

Abstract
Alcoholic liver disease (ALD) is characterized by lipid accumulation and liver injury. However, how chronic alcohol consumption causes hepatic lipid accumulation remains elusive. The present study demonstrates that activation of the mechanistic target of rapamycin complex 1 (mTORC1) plays a causal role in alcoholic steatosis, inflammation and liver injury. Chronic-plus-binge ethanol feeding led to hyperactivation of mTORC1, as evidenced by increased phosphorylation of mTOR and its downstream kinase S6K1 in hepatocytes. Aberrant activation of mTORC1 was likely attributed to the defects of the DEP-domain containing mTOR-interacting protein (DEPTOR) and the NAD+-dependent deacetylase SIRT1 in the liver of chronic-plus-binge ethanol-fed mice and in the liver of patients with ALD. Conversely, adenoviral overexpression of hepatic DEPTOR suppressed mTORC1 signaling and ameliorated alcoholic hepatosteatosis, inflammation and acute-on-chronic liver injury. Mechanistically, the lipid-lowering effect of hepatic DEPTOR was attributable to decreased proteolytic processing, nuclear translocation, and transcriptional activity of the lipogenic transcription factor SREBP-1. DEPTOR-dependent inhibition of mTORC1 also attenuated alcohol-induced cytoplasmic accumulation of the lipogenic regulator lipin 1 and prevented alcohol-mediated inhibition of fatty acid oxidation. Pharmacological intervention with rapamycin alleviated the ability of alcohol to upregulate lipogenesis, to downregulate fatty acid oxidation, and to induce steatogenic phenotypes. Chronic-plus-binge ethanol feeding led to activation of SREBP-1 and lipin 1 through S6K1-dependent and independent mechanisms. Furthermore, hepatocyte-specific deletion of SIRT1 disrupted DEPTOR function, enhanced mTORC1 activity, and exacerbated alcoholic fatty liver, inflammation and liver injury in mice.
Conclusion
the dysregulation of SIRT1-DEPTOR-mTORC1 signaling is a critical determinant of ALD pathology. Targeting SIRT1 and DEPTOR and selectively inhibiting mTORC1-S6K1 signaling may have therapeutic potential for treating ALD in humans.
INTRODUCTION
Alcoholic liver disease (ALD) encompasses a wide spectrum of chronic liver diseases, ranging from hepatic steatosis to fibrosis, cirrhosis, and ultimately hepatocellular carcinoma (1, 2). More than 90% of heavy drinkers develop hepatic steatosis (fatty liver) and about 20-40% of them develop more severe forms of ALD (1). Hepatic steatosis is an early and reversible stage of ALD (1, 2). However, there are no effective therapies available for end-stage ALD except liver transplantation (1, 2). There is an urgent need to develop new pathophysiology-oriented therapies in human ALD.
The mechanistic target of rapamycin (mTOR) is a conserved protein kinase that forms two functional complexes, termed mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) (3). mTORC1 regulates cellular processes including cell growth, proliferation, and metabolism (3). While mTORC1 inhibition prevents apoptosis in cancer cells (4), the potential effect of mTORC1 on alcoholic liver injury has not been investigated. Since DEP domain-containing mTOR-interacting protein (DEPTOR) has recently been emerged as an mTOR binding protein that inhibits the mTOR kinase (5, 6), the role of DEPTOR in affecting the pathogenesis of ALD needs more investigation. Because overexpression of the NAD-dependent deacetylase SIRT1 suppresses mTORC1 activity and attenuates fatty liver in genetically obese ob/ob mice (7), we hypothesize that abnormal regulation of mTORC1 is functionally linked to the development of ALD. To test this hypothesis, we used a recently developed mouse model of chronic-plus-binge ethanol feeding, which is characterized by hepatic steatosis and acute-on-chronic liver injury and closely resembles the pathogenesis and drinking pattern of ALD in humans (8, 9).
Here we provide the first clinical evidence that mTORC1 functions as a central regulator of the ethanol sensing machinery that links lipid metabolic dysregulation to hepatocellular apoptosis in mice and in patients with ALD. Our data illustrate that (1) DEPTOR serves as a critical negative regulator of mTORC1 that protects mice from alcohol-induced fatty liver, inflammation, and liver damage; (2) mTORC1 is necessary for alcohol to activate hepatic lipogenesis and to inhibit fatty acid oxidation in mice; (3) Overexpression of the kinase inactive S6K1, the downstream kinase of mTORC1, ameliorates alcoholic hepatic steatosis largely through reduced SREBP-1-dependent de novo lipogenesis; and (4) Defective SIRT1 is coupled to the downregulation of DEPTOR and upregulation of mTORC1 and lipogenesis in patients with ALD. Therefore, targeting the SIRT1-DEPTOR-mTORC1 axis may have therapeutic potential in human ALD.
RESULTS
mTORC1 activation is evident in the mouse model of chronic-plus-binge ethanol feeding
Because many patients with ALD have a history of chronic drinking superimposed by excessive binge drinking (1), a mouse model of chronic-plus-binge ethanol feeding was used as described recently (9, 10). As depicted in Fig. 1A and fig. S1, H&E staining showed that chronic-binge ethanol administration resulted in steatosis containing small and large lipid droplet vacuoles within hepatocytes, compared with pair-fed mice. Liver and plasma triglyceride levels were significantly higher in mice after chronic-binge ethanol feeding than those in pair-fed mice. Hepatic and plasma cholesterol levels, body weight, and food intake were comparable between two groups of the mice. Importantly, plasma alanine aminotransferase (ALT) levels were increased more than 3-fold in chronic-binge ethanol-fed mice.
