

Abstract
O Mannosylation is a vital protein modification involved in brain and muscle development whereas the biological relevance of O-mannosyl glycans has remained largely unknown owing to the lack of structurally defined glycoforms. An efficient scaffold synthesis/enzymatic extension (SSEE) strategy was developed to prepare such structures by combining gram-scale convergent chemical syntheses of three scaffolds and strictly controlled sequential enzymatic extension catalyzed by glycosyltransferases. In total, 45 O-mannosyl glycans were obtained, covering the majority of identified mammalian structures. Subsequent glycan micro-array analysis revealed fine specificities of glycan-binding proteins and specific antisera.
Keywords: carbohydrates, chemoenzymatic synthesis, glycosylation, microarrays, O-mannosyl glycans
Wide variety
An efficient scaffold synthesis/enzymatic extension (SSEE) strategy was developed to prepare 45 structurally well-defined O-mannosyl glycans by combining convergent chemistry and strictly programmed enzymatic extension catalyzed by glycosyltransferases. Glycan microarray analysis was also performed to mine fine specificities of glycan-binding proteins using these glycoforms.
O Mannosylation is a vital protein post-translational modification that plays essential roles in brain and muscle development and normal tissue function.[1] α-Dystroglycan (α-DG) is the most studied O-mannosyl protein, and is widely distributed in muscle and brain tissues. Abnormal O mannosylation on α-DG disrupts the receptor function of dystroglycan and leads to congenital muscular dystrophy.[2] In addition, O-mannosyl glycans have been reported to be involved in other human diseases, including arenaviral infections, cancer, and metastasis.[3]
In mammalian brain tissue, O-mannosyl glycans account for up to 30% of all O-linked glycans.[4] Despite their abundance, studies on mammalian O-mannosylation have been mainly focused on α-DG. Recent advances in glycoproteomics have enabled the discovery of an increasing number of O-mannosylated proteins and various O-mannosyl glycans.[1b,5] To date, more than 20 O-mannosyl glycans have been identified, which are classified into four core types.[5] As illustrated in Figure 1A, core M0, identified in cadherin family proteins,[6] represents a single structure of one mannose (Man) residue α-linked to serine (Ser) or threonine (Thr). Core M1 contains a linear β1,2-linked N-acetylglucosamine (GlcNAc), whereas the branched core M2 contains both β1,2- and β1,6-linked GlcNAc residues. Core M3 is a phosphorylated trisaccharide, to which glycosaminoglycan-like polysaccharides are usually attached.[5]
Figure 1.
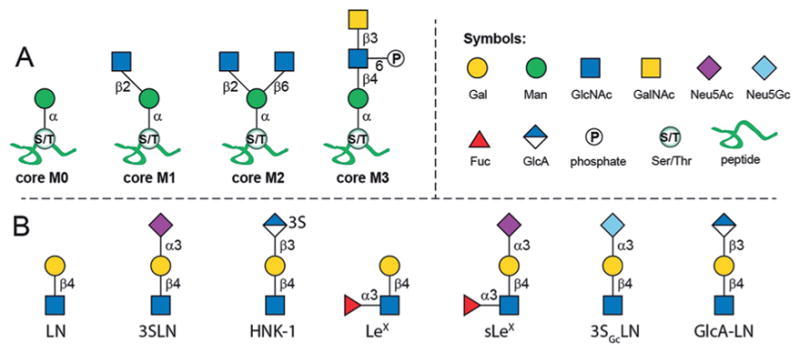
A) The four cores of mammalian O-mannosyl glycans and B) identified extensions on core M1 and M2. Gal =galactose, Man = mannose, GlcNAc =N-acetylglucosamine, GalNAc=N-acetylgalactos-amine, Neu5Ac=N-acetylneuraminic acid, Neu5Gc=N-glycolylneuraminic acid, Fuc =L-fucose, GlcA=glucuronic acid.
