

Abstract
During adolescence, the brain continues to undergo vital developmental processes. In turn, complex behavioral and cognitive skills emerge. Unfortunately, neurobiological development during adolescence can be influenced by environmental factors such as drug exposure. Engaging in drug use during adolescence has been a long-standing health concern, especially how it predicts or relates to drug using behavior later in life. However, recent findings suggest that other behavioral domains, such as learning and memory, are also vulnerable to adolescent drug use. Moreover, it is becoming increasingly apparent that deficits in learning and memory following adolescent drug use endure into adulthood, well after drug exposure has subsided. Although persistent effects suggest an interaction between drug exposure and ongoing development during adolescence, the exact acute and long-term consequences of adolescent drug exposure on substrates of learning and memory are not fully understood. Thus, this review will summarize human and animal findings on the enduring cognitive deficits due to adolescent drug exposure. Moreover, due to the fact that adolescents are more likely to consume drugs of abuse legally available to adults, this review will focus on alcohol, nicotine, and marijuana. Further, given the critical role of the frontal cortex and hippocampus in various learning and memory domains, the impact adolescent use of the previous listed drugs on the neurobiology within these regions will also be discussed.
Adolescence is marked by drastic changes in behavior and cognitive function (for reviews, see Spear 2000; Steinberg 2005). Research has demonstrated that the brain remains plastic through adolescence and displays developmental-based morphological changes in total white and gray matter volume (Giedd et al. 1999; Giedd 2004). Moreover, specific regions, such as the frontal cortex (FC), hippocampus (HC), and amygdala continue to develop into late adolescence (Suzuki et al. 2005; Wierenga et al. 2014). At the synaptic level, mass production of synaptic spines occurs in the prefrontal cortex during the neonatal period and peaks around school-age, after which large scale pruning occurs, extending through adolescence until the third decade of life (Petanjek et al. 2011). It is clear that the adolescent brain is highly plastic and undergoes developmental and biological changes that may be required for proper behavioral and cognitive development. Unfortunately, due to this highly malleable state, the adolescent brain is also vulnerable to environmental stimuli. For example, exposure to continuous stress during the adolescent period may lead to various biobehavioral consequences (Barha et al. 2011; Eiland and Romeo 2013). From this, it is important to note other environmental factors that may hinder or impact neurological and behavioral development.
Adolescent drug use has been a long-standing health concern. According to the monitoring the future study, in 2017 9.3% of 8th graders, 13.7% of 10th graders, and 19.5% of 12th graders have reported using any illicit drug, other than marijuana, in their life time (Johnston et al. 2018). Moreover, for legal substances, alcohol, cigarettes, and in some states marijuana, these numbers increase with 23.1% of 8th graders, 44.2% of 10th graders, and 61.5% of 12th graders reported alcohol use, 13.3 % of 8th graders, 19.9% of 10th graders, and 31.1% of 12th graders reported cigarette use, and 13.5% of 8th graders, 30.7% of 10th graders, and 45.0% of 12th graders reported using marijuana/hashish in their lifetime (Johnston et al. 2018). Although these substances are age restricted during most of the adolescent period, these findings clearly suggest that adolescent drug use is geared toward legal substances, which is most likely due to ease of obtainment. In adults, the long-term consequences of drug use have been thoroughly studied. However, the enduring impact of legal recreational drug use during adolescence remains understudied. Nevertheless, recent findings suggest that persistent deficits in behavior, such as learning and memory, may be present after adolescent drug use (Coleman et al. 2011; Gleason et al. 2012; Portugal et al. 2012).
The neuroanatomical contributors of learning and memory are abundant, as cognition is a multisystem behavior; however, large focus is given to the FC and HC. Animal studies have demonstrated that various sub regions within the FC play a critical role in behavioral flexibility and attention (Dias and Aggleton 2000; Chudasama et al. 2003; Chudasama and Robbins 2003), whereas sub regions of the HC appear to be vital in spatial and contextual learning and memory (Stubley-Weatherly et al. 1996; Logue et al. 1997). In adult rodents, acute administration of alcohol, nicotine, or marijuana alters aspects of learning and memory associated with the FC and HC (Hahn et al. 2002; Gould 2003; Arguello and Jentsch 2004; Pamplona and Takahashi 2006; Semenova et al. 2007; Portugal et al. 2012), indicating that mechanisms responsible for learning and memory are sensitive to those substances. From this, it is feasible that adolescent alcohol, nicotine, or marijuana use also disrupts FC and HC learning and memory systems acutely. However, due to the continuous development of these regions during the adolescent time period, these acute alterations may disturb typical development and ultimately manifest as persistent cognitive deficits.
This paper will provide a review of the literature examining the long-term consequences of adolescent drug use on learning and memory. Moreover, given the fact that alcohol, nicotine, and marijuana are the most widely abused substances during the adolescent period, this paper will exclusively focus on these substances and how they modulate learning and memory behaviors associated with the FC and HC.
Alcohol
Alcohol is a widely used recreational substance that alters neuronal activity and functioning, leading to a myriad of behavioral changes. In 2015, 56% of individuals 18 or older in the US reported using alcohol in the past month and 87% reported using alcohol within their lifetime (Johnston et al. 2016). Given the fact that alcohol can be legally purchased and consumed by individuals 21 yr of age or older in the US and has intrinsic rewarding properties, it is not surprising to see high rates of use among adults. However, as mentioned previously, the adolescent rate of alcohol use in the U.S. is of concern, with 18.2% of 8th graders, 37.7% of 10th graders, and 55.7% of 12th graders reporting alcohol use in the past year (Johnston et al. 2018). Moreover, 2.2% of 8th graders, 8.9% in 10th graders, and 19.1% of 12th graders reported being drunk within the last 30 d (Johnston et al. 2018), indicating that those who do decide to drink, consume multiple alcoholic drinks in one session.
From a pharmacology stand point, alcohol is a relatively “dirty drug” that impacts many neurotransmitter systems including but not limited to: glutamate, GABA, acetylcholine, and opioid (for review, see Deitrich et al. 1989). Behaviorally, in adult rodents, alcohol exposure 15–30 min prior to behavioral training/testing produces robust cognitive deficits in many aspects of learning and memory (Matthews et al. 2002; Gould 2003). Due to the breadth of systems impacted by alcohol, it is hard to pinpoint the exact mechanism by which alcohol produces cognitive impairments. However, in vivo and in vitro work in the rodent HC has demonstrated that exposure to ethanol 20 min prior to stimulation, completely inhibits or reduces the ability to induce long-term potentiation (LTP), a neural substrate of learning and memory (Givens and McMahon 1995; Ramachandran et al. 2015). Although previous findings indicate neurobiological and behavioral consequences of adult alcohol exposure, the adolescent brain is different and may be differentially sensitive to long-term outcomes following adolescent alcohol exposure. In support, current findings suggest that following adolescent alcohol exposure, specific FC and HC based cognitive deficits arise in adulthood, well beyond initial drug exposure.
Impact of adolescent alcohol exposure on the FC and behavior
The vulnerability of the adolescent FC to alcohol exposure has been examined through various techniques in both humans and rodent models. In humans, adolescents suffering from alcohol use disorder have significantly smaller prefrontal cortex volumes and white matter compared to healthy controls (De Bellis et al. 2005). Moreover, gray matter volume is significantly correlated with the maximum number of drinks per drinking episode, with greater number of drinks correlated with reduced prefrontal cortex gray matter. These findings are consistent with adult studies, which have shown reductions in FC gray matter in young and older adult alcoholics, however, only older alcoholics displayed reduced FC white matter (Pfefferbaum et al. 1997). Rodent models suggest that alterations in FC morphology following adolescent exposure to alcohol may be persistent, as rats exposed to chronic intermittent alcohol exposure during the adolescent period had a bilateral reduction in prefrontal cortex thickness and basal forebrain volume as adults (Coleman et al. 2011; Vetreno et al. 2017). Conversely, increased thickness in the anterior cingulate and increased volume of the orbital FC has also been observed in adult rodents exposed to alcohol during adolescence (Coleman et al. 2014; Vetreno et al. 2017).
At the molecular level, exposure to alcohol during adolescence produces persistent up-regulation of neuroinflammatory markers, high-mobility box group 1, and toll-like receptor 3 and 4 and glutamatergic binding in the FC (Sircar and Sircar 2006; Vetreno and Crews 2012). Moreover, recent findings also suggest that reductions in FC brain derived neurotrophic factor (BDNF) may also be a consequence of adolescent alcohol exposure, however the duration of this effect is inconsistent, with some reporting persistent reductions during abstinence (Fernandez et al. 2016) and others reporting reductions during alcohol exposure and acute withdrawal but not during abstinence (Fernandez et al. 2017). Differences in BDNF outcomes between studies may be attributed to differences in alcohol consumption. For example, Fernandez et al. (2016) used a chronic continuous administration method through drinking water, whereas Fernandez et al. (2017) utilized an intermittent binge-like exposure. Aside from methodological differences, long-term changes in BDNF may not be adolescent specific, as persistent reductions in FC BDNF have also been documented following adult alcohol exposure (Fernandez et al. 2016). Conversely, during acute withdrawal (24 h after last alcohol exposure), developmental differences arise in FC BDNF, with early-adolescent exposure leading to decreases in FC BDNF but adult exposure producing increases in FC BDNF (Fernandez et al. 2017). Based on these studies, it is possible that duration and type of exposure play a role in the biological outcomes following adolescent and adult alcohol exposure. Nevertheless, disturbances in FC neurobiology following adolescent alcohol exposure suggests that behavioral tasks mediated by the FC may also be vulnerable.