Fig.1. Chronic-plus-binge ethanol feeding leads to inhibition of DEPTOR and activation of mTORC1 signaling and promotes the development of fatty liver in mice.
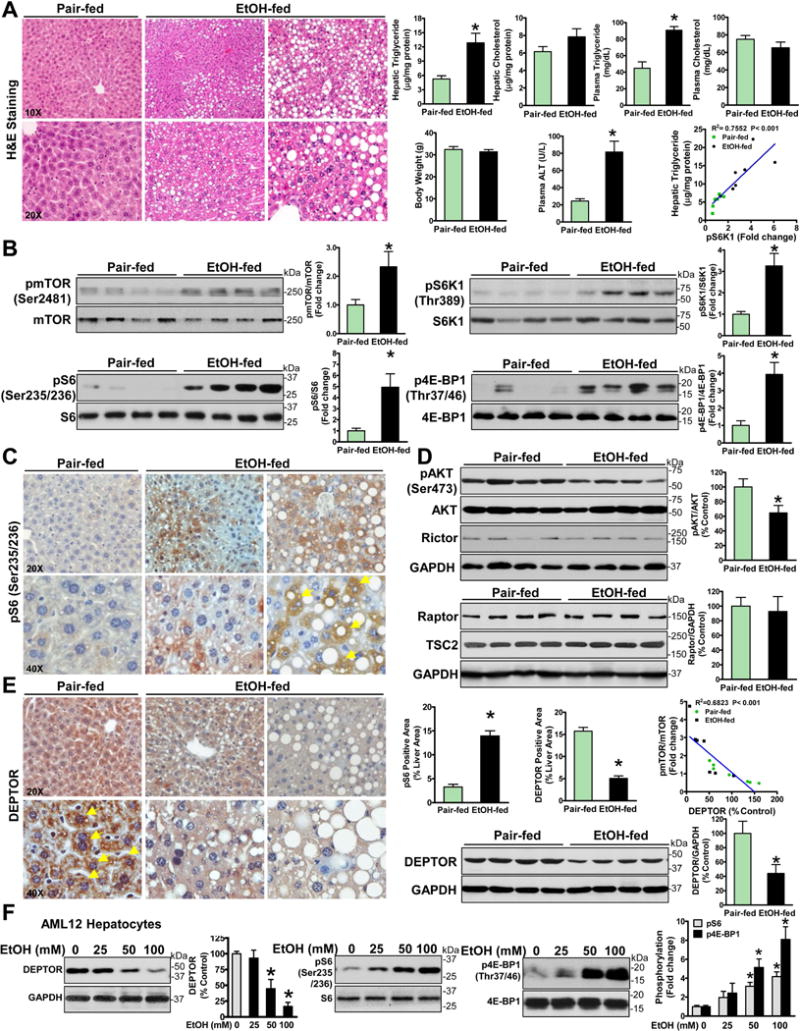
A. Effect of chronic-binge ethanol feeding on metabolic parameters and plasma alanine aminotransferase (ALT) levels. Representative H&E staining of hepatic steatosis in a mouse model of chronic-plus-binge ethanol feeding. Original magnification: 10X or 20X. B. Representative immunoblots for phosphorylation of mTOR (pmTOR), S6K1 (pS6K1), S6 (pS6), and 4E-BP1 (p4E-BP1) in livers from four mice each group. C. Positive immunostaining for phosphorylated S6 (a brown color) was predominantly located in lipid-rich hepatocytes (yellow arrows) in ethanol-fed mice. D. Effect of chronic-binge ethanol feeding on key regulators of mTORC1 and mTORC2. E. Chronic-binge ethanol feeding leads to an impairment of hepatic DEPTOR. Notably, positive staining for DEPTOR is visualized mainly in the cytoplasm of hepatocytes in pair-fed mice, and this effect is reduced in ethanol-fed mice. Original magnification: 20X or 40X. Linear regression between hepatic DEPTOR levels and mTOR phosphorylation in normal and ethanol-fed mice. The data are presented as mean ± SEM, n = 6–8 each group. *P < 0.05 vs. pair-fed mice. F. The effect of ethanol on DEPTOR and mTORC1 signaling in AML12 mouse hepatocytes.
To determine whether the mTORC1 is involved in ALD pathology, multiple markers of mTORC1 signaling were determined. As depicted in Fig. 1B, autophosphorylation of mTOR at Ser2481 was increased twofold in chronic-binge ethanol-fed mice compared with pair-fed mice. The Thr389 phosphorylation of S6K1, which is modulated by mTOR (11, 12), was increased 3.3-fold. The phosphorylation of the S40 ribosomal protein S6 at Ser235/Ser236, the best-characterized substrate of S6K1, was increased fivefold. The Thr37/46 phosphorylation of 4E-BP1, another substrate of mTORC1, was also enhanced threefold, further validating the inappropriate mTORC1 signaling in chronic-binge ethanol-fed mice. S6K1 activity was positively correlated with hepatic triglyceride content in mice. Furthermore, immunohistochemical analysis revealed that elevated positive signals of S6 phosphorylation were co-localized primarily in the cytoplasm of lipid droplet-rich hepatocytes of chronic-binge ethanol-fed mice (Fig. 1C).
We further observed that expression of Rictor, a major component of mTORC2, was unaffected. The Ser473 phosphorylation at the Akt hydrophobic site, which can be catalyzed by mTORC2 (3), was slightly decreased by ethanol feeding (Fig. 1D and fig. S1C). It is possible that activation of mTORC1 by ethanol may trigger a S6K1-dependent negative feedback loop toward IRS-1-PI3K signaling, leading to dampened activation of Akt (3). Taken together, aberrant activation of mTORC1, but not mTORC2, is associated with alcoholic hepatic steatosis.
