Schwer B, Shuman S. 2005. Structure-function analysis of the yeast NAD+-dependent tRNA 2′-phosphotransferase Tpt1. RNA 11: 107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwer B, Sawaya R, Ho CK, Shuman S. 2004. Portability and fidelity of RNA-repair systems. Proc Natl Acad Sci 101: 2788–2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinelli SL, Consaul SA, Phizicky EM. 1997. A conditional lethal yeast phosphotransferase mutant accumulates tRNA with a 2′-phosphate and an unmodified base at the splice junction. RNA 3: 1388–1400. [PMC free article] [PubMed] [Google Scholar]
- Spinelli SL, Malik HS, Consaul SA, Phizicky EM. 1998. A functional homolog of a yeast tRNA splicing enzyme is conserved in higher eukaryotes and in Escherichia coli. Proc Natl Acad Sci 95: 14136–14141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinelli SL, Kierzek R, Turner DH, Phizicky EM. 1999. Transient ADP-ribosylation of a 2′-phosphate implicated in its removal from ligated tRNA during splicing in yeast. J Biol Chem 274: 2637–2644. [DOI] [PubMed] [Google Scholar]
- Steiger MA, Kierzek R, Turner DH, Phizicky EM. 2001. Substrate recognition by a yeast 2′-phosphotransferase involved in tRNA splicing and its Escherichia coli homolog. Biochemistry 40: 14098–14105. [DOI] [PubMed] [Google Scholar]
- Steiger MA, Jackman JE, Phizicky EM. 2005. Analysis of 2′-phosphotransferase (Tpt1p) from Saccharomyces cerevisiae: evidence for a conserved two-step reaction mechanism. RNA 11: 99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N, Chakravarty AK, Maughan B, Shuman S. 2011. Novel mechanism of RNA repair by RtcB via sequential 2′,3′-cyclic phosphodiesterase and 3′-phosphate/5′-hydroxyl ligation reactions. J Biol Chem 286: 43134–43143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumbale P, Appel CD, Kraehenbuehl R, Robertson PD, Williams JS, Krahn J, Ahel I, Williams RS. 2011. Structure of an aprataxin-DNA complex with insights into AOA1 neurodegenerative disease. Nat Struct Mol Biol 18: 1189–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unciuleac M, Shuman S. 2013a. Distinctive effects of domain deletions on the manganese-dependent DNA polymerase and DNA phosphorylase activities of Mycobacterium smegmatis polynucleotide phosphorylase. Biochemistry 52: 2967–2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unciuleac M, Shuman S. 2013b. Discrimination of RNA versus DNA by polynucleotide phosphorylase. Biochemistry 52: 6702–6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LK, Lima CD, Shuman S. 2002. Structure and mechanism of T4 polynucleotide kinase: an RNA repair enzyme. EMBO J 21: 3873–3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LK, Ho CK, Pei Y, Shuman S. 2003. Mutational analysis of bacteriophage T4 RNA ligase 1: different functional groups are required for the nucleotidyl transfer and phosphodiester bond formation steps of the ligation reaction. J Biol Chem 278: 29454–29462. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.