Efforts to characterize the persistent behavioral consequences of adolescent alcohol exposure point to disruptions in behavioral flexibility. Behavioral flexibility refers to the ability to ignore or adapt a previously learned condition and adjust to fit a new set of learning rules, which has been shown to be dependent on the FC (for review, see Ragozzino 2007). This type of learning can be assessed in laboratory animals through maze learning (i.e., Barnes maze or Morris water maze), in which a previously learned escape condition is changed and the organism must learn a new strategy to escape an aversive stimulus. Utilizing the Barnes maze, Vetreno and Crews (2012) assessed both spatial and reversal learning in adult rats that were exposed to chronic alcohol during the adolescent period. They found that previous adolescent ethanol exposure did not impact acquisition of the Barnes maze but did disrupt reversal learning in adulthood, with alcohol exposed mice exhibiting longer latencies to find the new escape hole and spending a longer time in the previously learned hole location when compared to controls. Similar adult deficits in behavioral flexibility, due to adolescent ethanol exposure, have also been observed using the Morris water maze (Coleman et al. 2011) and set shifting models (Gass et al. 2014). It is important to note, however, that behavioral flexibility has also been shown to be compromised in adult rodents following chronic exposure to alcohol, suggesting that FC-mediated behavioral deficits induced by alcohol may not be exclusive to the adolescent period (Fernandez et al. 2017). Interestingly, not all cognitive domains may be impaired. For example, adolescent alcohol exposure enhanced long-term attentional capabilities in one study (Boutros et al. 2017) but produced no change in another (Semenova 2012). Similar null findings on attention outcomes have been found in human alcoholic adults during prolonged abstinence (Fein and McGillivray 2007). Collectively, human and animal studies indicate that the FC, during the adolescent period, is sensitive to alcohol exposure, which in turn alters certain aspects of learning and memory.
Impact of adolescent alcohol exposure on the HC and behavior
Aside from FC-mediated behaviors, other adult regionally specific cognitive domains are influenced by adolescent alcohol exposure. Fear conditioning is a well-validated model that allows for the investigation of HC-dependent learning and memory by pairing an aversive stimulus with a specific context and then examining the animal's behavior when reexposed to the same context after a time delay (for reivew, see Maren 2001). Studies using variations of fear conditioning indicate that adult hippocampal function may be impaired following adolescent alcohol exposure (Broadwater and Spear 2013b). For instance, rodents exposed to alcohol during adolescence show impaired memory of a context paired with an aversive stimulus when trained and tested in adulthood. Conversely, when the aversive stimuli was paired with a tone and the context, adult animals previously exposed to alcohol during adolescence showed a heightened fear response when reexposed to the context only, suggesting augmented fear learning and memory. From these results, it is possible that alcohol exposure during adolescents alters the ability to process and retain information. That is, adolescent alcohol exposure may lead to an inability to focus or remember subtle cues but enhance attention in the presence of more salient cues such as the tone. From this, it is possible that exposure to alcohol at differing time points may lead to different outcomes for learning and memory. In an effort to examine this possibility, Broadwater and Spear (2013a) reported that adolescent alcohol exposure selectively impairs certain aspects of fear learning depending on the time of alcohol exposure. They found that animals exposed to chronic intermittent alcohol exposure during the early adolescent period, but not the mid-late adolescent period or adulthood, displayed significantly less freezing during contextual retention testing as adults, indicating that there are developmental sensitive periods for alcohol exposure and subsequent impacts on adult cognitive functioning.
In line with impaired HC contextual memory formation, markers of neuroinflammation and cell death are up-regulated 24 h after chronic intermittent alcohol exposure in adolescent rodents, suggesting alcohol-induced neuronal damage in the HC following acute withdrawal (Pascual et al. 2007). Moreover, persistent HC damage following intermittent alcohol exposure has also been documented, with alcohol exposure in rats during the adolescent period leading to decreases in doublecortin labeled neurons (marker of neurogenesis), Ki-67 labeled cells (marker of cell proliferation), and BDNF, and increases in cleaved caspase-3 (marker of cell death) in the HC in adulthood (Vetreno and Crews 2015; Sakharkar et al. 2016). Interestingly, adult reductions in doublecortin, as a result of adolescent alcohol exposure, are prevented by the inhibition of histone deacetylase, suggesting a role of epigenetic modification in the deficits produced by adolescent alcohol exposure (Sakharkar et al. 2016). It should be noted that 2 d of binge exposure in adults produces significant increases in cell death in the gyrus 3 h (Cippitelli et al. 2010) and 4 d later (Obernier et al. 2002). Further, neurogenesis and cell proliferation within the HC are both impaired in adults exposed to alcohol suggesting overlapping neurological consequences of alcohol exposure in adolescents and adults (He et al. 2005). However, findings from Broadwater et al. (2014) indicate that age-related changes in neurogenesis and cell death differ in the duration of the effect. That is adolescent animals displayed reduced neurogenesis, 26 d after exposure where adult exposed animals did not. Due to the fact that adult exposed animals did not display changes in neurogenesis, a similar comparison between adolescent and adult exposure for cell death was not made. Interestingly, Broadwater et al. (2014) also reported no change in hippocampal cell proliferation, which is inconsistent with previous findings of reduced hippocampal cell proliferation in adult rodents exposed to alcohol during adolescence (Vetreno and Crews 2015; Sakharkar et al. 2016). Despite similar methodologies in binge-like alcohol exposure, variation in dose and age of assessment may account for the differences in hippocampal cell proliferation reported. From these findings, it is possible that the proliferation of cells in the HC is more sensitive to the dose of alcohol exposure and duration of abstinence, whereas cell death and neurogenesis are not.
Although there is evidence for persistent neurological and cognitive deficits in HC function, it is important to note that certain aspects of HC-dependent learning and memory, such as spatial learning and memory, were not impacted following adolescent exposure (Coleman et al. 2011; Vetreno and Crews 2012) but were in adults (Cippitelli et al. 2010). These selective cognitive deficits may help pinpoint the exact regional and molecular impact of adolescent alcohol exposure on the development of the HC. The human and rodent findings discussed above provide evidence to suggest that both the FC and HC are altered following adolescent exposure to alcohol. Moreover, it is apparent that some of these neurobiological alterations are persistent and may manifest later in life as specific cognitive deficits mediated by the FC and HC.
Nicotine
Tobacco use accounts for six million deaths a year worldwide, with trends predicting this number to increase to eight million by 2030 (World Health Organization 2018). Historically, tobacco industries have targeted adolescent aged individuals and young adults as new customers (Ling and Glantz 2002; Kreslake et al. 2008), with the goal of obtaining life-long sales of an addicting substance. This most likely accounts for the fact that most smokers begin smoking before the age of 18 (Center for Disease Control and Prevention 2014). Currently, in the United States, cigarette use in adolescents has declined over the past 15 yr (Johnston et al. 2018). However, with the recent surge in popularity of electronic cigarettes among youths (Camenga et al. 2014), adolescent nicotine exposure remains a health concern. In fact, 30-d prevalence of use among 8th, 10th, and 12th graders are higher for electronic cigarettes than that for conventional tobacco products (Johnston et al. 2018). Aside from the unknown risks of electronic cigarettes, the increase in use among adolescents begs for continued examination of the impact of nicotine exposure during adolescence.
Nicotine, the psychoactive substance in tobacco, acts as an agonist on nicotinic acetylcholine receptors (nAChRs), which are abundant in the FC and HC. Nicotinic acetylcholine receptors are expressed either as homomeric or heteromeric, comprised of either α (2–10) or combined α and β subunits (2–4) (for review, see Gotti et al. 2006). Functionally, these receptors play a role in learning and memory (Hasselmo 2006). For example, acute nicotine exposure enhances fear conditioning in mice (Gould and Higgins 2003). Enhanced cognitive performance following acute nicotine exposure has also been seen in rodent models of attention (Hahn et al. 2002). Conversely, nicotine withdrawal produces impairments in both contextual learning and attention-based models (Semenova et al. 2007; Gould et al. 2012; Portugal et al. 2012; Poole et al. 2014; Yildirim et al. 2015). From these studies, it is apparent that nicotine alters cognitive function in adults depending on multiple factors including duration of treatment. An important question is the impact of nicotine on cognitive function in adolescents.
Impact of adolescent nicotine exposure on the FC and behavior
Given the plasticity of the FC during adolescence, this region and processes mediated by it may be sensitive to developmental nicotine exposure. In young adult humans aged 17–22, Li et al. (2015) demonstrated that chronic nicotine exposure produces a decrease in the volume of the orbitofrontal and anterior cingulate cortex when compared to nonsmoking aged matched controls. Moreover, after assessing the participants’ nicotine dependence rating, they found that both the dorsolateral prefrontal cortex and orbitofrontal cortex volume were negatively correlated with the severity of dependence. Despite most participants in this study being considered young adults, the FC is still developing during this period (Giedd et al. 1999) and thus these findings highlight the vulnerability of a plastic FC to nicotine exposure, a characteristic that is similar between young adults and adolescent aged individuals. Functionally, adolescent nicotine exposure is associated with a blunted blood oxygen-level dependent (BOLD) response in the medial frontal lobe during an attention task when compared to nonsmoking controls (Musso et al. 2007). To investigate potential persistent cognitive deficits following adolescent smoking, Treur et al. (2015) examined the impact of adolescent smoking on attentional problems in a longitudinal study of monozygotic twins. They report that both in adults and adolescents smoking status was a significant predicator of attentional problems (as measured by the Conners Adult ADHD Rating Scales and the Child Behavior Checklist and the Youth Self Report), with smokers having increased attentional issues. Moreover, attentional problems were associated with age of first cigarette, with less attention problems reported when the twins started smoking at a later age. In discordant twin pairs (one smoker and one nonsmoker) the smoking twin on average had greater attentional problems than the nonsmoking twin in adulthood. This relationship was also found for adolescent 16-yr-old discordant twins but not 10- or 12-yr-old discordant twins, suggesting a vulnerable period in the middle of adolescence. It should be pointed out that these are not the first findings to demonstrate a link between attention issues with adolescent nicotine use (Fuemmeler et al. 2007), however, it is unclear whether nicotine use is a byproduct of attention issues or vice versa.
Animal models have been instrumental in disentangling the impact of adolescent nicotine use and subsequent attentional deficits. Despite the inherent challenges of investigating attention in a rodent model, the 5-Choice Serial Reaction Time Task allows researchers to examine various components of attention (for review, see Robbins 2002). Briefly, animals are trained to start each trial with a lever press, which after a set delay produces a short light stimulus in one of five nose-poke apertures on the opposite wall from the lever. If attention is properly directed, the animal will nose poke the lit aperture, which results in a food pellet. This cycle of behavior is continued until a certain amount of trials or time subsides. Changes in behaviors measured can be attributed to different components of attention. For example, a decrease in correct responses would indicate attentional deficits while an increase in premature responding would be akin to impulsivity. Using this paradigm, Counotte et al. (2009) demonstrated that 10 constitutive days of nicotine exposure during the adolescent period resulted in a decrease in the number of correct responses and an increase in the number of premature and time-out responses tested 5 wk later, when compared to controls, indicating persistent deficits in attention and impulsivity. Interestingly, the same duration of nicotine exposure did not result in any behavioral alterations in adult animals, suggesting that long-term attentional deficits produced by nicotine are exclusive to the adolescent period. Similar deficits in the number of correct and premature responses have been observed elsewhere (Counotte et al. 2011).