Hepatic DEPTOR is suppressed by chronic-plus-binge ethanol feeding
To elucidate the mechanism underlying the upregulation of mTORC1 by ethanol, we sought which upstream regulator of mTORC1 is involved in ALD. As shown in Fig. 1D and fig. S1D–E, expression of regulatory-associated protein of mTOR (Raptor), a critical scaffold and activator that stimulates mTORC1 activity (11), was not significantly affected by ethanol feeding. Endogenous levels of tuberous sclerosis complex 2 (TSC2), a negative regulator of mTORC1 (3), and the activity of ERK1/2, the upstream kinases of the TSC complex, were comparable between the two groups. DEPTOR has been recently identified as an endogenous inhibitor of the mTOR kinase (5). While DEPTOR activity is largely modulated through the control of DEPTOR protein levels in cultured cells (5), we are the first to explore the function of DEPTOR on lipid metabolism. As shown in Fig. 1E–F, DEPTOR was highly expressed in the cytoplasm of hepatocytes in pair-fed mice and it was markedly reduced by ethanol feeding. Strikingly, ethanol exposure decreased DEPTOR and increased phosphorylation of S6 and 4E-BP1 in a dose-dependent manner in AML-12 mouse hepatocytes. In contrast to hyperphosphorylated mTOR, hepatic DEPTOR levels were decreased ~50% in chronic-binge ethanol-fed mice. DEPTOR levels inversely correlated with mTOR phosphorylation in ethanol-fed mice, suggesting that alcohol-induced mTORC1 activation may be attributed to the downregulation of DEPTOR. Although other mechanisms may contribute to alcohol-induced activation of mTORC1, impaired DEPTOR in hepatocytes may be a key determinant of early stage ALD.
Adenoviral overexpression of DEPTOR inhibits mTORC1 signaling and ameliorates alcoholic fatty liver
To gain direct evidence for the in vivo functional relevance of decreased DEPTOR to ALD pathology, we generated an adenoviral vector expressing DEPTOR (Ad-DEPTOR) and confirmed adenoviral overexpression of DEPTOR in human HepG2 cells (Fig. 2A). Adenovirus-mediated overexpression of DEPTOR was achieved by tail vein injection of Ad-DEPTOR into chronic-binge ethanol-fed mice, as reflected by a 3.7-fold increase in hepatic DEPTOR (Fig. 2B and fig. S2A). We further determined whether alcohol-mediated hyperactivation of mTORC1 in the liver is a reversible process. Mice overexpressing DEPTOR displayed much less hepatic lipid vacuolization and triglyceride accumulation compared with control mice overexpressing GFP. Hepatic and plasma levels of cholesterol, body weight, and food intake were comparable between the two groups (Fig. 2C and fig. S2D–E). Because of no significant changes in TSC2 and Raptor in ethanol-fed mice (Fig. 1D), the results indicate that excessive alcohol consumption inhibits DEPTOR, which may stimulate mTORC1 and promote hepatic steatosis.
Fig. 2. Adenoviral overexpression of hepatic DEPTOR inhibits mTORC1 and improves hepatic steatosis in chronic-binge ethanol-fed mice.
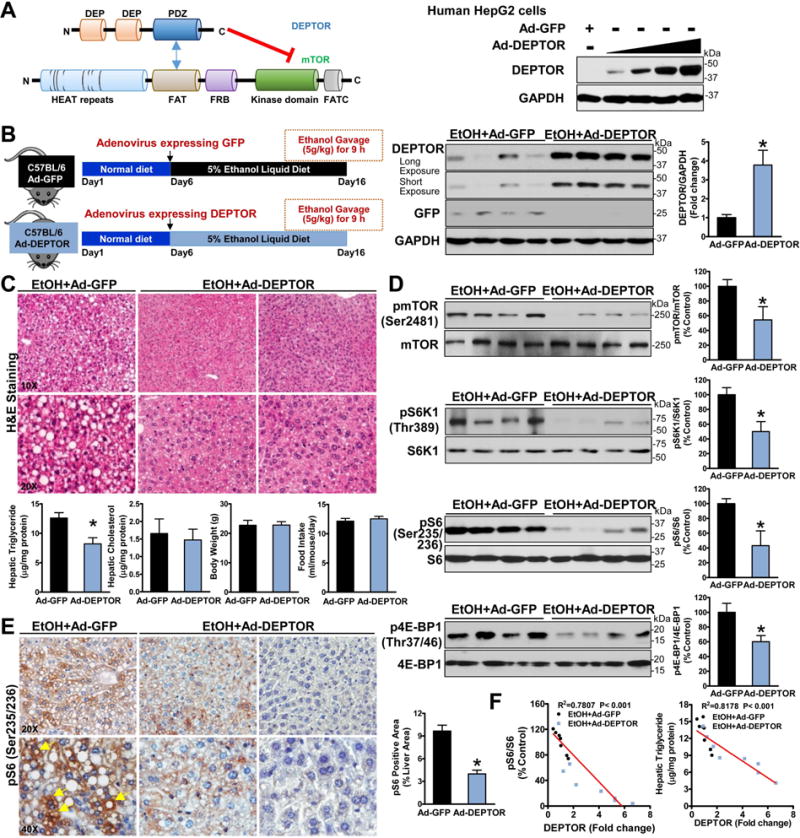
A. Schematic representation of the interaction regions between mTOR and DEPTOR. DEPTOR inhibits mTOR activity by binding the PDZ domain of DEPTOR to the FAT domain of the mTOR kinase. Immunoblotting analyses confirmed adenovirus-mediated overexpression of DEPTOR in human HepG2 cells. B. Hepatic overexpression of either DEPTOR protein (∼46 kDa) or GFP protein (∼27 kDa) in mice is confirmed. C. Overexpression of DEPTOR improves hepatic steatosis and lowers triglyceride accumulation in ethanol-fed mice. D. Overexpression of DEPTOR represses the induction of mTORC1 toward the downstream signaling in ethanol-fed mice. E. Positive staining for phosphorylated S6 in hepatocytes (yellow arrows) in ethanol-fed mice is reduced by overexpressing DEPTOR. F. Increased DEPTOR levels correlate with decreased S6 phosphorylation and lowered hepatic triglyceride content in mice. The data are presented as mean ± SEM, n = 6–8. *P < 0.05 vs. ethanol-fed mice with Ad-GFP injection.
