

Abstract
Methods to efficiently deliver fluorophores across the cell membrane are crucial for imaging the dynamics of intracellular proteins using fluorescence. Here we describe a simple protocol for permeabilizing living cells using Streptolysin O, a bacterial toxin, which allows transient uptake of fluorescent probes for labeling specific intracellular proteins. The technique is applicable for delivering different classes of fluorescent probes with a molecular weight of <150 kDa, and it is also applicable to a variety of different cell lines. The technique enables the utilization of a broad range of fluorophores for live cell imaging of intracellular proteins. Extended observation of intracellular fluorescence bound to specific proteins is now possible through super-resolution microscopy by using fluorophores that are photostable in “cell-friendly” de-oxygenating and reducing conditions.
INTRODUCTION
Recent advances in super-resolution fluorescence microscopy have made it possible to obtain sub-diffraction-limited images of cellular structures and protein-protein complexes inside the cell (Betzig et al., 2006; Gustafsson, 2005; Rust et al., 2006; Yildiz et al., 2003). However, the application thus far has been mostly limited to imaging in vitro or fixed samples, except for techniques that rely on photoactivatable or photoswitchable fluorescent proteins (Betzig et al., 2006; Hofmann et al., 2005). Fluorescent photoactivatable proteins, while frequently used in super-resolution microscopy has its limitations, such as photostability and limited choice of colors. Furthermore, when plasmid DNA encoding the fluorescent fusion protein is incorporated into the cell by transient transfection, it causes the overexpression of the target protein, which may influence the interpretation of experimental results. Availability of a wide array of fluorescent dyes would be extremely helpful for the advance of (regular and) super-resolution fluorescence microscopy. However, at the moment there are only a few fluorescent probes that can reach inside the cytoplasm of a living cell. Permeability is either limited by the fluorophore or the entity that the fluorophore attaches to, whether it is a ligand or a protein. To name a few fluorophores that suffer from such limitations, ATTO647N has extraordinary photostability and is excellent for single particle tracking, but is cell-impermeant. Another example of a cell-impermeant fluorophore commonly used in blinking-based super-resolution imaging is Alexa Fluor 647. Furthermore, fluorophores that are cell-permeant when attached to small molecules or by itself will become membrane-impermeant when attached to proteins, such as single-domain nanobodies, antibody fragments, and antibodies; therefore they have limited utility. Since the success of super-resolution fluorescence experiments on living cell depends greatly on the ability to label the protein of interest with a fluorescent probe, it becomes important to explore simple methods to deliver these cell-impermeant fluorescent probes. There are numerous established techniques for delivering small fluorescent probes to macromolecules intracellularly. These include microinjection, bead loading, electroporation, cell squeezing, pore-forming toxin-based techniques and microfabrication-based techniques (Betzig et al., 2006; Hennig et al., 2015; Kim et al., 2008; Kollmannsperger et al., 2016; Lyon and Stasevich, 2017; McNeil and Warder, 1987; Okada and Rechsteiner, 1982; Walev et al., 2001; Wu et al., 2015). In this unit, we describe the use of pore-forming toxin, mainly Streptolysin O- (SLO) based techniques for delivering fluorescent probes, which has the following advantages over other cell-permeabilization techniques. 1) The procedure permeabilizes a monolayer of cells on the surface, as opposed to one cell at a time (as is used for microinjection). 2) The procedure can lead to permeabilization of >85% of the cells with less than 10% cell death; a good permeabilization to cell viability ratio. 3) The procedure is quick and straightforward without the need for special reagents and additional instrumentation, thus its relatively inexpensive to perform compared to other techniques. 4) The technique can be applied for imaging adherent cell lines without transferring cells between different surfaces, allowing the capture of protein dynamics within an hour of probe delivery. 5) The toxin makes a two-way opening that allows small, unbound fluorophore to escape (typically < 2kD), lowering the background from freely diffusive fluorophores in the cytoplasm. 6) The procedure requires incubation with 100 μL of a few hundred nanomolar of fluorescent probe per coverslip, which is significantly less than needed for other methods. Although the technique is robust, it does require optimization. Parameters to consider in the SLO-based loading technique includes concentration of SLO toxins to use for different cell lines, size and specificity of fluorescent probe, and concerns of cell health post-permeabilization due to over-permeabilization. Since the application of SLO loading technique described in this unit is geared towards imaging proteins inside living cells, strategies to extend the lifetime of fluorophore inside the living cell, and which dye to use for specific applications are also discussed.
