

Abstract
The rapid global evolution of influenza virus begins with mutations that arise de novo in individual infections, but little is known about how evolution occurs within hosts. We review recent progress in understanding how and why influenza viruses evolve within human hosts. Advances in deep sequencing make it possible to measure within-host genetic diversity in both acute and chronic influenza infections. Factors like antigenic selection, antiviral treatment, tissue specificity, spatial structure, and multiplicity of infection may affect how influenza viruses evolve within human hosts. Studies of within-host evolution can contribute to our understanding of the evolutionary and epidemiological factors that shape influenza virus’s global evolution.
Keywords: influenza, evolution, within-host, deep sequencing
Why study how influenza viruses evolve within human hosts?
Influenza viruses evolve rapidly on a global scale [1–4], and this evolution begins with mutations that arise de novo within infected hosts (Figure 1). As influenza viruses replicate during an infection, they quickly mutate [5–9] to form genetically diverse populations [10–13]. A small proportion of within-host variants transmit and found a new infection [14–16], and of those, a small number of variants may eventually fix in the global population of influenza viruses. Influenza virus’s evolution at the within-host scale is important because it provides the substrate for global evolution.
Figure 1. Within- and between-host evolutionary scales.
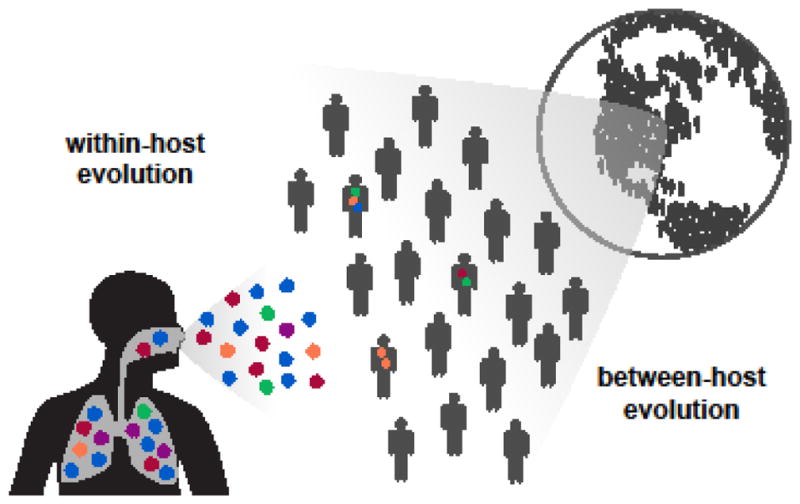
The rapid global evolution of influenza virus begins with de novo mutations that arise within individual infected hosts.
How do influenza viruses evolve within human hosts, and how does this within-host genetic variation give rise to influenza virus’s rapid global evolution? Within hosts, influenza viruses infect heterogeneous cell populations that are arranged in complex spatial structures [17,18]. Viruses encounter innate immune defenses like mucus barriers and interferon responses [19], as well as adaptive immune responses like antibodies that accumulate over the lifetime of the host [20,21]. In some cases, influenza viruses also encounter antiviral drugs like adamantanes and oseltamivir [22–24]. These factors can shape how influenza viruses evolve within humans as well as what viral variants arise and eventually transmit from one individual to another [25].
In this review, we summarize recent progress in understanding how and why influenza viruses evolve during the course of an infection, and how evolution within human hosts relates to the virus’s global evolution. High-throughput sequencing now makes it possible to “deep sequence” the viral population within a host to measure genetic diversity, so we begin by surveying current deep sequencing methods and their limitations. We then present studies that use deep sequencing to assess viral genetic variation during acute human influenza A infections as well as during chronic influenza infections in immunocompromised human hosts. We consider how factors like antigenic selection, antiviral treatment, tissue specificity, spatial structure, and multiplicity of infection may shape how influenza viruses evolve within hosts. Finally, we discuss how this within-host diversity might relate to global evolution.
How is deep sequencing used to measure within-host viral diversity?
Traditionally, the viral population within an influenza infection is summarized as a single consensus sequence, representing the most frequent nucleotide at each genome position. For instance, public databases contain tens of thousands of influenza virus sequences, nearly all of which are consensus sequences [26–28]. But in reality, each influenza infection generates a genetically diverse cloud of viral variants that are formed through de novo mutation, and variants can also be transmitted from host to host [10–13,29]. Most mutations in a viral population are expected to reach very low frequencies (Box 1), and very few of these viral variants ever reach majority status in an infection. But the genetic diversity within an infection can reveal important evolutionary dynamics—and provides the material on which Darwinian selection can act.
Box 1. Within-host diversity of influenza viruses under neutral evolution.
How much genetic diversity is expected to arise as influenza viruses replicate within human hosts? In evolutionary biology, it can be useful to estimate what variation would be observed if all mutations were purely neutral. Simple frameworks that model neutral evolution can establish basic expectations, even though purifying and positive selection clearly affect the mutation frequencies observed in real infections.
In human hosts, influenza virus populations expand exponentially at the beginning of an acute infection. Viral titers peak two to four days after the infection’s start, and afterwards, titers decline for three or four days until the virus reaches undetectable levels [57,111]. Mutations that arise early in the exponential expansion can reach high frequencies through Luria-Delbruck dynamics [112].
To estimate how many mutations are expected to reach detectable frequencies under neutral evolution, we use the stochastic birth-death model proposed by Bozic et al. to describe how neutral mutations accumulate as cells expand clonally during cancer evolution [113]. In this model, a viral population begins with a single starting genotype, although natural human infections begin with anywhere from one to several hundred initial genotypes [62,61]. Viruses reproduce at rate b and leave the population at rate d. Neutral mutations occur at a rate of u mutations per genome per replication cycle, and all sites are completely linked. Bozic et al. demonstrate that the expected number of mutations m above frequency α is
We estimate b and d using Beauchemin’s and Handel’s models of influenza-virus kinetics within human hosts [111]. If influenza viruses expand exponentially with rate b-d for the first phase of the infection and then decline exponentially with rate d after viral titers peak, then we estimate b ≈5.7/day and d≈3.2/day. Most studies in cell culture estimate mutation rates ranging from 10−6 to 10−5/site/generation depending on the type of mutation and exact method of estimation [5–8], although one recent study estimates a higher rate of 10−4/site/generation [9]. These per-site mutation rates correspond to u≈0.013 to u≈1.3 across the 1.3kB viral genome. Since the number of expected mutations is directly proportional to the viral mutation rate, this variation has a large effect on estimates of genetic diversity (Figure I).
Future work that refines estimates of mutation rate would help establish more confident expectations about within-host viral diversity. It will also be important to develop models with more realistic assumptions about initial within-host genetic diversity, as well as how purifying and positive selection would affect this variation. By comparing these models with empirical observations, we can improve our understanding of how influenza viruses evolve within human hosts.