As shown in Fig. 2D and fig. S2B–D, enforced expression of DEPTOR was sufficient to inhibit alcohol-induced activation of mTORC1, accompanied by reduced Ser2481 autophosphorylation of mTOR and Thr389 phosphorylation of S6K1, which is essential for mTOR-regulated activity of S6K1 (11, 12). Overexpression of DEPTOR also caused an approximately 50% reduction in phosphorylation of S6 and 4E-BP1 in ethanol-fed mice, largely owing to inactivation of mTOR and S6K1 kinases. The intensity and areas of positive staining for S6 phosphorylation present in lipid droplet-rich hepatocytes were reduced by DEPTOR overexpression (Fig. 2E). Increasing DEPTOR levels correlated with the reduction in S6 phosphorylation and triglyceride overproduction in chronic-binge ethanol-fed mice (Fig. 2F). Collectively, the dysregulation of DEPTOR-mTORC1 signaling represents a key determinant of ethanol sensing machinery that modulates lipid metabolism.
Hepatic overexpression of DEPTOR suppresses alcohol-induced activation of SREBP-1 in mice
The sterol regulatory element-binding protein-1 (SREBP-1), a critical transcription factor that activates the synthesis of fatty acids and triglycerides, has been implicated in hepatic steatosis in obesity-induced fatty liver (13, 14). While the role of ethanol in SREBP-1 appears controversial (15, 16), SREBP-1 null mice exhibit lowered hepatic triglyceride accumulation upon ethanol feeding (15). To delineate the mechanism by which overexpression of DEPTOR restores hepatic lipid homeostasis in ALD, the cleavage processing of SREBP-1 was examined by assessing amounts of the precursor (~125 kDa) and nuclear active form (~68 kDa) of SREBP-1. As shown in Fig. 3A–B, the active, nuclear form of SREBP-1 was significantly elevated in chronic-binge ethanol feeding, accompanied by increased SREBP-1 precursor, as was seen in earlier studies (15). Immunohistochemical analysis further confirmed that expression and nuclear translocation of SREBP-1 were increased by ethanol feeding and downregulated by DEPTOR overexpression. Notably, strong positive staining for SREBP-1 was primarily localized in the nucleus of lipid droplet-rich hepatocytes of ethanol-fed mice, and this effect was eliminated by DEPTOR overexpression. SREBP-1c has been demonstrated to be transcriptionally upregulated by nuclear SREBP via a feed-forward mechanism (13), because nuclear SREBP-1 can bind to the sterol regulatory element (SRE) motif present on its own promoters (13). Intriguingly, the accumulation of the active, nuclear form of SREBP-1 was suppressed by DEPTOR overexpression. The precursor form of SREBP-1 was slightly diminished possibly owning to the reduction of SREBP-1c mRNA by suppressing its feed-forward loop. Moreover, ethanol-mediated upregulation of SREBP-1c and its target genes, including acetyl-CoA carboxylase (ACC1), fatty acid synthase (FAS), and stearoyl CoA desaturase 1 (SCD1), was significantly decreased by DEPTOR overexpression. Consequently, ethanol-mediated induction of FAS was reduced by DEPTOR overexpression, which coincided with lowered triglyceride content in mice and in AML-12 hepatocytes (Fig. 3C, fig. S3, and Fig. 2F). Thus, hepatic overexpression of DEPTOR protects against alcohol-induced de novo lipogenesis and steatosis at least in part through the downregulation of the proteolytic processing, nuclear translocation, and transcriptional activity of SREBP-1.
Fig. 3. Hepatic overexpression of DEPTOR ameliorates alcohol-mediated dysregulation of lipid metabolism in chronic-binge ethanol-fed mice.