BASIC PROTOCOL 1: Reversible Permeabilization of Living Cell with SLO
SLO-based probe delivery involves only two steps: permeabilization of the cells by SLO, and recovery. The amount of SLO to use for permeabilization depends on both the confluency of the cell and the cell line. It is necessary to first perform a titration experiment every time a new cell line is used, or new stock solution of SLO is prepared (see Support Protocol 1). In the Imaging section (Support Protocol 2), details on how to extend the lifetime of fluorophore using Oxyrase, and how to efficiently induce blinking using glutathione is explained.
Materials
Streptolysin O (S5265 Sigma, dissolve the entire contents of the vial (25,000–50,000 U) in 1 mL molecular biology grade water. Place 10 uL of dissolved Streptolysin O in 500 μL Protein Low Bind tubes (Eppendorf, Catalogue No. 0030108094) and store in aliquots, flash freeze the aliquots in liquid nitrogen, store at −80°C)
0.5 M TCEP (dissolve appropriate amount of TCEP-HCl in 10x PBS, prepare 10-μL aliquots in 0.6 mL microfuge tubes and store in −80°C)
DPBS, no calcium, no magnesium, 37°C
1 M MgCl2 (in ddH2O sterile filtered)
DPBS 1 mM MgCl2
Tyrode’s solution
DMEM (phenol red free) with 4.5g/L glucose, L-glutamine, and sodium pyruvate
FBS (heat inactivated)
50x Nucleotide glucose mixture (100 mM ATP, 100 mM GTP, 100 mg/mL Glucose)
Recovery buffer (10% FBS, DMEM (phenol red free), 1x Nucleotide glucose mixture)
10X Casein (Vector Lab, optional)
Fluorescent Probes (at least a 5 μM stock)
35 mm glass bottom dish with 14 mm micro well center (Cellvis #D35-14-1-N)
Adherent cells grown to ~75% confluency. Adherent cells are typically sub-cultured on a 35 mm glass bottom dish 2 days before the permeabilization experiment. The optimal degree of confluency of cells depends on the experiment, but in general, the result of permeabilization is more reproducible at higher confluency. Therefore, we recommend no less than 75% confluency of cells.
Activation of SLO:
-
1
Immediately before experiments thaw one aliquot of 10 μL SLO stock and 0.5 M TCEP stored in −80°C freezer.
-
2
Add 0.2 μL of 0.5 M TCEP to the SLO aliquot, incubate at 37°C for 20 minutes.
-
3
Warm DPBS, DPBS 1 mM MgCl2, Tyrode’s solution, and recovery buffer in 37°C water bath.
-
4
Prepare appropriate dilutions of fluorescent probes, typically at a concentration of 200–600 nM in 100 μL Tyrode’s Solution. A 40x diluted 10x casein [*Author: the previous phrase is unclear. Please clarify how much casein should be added.] can be added to the dye mixture to prevent non-specific binding of fluorescent probes to cells and coverslip.
Permeabilization of cells:
-
5
Dilute activated SLO from Step 2 to appropriate concentration in DPBS 1 mM MgCl2 (see Support Protocol 1 for detail).
-
6
Completely remove cell culture media and wash three times with DPBS.
-
7
Incubate cells with 100 μL of diluted SLO solution. Make sure the SLO solution completely covers the glass part of the glass bottom dish. Put the cells with SLO solution back into the 37°C 5% CO2 incubator for 5–10 minutes. Optional: At 5 minute mark, check the cell morphology under phase contrast microscope at 20x magnification. Depending on the cell line tested, successful permeabilization will show signs of enlarged nucleolus immediately after SLO incubation (Figure 1).
-
8
Remove the SLO solution from cells, gently wash the cells three times with DPBS 1 mM MgCl2.