Figure I.

Expected number of within-host variants m above a given variant frequency α under neutral evolution. Expectations are displayed for different values of the per-site, per-generation mutation rate μ, which is multiplied by the number of base pairs in the genome of influenza virus to obtain the per-genome mutation rate u.
Recent advances in high-throughput sequencing have made it possible to assess mutation frequencies and measure within-host genetic diversity (Figure 2a) [30,31]. Common deep-sequencing approaches can detect viral mutations above frequencies of approximately 1% in the total within-host viral population [32,33], though it remains difficult to determine linkage among these mutations [30,31]. But despite its power, deep sequencing is subject to important technical limitations that are essential to consider when designing experiments and analyzing data [31–33].
Figure 2. Deep-sequencing approaches to measuring within-host genetic diversity.
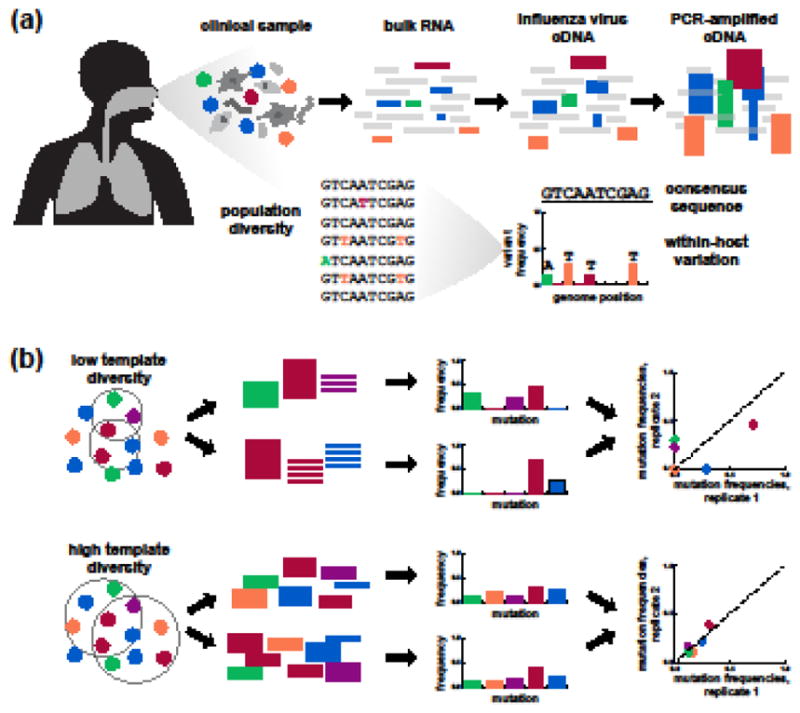
(a) Common deep-sequencing workflows can identify variants that make up approximately 1% of the within-host population. (b) Most studies amplify viral genetic material prior to deep sequencing. Low template diversity, typically due to low viral load, can distort the variant frequencies measured by deep sequencing. Replicate libraries are important for identifying and excluding samples with low viral load that should be excluded from downstream analyses.
Experimental design
A fundamental challenge of viral deep sequencing is the fact that in clinical samples, viral genetic material is often dwarfed by that of the host and co-occurring microbes. To compensate, most studies rely on PCR amplification to enrich for viral genetic material [32,34–36]. This amplification is relatively straightforward for the influenza-virus genome, which contains conserved regions at the ends of each gene segment [37]. Following reverse transcription, the entire genome can be amplified using a single set of PCR primers complementary to these conserved regions [38–40] or a primer cocktail that is complementary to the conserved regions along with non-coding sequence specific to each gene [35,37].
Various aspects of the sample and its preparation affect how accurately deep sequencing measures the actual viral variant frequencies within an infected individual [31–33,41,42]. Of these factors, the most important by far is viral load (Figure 2b) [32,43]. During whole-genome amplification, anywhere from 20 to 35 cycles of PCR may be required to produce sufficient material for sequencing. When the number of starting viral template molecules is low, below about 1000 copies per uL total RNA [32], this amplification can significantly distort variant frequencies [32,43,44]. By comparison, errors that accumulate during reverse transcription, PCR, and Illumina sequencing have smaller effects for samples with typical low viral loads [32,43].
It is therefore essential to maximize the amount of viral genetic material used in each RNA extraction, reverse-transcription, and PCR reaction to ensure that deep sequencing accurately measures variant frequencies in the viral population. It is also important to prepare and sequence replicate libraries [41], preferably beginning from independent reverse-transcription reactions [32]. Replicate libraries make it possible to identify samples with low viral load [32,35] or effective sequencing depth [41] that should be excluded from downstream analyses (Figure 2b). They also make it possible to empirically set variant-calling thresholds and exclude specific low-confidence viral variants whose frequencies vary extensively between replicates in an otherwise high-quality sample [32].
Limitations
Deep sequencing can identify rare mutations in a viral population, but it has limited power to determine patterns of linkage between mutations, which can reveal patterns of epistasis [45] and clonal competition [35]. Short reads can sometimes reveal linkage between closely spaced mutations [35,46–48], but the reads produced by Illumina sequencing are unable to span even the smallest influenza-virus genes. Several groups have successfully assembled viral haplotypes and assessed their frequencies by combining low-coverage PacBio sequencing, which produces long reads, with high-coverage Illumina sequencing [36]. But even these methods cannot directly determine linkage between mutations on different gene segments, even though intergenic epistasis [49–52] and gene reassortment [53] both affect influenza-virus evolution. In the absence of sequencing data that directly observe patterns of linkage between mutations, computational methods can sometimes infer longer haplotypes by assembling multiple short-read haplotypes [30,31,45,48] and tracking concordant changes in allele frequencies between mutations located on different genes [45,48]. Even with current technical limitations, deep-sequencing approaches to measure viral variation can still shed light on important within-host evolutionary dynamics.
How do influenza viruses evolve within human hosts?
Several recent studies have used deep sequencing to characterize the spectrum of genetic diversity within natural human influenza A infections, and we summarize their findings here. Most studies focus on typical acute infections in immunocompetent hosts, but some studies also examine viral evolution during the lengthy infections experienced by immunocompromised patients.
Acute infections
Viruses like HIV and hepatitis C virus establish long-term infections and evolve over years or decades to avoid the immune system and develop antiviral resistance [54–56]. In contrast, influenza infections typically last five to seven days, and viral shedding peaks two to four days after infections begin [57,58]. These short infections provide little time for de novo mutations to arise, for selection to act on these mutations, and for selected mutations to reach frequencies at which they are detectable by deep sequencing (Box 1).