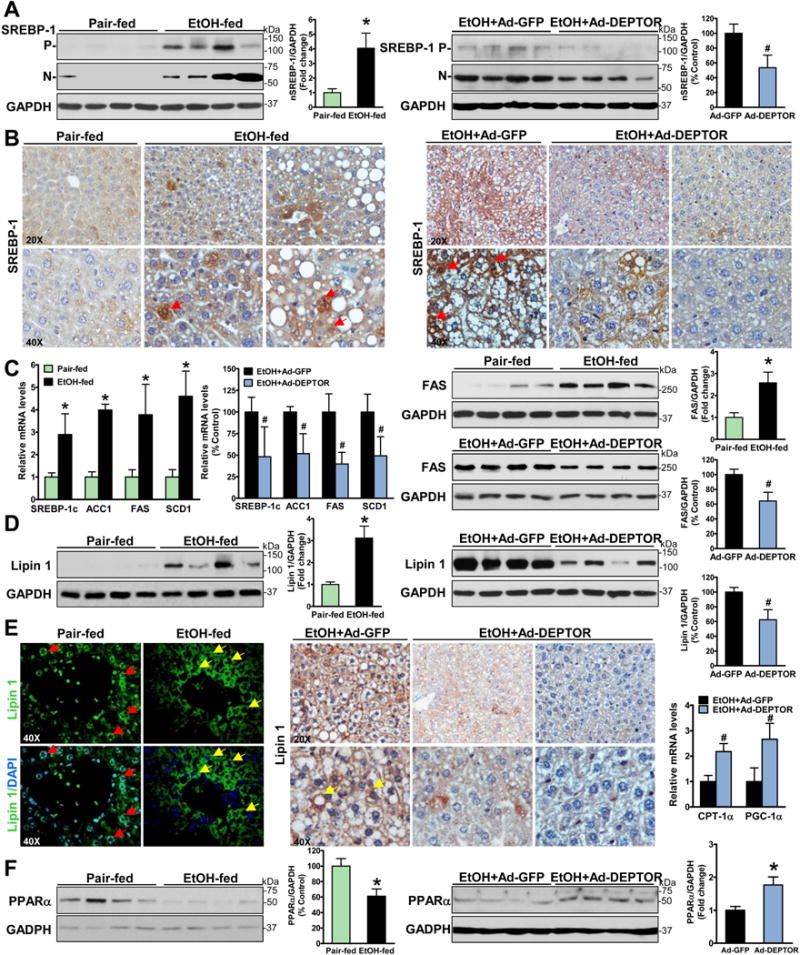
A. The active, nuclear form of SREBP-1 is increased in ethanol-fed mice and decreased by DEPTOR overexpression. P and N denote the precursor (~125 kDa) and cleaved nuclear (~68 kDa) forms of SREBP-1. B-C. The nuclear translocation of SREBP-1 as well as expression of SREBP-1c and its targets including ACC1, FAS, and SCD1 are reduced by DEPTOR overexpression. Notable, positive staining for SREBP-1 is primarily located in the nuclei of the hepatocytes of ethanol-fed mice (red arrows). D–E. Overexpression of DEPTOR represses the expression and cytoplasmic translocation of lipin-1 in chronic-binge ethanol-fed mice. Immunofluorescent staining showed that strong staining for lipin 1 (green) was predominantly located in the nuclear (blue) of hepatocytes (red arrows) of pair-fed mice, but it was elevated and mainly presented in the cytoplasm in hepatocytes (yellow arrows) surrounding central or portal veins of ethanol-fed mice. Notably, strong staining for lipin 1 is visualized mainly in the cytoplasm of hepatocytes (yellow arrows) of control mice on the ethanol diet, and this induction is inhibited by overexpression of DEPTOR. F. Expression of key genes involving fatty acid oxidation such as PPARα, CPT-1α and PGC-1α is analyzed. The data are presented as mean ± SEM, n = 6–8. *P < 0.05 vs. pair-fed mice. #P < 0.05 vs. ethanol-fed mice with Ad-GFP injection.
Hepatic overexpression of DEPTOR reduces alcohol-induced cytoplasmic accumulation of lipin 1 in mice
We next tested the hypothesis that DEPTOR might be functionally linked to the regulation of lipin 1, the key metabolic enzyme that dephosphorylates phosphatidic acid (PA) to form diacylglycerol (PAP activity) in triglyceride synthetic pathway (17). As shown in Fig. 3D–E, chronic-binge ethanol exposure caused a robust increase in lipin 1 and the cytoplasmic localization of lipin 1, consistent with increased PAP activity of cytoplasmic lipin 1 in the mouse model of chronic ethanol feeding (18). Notably, positive staining areas for lipin 1 were overlapped with those of lipid droplet-rich hepatocytes in ethanol-fed mice. Conversely, enforced expression of DEPTOR reduced the expression and cytoplasmic accumulation of lipin 1 caused by ethanol. Taken together, DEPTOR inhibits alcohol-induced lipogenesis at least partially by reducing cytoplasmic translocation of lipin 1.
Hepatic overexpression of DEPTOR stimulates expression of key genes involved in fatty acid oxidation in chronic-binge ethanol-fed mice
Real-time PCR analysis showed that chronic-binge ethanol exposure caused a significant impairment in PPARα-mediated fatty acid oxidation, as evidenced by decreased expression of the nuclear receptor PPARα (Fig. 3F). Complementary to the upregulation of PPARα by DEPTOR overexpression, expression of its major target gene, carnitine palmitoyl transferase 1α (CPT-1α) that is the key rate-limiting enzyme of fatty acid oxidation, was also induced. The lipid-lowering effect of DEPTOR was further supported by elevated PPARα coactivator 1α (PGC-1α), a transcription cofactor known to activate PPARα. Collectively, alcoholic disruption of DEPTOR signaling causes a metabolic switch from fatty acid oxidation toward fatty acid synthesis, which can be reversed by overexpression of DEPTOR.
mTORC1 is essential for alcohol-induced lipid metabolic abnormalities and hepatic steatosis in mice
To explore the therapeutic potential of a pharmacological inhibitor of mTORC1 on ALD, we took advantage of rapamycin, because pharmacological inhibitors of mTOR such as rapamycin are FDA-approved drugs for immunosuppression and anticancer treatment in humans. Rapamycin forms a complex with FKBP12 (12 kDa FK506-binding protein); this complex specifically binds to FKBP12-rapamycin binding (FRB) domain of mTORC1 that allosterically inhibits its kinase activity (3) (Fig. 4A). As shown in Fig. 4B, the induction of Ser2481 phosphorylation of mTOR and Thr389 phosphorylation of S6K1 in ethanol-fed mice was almost completely blocked by rapamycin treatment. Suppression of hepatic mTOR and S6K1 kinases was evidenced by markedly decreased phosphorylation of 4E-BP1 and S6, respectively. Rapamycin administration attenuated hepatic steatosis with a ~40% reduction in liver triglyceride content, compared with vehicle-treated, ethanol-fed mice (Fig. 4C and F). No significant alterations in hepatic cholesterol content, body weight, and food intake were noted in rapamycin-treated mice (fig. S4). Furthermore, the number and density of hepatocytes stained with S6 phosphorylation were much less in rapamycin-treated mice than those in vehicle-treated mice (Fig. 4C). This suggests that ethanol stimulates S6K1 activity and hepatic steatosis by acting upstream of mTORC1.