-
9
Add fluorescent probes solution prepared in step 4 to the cells, incubate the cells with fluorescent probe on ice for 5 minutes.
-
10
Discard the fluorescent probe solution, wash cells three times with Tyrode’s solution.
-
11
Add sufficient recovery buffer to cover the glass bottom dish (>100 μL). Incubate cells in recovery buffer for at least 20 minutes in a 37°C 5% CO2 incubator.
-
12
Remove recovery buffer from cells, and replace with imaging buffer of choice (e.g., DPBS or 10% DMEM Phenol red-free). The nucleolus of the cells should now appear less distinctive. (Figure 1).
Figure 1. Morphology Change of the Cells During SLO Permeabilization and After Recovery.
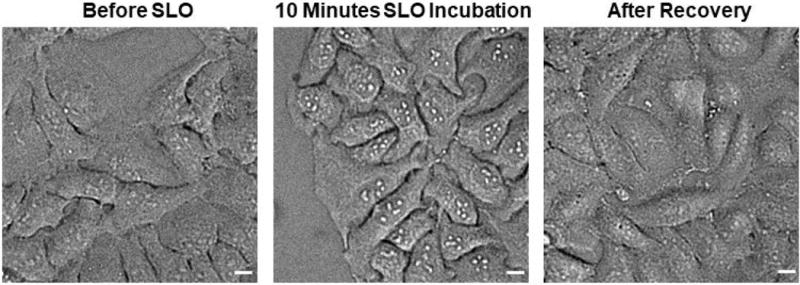
Before SLO permeabilization, the nucleolus of HeLa cell does not appear very distinctive under phase contrast microscope at 10× magnification. After exposing the cells to SLO for 10 minutes, the nucleolus of HeLa cells becomes much more distinctive, possibly due to stress response caused by a compromised cell membrane. After recovery, the nucleolus returns to the state that is visibly similar to cells pre-treated with SLO. Scale bar denotes 10 μm.
Support Protocol 1: Optimization of SLO Concentration for Permeabilization
This protocol describes how to determine the optimal SLO concentration for the experiment. All the cell lines the authors have tested thus far have been permeabilized using the concentration gradient 200–0 U/mL. The degree of permeabilization can be determined quantitatively or qualitatively by examining the fluorescence microscopy images.
Additional Materials (See also Basic Protocols)
Cultured adherent cells
Fluorescent probes for marking permeabilized cells, we use either phalloidin- ATTO488/Alexa Fluor 647, or various sized dextrans (10, 40, 70 kDa FITC-Dextran)
Permeabilization of cells:
-
1
Culture adherent cells in a 8-well chamber until ~75% confluency is reached.
-
2
Activate SLO following the step 1 and 2 outlined in Basic Protocol.
-
3
After SLO activation, dilute SLO toxins into a gradient of concentration, we use 200 U/mL, 175 U/mL, 150 U/mL, 100 U/mL, 75 U/mL, 50 U/mL, 25 U/mL, 0 U/mL, assuming the stock is at 25,000 U/mL.
-
4
Withdraw and discard all culture media in 8-well chamber.
-
5
Wash the cells in the 8-well chamber three times with 300 uL of DPBS.
-
6
Add solution with different SLO concentration into each of the 8-well chamber.
-
7
Incubate the 8-well chamber at 37°C for 10 minutes.
-
8
Dilute the fluorescent marker to 0.5–1.0 μM in Tyrode’s solution.
-
9
Remove the SLO solution, wash the cells three times with DPBS 1 mM MgCl2.
-
10
Add fluorescent probe marker for cell permeabilization. Incubate on ice for 5 minutes.
-
11
Remove fluorescent marker solution. Wash the cells three times with Tyrode’s solution.
-
12
Removed the last wash with Tyrode’s buffer. Recover the cells using 100 μL of the recovery solution for 15 minutes. Keep the cells in 37 °C, 5% CO2 incubator during the recovery.
-
13
Observe the cells under the microscope.