It is possible that alterations within the acetylcholine system in the FC account for the enduring attention deficits following adolescent nicotine. For example, 1-wk nicotine exposure in adolescent mice significantly increased nAChR binding in the cerebral cortex 1 d and 1 wk but not 1 mo after exposure (Abreu-Villaça et al. 2003). In line with these findings, Counotte et al. (2012) found that chronic nicotine exposure in adolescents but not adults produced an increase in nAChR binding in the medial prefrontal cortex 1 d after exposure but not after 5 wk. Moreover, they also found a significant increase in both α4 and β2 nAChR specific binding after 1 d but not after 5 wk in adolescent animals. Although these studies clearly demonstrate that nicotine alters FC acetylcholine signaling in adolescent animals, the effects are not persistent, which suggest that they are not completely responsible for the attentional deficits observed in adulthood or that alterations in downstream signaling cascades produced by the initial acetylcholine activation facilitate the attentional deficits observed in adulthood.
Interestingly, recent findings suggest that altered neuronal substrates of learning and memory mediated by excitatory/inhibitory input may be responsible for adult attentional deficits following adolescent nicotine exposure (Counotte et al. 2012). Goriounova and Mansvelder (2012) found that adolescent nicotine exposure produced a significant reduction in medial prefrontal cortex LTP 1–4 d after the last exposure. Conversely, 5 wk after nicotine exposure, LTP was enhanced in the medial prefrontal cortex in animals exposed to nicotine during adolescence, but not adulthood, indicating an inhibitory/excitatory switch produced by adolescent nicotine exposure. Further research has pointed to the inhibitory Type 2 metabotropic glutamate receptor (mGluR2) as a potential mechanism responsible for this developmental dichotomy (Counotte et al. 2011). Specifically, mGluR2 levels were significantly reduced following 1 d of abstinence but increased following 5 wk of abstinence in adolescent rats exposed to continuous nicotine. In HC slices, blockade of the mGluR2 prevented the acute reduction in LTP found in adolescent nicotine exposed animals, whereas activation of the mGluR2 prevented the excessive LTP observed in adult rats previously exposed to nicotine during adolescence (Goriounova and Mansvelder 2012). These results are in line with behavioral findings that showed that the blockade of mGluR2 prevents persistent adult attentional deficits in the 5-Choice Serial Reaction Time Task following adolescent nicotine exposure (Counotte et al. 2011). As the authors suggested, the long-term aberrant attentional issues driven by adolescent nicotine exposure may stem from an overexcited system, which was represented by increased adult LTP, leading to an inability to focus on a desired stimulus. It is important to note, however, that impairments in impulsivity were not prevented with the blockade of mGluR2, suggesting that multiple systems contribute to FC-mediated deficits due to adolescent nicotine exposure. When taken together, these findings demonstrate that nicotine exposure during the adolescent period modulates the FC acetylcholine system acutely, however, the persistent cognitive deficits appear to be facilitated by downstream mechanisms.
Impact of adolescent nicotine exposure on the HC and behavior
In both adolescent and adult rodents, nicotine exposure leads to alterations of the cholinergic system in the HC (Trauth et al. 2000; Doura et al. 2008; Portugal et al. 2012). Recent evidence suggests these alterations may depend on the age of exposure. For example, both adults and adolescents displayed significant up-regulation of α4β2 nAChRs in the dentate gyrus and CA1 region of the HC during chronic nicotine exposure but only adults displayed significant differences in α7 nAChR in the same regions (Doura et al. 2008). These findings are in-line with Trauth et al. (1999) who found significant increases in α4β2 nAChR binding in the HC of both adolescent and adult animals during chronic nicotine exposure. Moreover, during withdrawal, α4β2 nAChRs remained up-regulated in both adolescents and adults 2 wk after nicotine exposure subsided. Interestingly, adolescent male, but not female, animals displayed significant up-regulation of α4β2 nAChRs in the HC 1 mo after nicotine exposure, an effect that was not present in adults exposed to nicotine. These results suggest that both adult and adolescent nAChRs composition is sensitive to chronic nicotine exposure, however, in the HC of male adolescent animals this effect was persistent, lasting well beyond the period of drug exposure. It must be pointed out however, that not all findings have reported alterations in nAChR binding following adolescent nicotine exposure (Portugal et al. 2012). Nevertheless, it is possible that age of exposure may lead to differences in nAChR binding following chronic nicotine exposure. For example, onset of chronic nicotine exposure on postnatal day 23 produced no differences in nAChR binding during withdrawal (Portugal et al. 2012), whereas onset of chronic nicotine exposure on postnatal day thirty produced up-regulation of nAChRs during withdrawal (Trauth et al. 1999). These findings suggest that different developmental periods of sensitivity to the effects of nicotine exist during adolescence; some that may result in long-term changes in nAChRs.
Cellularly, nicotine produces long-term changes in neuron morphology within the HC based on age. For example, in the CA1 region, nicotine exposure during the adolescent period, but not in adulthood, reduced apical and total dendritic length 30 d later (Holliday et al. 2016). Conversely, CA3 basal and total dendritic length were reduced in adults exposed to nicotine but not adolescents when examined 30 d later. Moreover, adolescent nicotine exposure but not adult nicotine exposure produced a significant reduction in AMPA GluR2/3 subunits in the HC regardless of sex (Adriani et al. 2004). From these biological alterations, it could be expected that similar age-specific outcomes would be found in behavior.
Using the previously described fear conditioning paradigm, Portugal et al. (2012) reported age, duration, and dose specific alterations of nicotine exposure. They found that acute nicotine enhanced fear conditioning in preadolescent, adolescent, and adult mice but that preadolescent mice were the most sensitive to the acute effects. Conversely, when fear conditioning was assessed on the last day of chronic nicotine exposure, only the preadolescent group displayed enhanced fear conditioning but only at the highest dose tested. During withdrawal from nicotine (1 d of abstinence after chronic nicotine exposure), adolescent mice exhibited deficits at all dosages tested, whereas preadolescent showed a withdrawal deficit only at the highest dose, suggesting that preadolescent mice are less sensitive to nicotine withdrawal when compared to adolescent mice. Adult mice had withdrawal deficits at the two highest doses. Interestingly, when fear conditioning was assessed after a 30-d abstinence period following chronic nicotine exposure, preadolescent and adolescent exposed but not adult exposed animals displayed persistent cognitive impairments, indicating that adult deficits in fear conditioning during prolonged nicotine abstinence are specific to preadolescent and adolescent nicotine exposure. In line with these findings, similar deficits in adult contextual memory following chronic adolescent nicotine exposure have been documented elsewhere (Spaeth et al. 2010; Holliday et al. 2016). Further, exposure to chronic nicotine during adolescence, but not adulthood, disrupted trace fear conditioning when assessed in adulthood (Connor and Gould 2017), a cognitive model that encompasses working memory and is dependent on the HC as well as the prefrontal cortex (Connor and Gould 2016). In support, other cognitive tasks of working memory have also been shown to be sensitive to adolescent nicotine exposure when tested in adulthood (Fountain et al. 2008; Pickens et al. 2013). These results suggest that the preadolescent or adolescent HC and prefrontal cortex are susceptible to nicotine exposure, which leads to persistent cognitive deficits. Moreover, these effects are most likely driven by alterations in the development of these regions since persistent deficits are not observed in adults following nicotine exposure.
Although the literature is consistent surrounding the long-term deficits in contextual fear conditioning following adolescent nicotine exposure, no study, to the best of our knowledge, has examined the impact of sex on these effects. However, studies suggest that there may be sex differences. Up-regulation of membrane protein levels (marker of cell damage) and down-regulation of glial fibrillary acidic protein (marker of astrocyte), neurofilament 68-kDA protein, and neurofilament 200-kDA protein are found in the HC of adult female but not male rodents that were previously exposed to nicotine during adolescence (Trauth et al. 1999; Xu et al. 2003). Moreover, adolescent males but not females displayed prolonged up-regulation of nAChR α4β2 binding in the HC following nicotine exposure (Trauth et al. 1999). Thus, the behavioral outcomes following nicotine exposure during adolescence may differ based on sex. In line with these biological differences, Mateos et al. (2011) found that chronic nicotine exposure during the adolescent period impaired performance in the object in place test, which is dependent on the HC, in adult females but not males. Conversely, persistent impairments in fear conditioning following adolescent nicotine exposure have been reported in males, though females were not tested (Spaeth et al. 2010; Portugal et al. 2012; Holliday et al. 2016). Future work examining the role of sex on long-term cognitive function following adolescent nicotine exposure is needed.
The adolescent brain, specifically the HC and FC, responds differently to nicotine exposure than the adult brain. This should not be surprising given that the adolescent brain is still developing. Results from human studies indicate that adolescent nicotine exposure results in persistent attention deficits, a finding that is supported with findings from rodent models. Moreover, HC based learning and memory is altered following adolescent nicotine exposure well beyond cessation of drug exposure. Additionally, changes in cell physiology and structure have been reported in the FC and HC in adults previously exposed to nicotine as adolescents. Largely, the behavioral and neurobiological changes appear specific to adolescent exposure. Attempts to uncover a biological mechanism responsible for these physiological and cognitive deficits indicate that the acetylcholine and glutamatergic system may be involved, however, continued examination is needed to elucidate how the changes in these systems or others lead to persistent cognitive deficits.
Marijuana
Currently, attitudes and beliefs toward marijuana use are shifting, which is reflected in the recent legalization of marijuana in a few states in the US. Further, these attitude shifts are also seen in adolescent aged individuals, with 8th, 10th, and 12th graders all showing a decline in disapproval of experimental marijuana use from 2016 to 2017 (Johnston et al. 2018). From this, it is not surprising that marijuana use is slightly increased from 2016 to 2017, when all three age groups are collapsed (Johnston et al. 2018).