Fig. 4. Pharmacologic intervention via mTORC1 inhibition reduces hepatic lipogenesis, stimulates fatty acid oxidation, and ameliorates hepatic steatosis in chronic-binge ethanol-fed mice.
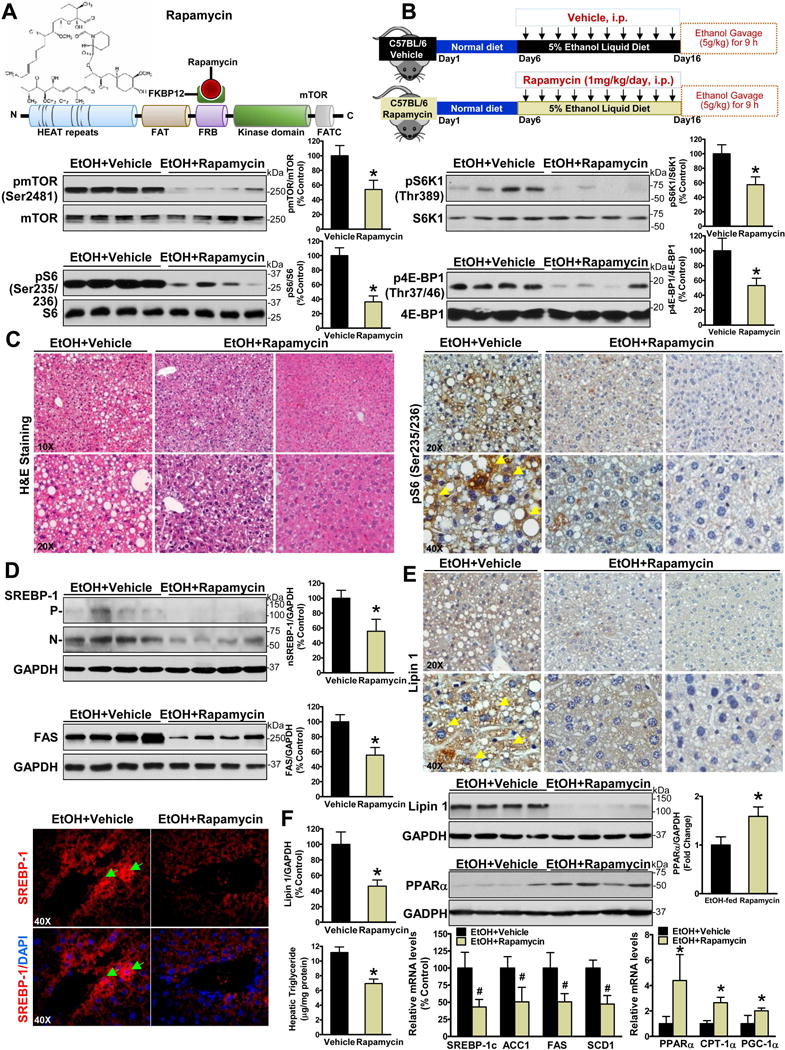
A. Schematic structure and rapamycin binding site of the mTOR kinase. The FRB domain is the docking site of the FK506-binding protein 12 (FKBP12)–rapamycin complex. B. Phosphorylation of mTOR and its downstream effectors was sensitive to treatment with rapamycin in ethanol-fed mice. C. Positive staining for phosphorylated S6 was primarily localized in the cytoplasm of lipid-laden hepatocytes (yellow arrows) in vehicle control mice, and the staining intensity is reduced in rapamycin-treated mice. D. The cleavage and nuclear translocation of SREBP-1 and induction of FAS are diminished by rapamycin treatment. Representative photomicrographs of immunofluorescence for SREBP-1 (red) and DAPI (blue) in liver sections of mice. E-F. mTORC1 is required for alcohol to induce hepatic lipogenesis and to inhibit PPARα-mediated fatty acid oxidation in mice. The data are presented as the mean ± SEM, n = 6–8. *P < 0.05, vs. ethanol-fed mice with vehicle administration.
Ethanol-mediated accumulation of nuclear SREBP and induction of FAS were abolished in a rapamycin-sensitive manner (Fig. 4D). Immunofluorescence analysis revealed that ethanol-induced nuclear translocation of SREBP-1 was eliminated by rapamycin treatment. Newly generated SREBP-1 precursor was also downregulated possibly due to reduced SREBP-1c transcription. Moreover, expression and cytoplasmic location of lipin 1 were suppressed by rapamycin administration (Fig. 4E). Consequently, mRNA abundance of SREBP-1c and its target genes including ACC1, FAS and SCD1 was reduced. The downregulation of both SREBP-1 and lipin 1 and upregulation of PPARα coincided with triglyceride reduction in ethanol-fed mice upon rapamycin treatment (Fig. 4F). Our studies with pharmacological and molecular inhibition of mTORC1 demonstrate that mTORC1 is necessary to activate lipogenesis and inhibit fatty acid oxidation in response to alcohol challenge.