The M1 and M2 cores are typically extended and have been identified with structures including N-acetyllactosamine (Gal-β1,4-GlcNAc, LN), 3-sialyl-N-acetyllactosamine (Neu5Ac-α2,3-Gal-β1,4-GlcNAc, 3SLN), Lewis X [Gal-β1,4-(Fuc-α1,3-)GlcNAc, LeX], sialyl-LeX (sLeX), and the human natural killer 1 (3-sulfo-GlcA-β1,3-Gal-β1,4-Glc, HNK-1) epitopes (Figure 1B).[1b] These terminal epitopes largely diversify core M1 and M2 based glycans, and in fact, the majority of identified structures belong to these two core types.[1b] Additionally, it was found that the structures centered on M1 and M2 cores account for 15% and 5% of all brain O-linked glycans, respectively.[4] Despite the abundance of core M1 and M2 structures, very little is known about their functional relevance.[5] Therefore, structurally well-defined O-mannosyl glycans not only serve as ideal standards for the identification and characterization of such glycoforms, but also provide unique probes to uncover their biological roles.
To date, only a few O-mannosyl glycans have been prepared.[7] A core M1 tetrasaccharide (Figure 2, M102) was first synthesized in 1999,[7c,d] and later two lower homologues were obtained (M100, M101). Most recently, Cao and coworkers prepared six core M1 structures (M100, M101, M102, M104, M105, and M102G) by employing a chemoenzymatic approach.[7a] Glycopeptides harboring basic O-mannosyl structures (e.g., M101,[8] M102,[9] core M0,[7e] M000,[10] or core M3[11]) have also been generated. Nevertheless, the majority of O-mannosyl glycans (especially core M2 branched structures) are still not accessible, which has hampered our in-depth understanding of O mannosylation. Herein, an efficient strategy is described that employs scaffold synthesis/enzymatic extension (SSEE) to prepare 45 structurally well-defined core M1 and M2 based O-mannosyl glycans (Figure 2), covering all identified glycoforms with the exception of the HNK-1 epitope.[1b]
Figure 2.

The 45 core M1 and M2 O-mannosyl glycans prepared in this study. The three scaffold structures are shown in boxes. TF =Fmoc-protected threonine.
Considering the structural signatures of O-mannosyl glycans, we concluded that all core M1 and M2 structures can be enzymatically elaborated from the three scaffolds M100, M201, and M301 (Figure 2). We envisioned that the protected Man derivative 1 (Figure 3A) would serve as a versatile precursor for the synthesis of these scaffolds. It has a free hydroxy group at the C2 position, and the 4,6-hydroxy groups are protected as the benzylidene acetal. This arrangement allows for chemical glycosylation at the C2 position followed by chemical glycosylation at the C6 position upon removal of the acetal protecting group.
Figure 3.
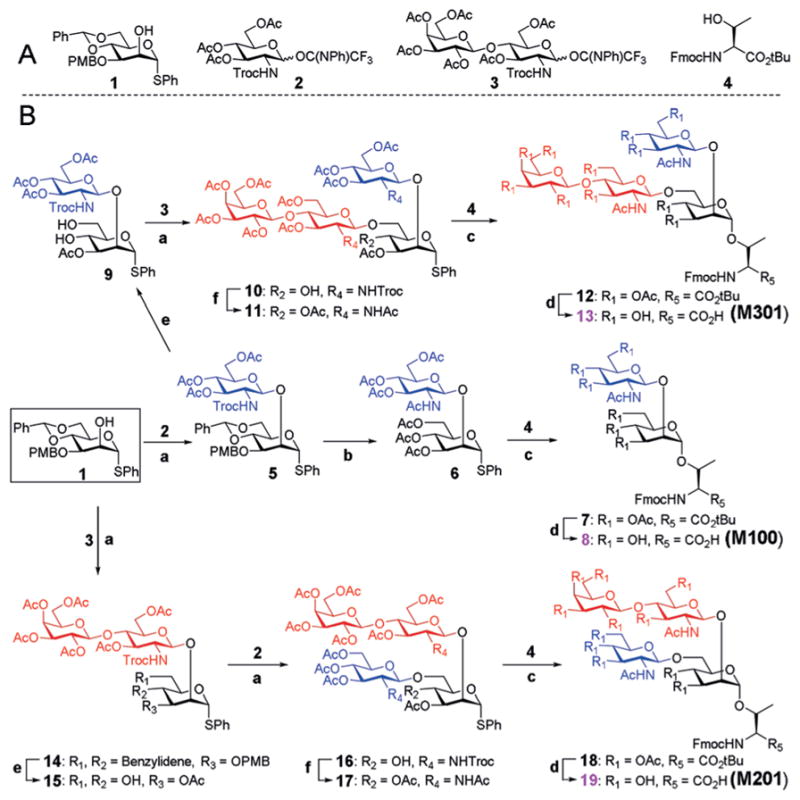