The permeabilization may be un-even at the edge of the well. Assess the degree of permeabilization at the center of the well. An optimal SLO concentration to use for the experiment would show >50% of the cells stained by the fluorescent probes, while dead cells stained by propidium iodide is at a minimum (<10%). An example of cell permeabilized at different concentrations of SLO is shown in Figure 2.
Figure 2. Titration Experiment Using Different Concentration of SLO.
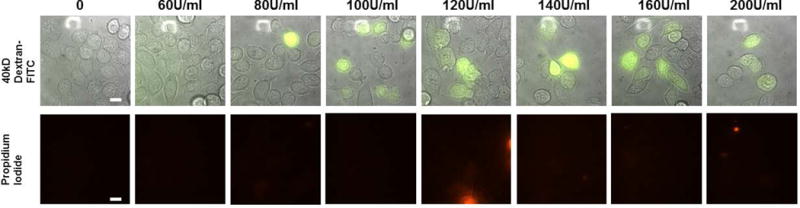
HCT116 cells were incubated with a gradient of SLO concentration (0, 60, 80, 100, 120, 140, 160, 200 U/mL) before exposing to 40kD-Dextran-FITC. Post-recovery the cells were exposed to propidium iodide to access viability. Significant permeabilization to FITC Dextran-FITC started appearing at 100 U/mL (green fluorescence, top row). SLO concentration above 100 U/mL and below 200 U/mL did not appear to harm the cells as indicated by propidium iodide staining (red fluorescence, bottom row). Scale bar denotes 10 μm.
-
14
If the probe did not enter the cell even at 200 U/mL see section on troubleshooting.
Support Protocol 2: Improving the Photostability of Delivered Fluorescent Probes During Imaging
The use of SLO opens up the selection of dyes that can be used for fluorescence microscopy experiments. Some of these dyes may benefit greatly from de-oxygenating additives (Cy3, Alexa Fluor 647, etc.). Below, we describe strategies to prolong the photostability of fluorescent dyes in living cells under continuous illumination or stochastic activation.
Materials
Permeabilized cells (output of Basic Protocol 1)
Oxyrase (Oxyrase Inc.)
Sodium D-Lactate
Glutathione, reduced
Prepare Oxyrase and sodium lactate stock solutions:
-
1
Thoroughly mix Oxyrase and flash freeze in 400 μL aliquots, store up to 18 months in −20°C.
-
2
Prepare 1 M sodium D-lactate solution in ddH2O, aliquot in 80μL volume and store at −20°C.
Prepare Imaging Buffer for Live Cell on a 35 mm glass bottom dish:
-
3
Centrifuge Oxyrase stock solution at 3,830×g for 5 minutes to remove/pellet large aggregates.
-
4
Collect the supernatant, add the entire 400 μL of supernatant to 4 mL of phenol red free DMEM with 10% FBS.
-
5
Add sodium lactate solution to the Oxyrase media mixture enough to make the final concentration of lactate 20 mM.
Prepare cells for imaging:
-
6
Remove the recovery media at the end of the Basic Protocol from the dish.
-
7
Add Imaging Buffer to the dish, fill it almost to the top of the dish.
-
8Put the lid on the dish and seal with parafilm, if possible. Make sure the parafilm does not touch the sample holder to avoid focus drift, which is the slow shifting of x-y-z position of the coverslip due to elasticity of the parafilm making contact with the microscope sample holder.The enzyme in Oxyrase removes oxygen from the imaging buffer, lengthening the photostability of the fluorophores. After 5 minutes of incubation with the imaging buffer, the cells are now ready to be imaged in deoxygenated buffer.The fluorophore should demonstrate prolonged photostability (Figure 3a).
Figure 3. Improvement in photostability of dyes inside living cell by using Oxyrase.