Hepatic S6K1 activity is required for alcohol to induce SREBP-1-dependent lipogenesis in mice
As mentioned above, enhanced phosphorylation and activity of S6K1, the major downstream kinase of mTORC1, are associated with hepatic lipid accumulation in ethanol-fed mice (Fig. 1B). If this kinase is important for the development of ALD, inactivation of S6K1 would mimic the protective effect of DEPTOR on ALD. To this end, a dominant negative form of S6K1 (DN-S6K1) containing the K100R mutation in the ATP binding site on the kinase domain (Fig. 5A), which is catalytically inactive form of S6K1 (11, 12, 19), was used. As shown in Fig. 5B, in vivo adenoviral gene transfer of DN-S6K1 into chronic-binge ethanol-fed mice was successfully accomplished through tail vein injection, as evidenced by a ~6-fold elevation of S6K1 and a remarkable reduction of S6 phosphorylation. No significant alteration in 4E-BP1 phosphorylation was noted. To define the functional consequence of DN-S6K1, mice expressing DN-S6K1 displayed less ethanol-caused steatosis, as revealed by liver histology and reduced liver triglyceride levels (Fig. 5C and E). No difference in hepatic cholesterol, body weight, and food intake between the two groups were evident (fig. S5). Immunohistochemical staining showed significantly less positive staining for S6 phosphorylation in hepatocytes of mice expressing DN-S6K1 than those of control mice expressing GFP (Fig. 5C). Furthermore, alcohol-induced accumulation of nuclear SREBP-1 and expression of key lipogenic genes were counteracted by overexpressing DN-S6K1 (Fig. 5D). Surprisingly, unlike the downregulation of SREBP-1 by DN-S6K1, hepatic levels of lipin 1 appeared indistinguishable between the two groups (Fig. 5E–F), suggesting that alcohol upregulates lipin 1 via a S6K-independent mechanism. These studies support a model, in which uncontrolled mTORC1 activity by ethanol promotes lipid biosynthesis through S6K1-dependent and independent mechanisms.
Fig. 5. Hepatic S6K1 activity is required for the proteolytic activation of SREBP-1 and its lipogenic process in mice after chronic-binge ethanol feeding.
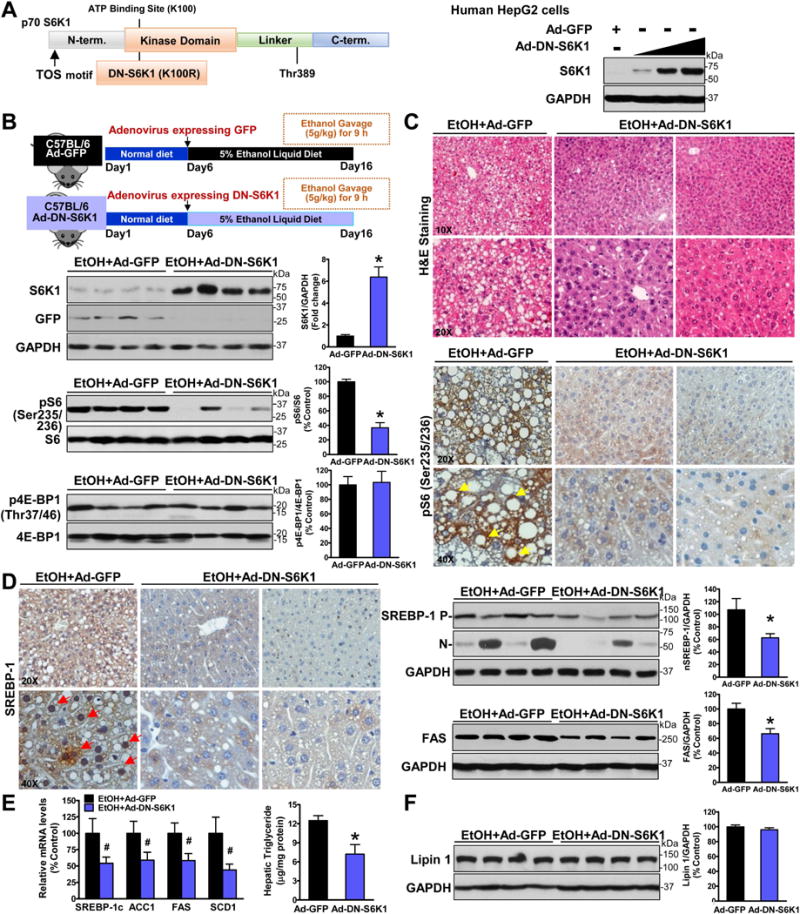
A. Schematic structure and phosphorylation sites of p70S6K1. The dominant negative form of S6K1 bears a mutation of lysine 100 to arginine (K100R) in the ATP binding site of the kinase domain. Immunoblots confirmed adenovirus-mediated overexpression of the dominant negative form of S6K1 (DN-S6K1) in HepG2 hepatocytes. B–C. Overexpression of DN-S6K1 inhibits S6K1 activity and attenuates hepatic steatosis in ethanol-fed mice. Notably, strong positive staining for phosphorylated S6 in the cytoplasm of hepatocytes (yellow arrows) in ethanol-fed mice is markedly decreased by overexpression of DN-S6K1. D–E. Overexpression of DN-S6K1 suppresses the accumulation of nuclear SREBP-1 and induction of lipogenic genes in response to ethanol feeding. Notably, positive staining for the nuclear translocation of SREBP-1 was presented in hepatocytes (red arrows) around central and portal veins in GFP-expressed mice. F. Alcohol-mediated induction of lipin 1 is unaffected by overexpression of DN-S6K1. The data are presented as mean ± SEM, n = 6–8. *P < 0.05 vs. ethanol-fed mice with Ad-GFP injection.