Most studies of natural, acute influenza infections analyze one or two nasal swab or nasal wash samples from each patient by deep sequencing the hemagglutinin gene [59,60] or the entire viral genome [34,61,62]. The exact number of viral variants identified is highly dependent on sample quality and sequencing methodology. But several studies have observed relatively limited genetic diversity within acute human influenza infections [34,60,62], identifying fewer than ten variants per infection across the influenza-virus genome at a limit of detection of approximately 1–2% [34,62]. Most of these mutations are rare, present in less than 10% of the viruses within a host [34,60,62], and the number and frequency of within-host viral variants does not seem to correlate with how many days post-infection the samples were collected [34]. However, some acute infections harbor high genetic diversity due to apparent co-infection by multiple, related viral strains [61,62]. One study has found evidence of mixed infections in approximately half of the patients sequenced [61], and the contribution of co-infection to within-host genetic diversity requires further careful study. Overall, the limited genetic diversity found in many acute human influenza infections agrees with prior studies in dogs and horses that sequenced viral clones to measure within-host viral variation [63–66].
It remains unclear what influences the patterns of observed variation, although we discuss potential biological factors below. Generally, within-host variants tend to be dispersed across the viral genome [34,62], though one study observes some low-frequency variation in putative antigenic sites [60]. Another study estimated that the ratio of nonsynonymous to synonymous within-host variants is about 0.64 and suggested that purifying selection removes some deleterious variants in human infections [62]. Even if most acute human infections do not contain high-frequency mutations, the sheer number of influenza infections every year may allow the rapid global evolution of influenza virus to arise from limited within-host genetic diversity.
Chronic infections
The vast majority of influenza infections are acute, but immunocompromised patients can experience severe infections lasting multiple weeks or months [67–69]. These chronic infections differ from acute infections in that host immune responses may be weakened or absent, infections are commonly treated with long courses of antiviral drugs, and influenza virus commonly co-occurs with other respiratory pathogens [67–69].
Nevertheless, chronic infections provide unusual opportunities to observe how influenza viruses evolve within humans over longer spans of time, when selection has more opportunities to shape viral variation. Immunocompromised patients often receive close clinical monitoring, and several studies have tracked within-host evolution longitudinally by deep sequencing clinical samples taken from different time points in an infection [35,36,70]. In these chronic infections, influenza viruses can display extensive evolution. Putative antigenic variants can arise and reach high within-host frequencies [35,71–73]. Multiple drug-resistant variants can also arise during these lengthy infections [35,36,70,73]. It is common for multiple beneficial mutations to compete with one another within a patient [35,36], displaying clonal interference dynamics commonly observed in experimental evolution [74–76].
The relatively weak immune responses mounted by immunocompromised hosts can have important evolutionary consequences, regardless of the exact underlying medical conditions. Small viral populations can survive and replicate in the presence of weak selection, making it easier for multiple adaptive mutations to emerge simultaneously [77]. In chronic influenza infections, relatively weaker immune responses can lead to much longer viral infections, enabling putative antigenic variants to arise in ways that sometimes parallel global evolutionary trends [35]. Overall, though, it remains unclear how much the evolutionary forces that act within chronic infections resemble selective pressures within more common, acute infections.
What affects how influenza viruses evolve within humans?
Here, we consider evidence for how antigenic selection, antiviral treatment, tissue specificity, spatial structure, and multiplicity of infection may shape how influenza viruses evolve within humans (Figure 3).
Figure 3. Factors affecting the evolution of influenza virus within human hosts.
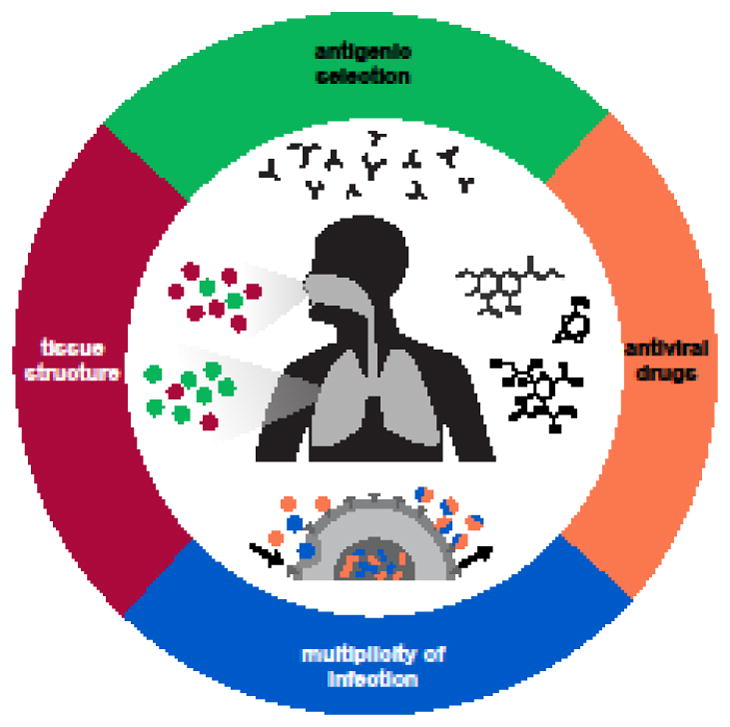
Antigenic selection, antiviral drugs, tissue structure, and multiplicity of infection can affect how influenza viruses evolve within hosts.
Antigenic selection
Human influenza viruses undergo constant antigenic drift and occasional antigenic shift on a global evolutionary scale [20,78,79], but it is unclear how much immune selection takes place within a typical human infection. Recent deep-sequencing studies have identified few antigenic variants within acute infections [34,60,62], though it remains unclear whether antigenic variants are enriched or depleted relative to the frequency of within-host variants as a whole. In immunocompromised patients, putative antigenic variants can arise, display complex clonal dynamics, and even fix during an infection [35,71–73]. Some of the putative antigenic variants that arise in immunocompromised patients also reach a high frequency in the global population of influenza viruses [35].
Another source of antigenic selection might be vaccination, which boosts immune responses against influenza viruses. Two recent studies deep sequenced viral populations from vaccine recipients and control groups [34,60]. They found that vaccination status did not seem to affect consensus viral sequences, suggesting that infections in vaccinated individuals are not caused by specific resistant viral strains [34,60]. Moreover, they found that vaccination had no detectable effect on the number or population frequency of within-host variants [34,60]. One interpretation is that antigenic selection does not act detectably in most infections. An alternative explanation is that many unvaccinated individuals may already have strong immunity from natural infections, and vaccination may not alter immunity enough to exert additional antigenic selection.