A) The building blocks used for the assembly of the three O-mannosyl scaffold structures. B) Synthetic scheme. Reagents and conditions: a) TMSOTf, DCM, −60°C, 5: 85%; 10: 79%; 14: 75%; 16: 82%; b) i) TFA, DCM; ii) Zn, AcOH; iii) Ac2O, Py, 80% over three steps; c) NIS, AgOTf, DCM/Et2O = 1:1, 0 °C, 7: 93%; 12 : 89%; 18 : 83%; d) i) TFA, DCM; ii) NaOMe, MeOH; iii) FmocOSu, NaHCO3, acetone/H2O = 3:1, 8 : 90%; 13 : 85%; 19 : 87%; e) i) DDQ, DCM/PBS buffer =9:1; ii) Ac2O, Py; iii) EtSH, TsOH, DCM, 9 : 77%; 15 : 81%; f) i) Zn, AcOH; ii) Ac2O, Py, 11: 87%; 17: 82%.
As shown in Figure 3, the three scaffolds required for the enzymatic extension were synthesized in a convergent approach and were assembled from three simple building blocks 1, 2, and 3 (see the Supporting Information for details) and Fmoc-Thr(OH)-OtBu (4). To prepare the glycoamino acid M100, donor 2 was chemically glycosylated to the glycosyl acceptor 1 at the C2 position to obtain the disaccharide 5 in 85% yield. An N-phenyltrifluoroacetimi-date donor was used instead of the typical trichloroacetimi-date as the latter can generate over 50% of a glucosamine thioether side product through thioether migration. Typical deprotection and protection strategies were employed to convert the benzylidene acetal, 4-methoxybenzyl ether (PMB), and trichloroethoxycarbonyl (Troc) protecting groups on 5 into acetyl (Ac) groups to afford 6 over three steps, with a total yield of 80%. The obtained thiophenyl glycosyl donor 6 was coupled to the protected threonine amino acid 4 at 0°C in the presence of N-iodosuccinimide (NIS) and AgOTf to give the protected glycoamino acid derivative 7 in excellent yield. The t-butyl (tBu) and Ac groups were then removed successively, followed by reintroduction of the partially cleaved Fmoc to provide the final product 8 (M100) in gram amounts, with a total yield of 90% over three steps.
To synthesize M301, the PMB group on disaccharide 5 was initially converted into an Ac moiety, followed by deprotection of the benzylidene acetal to give diol 9. Glycosylation of 9 with the glycosyl donor 3 yielded the protected target tetrasaccharide 10 in 79% yield. Successively, the Troc protecting groups were converted into Ac groups to provide compound 11, which was used as the glycosyl donor for coupling with the amino acid 4 to obtain the tetrasaccharide 12 in 89% yield. Complete removal of the tBu and Ac protecting groups was performed under standard reaction conditions, and reprotection of the partially deprotected Fmoc amine afforded gram amounts of compound 13 (M301), in a total yield of 85% over three steps. Similarly, 19 (M201) was prepared by coupling of disaccharide donor 3 to the C2 position of acceptor 1 followed by coupling of 2 to the C6 position. Then coupling of the obtained oligosaccharide thioglycoside 18 with amino acid 4 gave the desired glycosylated amino acid 19 in a total yield of 29% over eleven steps.