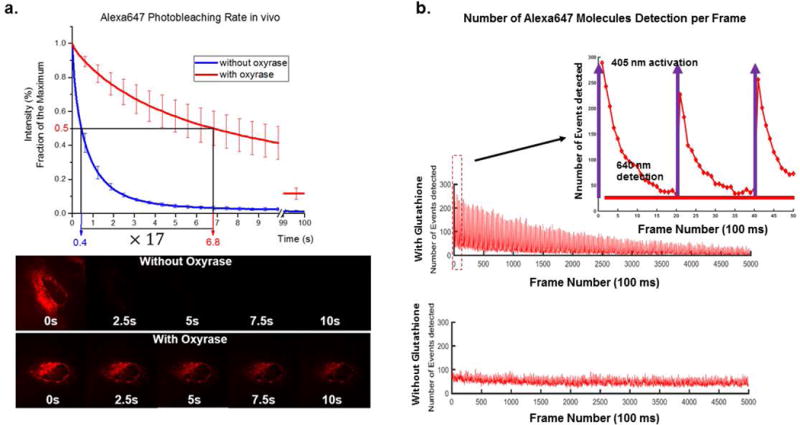
a) HeLa cells expressing mitochondria-Halotag was labeled with Alexa Fluor 647 conjugated to Halotag ligand, which was delivered by SLO. Oxyrase was added to imaging media, and time series image was recorded under continuously illumination. The photobleaching rate of Alexa Fluor 647 improved by roughly 17 times under the influence of Oxyrase. b) For the application of super-resolution imaging where de-activation and activation of fluorophore is required, glutathione is added to the recovery media. The fluorophore, in this case Alexa Fluor 647, was deactivated by intense illumination of 640 nm laser, and the fluorophore is re-activated by using pulses of 405 nm laser in the presence of glutathione in the cytosol (adapted from (Teng et al., 2016)).
SUPPORT PROTOCOL 3: Prepare Recovery and Imaging Buffer for Live Cell dSTORM experiment
Streptolysin O enables the delivery of otherwise cell-impermeant dyes, including dyes that are used for super-resolution imaging (dSTORM). Below is an optional protocol on how to perform a dSTORM experiment using delivered fluorophores capable of photoactivation and deactivation. For a list of fluorophores capable for dSTORM please see the Commentary section.
Materials (see Basic Protocol)
Prepare 100 mM of glutathione solution in ddH2O and keep on ice during the SLO activation step in Basic Protocol.
Add 4 uL of 100 mM glutathione to the recovery buffer.
Follow the rest of the Basic and Imaging protocol [*Author: Please specify which protocol(s) and the step numbers for each.] in the presence of Oxyrase in the imaging buffer.
- For super-resolution imaging (dSTORM), use the lowest 405 nm laser power necessary to activate ~300 events per frame (Figure 3b).The number of molecules that turn on in response to 405 nm laser will decrease over time due to photobleaching. The power of 405 nm laser can then be increased to turn on more molecules, until there are no more molecules left to be turned on.
Reagents and Solution
Tyrode’s Solution:
140 mM NaCl
5 mM KCl
1 mM MgCl2
10 mM HEPES
10 mM glucose
pH 7.4
sterile filtered, store at 4°C for one year.
COMMENTARY
Background Information
Streptolysin O and other pore-forming toxins have been used to permeabilize cells to DNA, RNA, and ligands, and inert proteins for many decades (Nitin and Bao, 2008; Rajapakse et al., 2010; Teng et al., 2016; Walev et al., 2001). However, the method to recover cells varies greatly among publications. The recovery method has been reported to be as simple as using calcium-containing buffer to as complicated as addition of mammalian cell extracts with additives (Boyle and Lieberman, 1999; Kano et al., 2012). There is also great variation in recovery time for the cells to return to a healthy state, from minutes to couple of hours (Kano et al., 2012; Nitin and Bao, 2008; Walev et al., 2001). In our experience, we found that calcium, FBS, and cell culture media separately was not able to induce cell recovery (Teng et al., 2016). On the other hand, 15 minutes of incubation in complete medium (phenol red-free medium plus 10% FBS) was enough to induce recovery in the adherent cell lines that were tested (CHO-K1, COS7, HeLa, U2OS, etc.). If the cells do not recover within the given time using the recovery buffer, most likely the cells were over-permeabilized, and the cells are now beyond repair. Cells that fail to recover after permeabilization will remain permeable to propidium iodide and a variety of large sized molecules, but they will eventually lyse and detach from the surface.