Delta-9-tetrahydrocannainol (THC) is the psychoactive substance in marijuana that is responsible for its rewarding and reinforcing effects. Within the brain, THC works on the endogenous endocannabinoid system that is comprised of two metabotropic receptors CB1 & CB2. Functionally, this system plays a modulatory role for many neurotransmitter systems, which in turn facilitate many physiological and behavioral outcomes including pain, inflammation, and stress (Walker et al. 1999; Marchalant et al. 2007; Hill et al. 2010). Moreover, in adult rodents, activation of the endocannabinoid system leads to acute deficits in FC and HC mediated learning and memory (Arguello and Jentsch 2004; Pamplona and Takahashi 2006; Suenaga and Ichitani 2008). Further, other aspects of learning and memory, such as cognitive effort, appear to be compromised following adult THC administration (Silveira et al. 2017). Collectively, these findings indicate that adult cognitive functioning is disrupted following exposure to a CB1 agonist, however, less is known about the cognitive consequences following adolescent exposure to a CB1 agonist. Nevertheless, recent research suggests that adolescent exposure to a CB1 agonist are associated with enduring cognitive deficits in adulthood.
Impact of adolescent marijuana exposure on the FC and behavior
Historically, concern around adolescent marijuana use focused on the development of psychosis (for review, see Malone et al. 2010), however, recent findings indicate that cognitive domains mediated by the FC may also be at risk following adolescent marijuana use. Results from human studies demonstrate that adolescent marijuana use leads to altered FC connectivity and volume (Churchwell et al. 2010; Lopez-Larson et al. 2011, 2015; Camchong et al. 2017); many of these outcomes being more severe with earlier onset of marijuana use. Likewise, adolescent marijuana exposure results in acute (Jacobsen et al. 2004; Harvey et al. 2007) and persistent (Ehrenreich et al. 1999; Meier et al. 2012) impaired attentional capabilities. However, it should be noted that attention deficits may be attributed to global cognitive difficulties, as attentional deficits are not present when controlling for overlapping variance between cognitive tests (i.e., short-term memory and implusivity; Dougherty et al. 2013). Moreover, there is conflicting evidence that abstinence from cannabis use during adulthood may produce multiple cognitive impairments including attention (Bolla et al. 2002) or no change in cognitive function (Pope et al. 2001) 28 d later, suggesting that enduring cognitive deficits following exposure to a CB1 agonist may not be specific to the adolescent time period.
In rodent models, these confounding variables can be more easily controlled and certain aspects of learning and memory can be disentangled. In an active place avoidance model, where an animal learns to avoid a specific environment that is paired with an unpleasant stimulus, repetitive THC exposure during early adolescence does not hinder task acquisition or retention in adulthood, but does impair reversal learning when the location of the conditioned environment is changed, an effect that was not observed following chronic THC exposure in late adolescence (Harte and Dow-Edwards 2010). Moreover, in males, administration of a synthetic cannabinoid agonist during adolescence disrupted reversal learning in the attentional set-shifting paradigm but did not impact other complex behaviors associated with attention in adulthood (Gomes et al. 2014). Interestingly, there is evidence indicating that a single, extremely low dose (0.001 mg/kg) exposure of THC in adulthood produces cognitive deficits in reversal learning 3 wk after exposure (Senn et al. 2008). Based on these findings it is possible that THC exposure produces long-term cognitive deficits in reversal learning for both adolescent and adult rats, however, these outcomes are dose dependent. Long-term disruption of recognition memory has also been observed following CB1 agonist exposure in female adolescent but not adult rodents (O'Shea et al. 2004). Interestingly, conflicting reports are found for males, with some finding deficits only following adolescent exposure (Quinn et al. 2008) and others finding deficits following both adolescent and adult exposure (O'Shea et al. 2006). It is possible that differences in drug exposure (THC and CB1 synthetic agonist) and washout length may account for these discrepancies. Nevertheless, these findings suggest that neural substrates of recognition memory may be sensitive to CB1 agonist exposure. It is worth mentioning that the exact circuitry of recognition memory is debated as both the FC and HC have been suggested to play a role in certain aspects (Cohen and Stackman 2015; Warburton and Brown 2015), while other evidence suggests that recognition memory may predominantly involve the perirhinal cortex (Winters et al. 2008). While studies in humans have found impairments in attention, these studies suggest that adolescent and maybe adult marijuana exposure produces persistent impairments in FC-mediated behaviors such as behavioral flexibility.
Impairments in cognitive function are mirrored with findings of long-term alterations in FC biology and function. For instance, repetitive THC exposure in adolescent female rodents led to reductions in pre- and postsynaptic markers of neuroplasticity and total and basal dendritic length in the prefrontal cortex in adulthood, indicating multiple potential changes in cortical organization (Rubino et al. 2009a). Conversely, reports examining similar outcomes in male adolescents are conflicting, with some reporting reductions in postsynaptic markers and dendritic count and length (Renard et al. 2016) and others reporting no change (Zamberletti et al. 2016). It should be noted, however, that the discrepancies in male prefrontal cortex outcomes may be a byproduct of different drugs used, as Renard et al. (2016) administered a synthetic cannabinoid agonist and Zamberletti et al. (2016) administered THC. Moreover, in regard to potential sex differences, both Rubino et al. (2009a) and Zamberletti et al. (2016) utilized an increasing dose regimen throughout the adolescent period but did not investigate a range of dosages, indicating that sex may contribute to differences in sensitivity to THC.
From a molecular prospective, the mechanism driving adult brain and behavioral alterations following adolescent exposure to THC, or a CB1 agonist, remains unclear but changes in GABAergic and glutamatergic systems may contribute. For instance, reductions in FC basal GABA, GAD67 (enzyme responsible for GABA synthesis), total Type 2/3 metabotropic glutamate receptors (mGluR2/3), and inhibitory tone after administration of a mGluR2/3 agonist have been observed in female adult rodents that were exposed to a CB1 agonist during adolescence (Zamberletti et al. 2014; Lovelace et al. 2015). Interestingly, total FC CB1 receptors remain unchanged in adult female rodents following adolescent exposure to a CB1 receptor agonist (Lovelace et al. 2015), suggesting that persistent cognitive deficits in FC-mediated tasks produced by adolescent exposure to a CB1 agonist may be mediated by downstream systems such as glutamate and GABA and not the endocannabinoid system. Moreover, recent findings indicate that repetitive THC exposure during adolescence, but not adulthood, alters adult prefrontal cortex gene expression associated with synaptic plasticity and GABAergic and glutamatergic systems in female rats (Prini et al. 2018). Nevertheless, alterations in long-term depression in the FC of adult female rodents exposed to a CB1 agonist during adolescence is prevented by inhibiting the breakdown of the endogenous endocannabinoid 2-arachidonyglycerol (Lovelace et al. 2015). Moreover, reductions in CB1 receptors were present, but only when colocalized with vesicular glutamate transporter 1 in adults exposed to a CB1 agonist during the adolescent period. From these results, it is possible that persistent FC-based behavioral deficits following adolescent exposure to a CB1 agonist may be the product of a down regulation of the endocannabinoid system which in turn leads to altered modulation of specific systems such as GABA and glutamate. In line with the latter portion of this hypothesis, Cass et al. (2014) found that repetitive exposure to a CB1 agonist during adolescence but not adulthood produced aberrant GABAergic mediated local field potentials in the medial prefrontal cortex. Specifically, adults with previous exposure to a CB1 agonist during adolescence displayed reduced spontaneous GABAergic inhibitory postsynaptic currents in the medial prefrontal cortex. Interestingly, the reductions in GABAergic inhibitory tone in adults produced by adolescent exposure to a CB1 agonist mirrored typical GABAergic inhibitory tone observed in adolescent animals, suggesting blunted development of the prefrontal cortex GABAergic system.
It must be pointed out that many of the findings reporting altered prefrontal cortex biological outcomes discussed above were only found in females. In fact, Zamberletti et al. (2016) found no changes in various glutamatergic receptors or pre- and postsynaptic markers in males, suggesting that the prefrontal cortex may be more vulnerable in females than in males to adolescent exposure to a CB1 agonist. However, deficits in reversal learning were found in both sexes (Harte and Dow-Edwards 2010) indicating deficits in FC-mediated cognitive functioning. From these results, it is possible that many of the biological outcomes unique to females following adolescent exposure to a CB1 agonist do not relate to deficits in reversal learning. Nevertheless, inferences between biology and behavior based on the studies discussed above must be done with caution as many experiential design differences, such as dose, exist.
Impact of adolescent marijuana exposure on the HC and behavior
In adult rodents, exposure to a CB1 agonist produces impaired HC learning and memory in various paradigms (Lichtman et al. 1995; Varvel et al. 2001; Da and Takahashi 2002; Pamplona and Takahashi 2006; Suenaga and Ichitani 2008; Wegener et al. 2008). Learning is associated with neurogenesis and disruption of neurogenesis can hinder learning (Gould et al. 1999; Hernandez-Rabaza et al. 2009). However, activation of the endocannabinoid system (CB1 or CB2) is associated with increased neurogenesis (for review, see Prenderville et al. 2015). These findings suggest that in some cases increased neurogenesis may not be adaptive for learning or that cognitive deficits produced by exposure to a CB1 agonist are not related to changes in neurogenesis. This area warrants further investigation. Nevertheless, Abboussi et al. (2014) found that chronic exposure to a CB1 agonist followed by a washout period produced no differences in HC neurogenesis in adults when compared to controls, indicating a transient effect. Conversely, when animals were exposed chronically to a CB1 agonist during adolescence, long-term reductions in HC neurogenesis were found in adulthood, indicating developmental sensitivity to a CB1 agonist exposure.
Further characterization of the long-term biological consequences of adolescent exposure to a CB1 receptor agonist demonstrates that multiple processes are altered. Indeed, Moretti et al. (2015) reported significant increases in HC inflammatory markers in both adolescent and adult mice exposed to subchronic THC, however, adolescent but not adult mice had persistent up-regulation of HC inflammatory markers almost 7 wk after subchronic exposure. Moreover, reductions in HC dendritic length and number of proximal and distal dendrites in adults previously exposed to THC as adolescents has been reported (Rubino et al. 2009b). Within the glutamatergic system, adults previously exposed to a CB1 agonist during adolescence had increased GluA1, GluA2, and GluN2B receptors, increased synaptosomal stimulated glutamate release (Zamberletti et al. 2016), and reduced type 5 metabotropic glutamate receptors (Gleason et al. 2012). Collectively, these results indicate that the neurobiological impact of CB1 agonist exposure during adolescents is diverse, resulting in alterations in multiple neurotransmitter systems.