Antiviral resistance
Antiviral agents are used to treat only a minority of acute influenza infections, but they can still exert important influences on viral evolution [22–24]. For instance, many influenza A strains are resistant to adamantanes [24,80], and resistance to oseltamivir swept to fixation in seasonal H1N1 influenza viruses before they were replaced by pandemic H1N1 [24,81,82]. For antivirals like oseltamivir, where drug resistance is not yet widespread in current strains, influenza viruses can gain resistance within individual infections by acquiring one or more de novo mutations [24]. As with antigenic selection, it is unclear how frequently drug resistance arises within typical, acute infections. In one case report, resistance arose even when oseltamivir was used for prophylaxis [83], but deep sequencing of viral populations from thirteen individuals in a human challenge study detected no drug-resistant variants following early or standard oseltamivir treatment [48]. There is ample evidence, however, that resistance can arise rapidly during longer infections [35,36,67–69,73,84,85]. In some cases, multiple drug-resistant variants may even compete within a single patient [35,36]. Since the mutations and molecular mechanisms underlying antiviral resistance are well established, antiviral resistance can serve as a useful comparison for studying how other selective pressures may act within hosts.
Tissue specificity and spatial structure
Influenza viruses infect heterogeneous, spatially structured populations of cells in the human airways. Differences between tissues, along with neutral processes of migration and genetic drift, may have important effects on viral evolution. One major difference between the upper and lower human airways is their distribution of sialic acid receptors, which influenza viruses use to enter host cells. Most human influenza infections primarily take place in the upper human respiratory tract, which contains a higher proportion of α2,6-linked sialic acids than the lower airways, which contain a higher proportion of α2,3-linked sialic acids [17,18]. These histological differences may affect which viruses are transmitted. In ferrets, for example, viruses tend to transmit from the upper respiratory tract [15], and viral variants that preferentially bind to α2,6-linked receptors transmit more frequently than variants that bind α2,3-linked receptors [86]. This combination of spatial structure and tissue specificity provides one possible explanation for why avian-derived viruses, which tend to be adapted to the α2,3-linked sialic acid receptors in avian airways, can cause severe, lower lung infections in humans but rarely transmit from one human host to another [17,18,87]. Within these two broad linkage categories, sialic acid chains also vary extensively in length and chemical linkages and are distributed differently in the airways of avian and mammalian host species, potentially affecting influenza virus binding [88–90].
Even in the absence of tissue-specific selection, spatial structure can also limit genetic exchange between different parts of the human airways. For instance, one case report of a human infection documented the presence of distinct viral populations in the right and left lungs [91]. More generally, though, no deep sequencing studies have systematically compared viral populations sampled from different parts of the human airways, and the extent of tissue-specific selection remains an important open question.
Multiplicity of infection
Spatial structure affects how densely viruses populate different parts of the human airways, and in turn, this within-host multiplicity of infection (MOI) determines how often two or more viruses co-infect the same host cell. When multiple viruses co-infect the same cell, viral gene segments have an opportunity to reassort, and they do so readily in cell culture and animal models [53,92,93]. New combinations of gene segments are important for purging deleterious mutations in an otherwise clonal population and for forming new, potentially advantageous combinations between mutations [53]. It is usually difficult to estimate rates of within-host reassortment because current deep sequencing techniques are unable to establish linkage across multiple gene segments. But one group has developed a population-genetics framework to infer recombination from longitudinal, short-read sequencing data and estimated that the rate of effective within-host reassortment is low in human infections [48]. Rates of effective reassortment may be low even when viral load is high because spatial structure limits viral exchange so that most co-infection and reassortment occurs between genetically similar viruses.
Viral co-infection also provides opportunities for genetic complementation, which can decrease the efficacy of selection. If a wild-type virus and a virus carrying a deleterious mutation co-infect the same cell, the progeny virions can package both viral genomes, allowing the deleterious mutation to persist. The effects of complementation are especially clear in cell culture, where most influenza viruses are grown at high MOIs: defective viruses that carry large gene deletions quickly arise and spread through the population [94,95]. Large internal deletions have been documented in human influenza infections [96,97], and studies of influenza outbreaks in pigs and horses have documented the transmission of nonsense variants as well [66,98]. However, the overall prevalence of defective viral particles and their association with infection length and severity remain poorly understood.
Altogether, studies in cell culture and animal models suggest various biochemical and morphological factors that may affect how influenza viruses evolve within human hosts, but few deep sequencing studies so far have had the power to detect their effects. Additional sequencing of viral populations collected from different human hosts and tissues will improve our understanding of how influenza viruses evolve within a complex host environment.
How does influenza virus’s diversity within hosts relate to its global evolution?
The within-host evolution of influenza virus ultimately provides the substrate for the virus’s rapid global evolution, but the forces that transform within-host genetic diversity into global variation are largely unknown. Selection and drift can operate within hosts, but they also shape viral variation at transmission and at the host-population level.
Transmission
Only a small fraction of the influenza viruses within an infected individual go on to initiate subsequent infections (Figure 4). Transmission bottlenecks can limit the genetic diversity passed from one host to another and introduce stochasticity in variant frequencies along a transmission chain [29,62,61]. Transmission bottleneck sizes also affect how often genetically distinct strains of influenza virus infect the same individual [66,93], and looser bottlenecks increase the chance for beneficial reassortment [53,93].
Figure 4. Transmission bottlenecks shape viral evolution.

The size (a) and randomness (b) of transmission bottlenecks affect how much of the viral genetic diversity generated within one host survives to initiate another infection.
Deep sequencing of contact and recipient viral populations can help estimate transmission bottleneck sizes in natural infections. Narrower transmission bottlenecks increase the variance with which viral variants are transmitted [99]. Animal studies suggest that vaccination status [64] and route of transmission [15,16] can affect transmission bottleneck size, which appears to be looser for direct contact than for aerosol transmission [15,16]. Studies of influenza outbreaks in pigs and horses have suggested that transmission bottlenecks can be loose, with frequent mixed infections [65,66,98].
In human influenza infections, few studies have had the power to estimate transmission bottleneck sizes, and the two recent studies to do so have disagreed considerably in their results. Poon et al. estimate a relatively loose bottleneck size of approximately 200 distinct genomes for both H3N2 and pandemic H1N1 influenza virus based on a household cohort study performed during the first wave of the 2009 H1N1 pandemic [61], and a recent re-analysis of the same data supports these estimates [99]. More recently, McCrone et al. use similar analytical methods to infer a very narrow transmission bottleneck of 1 or 2 distinct genomes in a household cohort study that primarily sampled seasonal H3N2 influenza viruses from 2010 to 2015 [62].