To obtain reproducible results with SLO, it is necessary to optimize the concentration of SLO used for permeabilizing the cells. There have been reports of using flow cytometry to optimize SLO permeabilization and minimize cell death (Spiller and Tidd, 1995; Walev et al., 2001). The method of optimization depends on what instrument the lab has access to. In our laboratory, assessing the degree of permeabilization by imaging is convenient and directly translates to what we would observe in the actual experiment.
Our goal of applying the SLO technique was to enable the delivery of fluorescent probes. However, one should be aware that some fluorescent probes are toxic to cells. For example, dye-conjugated phalloidin and DAPI were efficient molecules for proof of concept demonstration of SLO permeabilization but these molecules could be toxic to the cell. Therefore, toxicity should be considered when interpreting the biological data obtained after delivering these probes.
Critical Parameters
Cell line
The cholesterol content of the cell membrane will influence the concentration of SLO needed to induce permeabilization. In addition, there are various wash steps in the permeabilization protocol that are not suitable for weakly adherent cells. We have encountered cell detachment when using HEK cells due to it not being able to withstand repetitive washes. For non-adherent cells, it is theoretically possible to exchange buffers outlined in this protocol by centrifugation, but it has not been extensively tested. Lastly, at high confluency, higher concentrations of SLO will be required to permeabilize a monolayer of cells. We found that highly confluent (>80% confluent) cells were able to tolerate a greater range of SLO concentration and permeabilization time at high SLO concentration, thus yielding overall more reproducible results. Cells respond differently to excess amounts of SLO; some cells will detach when severely permeabilized (3T3 cells), and some cells will remain adherent, but round up (CHO-K1 cells). In certain cell lines, the effects of permeabilization can be observed morphologically immediately after 5–10 minutes of SLO incubation, and the change in morphology will disappear after the recovery step.
Concentration of the fluorescent probe
The upper limit of probe concentration that can be used depends on the degree of nonspecific binding of the fluorescent probe to the cell membrane and coverslip. Adding excess fluorophore may cause it to stick non-specifically, causing high background. When using small ligand probes (<3 kDa), the concentration of the probe can be up to few μM if it does not cause non-specific binding. Since small ligand probe eventually leaks out during cell recovery, unlabeled dyes are not a cause of concern. However, when using protein-based fluorescent probes, one should be aware of the freely diffusive fluorescent probes inside the cell. With a transfected cell system, the amount of expressed protein is often very large and likely will exceed that of the delivered fluorescent probes, and therefore no unlabeled protein will be observed. For labeling native protein, the amount of excess fluorescent probe in the cell will depend on the abundance of the native protein of interest. It is very important to have the highest purity of nanobody/antibody for intracellular delivery in order to avoid unnecessary background of floating dyes.
Fluorescent Dye of choice
The choice of fluorescent dye should be made based on the type of fluorescence microscopy experiment to be performed. For long-term tracking of individual protein dynamics, photostable dyes like ATTO647N can be used without a de-oxygenation system. A de-oxygenation environment without proper triplet state quencher may reduce the fluorescence intensity of ATTO647N dye. For observing cellular structure using activation/de-activation-based super-resolution technique, Alexa Fluor 647, Cy3B or other photo-activatable dyes should be used. When using photo-activatable dyes, it is often necessary to include de-oxygenation system and reducing environment.
Troubleshooting
No permeabilization is observed
When no permeabilization is observed, make sure that SLO has been activated with a reducing agent. The SLO should also not be left activated for longer than 3 hours before use, as it will be less potent. Observing cell death by the addition of SLO in the optimization experiment is the first step to make sure that the SLO is effective on the particular cell line of choice. Once cell death is observed, one simply needs to dilute the SLO concentration to reach an optimal balance between permeabilization and cell death. The presence of calcium and serum in the permeabilization buffer will also inhibit permeabilization. Lastly, the activity of SLO is a function of temperatures, for instance at 4°C we observed no permeabilization of cells, so it is vital to keep the incubation of SLO and the cells at 37 °C for consistency.