The Fmoc group was reintroduced to facilitate product detection and purification, as Fmoc both absorbs in the UV region (λ =260 nm) and fluoresces (λex = 260 nm, λem = 310 nm).[12] The bulky hydrophobic group also enhanced retention and separation of hydrophilic O-mannosyl glycans on reverse-phase chromatography. Herein, HPLC was used to detect, purify, and quantify enzymatically extended O-mannosyl glycans (see the Supporting Information, part II).
Four robust bacterial GTs and a human β1,3-glucuronyl-transferase (GlcAT-P) were used to extend the scaffold M100 to eight core M1 O-mannosyl glycans (Figure 4A). The bacterial GTs included β1,4-galactosyltransferase from Neisseria meningitidis (NmLgtB),[13] mutant M144D of α2,3-sialyltransferase 1 from Pasteurella multocida (PmST1-M144D) with decreased donor hydrolysis plus reduced sialidase activity,[14] α2,6-sialyltransferase from Photobacterium damselae (Pd26ST),[15] and C-terminal 66 amino acid truncated α1,3-fucosyltransferase from Helicobacter pylori (Hp3FT) with increased solubility.[16] These GTs are highly active and recognize minimal motifs (e.g., GlcNAc for NmLgtB, LN for Pd26ST and Hp3FT, LN or LeX for PmST1-M144D) on oligosaccharide acceptors, and have been previously applied for preparing complex glycans.[17]
Figure 4.

Enzymatic extension of A) core M1 and B) core M2 structures. Reagents and conditions: a) NmLgtB, UDP-Gal, Mg2+; b) PmST1-M144D, NmCSS, Neu5Ac, CTP, Mg2+; c) Pd26ST, NmCSS, Neu5Ac, CTP, Mg2+; d) Hp3FT, GDP-Fuc, Mg2+; e) PmST1-M144D, NmCSS, Neu5Gc, CTP, Mg2+; f) Pd26ST, NmCSS, Neu5Gc, CTP, Mg2+; g) GlcAT-P, UDP-GlcA, Mg2+; h) PmST1m, NmCSS, Neu5Ac, CTP, Mg2+; i) βGalD.
As illustrated in Figure 4, M101 was prepared by an NmLgtB-catalyzed reaction, containing M100 (10 mM), uridine 5′-diphosphogalactose (UDP-Gal; 15 mM), MgCl2 (10 mM), and an appropriate amount of NmLgtB. After overnight incubation, an m/z peak of 867.2972 was observed in the ESI mass spectrum, corresponding to M101 [M–H]−. Meanwhile, on the HPLC profile, a new peak (TR = 21.71 min) was observed, of which the area underneath increased while the peak corresponding to M100 (TR = 23.32 min) became smaller over time. After over 90% conversion (monitored by HPLC with a 4.6 × 250 mm Inertsil ODS-4 column), M101 was isolated by semipreparative HPLC (Inertsil ODS-4, 20 × 250 mm). The purified M101 was then lyophilized, and further extended to afford M102 and M104 in reactions catalyzed by PmST1-M144D and Hp3FT, respectively (Figure 4). To make glycosylation reactions more efficient and less expensive, the one-pot two-enzyme (OPTE)[18] approach was adopted when adding sialic acid residues. For example, in the PmST1-M144D-catalyzed reaction to generate M102, Neu5Ac, cytidine 5′-triphosphate (CTP), MgCl2, and N. meningitidis CMP-sialic acid synthetase (NmCSS)[19] were added to generate the sugar donor CMP-Neu5Ac in situ. Finally, M105 was prepared from M104 by the same OPTE approach. By employing a bacterial α2,6-sialyltransferase (Pd26ST), we were also able to generate a not yet identified structure, M103, harboring the 6-sialyl-N-acetyllactosamine (Neu5Ac-α2,6-Gal-β1,4-GlcNAc, 6SLN) epitope (Figure 4), which may serve as an ideal standard for mining such structures.