It is unclear what accounts for the differences between these two estimates, although differences in study populations may contribute. For instance, influenza virus transmission depends on temperature and humidity [100]. The Poon et al. cohort was recruited in sub-tropical Hong Kong, while the McCrone et al. cohort was recruited in temperate Michigan, in the northern United States. Moreover, the Poon et al. study recruited index patients with acute respiratory illnesses and then prospectively followed their family members, whereas the McCrone et al. study prospectively enrolled households and queried participants weekly about symptoms of illness. Furthermore, estimates of transmission bottleneck size may also be highly sensitive to sample quality, library preparation and sequencing methods, and variant-calling thresholds.
Most studies assume that transmission bottlenecks act neutrally, but certain influenza-virus variants may be more likely than others to transmit and found new infections. For instance, one ferret study found that transmitted viruses tended to preferentially bind α2,6-linked sialic acid receptors and most closely resembled viral populations in the soft palate [86]. Selection can also affect maladapted strains of human influenza virus. In one recent human challenge study, volunteers were inoculated with viral stocks that had acquired passage-adaptation mutations during growth in eggs and cell culture. Many of these passage-adaptation mutations in the viral inoculum were purged from the viral population during or shortly after inoculation [101]. Selection may also act at transmission to promote global antigenic evolution if novel antigenic variants transmit and found new infections more frequently when host populations are mostly resistant to circulating strains. The strength and evolutionary effects of transmission bottlenecks remain important areas of study for understanding how the genetic diversity of influenza virus within hosts relates to its global genetic variation.
Comparing evolutionary scales
New mutations must arise and fix in individual hosts before they can spread through a large host population, linking within-host evolutionary dynamics to global evolution [102]. How do drift, positive selection, and purifying selection act within and between hosts? Studies of Ebola virus [103], Lassa virus [104], and dengue virus [105] have compared the proportions of nonsynonymous to synonymous within- and between-host variants to argue that purifying selection acts at within- and between-host scales to eliminate deleterious variants. However, the dN/dS ratio was originally developed to compare fixed variation between distantly diverged lineages, and within-host population dynamics can complicate its interpretation [106,107]. In cases where longitudinal deep-sequencing data is available, standard population-genetics models can be used to infer the influence of selection upon particular variants based on the changes in their allele frequencies over time [45,46,48]. But for most studies of within-host evolution, which lack longitudinal data, it remains a major challenge to develop appropriate methods that make use of deep-sequencing data to distinguish what evolutionary forces act on viral populations within hosts.
Concluding Remarks
By studying how influenza viruses evolve within humans, we can observe what biological factors affect the virus within its natural host environment (see Outstanding Questions). We can also determine what evolutionary and epidemiological forces transform within-host genetic diversity into global viral variation. As deep sequencing makes it easier to survey genetic diversity within hosts, it will be important to develop methodologies to systematically analyze within-host evolutionary dynamics and their relationship to global evolution.
Outstanding Questions.
What experimental designs and analytical approaches best identify rare viral variants in clinical samples?
How much does host immunity shape within-host viral genetic diversity?
How do viral populations evolve in different parts of the human airways?
What is the effective multiplicity of infection within human hosts?
How often do related viral strains co-infect the same host?
How do transmission bottlenecks shape the genetic diversity of founding viral populations?
How does host population immunity help transform within-host viral diversity into global genetic variation?
Trends.
Influenza viruses experience selection at the within- and between-host evolutionary scales.
Deep sequencing measures the genetic diversity of influenza viruses within human hosts.
Influenza virus accumulates relatively little diversity within typically short, acute human infections, although it can undergo substantial evolution during long-term human infections.
Influenza viruses replicate in a heterogeneous, spatially structured environment within hosts.
Transmission bottlenecks limit the genetic diversity that is passed from human to human.
Evolutionary and epidemiological factors shape how the within-host diversity of influenza virus relates to its global genetic change.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fitch WM, et al. Positive Darwinian evolution in human influenza A viruses. Proc Natl Acad Sci U S A. 1991;88:4270–4274. doi: 10.1073/pnas.88.10.4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ghedin E, et al. Large-scale sequencing of human influenza reveals the dynamic nature of viral genome evolution. Nature. 2005;437:1162–1166. doi: 10.1038/nature04239. [DOI] [PubMed] [Google Scholar]
- 3.Rambaut A, et al. The genomic and epidemiological dynamics of human influenza A virus. Nature. 2008;453:615–619. doi: 10.1038/nature06945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhatt S, et al. The genomic rate of molecular adaptation of the human influenza A virus. Mol Biol Evol. 2011;28:2443–51. doi: 10.1093/molbev/msr044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanjuán R, et al. Viral mutation rates. J Virol. 2010;84:9733–48. doi: 10.1128/JVI.00694-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nobusawa E, Sato K. Comparison of the mutation rates of human influenza A and B viruses. J Virol. 2006;80:3675–8. doi: 10.1128/JVI.80.7.3675-3678.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suarez-Lopez P, Ortin J. An estimation of the nucleotide substitution rate at defined positions in the influenza virus haemagglutinin gene. J Gen Virol. 1994;75:389–393. doi: 10.1099/0022-1317-75-2-389. [DOI] [PubMed] [Google Scholar]
- 8.Bloom JD. An experimentally determined evolutionary model dramatically improves phylogenetic fit. Mol Biol Evol. 2014;31:1956–78. doi: 10.1093/molbev/msu173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pauly MD, et al. A novel twelve class fluctuation test reveals higher than expected mutation rates for influenza A viruses. Elife. 2017;6:e26437. doi: 10.7554/eLife.26437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eigen M. Selforganization of matter and the evolution of biological macromolecules. Naturwissenschaften. 1971;58:465–523. doi: 10.1007/BF00623322. [DOI] [PubMed] [Google Scholar]