In concordance with alterations in HC morphology and neurochemistry, adolescent exposure to cannabinoids produces adult deficits in learning. For example, Gleason et al. (2012), found that adolescent rodents exposed to 10 consecutive days of a CB1 agonist displayed impaired contextual memory during fear conditioning when tested in adulthood, an effect that was not found when animals were exposed to a CB1 agonist during early adulthood. Further, adult HC-based cognitive deficits following adolescent exposure to a CB1 agonist have also been reported in the radial arm maze (Rubino et al. 2009a) and novel spatial object location (Abush and Akirav 2012; Zamberletti et al. 2016). However, studies utilizing the Morris water maze yield inconsistent results, with some displaying no change in spatial learning (Cha et al. 2007; Abush and Akirav 2012) or slight changes (Higuera-Matas et al. 2009), and others reporting deficits (Abboussi et al. 2014). Methodological variations may account for differences in Morris water maze findings, as age of drug exposure, type of drug used, duration of drug exposure, and washout period until behavioral testing all differed. It should be noted that similar inconsistent results for long-term spatial learning have also been observed following adult exposure to a CB1 agonist (Cha et al. 2007; Tselnicker et al. 2007). From these results it is possible that only certain aspects of adult HC learning and memory (contextual and working spatial memory) are vulnerable to adolescent marijuana use.
Given the modulatory role of the endocannabinoid system in central nervous system signaling, it is not surprising that adolescent exposure to endocannabinoid substances leads to changes in learning and memory in adults. The results discussed indicate that activation of the endocannabinoid system via THC or synthetic cannabinoids in the developing adolescent leads to persistent alterations in multiple neurochemical systems in the FC and HC even after prolonged periods of abstinence. Moreover, these persistent alterations in neurological morphology and neurochemistry may mediate cognitive deficits in multiple aspects of learning and memory.
Conclusion
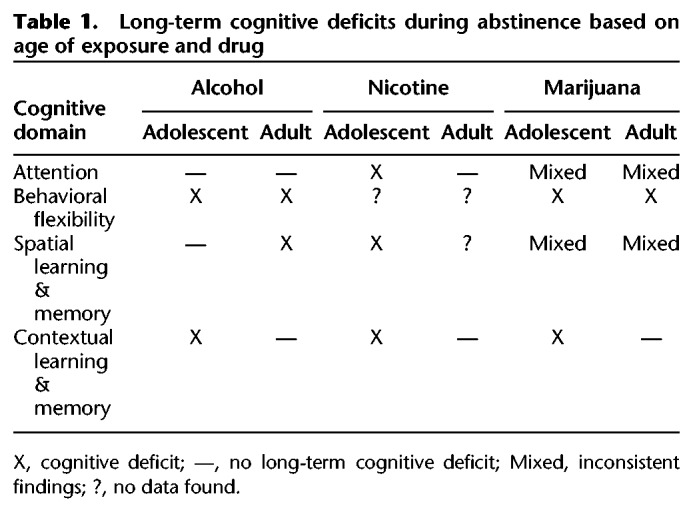
The adolescent time period is marked by increased risky-behavior, which includes drug use (Johnston et al. 2018). In the United States, legal recreational use is limited to alcohol, nicotine, and in some states marijuana and is age restricted to adults. Despite these age restrictions, use of these substances is high among the adolescent population (Johnston et al. 2018), which draws concern about the long-term consequences of exposure to psychoactive substances during a critical developmental period. Human and animal findings validate these concerns by demonstrating that exposure to these substances can lead to long-term cognitive dysfunction into adulthood, even after extended abstinence periods. However, it is important to note that certain aspects of learning and memory are compromised depending on the drug. For example, adult attention capabilities were compromised following adolescent nicotine exposure, but enhanced or not changed following adolescent alcohol exposure. Moreover, adolescent marijuana exposure produces attentional deficits in human studies. Further, adolescent alcohol and marijuana exposure compromised behavioral flexibility, whereas the exact impact of nicotine on behavioral flexibility remains unknown. Interestingly, context learning and memory was compromised following exposure to all three drugs. These findings suggest that certain deficits are produced by or through entirely unique biological pathways, while others may overlap (i.e., GABA or glutamate, see Fig. 1). It must be pointed out, however, that the relationship between each of these drugs and their prolonged outcomes following adolescent exposure is not simple. For instance, onset and duration of exposure varies greatly depending on experimental design, which may lead to different behavioral and biological outcomes. Moreover, there is evidence from animal studies that some of the long-term cognitive consequences following adolescent drug exposure are not unique to the adolescent time period. That is, prolonged drug exposure during adulthood can also lead to enduring cognitive deficits following abstinence (see Table 1). For example, Fernandez et al. (2017) found that chronic intermittent exposure to alcohol produced long-term deficits in reversal learning following adolescent or adult exposure. While in contrast, Broadwater and Spear (2013a) found long-term deficits in contextual fear conditioning in adult rats exposed to alcohol in early adolescence but not exposed in adulthood. From these findings, it could be argued that repeated alcohol exposure produces permanent alterations in the neurobiology of the HC regardless of the age of exposure. Conversely, nicotine exposure during adolescence but not adulthood produces long-term cognitive deficits in contextual learning and memory (Portugal et al. 2012). These findings suggest that the persistent cognitive consequences of nicotine use are most likely based on disrupted development of the HC as they are only present following adolescent exposure. Continued study of the similarities and differences between the effects of drugs of abuse and age of exposure on cognitive function can provide valuable information toward uncovering mechanisms responsible for prolonged cognitive deficits that persist after abstinence.
Figure 1.

Neurobiological consequences in the FC and HC and long-term cognitive impairments following adolescent exposure to alcohol, nicotine, or marijuana (CB1 agonist).
Table 1.
Long-term cognitive deficits during abstinence based on age of exposure and drug
The studies reviewed here highlight the importance of further examining the long-term biobehavioral deficits of adolescent drug use. Several important issues could be further explored. For instance, drug initiation during the adolescent time period has been linked with heightened levels of stress exposure (Wills et al. 2001; Charles et al. 2017), thus studies using rodent models examining the impact of the interaction effects of adolescent stress and drug use on adult brain and behavioral outcomes associated with learning and memory are needed. Moreover, given the relatively high rate of poly drug use (Collins et al. 1998), continued investigation of the long-term cognitive consequences of multidrug exposure during adolescence in an animal model may be useful (Mateos et al. 2011). Aside from these suggestions, current findings indicate that the development of the FC and HC is vulnerable to drug abuse during adolescence, which in turn manifest as cognitive deficits later in life. A deeper understanding of the biological mechanisms responsible for these deficits may help mitigate the consequences of adolescent drug use.
Acknowledgments
This work was funded by 1U01DA041632 (T.J.G), the Jean Phillips Shibley Endowment (T.J.G.), and Penn State University (T.J.G.).
Footnotes
Article is online at http://www.learnmem.org/cgi/doi/10.1101/lm.046672.117.
References
- Abboussi O, Tazi A, Paizanis E, El Ganouni S. 2014. Chronic exposure to WIN55,212-2 affects more potently spatial learning and memory in adolescents than in adult rats via a negative action on dorsal hippocampal neurogenesis. Pharmacol Biochem Behav 120: 95–102. [DOI] [PubMed] [Google Scholar]
- Abreu-Villaça Y, Seidler FJ, Qiao D, Tate CA, Cousins MM, Thillai I, Slotkin TA. 2003. Short-term adolescent nicotine exposure has immediate and persistent effects on cholinergic systems: critical periods, patterns of exposure, dose thresholds. Neuropsychopharmacology 28: 1935–1949. [DOI] [PubMed] [Google Scholar]
- Abush H, Akirav I. 2012. Short- and long-term cognitive effects of chronic cannabinoids administration in late-adolescence rats. PLoS One 7: e31731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adriani W, Granstrem O, Macri S, Izykenova G, Dambinova S, Laviola G. 2004. Behavioral and neurochemical vulnerability during adolescence in mice: studies with nicotine. Neuropsychopharmacology 29: 869–878. [DOI] [PubMed] [Google Scholar]
- Arguello PA, Jentsch JD. 2004. Cannabinoid CB1 receptor-mediated impairment of visuospatial attention in the rat. Psychopharmacology (Berl) 177: 141–150. [DOI] [PubMed] [Google Scholar]
- Barha CK, Brummelte S, Lieblich SE, Galea LA. 2011. Chronic restraint stress in adolescence differentially influences hypothalamic-pituitary-adrenal axis function and adult hippocampal neurogenesis in male and female rats. Hippocampus 21: 1216–1227. [DOI] [PubMed] [Google Scholar]
- Bolla KI, Brown K, Eldreth D, Tate K, Cadet JL. 2002. Dose-related neurocognitive effects of marijuana use. Neurology 59: 1337–1343. [DOI] [PubMed] [Google Scholar]
- Boutros N, Der-Avakian A, Markou A, Semenova S. 2017. Effects of early life stress and adolescent ethanol exposure on adult cognitive performance in the 5-choice serial reaction time task in Wistar male rats. Psychopharmacology (Berl) 234: 1549–1556. [DOI] [PubMed] [Google Scholar]
- Broadwater M, Spear LP. 2013a. Consequences of ethanol exposure on cued contextual fear conditioning and extinction differ depending on timing of exposure during adolescence or adulthood. Behav Brain Res 256: 10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadwater M, Spear LP. 2013b. Tone conditioning potentiates rather than overshadows context fear in adult animals following adolescent ethanol exposure. Dev Psychobiol 56: 1150–1155. [DOI] [PubMed] [Google Scholar]