Core M1 glycans terminated with N-glycolylneuraminic acid (Neu5Gc; e.g., 3SGcLN, Figure 1) were also observed on animal α-DG.[4,20] Structures with a Neu5Gc residue (M102G and M103G) were thus prepared by using PmST1-M144D-and Pd26ST-catalyzed OPTE systems similar to those employed for the synthesis of M102 and M103. The HNK-1 epitope is a unique sulfated trisaccharide that is highly expressed in the nervous system and plays critical roles in neuronal plasticity and diseases.[21] We optimized the codon and cloned the β-glucuronosyltransferase gene (GlcAT-P) for heterogeneous expression in E. coli. Even though pure, soluble proteins were not obtained, we were able to synthesize a precursor structure of the HNK-1 epitope on core M1 (Figure 4, M106) by using GlcAT-P-containing cell lysate (see the Supporting Information, part II). Research towards obtaining a 3-sulfotransferase for generating HNK-1 epitopes is in progress.
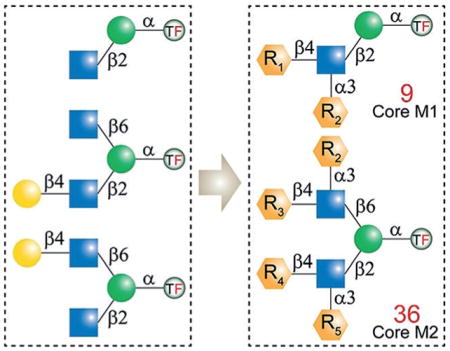
Core M2 O-mannosyl glycans can be classified into symmetric (with the same motifs on the β1,2- and β1,6-branches; e.g., M0X0) and asymmetric (with different motifs on the branches; e.g., M2XX and M3XX) structures. To generate symmetric structures, chemically prepared M201 was first galactosylated by NmLgtB to afford M010. Similar as described for the synthesis of core M1 glycans, M020, M030, and M040 were then prepared starting from M010 in reactions catalyzed by PmST1-M144D, Pd26ST, and Hp3FT, respectively. On the other hand, M050 was prepared starting from M040 via the PmST1-M144D-catalyzed reaction (Figure 4B). Specifically, M000 was generated by treating M201 with a β-galactosidase from Streptococcus pneumoniae (βGalD).[20]
The synthesis of asymmetric core M2 O-mannosyl glycans was performed from M201 or M301, by enzymatic extension of the Gal-containing branch first and then the other, in a strictly controlled sequential manner. The synthesis of M2XX is illustrated in Figure 4B. These synthetic routes were designed according to the substrate specificities of the corresponding GTs to avoid undesirable glycosylation. With M234 as an example, the scaffold M201 was first fucosylated to form M204, followed by galactosylation on the β1,6-branch to provide M214. Such a synthetic sequence avoids the addition of Fuc onto the β1,6-branch as Hp3FT requires an LN disaccharide motif for its activity.[16] Lastly, M234 was formed by a Pd26ST-catalyzed reaction, which attached a Neu5Ac residue selectively onto the terminal Gal on the β1,6-branch as α1,3-fucosylation prevents Pd26ST-catalyzed α2,6-sialylation[22] on the β1,2-branch. The synthetic scheme of M3XX is shown in Figure S1. It should be noted that M224 and M324 were synthesized from M214 and M314 (Figure 4B and Figure S1) by using mutant E271F/R313Y of PmST1 (PmST1m) instead of PmST1-M144D, which greatly prefers the LN disaccharide over the LeX trisaccharide,[23] to avoid undesired sialylation. Collectively, all 36 possible combinations of core M2 glycans that harbor LN, 3SLN, 6SLN, LeX, or sLeX motifs were prepared. These glycans were purified by HPLC and characterized by ESI/MALDI-MS and NMR spectroscopy to confirm the structures (Supporting Information, parts IV and V).