Excessive cell death or detachment
As mentioned, observing cell death means SLO is effective on the cell line of choice. The concentration of SLO will need to be further diluted to prevent cells from dying. Also make sure that the recovery buffer has the correct formulation. The volume we recommend for various incubation steps is just 100 μL, which is barely enough to cover the center of the glass bottom dish. Make sure that the center is not dried up or fluid has leaked into the surrounding, causing the cells to be exposed to air. Another possible solution for cell lines that are sensitive to temperature change is to incubate the non-calcium/casein-containing fluorescent probe along with SLO during the permeabilization step to avoid putting cells on ice during recovery.
No improvement in photostability after adding Oxyrase
The time it takes for Oxyrase to deplete oxygen depends on the concentration of the enzyme (Oxyrase) and the concentration of sodium lactate, as well as the dimensions of the imaging chamber. More Oxyrase and sodium lactate can be added to increase the efficiency of oxygen depletion. Keep in mind that excess sodium lactate can increase the osmotic pressure of the buffer, which may cause the cells to adopt hyperosmotic morphology. In addition, it is also possible that the dye is insensitive to the amount of oxygen in the solution. Alexa Fluor 647 shows great contrast with and without oxygen, and is currently our dye of choice to test the efficiency of the oxygen-scavenging system.
Anticipated Results
In a successful experiment involving SLO permeabilization, most of the cells (85%) appear permeabilized as exhibited by morphology changes or enlarged nucleolus under phase contrast microscope after 5–10 minutes of SLO incubation at 37 °C. These changes disappear after the recovery step, and the cells appear “normal” once again. Under a fluorescence microscope, the delivered probe will appear co-localized with the target (if a fluorescent protein is attached to the target). If the labeled protein is a part of the cellular structure, (e.g., mitochondria, cytoskeleton, etc.), the fluorescence image of the delivered probe should resemble the cellular structure. The labeled protein should ideally resume normal cellular functions. For example, transcription factors should properly respond to external stimuli, and molecule motors should show directed motion. When performing the experiment in imaging buffer, the fluorophore should last much longer in the absence of oxygen, and should be photoactivatable in the presence of a reducing environment. The improvement in photostability depends on the fluorophore used, and the duration of photoactivation could last close to one hour.
Time Consideration
The permeabilization experiment takes roughly 1 hour to perform, including the activation of SLO and recovery of the cells. The imaging experiment typically takes 1 hour, which depends on several factors, such as the sensitivity of the cell to pH change, temperature, and phototoxicity. If de-oxygenating condition is required, we have been able to successfully reactivate and deactivate fluorophore for up to 1 hour in a de-oxygenated environment created by Oxyrase.
Acknowledgments
The author would like to acknowledge Professor Carol Prives at Columbia University for her generous donation of HCT116 cells. Mitochondria HaloTag (pERB254) was a gift from Michael Lampson from University of Pennsylvania (Addgene plasmid # 67762)(Ballister et al., 2015). This work was in part supported by NSF PHY-1430124, and NIH GM108578 to PRS.