- Broadwater MA, Liu W, Crews FT, Spear LP. 2014. Persistent loss of hippocampal neurogenesis and increased cell death following adolescent, but not adult, chronic ethanol exposure. Dev Neurosci 36: 297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camchong J, Lim KO, Kumra S. 2017. Adverse effects of cannabis on adolescent brain development: a longitudinal study. Cereb Cortex 27: 1922–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camenga DR, Delmerico J, Kong G, Cavallo D, Hyland A, Cummings KM, Krishnan-Sarin S. 2014. Trends in use of electronic nicotine delivery systems by adolescents. Addict Behav 39: 338–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cass DK, Flores-Barrera E, Thomases DR, Vital WF, Caballero A, Tseng KY. 2014. CB1 cannabinoid receptor stimulation during adolescence impairs the maturation of GABA function in the adult rat prefrontal cortex. Mol Psychiatry 19: 536–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Center for Diesase Control and Prevention. 2014. Smoking and Youth. (ed. CfDCa Prevention), https://www.cdc.gov/tobacco/data_statistics/sgr/50th-anniversary/index.htm
- Cha YM, Jones KH, Kuhn CM, Wilson WA, Swartzwelder HS. 2007. Sex differences in the effects of Δ9-tetrahydrocannabinol on spatial learning in adolescent and adult rats. Behav Pharmacol 18: 563–569. [DOI] [PubMed] [Google Scholar]
- Charles NE, Mathias CW, Acheson A, Dougherty DM. 2017. Preadolescent sensation seeking and early adolescent stress relate to at-risk adolescents’ substance use by age 15. Addict Behav 69: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudasama Y, Robbins TW. 2003. Dissociable contributions of the orbitofrontal and infralimbic cortex to Pavlovian autoshaping and discrimination reversal learning: further evidence for the functional heterogeneity of the rodent frontal cortex. J Neurosci 23: 8771–8780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudasama Y, Baunez C, Robbins TW. 2003. Functional disconnection of the medial prefrontal cortex and subthalamic nucleus in attentional performance: evidence for corticosubthalamic interaction. J Neurosci 23: 5477–5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchwell JC, Lopez-Larson M, Yurgelun-Todd DA. 2010. Altered frontal cortical volume and decision making in adolescent cannabis users. Front Psychol 1: 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cippitelli A, Zook M, Bell L, Damadzic R, Eskay RL, Schwandt M, Heilig M. 2010. Reversibility of object recognition but not spatial memory impairment following binge-like alcohol exposure in rats. Neurobiol Learn Mem 94: 538–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SJ, Stackman RW Jr. 2015. Assessing rodent hippocampal involvement in the novel object recognition task. A review. Behav Brain Res 285: 105–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman LG Jr, He J, Lee J, Styner M, Crews FT. 2011. Adolescent binge drinking alters adult brain neurotransmitter gene expression, behavior, brain regional volumes, and neurochemistry in mice. Alcohol Clin Exp Res 35: 671–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman LG Jr, Liu W, Oguz I, Styner M, Crews FT. 2014. Adolescent binge ethanol treatment alters adult brain regional volumes, cortical extracellular matrix protein and behavioral flexibility. Pharmacol Biochem Behav 116: 142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins RL, Ellickson PL, Bell RM. 1998. Simultaneous polydrug use among teens: prevalence and predictors. J Subst Abuse 10: 233–253. [DOI] [PubMed] [Google Scholar]
- Connor DA, Gould TJ. 2016. The role of working memory and declarative memory in trace conditioning. Neurobiol Learn Mem 134(Pt B): 193–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor DA, Gould TJ. 2017. Chronic fluoxetine ameliorates adolescent chronic nicotine exposure-induced long-term adult deficits in trace conditioning. Neuropharmacology 125: 272–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counotte DS, Spijker S, Van de Burgwal LH, Hogenboom F, Schoffelmeer AN, De Vries TJ, Smit AB, Pattij T. 2009. Long-lasting cognitive deficits resulting from adolescent nicotine exposure in rats. Neuropsychopharmacology 34: 299–306. [DOI] [PubMed] [Google Scholar]
- Counotte DS, Goriounova NA, Li KW, Loos M, van der Schors RC, Schetters D, Schoffelmeer AN, Smit AB, Mansvelder HD, Pattij T, et al. 2011. Lasting synaptic changes underlie attention deficits caused by nicotine exposure during adolescence. Nat Neurosci 14: 417–419. [DOI] [PubMed] [Google Scholar]
- Counotte DS, Goriounova NA, Moretti M, Smoluch MT, Irth H, Clementi F, Schoffelmeer AN, Mansvelder HD, Smit AB, Gotti C, et al. 2012. Adolescent nicotine exposure transiently increases high-affinity nicotinic receptors and modulates inhibitory synaptic transmission in rat medial prefrontal cortex. FASEB J 26: 1810–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da S, Takahashi RN. 2002. SR 141716A prevents Δ9-tetrahydrocannabinol-induced spatial learning deficit in a Morris-type water maze in mice. Prog Neuropsychopharmacol Biol Psychiatry 26: 321–325. [DOI] [PubMed] [Google Scholar]
- De Bellis MD, Narasimhan A, Thatcher DL, Keshavan MS, Soloff P, Clark DB. 2005. Prefrontal cortex, thalamus, and cerebellar volumes in adolescents and young adults with adolescent-onset alcohol use disorders and comorbid mental disorders. Alcohol Clin Exp Res 29: 1590–1600. [DOI] [PubMed] [Google Scholar]
- Deitrich RA, Dunwiddie TV, Harris RA, Erwin VG. 1989. Mechanism of action of ethanol: initial central nervous system actions. Pharmacol Rev 41: 489–537. [PubMed] [Google Scholar]
- Dias R, Aggleton JP. 2000. Effects of selective excitotoxic prefrontal lesions on acquisition of nonmatching- and matching-to-place in the T-maze in the rat: differential involvement of the prelimbic-infralimbic and anterior cingulate cortices in providing behavioural flexibility. Eur J Neurosci 12: 4457–4466. [DOI] [PubMed] [Google Scholar]
- Dougherty DM, Mathias CW, Dawes MA, Furr RM, Charles NE, Liguori A, Shannon EE, Acheson A. 2013. Impulsivity, attention, memory, and decision-making among adolescent marijuana users. Psychopharmacology (Berl) 226: 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doura MB, Gold AB, Keller AB, Perry DC. 2008. Adult and periadolescent rats differ in expression of nicotinic cholinergic receptor subtypes and in the response of these subtypes to chronic nicotine exposure. Brain Res 1215: 40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenreich H, Rinn T, Kunert HJ, Moeller MR, Poser W, Schilling L, Gigerenzer G, Hoehe MR. 1999. Specific attentional dysfunction in adults following early start of cannabis use. Psychopharmacology (Berl) 142: 295–301. [DOI] [PubMed] [Google Scholar]
- Eiland L, Romeo RD. 2013. Stress and the developing adolescent brain. Neuroscience 249: 162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fein G, McGillivray S. 2007. Cognitive performance in long-term abstinent elderly alcoholics. Alcohol Clin Exp Res 31: 1788–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez GM, Stewart WN, Savage LM. 2016. Chronic drinking during adolescence predisposes the adult rat for continued heavy drinking: neurotrophin and behavioral adaptation after long-term, continuous ethanol exposure. PLoS One 11: e0149987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez GM, Lew BJ, Vedder LC, Savage LM. 2017. Chronic intermittent ethanol exposure leads to alterations in brain-derived neurotrophic factor within the frontal cortex and impaired behavioral flexibility in both adolescent and adult rats. Neuroscience 348: 324–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fountain SB, Rowan JD, Kelley BM, Willey AR, Nolley EP. 2008. Adolescent exposure to nicotine impairs adult serial pattern learning in rats. Exp Brain Res 187: 651–656. [DOI] [PubMed] [Google Scholar]
- Fuemmeler BF, Kollins SH, McClernon FJ. 2007. Attention deficit hyperactivity disorder symptoms predict nicotine dependence and progression to regular smoking from adolescence to young adulthood. J Pediatr Psychol 32: 1203–1213. [DOI] [PubMed] [Google Scholar]
- Gass JT, Glen WB Jr, McGonigal JT, Trantham-Davidson H, Lopez MF, Randall PK, Yaxley R, Floresco SB, Chandler LJ. 2014. Adolescent alcohol exposure reduces behavioral flexibility, promotes disinhibition, and increases resistance to extinction of ethanol self-administration in adulthood. Neuropsychopharmacology 39: 2570–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giedd JN. 2004. Structural magnetic resonance imaging of the adolescent brain. Ann N Y Acad Sci 1021: 77–85. [DOI] [PubMed] [Google Scholar]
- Giedd JN, Blumenthal J, Jeffries NO, Castellanos FX, Liu H, Zijdenbos A, Paus T, Evans AC, Rapoport JL. 1999. Brain development during childhood and adolescence: a longitudinal MRI study. Nat Neurosci 2: 861–863. [DOI] [PubMed] [Google Scholar]
- Givens B, McMahon K. 1995. Ethanol suppresses the induction of long-term potentiation in vivo. Brain Res 688: 27–33. [DOI] [PubMed] [Google Scholar]
- Gleason KA, Birnbaum SG, Shukla A, Ghose S. 2012. Susceptibility of the adolescent brain to cannabinoids: long-term hippocampal effects and relevance to schizophrenia. Transl Psychiatry 2: e199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes FV, Guimarães FS, Grace AA. 2014. Effects of pubertal cannabinoid administration on attentional set-shifting and dopaminergic hyper-responsivity in a developmental disruption model of schizophrenia. Int J Neuropsychopharmacol 18: pyu018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goriounova NA, Mansvelder HD. 2012. Short- and long-term consequences of nicotine exposure during adolescence for prefrontal cortex neuronal network function. Cold Spring Harb Perspect Med 2: a012120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotti C, Zoli M, Clementi F. 2006. Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol Sci 27: 482–491. [DOI] [PubMed] [Google Scholar]
- Gould TJ. 2003. Ethanol disrupts fear conditioning in C57BL/6 mice. J Psychopharmacol 17: 77–81. [DOI] [PubMed] [Google Scholar]