- 11.Holland J, et al. Rapid evolution of RNA genomes. Science (80-) 1982;215:1577–1585. doi: 10.1126/science.7041255. [DOI] [PubMed] [Google Scholar]
- 12.Lauring AS, Andino R. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog. 2010;6:e1001005. doi: 10.1371/journal.ppat.1001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andino R, Domingo E. Viral quasispecies. Virology. 2015;479–480:46–51. doi: 10.1016/j.virol.2015.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brankston G, et al. Lancet Infectious Diseases. Vol. 7. Elsevier; Apr 01, 2007. Transmission of influenza A in human beings; pp. 257–265. [DOI] [PubMed] [Google Scholar]
- 15.Varble A, et al. Influenza A virus transmission bottlenecks are defined by infection route and recipient host. Cell Host Microbe. 2014;16:691–700. doi: 10.1016/j.chom.2014.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frise R, et al. Contact transmission of influenza virus between ferrets imposes a looser bottleneck than respiratory droplet transmission allowing propagation of antiviral resistance. Sci Rep. 2016;6:29793. doi: 10.1038/srep29793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shinya K, et al. Avian flu: influenza virus receptors in the human airway. Nature. 2006;440:435–6. doi: 10.1038/440435a. [DOI] [PubMed] [Google Scholar]
- 18.van Riel D, et al. H5N1 Virus Attachment to Lower Respiratory Tract. Science. 2006;312:399. doi: 10.1126/science.1125548. [DOI] [PubMed] [Google Scholar]
- 19.Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nat Rev Immunol. 2014;14:315–328. doi: 10.1038/nri3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith DJ, et al. Mapping the Antigenic and Genetic Evolution of Influenza Virus. Science (80-) 2004;305:371–376. doi: 10.1126/science.1097211. [DOI] [PubMed] [Google Scholar]
- 21.Fonville JM, et al. Antibody landscapes after influenza virus infection or vaccination. Science (80-) 2014;346:996–1000. doi: 10.1126/science.1256427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McKimm-Breschkin JL. Resistance of influenza viruses to neuraminidase inhibitors — a review. Antiviral Res. 2000;47:1–17. doi: 10.1016/s0166-3542(00)00103-0. [DOI] [PubMed] [Google Scholar]
- 23.De Clercq E. Antiviral agents active against influenza A viruses. Nat Rev Drug Discov. 2006;5:1015–1025. doi: 10.1038/nrd2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van der Vries E, et al. Chapter Six Influenza Virus Resistance to Antiviral Therapy. Advances in Pharmacology. 2013;67:217–246. doi: 10.1016/B978-0-12-405880-4.00006-8. [DOI] [PubMed] [Google Scholar]
- 25.Grenfell BT. Unifying the Epidemiological and Evolutionary Dynamics of Pathogens. Science (80-) 2004;303:327–332. doi: 10.1126/science.1090727. [DOI] [PubMed] [Google Scholar]
- 26.Bogner P, et al. A global initiative on sharing avian flu data. Nature. 2006;442:981–981. [Google Scholar]
- 27.Bao Y, et al. The influenza virus resource at the National Center for Biotechnology Information. J Virol. 2008;82:596–601. doi: 10.1128/JVI.02005-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Squires RB, et al. Influenza Research Database: an integrated bioinformatics resource for influenza research and surveillance. Influenza Other Respi Viruses. 2012;6:404–416. doi: 10.1111/j.1750-2659.2011.00331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCrone JT, Lauring AS. Genetic bottlenecks in intraspecies virus transmission. Curr Opin Virol. 2018;28:20–25. doi: 10.1016/j.coviro.2017.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beerenwinkel N, et al. Challenges and opportunities in estimating viral genetic diversity from next-generation sequencing data. Front Microbiol. 2012;3:329. doi: 10.3389/fmicb.2012.00329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Posada-Cespedes S, et al. Recent advances in inferring viral diversity from high-throughput sequencing data. Virus Research. 2017;239:17–32. doi: 10.1016/j.virusres.2016.09.016. [DOI] [PubMed] [Google Scholar]
- 32.McCrone JT, Lauring AS. Measurements of Intrahost Viral Diversity Are Extremely Sensitive to Systematic Errors in Variant Calling. J Virol. 2016;90:6884–6895. doi: 10.1128/JVI.00667-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kugelman JR, et al. Error baseline rates of five sample preparation methods used to characterize RNA virus populations. PLoS One. 2017;12:e0171333. doi: 10.1371/journal.pone.0171333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Debbink K, et al. Vaccination has minimal impact on the intrahost diversity of H3N2 influenza viruses. PLOS Pathog. 2017;13:e1006194. doi: 10.1371/journal.ppat.1006194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xue KS, et al. Parallel evolution of influenza across multiple spatiotemporal scales. Elife. 2017;6:e26875. doi: 10.7554/eLife.26875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rogers MB, et al. Intrahost dynamics of antiviral resistance in influenza A virus reflect complex patterns of segment linkage, reassortment, and natural selection. MBio. 2015;6:e02464–14. doi: 10.1128/mBio.02464-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoffmann E, et al. Universal primer set for the full-length amplification of all influenza A viruses. Arch Virol. 2001;146:2275–2289. doi: 10.1007/s007050170002. [DOI] [PubMed] [Google Scholar]
- 38.Zhou B, et al. Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and Swine origin human influenza a viruses. J Virol. 2009;83:10309–13. doi: 10.1128/JVI.01109-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McGinnis J, et al. Next generation sequencing for whole genome analysis and surveillance of influenza A viruses. J Clin Virol. 2016;79:44–50. doi: 10.1016/j.jcv.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 40.Zhou B, et al. Universal influenza B virus genomic amplification facilitates sequencing, diagnostics, and reverse genetics. J Clin Microbiol. 2014;52:1330–7. doi: 10.1128/JCM.03265-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Illingworth CJR, et al. On the effective depth of viral sequence data. Virus Evol. 2017;3:vex030. doi: 10.1093/ve/vex030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zanini F, et al. Error rates, PCR recombination, and sampling depth in HIV-1 whole genome deep sequencing. Virus Res. 2017;239:106–114. doi: 10.1016/j.virusres.2016.12.009. [DOI] [PubMed] [Google Scholar]
- 43.Gallet R, et al. The number of target molecules of the amplification step limits accuracy and sensitivity in ultra deep sequencing viral population studies. J Virol. 2017 doi: 10.1128/JVI.00561-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kanagawa T. Bias and artifacts in multitemplate polymerase chain reactions (PCR) J Biosci Bioeng. 2003;96:317–323. doi: 10.1016/S1389-1723(03)90130-7. [DOI] [PubMed] [Google Scholar]