The synthesized O-mannosyl glycans are well-defined and closely related glycoforms (Figure 2), providing unique probes for mining fine specificities of glycan-binding proteins (GBPs). As shown in Figure 5A, microarray analysis (Supporting Information, part III) showed that both Ricinus communis lectin I (RCA-I) and Erythrina cristagalli lectin (ECA) strongly bound to M010, which is consistent with its primary specificity towards terminal LN epitopes.[24] Moreover, ECA exhibited a broader specificity towards all O-mannosyl glycans harboring a free terminal LN (M101, M201, M21X, M301, and M31X), whereas RCA-I seems to prefer terminal LN on the β1,6-branch (M301 and M21X) over the β1,2-branch (M101, M201, M314, and M215; Figure 5A). Such a branch preference was also found for the anti-CD15s antibody (specific to sLeX epitopes), which bound to glycans that contain sLeX on the β1,6-branch (M050, M3X5) but not to those with sLeX only on the β1,2-branch (M105, M2X5; Figure 5B). On the other hand, Aleuria aurantia lectin (AAL, specific to α-Fuc) exhibited a preference towards the β1,2-branch, as well as other fine specificities (see the Supporting Information, part III and Figure S2).
Figure 5.

Microarray analysis and evaluation of binding to spotted O-mannosyl glycans against Ricinus communis lectin I and Erythrina crystagalli lectin (A), anti-CD15s antibody (B), and core M2 polyclonal rabbit sera 26560 (C). The x axis shows the glycans, and the y axis shows the relative fluorescence. Readout by Cy3-streptavidin (A), goat anti-mouse IgG–Alexa Fluor 647 conjugate (B), and goat anti-rabbit IgG–Alexa Fluor 647 conjugate (C). NC =printing buffer, PC1 =biotin-PEG2-amine, PC2 =rabbit IgG, M =human IgG–Cy3 conjugate and human IgG–Alexa647 conjugate.
ConA, an α-Man-specific lectin commonly used for enriching tryptic O-mannosyl peptides, strongly bound to M100 but not to other natural core M1 or any core M2 structures (Figure S3). It can thus be speculated that detection or enrichment of O-mannosyl glycans using ConA may miss a substantial amount of complex structures. Interestingly, weak binding of ConA to unnatural α2,6-sialylated core M1 glycans (M103, M103G) was observed (Figure S3), suggesting that such modifications may result in conformational changes that facilitate ConA binding.
Anti-glycan antibodies, on the other hand, may be better detection tools. For example, antiserum from two rabbits (26559, 26560) that had been immunized with a core M2 glycan (M000) conjugate bound to a broad range of core M2 containing glycopeptides regardless of varied peptide sequences.[10] We further evaluated the antisera towards synthesized O-mannosyl glycans. Both sera exhibited binding specificities towards core M2 O-mannosyl glycans containing at least one free terminal GlcNAc residue (M000, M20X, M30X) as well as the core M1 disaccharide M100 (Figure 5C and Figure S4). In addition, antisera from rabbit 26560 exhibited comparable bindings to all those glycans, whereas antisera from rabbit 26559 showed stronger binding to glycans contain a free GlcNAc on the β1,6-branch (M20X) compared with binding to the β1,2-branch (M100, M30X; Figure S4). These results imply that antibodies generated from different hosts may possess certain individual heterogeneities (or individual differences). Nevertheless, our results reveal fine specificities of the antisera towards O-mannosyl glycans. Compared with ConA, these antisera are advantageous in the detection of branched O-mannosyl glycans.
In summary, by combining convergent chemical synthesis with strictly programmed enzymatic synthesis in a stepwise manner, an efficient scaffold synthesis/enzymatic extension (SSEE) strategy was developed to generate 45 mammalian O-mannosyl glycans. Such unique glycoforms provide not only standards for identifying O-mannosyl glycans and revealing their biological roles, but also constitute ideal probes for mining fine details of protein–glycan interactions.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (U01GM116263 to P.G.W. and L.L.). We are grateful to Z Biotech LLC (Aurora, CO) for printing glycan microarrays (supported by the National Institute of Health, R43GM123820). Mab(IIH6) was a kind gift from Dr. Kevin Campbell (HHMI, University of Iowa). We thank Dr. Xi Chen from the University of California, Davis, for providing sialyltransferases. U.W. is grateful to the Ministerium für Kultur und Wissenschaft des Landes Nordrhein-Westfalen and the Bundesministerium für Bildung und Forschung.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under: https://doi.org/10.1002/anie.201803536.