- Gould TJ, Higgins JS. 2003. Nicotine enhances contextual fear conditioning in C57BL/6J mice at 1 and 7 days post-training. Neurobiol Learn Mem 80: 147–157. [DOI] [PubMed] [Google Scholar]
- Gould E, Beylin A, Tanapat P, Reeves A, Shors TJ. 1999. Learning enhances adult neurogenesis in the hippocampal formation. Nat Neurosci 2: 260–265. [DOI] [PubMed] [Google Scholar]
- Gould TJ, Portugal GS, Andre JM, Tadman MP, Marks MJ, Kenney JW, Yildirim E, Adoff M. 2012. The duration of nicotine withdrawal-associated deficits in contextual fear conditioning parallels changes in hippocampal high affinity nicotinic acetylcholine receptor upregulation. Neuropharmacology 62: 2118–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn B, Shoaib M, Stolerman IP. 2002. Nicotine-induced enhancement of attention in the five-choice serial reaction time task: the influence of task demands. Psychopharmacology (Berl) 162: 129–137. [DOI] [PubMed] [Google Scholar]
- Harte LC, Dow-Edwards D. 2010. Sexually dimorphic alterations in locomotion and reversal learning after adolescent tetrahydrocannabinol exposure in the rat. Neurotoxicol Teratol 32: 515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey MA, Sellman JD, Porter RJ, Frampton CM. 2007. The relationship between non-acute adolescent cannabis use and cognition. Drug Alcohol Rev 26: 309–319. [DOI] [PubMed] [Google Scholar]
- Hasselmo ME. 2006. The role of acetylcholine in learning and memory. Curr Opin Neurobiol 16: 710–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Nixon K, Shetty AK, Crews FT. 2005. Chronic alcohol exposure reduces hippocampal neurogenesis and dendritic growth of newborn neurons. Eur J Neurosci 21: 2711–2720. [DOI] [PubMed] [Google Scholar]
- Hernandez-Rabaza V, Llorens-Martin M, Velazquez-Sanchez C, Ferragud A, Arcusa A, Gumus HG, Gomez-Pinedo U, Perez-Villalba A, Rosello J, Trejo JL, et al. 2009. Inhibition of adult hippocampal neurogenesis disrupts contextual learning but spares spatial working memory, long-term conditional rule retention and spatial reversal. Neuroscience 159: 59–68. [DOI] [PubMed] [Google Scholar]
- Higuera-Matas A, Botreau F, Miguens M, Del Olmo N, Borcel E, Perez-Alvarez L, Garcia-Lecumberri C, Ambrosio E. 2009. Chronic periadolescent cannabinoid treatment enhances adult hippocampal PSA-NCAM expression in male Wistar rats but only has marginal effects on anxiety, learning and memory. Pharmacol Biochem Behav 93: 482–490. [DOI] [PubMed] [Google Scholar]
- Hill MN, McLaughlin RJ, Bingham B, Shrestha L, Lee TT, Gray JM, Hillard CJ, Gorzalka BB, Viau V. 2010. Endogenous cannabinoid signaling is essential for stress adaptation. Proc Natl Acad Sci 107: 9406–9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holliday ED, Nucero P, Kutlu MG, Oliver C, Connelly KL, Gould TJ, Unterwald EM. 2016. Long-term effects of chronic nicotine on emotional and cognitive behaviors and hippocampus cell morphology in mice: comparisons of adult and adolescent nicotine exposure. Eur J Neurosci 44: 2818–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen LK, Mencl WE, Westerveld M, Pugh KR. 2004. Impact of cannabis use on brain function in adolescents. Ann N Y Acad Sci 1021: 384–390. [DOI] [PubMed] [Google Scholar]
- Johnston LD, O'Malley PM, Bachman JG, Schulenberg JE, Miech RA. 2016. Monitoring the future national survey results on drug use, 1975–2015: Volume 2, College students and adult ages 19–55. Institute for Social Research, The University of Michigan, Ann Arbor. [Google Scholar]
- Johnston LD, Miech RA, O'Malley PM, Bachman JG, Schulenberg JE, Patrick ME. 2018. Monitoring the future national survey results on drug use: 1975–2017: Overview, key findings on adolescent drug use. Institute for Social Research, The University of Michigan, Ann Arbor. [Google Scholar]
- Kreslake JM, Wayne GF, Alpert HR, Koh HK, Connolly GN. 2008. Tobacco industry control of menthol in cigarettes and targeting of adolescents and young adults. Am J Public Health 98: 1685–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Yuan K, Cai C, Feng D, Yin J, Bi Y, Shi S, Yu D, Jin C, von Deneen KM, et al. 2015. Reduced frontal cortical thickness and increased caudate volume within fronto-striatal circuits in young adult smokers. Drug Alcohol Depend 151: 211–219. [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Dimen KR, Martin BR. 1995. Systemic or intrahippocampal cannabinoid administration impairs spatial memory in rats. Psychopharmacology (Berl) 119: 282–290. [DOI] [PubMed] [Google Scholar]
- Ling PM, Glantz SA. 2002. Using tobacco-industry marketing research to design more effective tobacco-control campaigns. JAMA 287: 2983–2989. [DOI] [PubMed] [Google Scholar]
- Logue SF, Paylor R, Wehner JM. 1997. Hippocampal lesions cause learning deficits in inbred mice in the Morris water maze and conditioned-fear task. Behav Neurosci 111: 104–113. [DOI] [PubMed] [Google Scholar]
- Lopez-Larson MP, Bogorodzki P, Rogowska J, McGlade E, King JB, Terry J, Yurgelun-Todd D. 2011. Altered prefrontal and insular cortical thickness in adolescent marijuana users. Behav Brain Res 220: 164–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Larson MP, Rogowska J, Yurgelun-Todd D. 2015. Aberrant orbitofrontal connectivity in marijuana smoking adolescents. Dev Cogn Neurosci 16: 54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovelace JW, Corches A, Vieira PA, Hiroto AS, Mackie K, Korzus E. 2015. An animal model of female adolescent cannabinoid exposure elicits a long-lasting deficit in presynaptic long-term plasticity. Neuropharmacology 99: 242–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone DT, Hill MN, Rubino T. 2010. Adolescent cannabis use and psychosis: epidemiology and neurodevelopmental models. Br J Pharmacol 160: 511–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchalant Y, Rosi S, Wenk GL. 2007. Anti-inflammatory property of the cannabinoid agonist WIN-55212–2 in a rodent model of chronic brain inflammation. Neuroscience 144: 1516–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maren S. 2001. Neurobiology of Pavlovian fear conditioning. Annu Rev Neurosci 24: 897–931. [DOI] [PubMed] [Google Scholar]
- Mateos B, Borcel E, Loriga R, Luesu W, Bini V, Llorente R, Castelli MP, Viveros MP. 2011. Adolescent exposure to nicotine and/or the cannabinoid agonist CP 55,940 induces gender-dependent long-lasting memory impairments and changes in brain nicotinic and CB(1) cannabinoid receptors. J Psychopharmacol 25: 1676–1690. [DOI] [PubMed] [Google Scholar]
- Matthews DB, Morrow AL, Tokunaga S, McDaniel JR. 2002. Acute ethanol administration and acute allopregnanolone administration impair spatial memory in the Morris water task. Alcohol Clin Exp Res 26: 1747–1751. [DOI] [PubMed] [Google Scholar]
- Meier MH, Caspi A, Ambler A, Harrington H, Houts R, Keefe RS, McDonald K, Ward A, Poulton R, Moffitt TE. 2012. Persistent cannabis users show neuropsychological decline from childhood to midlife. Proc Natl Acad Sci 109: E2657–E2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti S, Franchi S, Castelli M, Amodeo G, Somaini L, Panerai A, Sacerdote P. 2015. Exposure of adolescent mice to Δ9-tetrahydrocannabinol induces long-lasting modulation of pro- and anti-inflammatory cytokines in hypothalamus and hippocampus similar to that observed for peripheral macrophages. J Neuroimmune Pharmacol 10: 371–379. [DOI] [PubMed] [Google Scholar]
- Musso F, Bettermann F, Vucurevic G, Stoeter P, Konrad A, Winterer G. 2007. Smoking impacts on prefrontal attentional network function in young adult brains. Psychopharmacology (Berl) 191: 159–169. [DOI] [PubMed] [Google Scholar]
- Obernier JA, Bouldin TW, Crews FT. 2002. Binge ethanol exposure in adult rats causes necrotic cell death. Alcohol Clin Exp Res 26: 547–557. [PubMed] [Google Scholar]
- O'Shea M, Singh ME, McGregor IS, Mallet PE. 2004. Chronic cannabinoid exposure produces lasting memory impairment and increased anxiety in adolescent but not adult rats. J Psychopharmacol 18: 502–508. [DOI] [PubMed] [Google Scholar]
- O'Shea M, McGregor IS, Mallet PE. 2006. Repeated cannabinoid exposure during perinatal, adolescent or early adult ages produces similar longlasting deficits in object recognition and reduced social interaction in rats. J Psychopharmacol 20: 611–621. [DOI] [PubMed] [Google Scholar]
- Pamplona FA, Takahashi RN. 2006. WIN 55212–2 impairs contextual fear conditioning through the activation of CB1 cannabinoid receptors. Neurosci Lett 397: 88–92. [DOI] [PubMed] [Google Scholar]
- Pascual M, Blanco AM, Cauli O, Minarro J, Guerri C. 2007. Intermittent ethanol exposure induces inflammatory brain damage and causes long-term behavioural alterations in adolescent rats. Eur J Neurosci 25: 541–550. [DOI] [PubMed] [Google Scholar]
- Petanjek Z, Judas M, Simic G, Rasin MR, Uylings HB, Rakic P, Kostovic I. 2011. Extraordinary neoteny of synaptic spines in the human prefrontal cortex. Proc Natl Acad Sci 108: 13281–13286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfefferbaum A, Sullivan EV, Mathalon DH, Lim KO. 1997. Frontal lobe volume loss observed with magnetic resonance imaging in older chronic alcoholics. Alcohol Clin Exp Res 21: 521–529. [DOI] [PubMed] [Google Scholar]
- Pickens LR, Rowan JD, Bevins RA, Fountain SB. 2013. Sex differences in adult cognitive deficits after adolescent nicotine exposure in rats. Neurotoxicol Teratol 38: 72–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole RL, Connor DA, Gould TJ. 2014. Donepezil reverses nicotine withdrawal-induced deficits in contextual fear conditioning in C57BL/6J mice. Behav Neurosci 128: 588–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope HG Jr, Gruber AJ, Hudson JI, Huestis MA, Yurgelun-Todd D. 2001. Neuropsychological performance in long-term cannabis users. Arch Gen Psychiatry 58: 909–915. [DOI] [PubMed] [Google Scholar]