- 45.Illingworth CJR. Fitness inference from short-read data: Within-host evolution of a reassortant H5N1 influenza virus. Mol Biol Evol. 2015;32:3012–3026. doi: 10.1093/molbev/msv171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Illingworth CJR, et al. Identifying Selection in the Within-Host Evolution of Influenza Using Viral Sequence Data. PLoS Comput Biol. 2014;10:e1003755. doi: 10.1371/journal.pcbi.1003755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Illingworth CJR. SAMFIRE: Multi-locus variant calling for time-resolved sequence data. Bioinformatics. 2016;32:2208–2209. doi: 10.1093/bioinformatics/btw205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sobel Leonard A, et al. The effective rate of influenza reassortment is limited during human infection. PLOS Pathog. 2017;13:e1006203. doi: 10.1371/journal.ppat.1006203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mitnaul LJ, et al. Balanced Hemagglutinin and Neuraminidase Activities Are Critical for Efficient Replication of Influenza A Virus. J Virol. 2000;74:6015–6020. doi: 10.1128/jvi.74.13.6015-6020.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wagner R, et al. Functional balance between haemagglutinin and neuraminidase in influenza virus infections. Rev Med Virol. 2002;12:159–166. doi: 10.1002/rmv.352. [DOI] [PubMed] [Google Scholar]
- 51.Kryazhimskiy S, et al. Prevalence of epistasis in the evolution of influenza A surface proteins. PLoS Genet. 2011;7:e1001301. doi: 10.1371/journal.pgen.1001301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Neverov AD, et al. Coordinated Evolution of Influenza A Surface Proteins. PLoS Genet. 2015;11:e1005404. doi: 10.1371/journal.pgen.1005404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lowen AC. Constraints, Drivers, and Implications of Influenza A Virus Reassortment. Annu Rev Virol. 2017;4:105–121. doi: 10.1146/annurev-virology-101416-041726. [DOI] [PubMed] [Google Scholar]
- 54.Rambaut A, et al. The causes and consequences of HIV evolution. Nat Rev Genet. 2004;5:52–61. doi: 10.1038/nrg1246. [DOI] [PubMed] [Google Scholar]
- 55.Lemey P, et al. HIV evolutionary dynamics within and among hosts. AIDS Rev. 2006;8:125–40. [PubMed] [Google Scholar]
- 56.Simmonds P. Genetic diversity and evolution of hepatitis C virus--15 years on. J Gen Virol. 2004;85:3173–88. doi: 10.1099/vir.0.80401-0. [DOI] [PubMed] [Google Scholar]
- 57.Carrat F, et al. Time Lines of Infection and Disease in Human Influenza: A Review of Volunteer Challenge Studies. Am J Epidemiol. 2008;167:775–785. doi: 10.1093/aje/kwm375. [DOI] [PubMed] [Google Scholar]
- 58.Baccam P, et al. Kinetics of influenza A virus infection in humans. J Virol. 2006;80:7590–9. doi: 10.1128/JVI.01623-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cushing A, et al. Emergence of Hemagglutinin Mutations During the Course of Influenza Infection. Sci Rep. 2015;5:16178. doi: 10.1038/srep16178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dinis JM, et al. Deep Sequencing Reveals Potential Antigenic Variants at Low Frequencies in Influenza A Virus-Infected Humans. J Virol. 2016;90:3355–65. doi: 10.1128/JVI.03248-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Poon LLM, et al. Quantifying influenza virus diversity and transmission in humans. Nat Genet. 2016;48:195–200. doi: 10.1038/ng.3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McCrone JT, et al. The evolutionary dynamics of influenza A virus within and between human hosts. 2017 doi: 10.1101/176362. doi.org. [DOI] [Google Scholar]
- 63.Hoelzer K, et al. Intrahost evolutionary dynamics of canine influenza virus in naive and partially immune dogs. J Virol. 2010;84:5329–35. doi: 10.1128/JVI.02469-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Murcia PR, et al. Evolution of equine influenza virus in vaccinated horses. J Virol. 2013;87:4768–71. doi: 10.1128/JVI.03379-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Murcia PR, et al. Intra- and interhost evolutionary dynamics of equine influenza virus. J Virol. 2010;84:6943–54. doi: 10.1128/JVI.00112-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hughes J, et al. Transmission of equine influenza virus during an outbreak is characterized by frequent mixed infections and loose transmission bottlenecks. PLoS Pathog. 2012;8:e1003081. doi: 10.1371/journal.ppat.1003081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nichols WG, et al. Influenza infections after hematopoietic stem cell transplantation: risk factors, mortality, and the effect of antiviral therapy. Clin Infect Dis. 2004;39:1300–6. doi: 10.1086/425004. [DOI] [PubMed] [Google Scholar]
- 68.Vigil KJ, et al. Viral pneumonias in immunocompromised adult hosts. J Intensive Care Med. 2010;25:307–26. doi: 10.1177/0885066610377969. [DOI] [PubMed] [Google Scholar]
- 69.Memoli MJ, et al. The natural history of influenza infection in the severely immunocompromised vs nonimmunocompromised hosts. Clin Infect Dis. 2014;58:214–24. doi: 10.1093/cid/cit725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ghedin E, et al. Deep sequencing reveals mixed infection with 2009 pandemic influenza A (H1N1) virus strains and the emergence of oseltamivir resistance. J Infect Dis. 2011;203:168–74. doi: 10.1093/infdis/jiq040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McMinn P, et al. Antigenic drift of influenza A (H3N2) virus in a persistently infected immunocompromised host is similar to that occurring in the community. Clin Infect Dis. 1999;29:456–8. doi: 10.1086/520243. [DOI] [PubMed] [Google Scholar]
- 72.Rocha E, et al. Antigenic and genetic variation in influenza A (H1N1) virus isolates recovered from a persistently infected immunodeficient child. J Virol. 1991;65:2340–2350. doi: 10.1128/jvi.65.5.2340-2350.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Baz M, et al. Characterization of multidrug-resistant influenza A/H3N2 viruses shed during 1 year by an immunocompromised child. Clin Infect Dis. 2006;43:1555–61. doi: 10.1086/508777. [DOI] [PubMed] [Google Scholar]
- 74.Hegreness M, et al. An Equivalence Principle for the Incorporation of Favorable Mutations in Asexual Populations. Science (80-) 2006;311:1615–1617. doi: 10.1126/science.1122469. [DOI] [PubMed] [Google Scholar]
- 75.Kao KC, Sherlock G. Molecular characterization of clonal interference during adaptive evolution in asexual populations of Saccharomyces cerevisiae. Nat Genet. 2008;40:1499–1504. doi: 10.1038/ng.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lang GI, et al. Pervasive genetic hitchhiking and clonal interference in forty evolving yeast populations. Nature. 2013;500:571–574. doi: 10.1038/nature12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Feder AF, et al. More effective drugs lead to harder selective sweeps in the evolution of drug resistance in HIV-1. Elife. 2016;5:e10670. doi: 10.7554/eLife.10670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hensley SE, et al. Hemagglutinin Receptor Binding Avidity Drives Influenza A Virus Antigenic Drift. Science (80-) 2009;326:734–736. doi: 10.1126/science.1178258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bedford T, et al. Integrating influenza antigenic dynamics with molecular evolution. Elife. 2014;2014:e01914. doi: 10.7554/eLife.01914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dong G, et al. Adamantane-resistant influenza A viruses in the world (1902–2013): Frequency and distribution of M2 gene mutations. PLoS One. 2015;10:e0119115. doi: 10.1371/journal.pone.0119115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Renaud C, et al. Emerging oseltamivir resistance in seasonal and pandemic influenza A/H1N1. J Clin Virol. 2011;52:70–8. doi: 10.1016/j.jcv.2011.05.019. [DOI] [PubMed] [Google Scholar]