Contributor Information
Shuaishuai Wang, Department of Chemistry & Center for Diagnostics & Therapeutics Georgia State University, Atlanta, GA 30303 (USA).
Qing Zhang, Department of Chemistry & Center for Diagnostics & Therapeutics Georgia State University, Atlanta, GA 30303 (USA).
CongCong Chen, National Glycoengineering Research Center, School of Pharmaceutical Science, Shandong University, Jinan 250012 (China).
Yuxi Guo, Department of Chemistry & Center for Diagnostics & Therapeutics Georgia State University, Atlanta, GA 30303 (USA).
Dr. Madhusudhan Reddy Gadi, Department of Chemistry & Center for Diagnostics & Therapeutics Georgia State University, Atlanta, GA 30303 (USA)
Dr. Jin Yu, Leibniz-Institut für Analytische Wissenschaften—ISAS—e.V. 44227 Dortmund (Germany)
Prof. Dr. Ulrika Westerlind, Leibniz-Institut f4r Analytische Wissenschaften—ISAS—e.V. 44227 Dortmund (Germany). Department of Chemistry, Umeå University, 901 87 Umeå (Sweden)
Dr. Yunpeng Liu, Department of Chemistry & Center for Diagnostics & Therapeutics Georgia State University, Atlanta, GA 30303 (USA)
Dr. Xuefeng Cao, Department of Chemistry & Center for Diagnostics & Therapeutics Georgia State University, Atlanta, GA 30303 (USA)
Prof. Dr. Peng G. Wang, Department of Chemistry & Center for Diagnostics & Therapeutics Georgia State University, Atlanta, GA 30303 (USA)
Dr. Lei Li, Department of Chemistry & Center for Diagnostics & Therapeutics Georgia State University, Atlanta, GA 30303 (USA)
References
- 1.a) Endo T. J Biochem. 2015;157:1. doi: 10.1093/jb/mvu066. [DOI] [PubMed] [Google Scholar]; b) Praissman JL, Wells L. Biochemistry. 2014;53:3066. doi: 10.1021/bi500153y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yoshida-Moriguchi T, Willer T, Anderson ME, Venzke D, Whyte T, Muntoni F, Lee H, Nelson SF, Yu L, Campbell KP. Science. 2013;341:896. doi: 10.1126/science.1239951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dobson CM, Hempel SJ, Stalnaker SH, Stuart R, Wells L. Cell Mol Life Sci. 2013;70:2849. doi: 10.1007/s00018-012-1193-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stalnaker SH, Aoki K, Lim JM, Porterfield M, Liu M, Satz JS, Buskirk S, Xiong Y, Zhang P, Campbell KP, Hu H, Live D, Tiemeyer M, Wells L. J Biol Chem. 2011;286:21180. doi: 10.1074/jbc.M110.203281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sheikh MO, Halmo SM, Wells L. Glycobiology. 2017;27:806. doi: 10.1093/glycob/cwx062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vester-Christensen MB, Halim A, Joshi HJ, Steentoft C, Bennett EP, Levery SB, Vakhrushev SY, Clausen H. Proc Natl Acad Sci USA. 2013;110:21018. doi: 10.1073/pnas.1313446110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Zhang Y, Meng C, Jin L, Chen X, Wang F, Cao H. Chem Commun. 2015;51:11654. doi: 10.1039/c5cc02913a. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Seifert J, Ogawa T, Kurono S, Ito Y. Glycoconjugate J. 2000;17:407. doi: 10.1023/a:1007112232131. [DOI] [PubMed] [Google Scholar]; c) Seifert J, Ogawa T, Ito Y. Tetrahedron Lett. 1999;40:6803. [Google Scholar]; d) Matsuo I, Isomura M, Ajisaka K. Tetrahedron Lett. 1999;40:5047. [Google Scholar]; e) Live D, Wells L, Boons GJ. ChemBioChem. 2013;14:2392. doi: 10.1002/cbic.201300417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu J, Westerlind U. ChemBioChem. 2014;15:939. doi: 10.1002/cbic.201300537. [DOI] [PubMed] [Google Scholar]