- Portugal GS, Wilkinson DS, Turner JR, Blendy JA, Gould TJ. 2012. Developmental effects of acute, chronic, and withdrawal from chronic nicotine on fear conditioning. Neurobiol Learn Mem 97: 482–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prenderville JA, Kelly AM, Downer EJ. 2015. The role of cannabinoids in adult neurogenesis. Br J Pharmacol 172: 3950–3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prini P, Rusconi F, Zamberletti E, Gabaglio M, Penna F, Fasano M, Battaglioli E, Parolaro D, Rubino T. 2018. Adolescent THC exposure in female rats leads to cognitive deficits through a mechanism involving chromatin modifications in the prefrontal cortex. J Psychiatry Neurosci 43: 87–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn HR, Matsumoto I, Callaghan PD, Long LE, Arnold JC, Gunasekaran N, Thompson MR, Dawson B, Mallet PE, Kashem MA, et al. 2008. Adolescent rats find repeated Δ9-THC less aversive than adult rats but display greater residual cognitive deficits and changes in hippocampal protein expression following exposure. Neuropsychopharmacology 33: 1113–1126. [DOI] [PubMed] [Google Scholar]
- Ragozzino ME. 2007. The contribution of the medial prefrontal cortex, orbitofrontal cortex, and dorsomedial striatum to behavioral flexibility. Ann N Y Acad Sci 1121: 355–375. [DOI] [PubMed] [Google Scholar]
- Ramachandran B, Ahmed S, Zafar N, Dean C. 2015. Ethanol inhibits long-term potentiation in hippocampal CA1 neurons, irrespective of lamina and stimulus strength, through neurosteroidogenesis. Hippocampus 25: 106–118. [DOI] [PubMed] [Google Scholar]
- Renard J, Vitalis T, Rame M, Krebs MO, Lenkei Z, Le Pen G, Jay TM. 2016. Chronic cannabinoid exposure during adolescence leads to long-term structural and functional changes in the prefrontal cortex. Eur Neuropsychopharmacol 26: 55–64. [DOI] [PubMed] [Google Scholar]
- Robbins TW. 2002. The 5-choice serial reaction time task: behavioural pharmacology and functional neurochemistry. Psychopharmacology (Berl) 163: 362–380. [DOI] [PubMed] [Google Scholar]
- Rubino T, Realini N, Braida D, Alberio T, Capurro V, Vigano D, Guidali C, Sala M, Fasano M, Parolaro D. 2009a. The depressive phenotype induced in adult female rats by adolescent exposure to THC is associated with cognitive impairment and altered neuroplasticity in the prefrontal cortex. Neurotox Res 15: 291–302. [DOI] [PubMed] [Google Scholar]
- Rubino T, Realini N, Braida D, Guidi S, Capurro V, Vigano D, Guidali C, Pinter M, Sala M, Bartesaghi R, et al. 2009b. Changes in hippocampal morphology and neuroplasticity induced by adolescent THC treatment are associated with cognitive impairment in adulthood. Hippocampus 19: 763–772. [DOI] [PubMed] [Google Scholar]
- Sakharkar AJ, Vetreno RP, Zhang H, Kokare DM, Crews FT, Pandey SC. 2016. A role for histone acetylation mechanisms in adolescent alcohol exposure-induced deficits in hippocampal brain-derived neurotrophic factor expression and neurogenesis markers in adulthood. Brain Struct Funct 221: 4691–4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenova S. 2012. Attention, impulsivity, and cognitive flexibility in adult male rats exposed to ethanol binge during adolescence as measured in the five-choice serial reaction time task: the effects of task and ethanol challenges. Psychopharmacology (Berl) 219: 433–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenova S, Stolerman IP, Markou A. 2007. Chronic nicotine administration improves attention while nicotine withdrawal induces performance deficits in the 5-choice serial reaction time task in rats. Pharmacol Biochem Behav 87: 360–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senn R, Keren O, Hefetz A, Sarne Y. 2008. Long-term cognitive deficits induced by a single, extremely low dose of tetrahydrocannabinol (THC): behavioral, pharmacological and biochemical studies in mice. Pharmacol Biochem Behav 88: 230–237. [DOI] [PubMed] [Google Scholar]
- Silveira MM, Adams WK, Morena M, Hill MN, Winstanley CA. 2017. Δ9-Tetrahydrocannabinol decreases willingness to exert cognitive effort in male rats. J Psychiatry Neurosci 42: 131–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sircar R, Sircar D. 2006. Repeated ethanol treatment in adolescent rats alters cortical NMDA receptor. Alcohol 39: 51–58. [DOI] [PubMed] [Google Scholar]
- Spaeth AM, Barnet RC, Hunt PS, Burk JA. 2010. Adolescent nicotine exposure disrupts context conditioning in adulthood in rats. Pharmacol Biochem Behav 96: 501–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear LP. 2000. The adolescent brain and age-related behavioral manifestations. Neurosci Biobehav Rev 24: 417–463. [DOI] [PubMed] [Google Scholar]
- Steinberg L. 2005. Cognitive and affective development in adolescence. Trends Cogn Sci 9: 69–74. [DOI] [PubMed] [Google Scholar]
- Stubley-Weatherly L, Harding JW, Wright JW. 1996. Effects of discrete kainic acid-induced hippocampal lesions on spatial and contextual learning and memory in rats. Brain Res 716: 29–38. [DOI] [PubMed] [Google Scholar]
- Suenaga T, Ichitani Y. 2008. Effects of hippocampal administration of a cannabinoid receptor agonist WIN 55,212–2 on spontaneous object and place recognition in rats. Behav Brain Res 190: 248–252. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Hagino H, Nohara S, Zhou SY, Kawasaki Y, Takahashi T, Matsui M, Seto H, Ono T, Kurachi M. 2005. Male-specific volume expansion of the human hippocampus during adolescence. Cereb Cortex 15: 187–193. [DOI] [PubMed] [Google Scholar]
- Trauth JA, Seidler FJ, McCook EC, Slotkin TA. 1999. Adolescent nicotine exposure causes persistent upregulation of nicotinic cholinergic receptors in rat brain regions. Brain Res 851: 9–19. [DOI] [PubMed] [Google Scholar]
- Trauth JA, McCook EC, Seidler FJ, Slotkin TA. 2000. Modeling adolescent nicotine exposure: effects on cholinergic systems in rat brain regions. Brain Res 873: 18–25. [DOI] [PubMed] [Google Scholar]
- Treur JL, Willemsen G, Bartels M, Geels LM, van Beek JH, Huppertz C, van Beijsterveldt CE, Boomsma DI, Vink JM. 2015. Smoking during adolescence as a risk factor for attention problems. Biol Psychiatry 78: 656–663. [DOI] [PubMed] [Google Scholar]
- Tselnicker I, Keren O, Hefetz A, Pick CG, Sarne Y. 2007. A single low dose of tetrahydrocannabinol induces long-term cognitive deficits. Neurosci Lett 411: 108–111. [DOI] [PubMed] [Google Scholar]
- Varvel SA, Hamm RJ, Martin BR, Lichtman AH. 2001. Differential effects of Δ9-THC on spatial reference and working memory in mice. Psychopharmacology (Berl) 157: 142–150. [DOI] [PubMed] [Google Scholar]
- Vetreno RP, Crews FT. 2012. Adolescent binge drinking increases expression of the danger signal receptor agonist HMGB1 and Toll-like receptors in the adult prefrontal cortex. Neuroscience 226: 475–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, Crews FT. 2015. Binge ethanol exposure during adolescence leads to a persistent loss of neurogenesis in the dorsal and ventral hippocampus that is associated with impaired adult cognitive functioning. Front Neurosci 9: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, Yaxley R, Paniagua B, Johnson GA, Crews FT. 2017. Adult rat cortical thickness changes across age and following adolescent intermittent ethanol treatment. Addict Biol 22: 712–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JM, Huang SM, Strangman NM, Tsou K, Sanudo-Pena MC. 1999. Pain modulation by release of the endogenous cannabinoid anandamide. Proc Natl Acad Sci 96: 12198–12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburton EC, Brown MW. 2015. Neural circuitry for rat recognition memory. Behav Brain Res 285: 131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegener N, Kuhnert S, Thuns A, Roese R, Koch M. 2008. Effects of acute systemic and intra-cerebral stimulation of cannabinoid receptors on sensorimotor gating, locomotion and spatial memory in rats. Psychopharmacology (Berl) 198: 375–385. [DOI] [PubMed] [Google Scholar]
- Wierenga L, Langen M, Ambrosino S, van Dijk S, Oranje B, Durston S. 2014. Typical development of basal ganglia, hippocampus, amygdala and cerebellum from age 7 to 24. Neuroimage 96: 67–72. [DOI] [PubMed] [Google Scholar]
- Wills TA, Sandy JM, Yaeger AM, Cleary SD, Shinar O. 2001. Coping dimensions, life stress, and adolescent substance use: a latent growth analysis. J Abnorm Psychol 110: 309–323. [DOI] [PubMed] [Google Scholar]
- Winters BD, Saksida LM, Bussey TJ. 2008. Object recognition memory: neurobiological mechanisms of encoding, consolidation and retrieval. Neurosci Biobehav Rev 32: 1055–1070. [DOI] [PubMed] [Google Scholar]
- World Health Organization. 2018. Tobacco Fact sheet. World Health Organization, http://www.who.int/mediacentre/factsheets/fs339/en/. [Google Scholar]
- Xu Z, Seidler FJ, Tate CA, Garcia SJ, Slikker W Jr, Slotkin TA. 2003. Sex-selective hippocampal alterations after adolescent nicotine administration: effects on neurospecific proteins. Nicotine Tob Res 5: 955–960. [DOI] [PubMed] [Google Scholar]
- Yildirim E, Connor DA, Gould TJ. 2015. ABT-089, but not ABT-107, ameliorates nicotine withdrawal-induced cognitive deficits in C57BL6/J mice. Behav Pharmacol 26: 241–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamberletti E, Beggiato S, Steardo L Jr, Prini P, Antonelli T, Ferraro L, Rubino T, Parolaro D. 2014. Alterations of prefrontal cortex GABAergic transmission in the complex psychotic-like phenotype induced by adolescent Δ9-tetrahydrocannabinol exposure in rats. Neurobiol Dis 63: 35–47. [DOI] [PubMed] [Google Scholar]
- Zamberletti E, Gabaglio M, Grilli M, Prini P, Catanese A, Pittaluga A, Marchi M, Rubino T, Parolaro D. 2016. Long-term hippocampal glutamate synapse and astrocyte dysfunctions underlying the altered phenotype induced by adolescent THC treatment in male rats. Pharmacol Res 111: 459–470. [DOI] [PubMed] [Google Scholar]