- 82.Bloom JD, et al. Permissive secondary mutations enable the evolution of influenza oseltamivir resistance. Science. 2010;328:1272–1275. doi: 10.1126/science.1187816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Baz M, et al. Emergence of oseltamivir-resistant pandemic H1N1 virus during prophylaxis. N Engl J Med. 2009;361:2296–7. doi: 10.1056/NEJMc0910060. [DOI] [PubMed] [Google Scholar]
- 84.Boivin G, et al. Prolonged excretion of amantadine-resistant influenza a virus quasi species after cessation of antiviral therapy in an immunocompromised patient. Clin Infect Dis. 2002;34:E23–5. doi: 10.1086/338870. [DOI] [PubMed] [Google Scholar]
- 85.van der Vries E, et al. Prolonged influenza virus shedding and emergence of antiviral resistance in immunocompromised patients and ferrets. PLoS Pathog. 2013;9:e1003343. doi: 10.1371/journal.ppat.1003343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lakdawala SS, et al. The soft palate is an important site of adaptation for transmissible influenza viruses. Nature. 2015;526:122–125. doi: 10.1038/nature15379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xu R, et al. Preferential recognition of avian-like receptors in human influenza A H7N9 viruses. Science. 2013;342:1230–5. doi: 10.1126/science.1243761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chandrasekaran A, et al. Glycan topology determines human adaptation of avian H5N1 virus hemagglutinin. Nat Biotechnol. 2008;26:107–113. doi: 10.1038/nbt1375. [DOI] [PubMed] [Google Scholar]
- 89.Thompson CI, et al. Infection of human airway epithelium by human and avian strains of influenza a virus. J Virol. 2006;80:8060–8. doi: 10.1128/JVI.00384-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Matrosovich MN, et al. Neuraminidase is important for the initiation of influenza virus infection in human airway epithelium. J Virol. 2004;78:12665–7. doi: 10.1128/JVI.78.22.12665-12667.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hamada N, et al. Intrahost emergent dynamics of oseltamivir-resistant virus of pandemic influenza A (H1N1) 2009 in a fatally immunocompromised patient. J Infect Chemother. 2012;18:865–71. doi: 10.1007/s10156-012-0429-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Marshall N, et al. Influenza Virus Reassortment Occurs with High Frequency in the Absence of Segment Mismatch. PLoS Pathog. 2013;9:e1003421. doi: 10.1371/journal.ppat.1003421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tao H, et al. Influenza A Virus Coinfection through Transmission Can Support High Levels of Reassortment. J Virol. 2015;89:8453–61. doi: 10.1128/JVI.01162-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Davis AR, et al. Influenza defective interfering viral RNA is formed by internal deletion of genomic RNA. Proc Natl Acad Sci U S A. 1980;77:215–9. doi: 10.1073/pnas.77.1.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Frensing T, et al. Continuous influenza virus production in cell culture shows a periodic accumulation of defective interfering particles. PLoS One. 2013;8:e72288. doi: 10.1371/journal.pone.0072288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Saira K, et al. Sequence analysis of in vivo defective interfering-like RNA of influenza A H1N1 pandemic virus. J Virol. 2013;87:8064–74. doi: 10.1128/JVI.00240-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Vasilijevic J, et al. Reduced accumulation of defective viral genomes contributes to severe outcome in influenza virus infected patients. PLOS Pathog. 2017;13:e1006650. doi: 10.1371/journal.ppat.1006650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Murcia PR, et al. Evolution of an Eurasian avian-like influenza virus in naïve and vaccinated pigs. PLoS Pathog. 2012;8:e1002730. doi: 10.1371/journal.ppat.1002730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sobel Leonard A, et al. Transmission Bottleneck Size Estimation from Pathogen Deep-Sequencing Data, with an Application to Human Influenza A Virus. J Virol. 2017;91:JVI.00171-17. doi: 10.1128/JVI.00171-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lowen AC, et al. Influenza Virus Transmission Is Dependent on Relative Humidity and Temperature. PLoS Pathog. 2007;3:e151. doi: 10.1371/journal.ppat.0030151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sobel Leonard A, et al. Deep Sequencing of Influenza A Virus from a Human Challenge Study Reveals a Selective Bottleneck and Only Limited Intrahost Genetic Diversification. J Virol. 2016;90:11247–11258. doi: 10.1128/JVI.01657-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Alizon S, et al. Epidemiological and clinical consequences of within-host evolution. Trends in Microbiology. 2011;19:24–32. doi: 10.1016/j.tim.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 103.Park DJ, et al. Ebola Virus Epidemiology, Transmission, and Evolution during Seven Months in Sierra Leone. Cell. 2015;161:1516–1526. doi: 10.1016/j.cell.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Andersen KG, et al. Clinical Sequencing Uncovers Origins and Evolution of Lassa Virus. Cell. 2015;162:738–750. doi: 10.1016/j.cell.2015.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Holmes EC. Patterns of intra- and interhost nonsynonymous variation reveal strong purifying selection in dengue virus. J Virol. 2003;77:11296–11298. doi: 10.1128/JVI.77.20.11296-11298.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kryazhimskiy S, Plotkin JB. The population genetics of dN/dS. PLoS Genet. 2008;4:e1000304. doi: 10.1371/journal.pgen.1000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mugal CF, et al. Why time matters: Codon evolution and the temporal dynamics of dN/dS. Mol Biol Evol. 2014;31:212–231. doi: 10.1093/molbev/mst192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Leslie AJ, et al. HIV evolution: CTL escape mutation and reversion after transmission. Nat Med. 2004;10:282–289. doi: 10.1038/nm992. [DOI] [PubMed] [Google Scholar]
- 109.Herbeck JT, et al. Human immunodeficiency virus type 1 env evolves toward ancestral states upon transmission to a new host. J Virol. 2006;80:1637–44. doi: 10.1128/JVI.80.4.1637-1644.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zanini F, et al. Population genomics of intrapatient HIV-1 evolution. Elife. 2015;4:e11282. doi: 10.7554/eLife.11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Beauchemin CA, Handel A. A review of mathematical models of influenza A infections within a host or cell culture: lessons learned and challenges ahead. BMC Public Health. 2011;11:S7. doi: 10.1186/1471-2458-11-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Luria SE, Delbrück M. Mutations of bacteria from virus sensitivity to virus resistance. Genetics. 1943;28:491–511. doi: 10.1093/genetics/28.6.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bozic I, et al. Quantifying Clonal and Subclonal Passenger Mutations in Cancer Evolution. PLOS Comput Biol. 2016;12:e1004731. doi: 10.1371/journal.pcbi.1004731. [DOI] [PMC free article] [PubMed] [Google Scholar]