- 9.Sardzik R, Green AP, Laurent N, Both P, Fontana C, Voglmeir J, Weissenborn MJ, Haddoub R, Grassi P, Haslam SM, Widmalm G, Flitsch SL. J Am Chem Soc. 2012;134:4521. doi: 10.1021/ja211861m. [DOI] [PubMed] [Google Scholar]
- 10.Yu J, Grant OC, Pett C, Stahl S, Woods RJ, Westerlind U. Chem Eur J. 2017;23:3466. doi: 10.1002/chem.201605627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Halmo SM, Singh D, Patel S, Wang S, Edlin M, Boons GJ, Moremen KW, Live D, Wells L. J Biol Chem. 2017;292:2101. doi: 10.1074/jbc.M116.764712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Einarsson S, Josefsson B, Lagerkvist S. J Chromatogr A. 1983;282:609. [Google Scholar]
- 13.Lau K, Thon V, Yu H, Ding L, Chen Y, Muthana MM, Wong D, Huang R, Chen X. Chem Commun. 2010;46:6066. doi: 10.1039/c0cc01381a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sugiarto G, Lau K, Qu J, Li Y, Lim S, Mu S, Ames JB, Fisher AJ, Chen X. ACS Chem Biol. 2012;7:1232. doi: 10.1021/cb300125k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu H, Huang S, Chokhawala H, Sun M, Zheng H, Chen X. Angew Chem Int Ed. 2006;45:3938. doi: 10.1002/anie.200600572. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2006;118:4042. [Google Scholar]
- 16.Lin SW, Yuan TM, Li JR, Lin CH. Biochemistry. 2006;45:8108. doi: 10.1021/bi0601297. [DOI] [PubMed] [Google Scholar]
- 17.a) Li L, Liu Y, Ma C, Qu J, Calderon AD, Wu B, Wei N, Wang X, Guo Y, Xiao Z, Song J, Sugiarto G, Li Y, Yu H, Chen X, Wang PG. Chem Sci. 2015;6:5652. doi: 10.1039/c5sc02025e. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wu Z, Liu Y, Ma C, Li L, Bai J, Byrd-Leotis L, Lasanajak Y, Guo Y, Wen L, Zhu H, Song J, Li Y, Steinhauer DA, Smith DF, Zhao B, Chen X, Guan W, Wang PG. Org Biomol Chem. 2016;14:11106. doi: 10.1039/c6ob01982j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu H, Chen X. Org Biomol Chem. 2016;14:2809. doi: 10.1039/c6ob00058d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu H, Yu H, Karpel R, Chen X. Bioorg Med Chem. 2004;12:6427. doi: 10.1016/j.bmc.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 20.Stalnaker SH, Hashmi S, Lim JM, Aoki K, Porterfield M, Gutierrez-Sanchez G, Wheeler J, Ervasti JM, Bergmann C, Tiemeyer M, Wells L. J Biol Chem. 2010;285:24882. doi: 10.1074/jbc.M110.126474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morise J, Takematsu H, Oka S. Biochim Biophys Acta Gen Subj. 2017;1861:2455. doi: 10.1016/j.bbagen.2017.06.025. [DOI] [PubMed] [Google Scholar]
- 22.Unger WW, Mayer CT, Engels S, Hesse C, Perdicchio M, Puttur F, Streng-Ouwehand I, Litjens M, Kalay H, Berod L, Sparwasser T, van Kooyk Y. Oncoimmunology. 2015;4:e970462. doi: 10.4161/21624011.2014.970462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sugiarto G, Lau K, Li Y, Khedri Z, Yu H, Le DT, Chen X. Mol Biosyst. 2011;7:3021. doi: 10.1039/c1mb05182b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Itakura Y, Nakamura-Tsuruta S, Kominami J, Sharon N, Kasai K, Hirabayashi J. J Biochem. 2007;142:459. doi: 10.1093/jb/mvm153. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.