Literature Cited
- Ballister ER, Ayloo S, Chenoweth DM, Lampson MA, Holzbaur ELF. Optogenetic control of organelle transport using a photocaged chemical inducer of dimerization. Curr Biol. 2015;25:R407–R408. doi: 10.1016/j.cub.2015.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Imaging Intracellular Fluorescent Proteins at Nanometer Resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- Boyle RT, Lieberman M. Permeabilization by streptolysin-o reveals a role for calcium-dependent protein kinase c isoforms alpha and beta in the response of cultured cardiomyocytes to hyposmotic challenge. Cell Biol Int. 1999;23:685–693. doi: 10.1006/cbir.1999.0435. [DOI] [PubMed] [Google Scholar]
- Gustafsson MGL. Nonlinear structured-illumination microscopy: Wide-field fluorescence imaging with theoretically unlimited resolution. Proc Natl Acad Sci U S A. 2005;102:13081–13086. doi: 10.1073/pnas.0406877102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig S, van de Linde S, Lummer M, Simonis M, Huser T, Sauer M. Instant Live-Cell Super-Resolution Imaging of Cellular Structures by Nanoinjection of Fluorescent Probes. Nano Lett. 2015;15:1374–1381. doi: 10.1021/nl504660t. [DOI] [PubMed] [Google Scholar]
- Hofmann M, Eggeling C, Jakobs S, Hell SW. Breaking the diffraction barrier in fluorescence microscopy at low light intensities by using reversibly photoswitchable proteins. Proc Natl Acad Sci U S A. 2005;102:17565–17569. doi: 10.1073/pnas.0506010102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano F, Nakatsu D, Noguchi Y, Yamamoto A, Murata M. A Resealed-Cell System for Analyzing Pathogenic Intracellular Events: Perturbation of Endocytic Pathways under Diabetic Conditions. PLOS ONE. 2012;7:e44127. doi: 10.1371/journal.pone.0044127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JA, Cho K, Shin MS, Lee WG, Jung N, Chung C, Chang JK. A novel electroporation method using a capillary and wire-type electrode. Biosens Bioelectron. 2008;23:1353–1360. doi: 10.1016/j.bios.2007.12.009. [DOI] [PubMed] [Google Scholar]
- Kollmannsperger A, Sharei A, Raulf A, Heilemann M, Langer R, Jensen KF, Wieneke R, Tampé R. Live-cell protein labelling with nanometre precision by cell squeezing. Nat Commun. 2016;7:10372. doi: 10.1038/ncomms10372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon K, Stasevich TJ. Imaging Translational and Post-Translational Gene Regulatory Dynamics in Living Cells with Antibody-Based Probes. Trends Genet. 2017;33:322–335. doi: 10.1016/j.tig.2017.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil PL, Warder E. Glass beads load macromolecules into living cells. J Cell Sci. 1987;88(Pt 5):669–678. doi: 10.1242/jcs.88.5.669. [DOI] [PubMed] [Google Scholar]
- Nitin N, Bao G. NLS peptide conjugated molecular beacons for visualizing nuclear RNA in living cells. Bioconjug Chem. 2008;19:2205–2211. doi: 10.1021/bc800322a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada CY, Rechsteiner M. Introduction of macromolecules into cultured mammalian cells by osmotic lysis of pinocytic vesicles. Cell. 1982;29:33–41. doi: 10.1016/0092-8674(82)90087-3. [DOI] [PubMed] [Google Scholar]
- Rajapakse HE, Gahlaut N, Mohandessi S, Yu D, Turner JR, Miller LW. Time-resolved luminescence resonance energy transfer imaging of protein–protein interactions in living cells. Proc Natl Acad Sci. 2010;107:13582–13587. doi: 10.1073/pnas.1002025107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat Methods. 2006;3:793–796. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiller DG, Tidd DM. Nuclear Delivery of Antisense Oligodeoxynucleotides Through Reversible Permeabilization of Human Leukemia Cells with Streptolysin O. Antisense Res Dev. 1995;5:13–21. doi: 10.1089/ard.1995.5.13. [DOI] [PubMed] [Google Scholar]
- Teng KW, Ishitsuka Y, Ren P, Youn Y, Deng X, Ge P, Belmont AS, Selvin PR. Labeling proteins inside living cells using external fluorophores for microscopy. eLife. 2016;5:e20378. doi: 10.7554/eLife.20378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walev I, Bhakdi SC, Hofmann F, Djonder N, Valeva A, Aktories K, Bhakdi S. Delivery of proteins into living cells by reversible membrane permeabilization with streptolysin-O. Proc Natl Acad Sci USA. 2001;98:3185–3190. doi: 10.1073/pnas.051429498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YC, Wu TH, Clemens DL, Lee BY, Wen X, Horwitz MA, Teitell MA, Chiou PY. Massively parallel delivery of large cargo into mammalian cells with light pulses. Nat Methods. 2015;12:439–444. doi: 10.1038/nmeth.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildiz A, Forkey JN, McKinney SA, Ha T, Goldman YE, Selvin PR. Myosin V Walks Hand-Over-Hand: Single Fluorophore Imaging with 1.5-nm Localization. Science. 2003;300:2061–2065. doi: 10.1126/science.1084398. [DOI] [PubMed] [Google Scholar]