Molecular and pharmacological inhibition of mTORC1 protects against alcohol-induced hepatic inflammation and injury in mice
As shown in Fig. 6A–D, hepatic mRNA expression of the pro-inflammatory cytokines and chemokines, including tumor necrosis factor alpha (TNF-α), interleukin (IL)−1β, IL-6, and monocyte chemotactic protein 1 (MCP-1), were significantly higher in chronic-binge–fed mice than those in pair-fed mice, and such an inflammatory response was attenuated by DEPTOR overexpression. A minimal alternation in hepatic expression of the macrophage marker F4/80 was evident, similar to the minimal effect of ethanol on DEPTOR in RAW264.7 mouse macrophages (fig. S6). However, consistent with neutrophil infiltration in human ALD (10), hepatic expression of neutrophil markers, such as Ly6G, MPO and CD11b, was upregulated by chronic-binge ethanol feeding, suggesting that neutrophil infiltration into liver tissue is a hallmark of local inflammation in mice and humans with early stage ALD. Importantly, overexpression of DEPTOR suppressed alcohol-induced liver neutrophil accumulation with a minimal effect on hepatic macrophages. To further evaluate the effect of DEPTOR on alcohol-induced cell death, decreased phosphorylation of Akt, a major regulator of hepatocyte survival, and increased caspase-3 cleavage, a specific marker of apoptosis, were observed in mice after chronic-binge ethanol feeding, and these pathological changes were prevented by DEPTOR overexpression. Accordingly, chronic-binge feeding-associated elevation of ALT levels was reduced. Hepatic DEPTOR levels were negative correlated with plasma ALT levels in chronic-binge ethanol-fed mice. Furthermore, rapamycin-treated mice were resistant to chronic-binge-feeding-induced hepatocellular damage and inflammation, as reflected by stimulation of Akt-mediated survival signaling, and inhibition of caspase-3 cleavage and neutrophil activation. Decreased levels of cleaved caspase-3 were correlated with lowered plasma ALT levels in rapamycin-treated mice (Fig. 6E). Finally, a similar reduction in hepatic apoptosis and plasma ALT levels was seen in mice expressing DN-S6K1 (Fig. 6F).
Fig. 6. Hepatic inhibition of mTORC1 protects against alcohol-induced hepatic inflammation, apoptosis, and liver injury in mice.
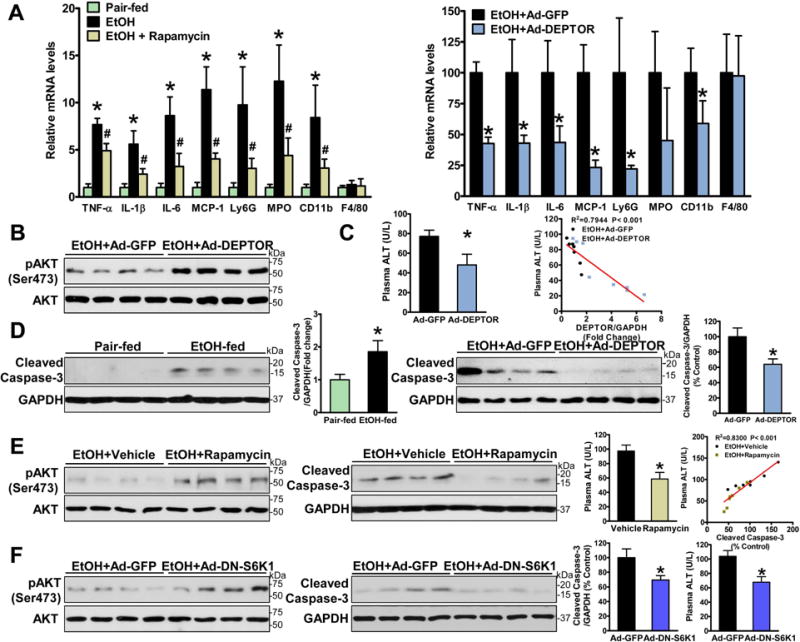
A. Liver mRNA levels of pro-inflammatory regulators, neutrophil markers (Ly6G, MPO and CD11b), and macrophage marker (F4/80) were analyzed by real-time PCR. B. Hepatic activity of Akt, the major hepatocyte survival regulator, is upregulated by DEPTOR in chronic-binge ethanol-fed mice. C. Liver injury is assessed by measuring serum ALT levels. D. Overexpressing DEPTOR protects against alcohol-induced hepatocyte apoptosis, as evidenced by reduced caspase-3 cleavage. E. Rapamycin administration alleviates alcohol-mediated inhibition of Akt and induction of hepatic apoptosis and injury. F. Overexpression of DN-S6K1 ameliorates alcohol-induced hepatic apoptosis and liver damage. The data are presented as the mean ± SEM, n = 4–7. *P < 0.05, vs. pair-fed mice; #P < 0.05, vs. corresponding ethanol-fed mice.
Hepatocyte-specific deletion of SIRT1 disrupts DEPTOR signaling, promotes mTORC1 activation, and exacerbates the development of alcoholic fatty liver and liver injury in mice
Our previous studies indicate that hepatic overexpression of SIRT1, a master nutrient sensor, inhibits mTORC1 activity and attenuates hepatic steatosis in obese ob/ob mice (7). However, there is little knowledge of the crosstalk between SIRT1 and mTORC1 signaling in ALD. As shown in Fig. 7A, protein levels of SIRT1 were dramatically reduced in AML-12 mouse hepatocytes and in the livers of mice in response to ethanol exposure. These results, along with the inhibitory effect of ethanol on DEPTOR (Fig. 1E–F), suggest that ethanol causes the concomitant downregulation of SIRT1 and DEPTOR in hepatocytes and in vivo.
Fig. 7. Hepatocyte-specific deletion of SIRT1 disrupts DEPTOR signaling, stimulates mTORC1 activity, and exacerbates the development of alcoholic fatty liver and liver injury in mice.
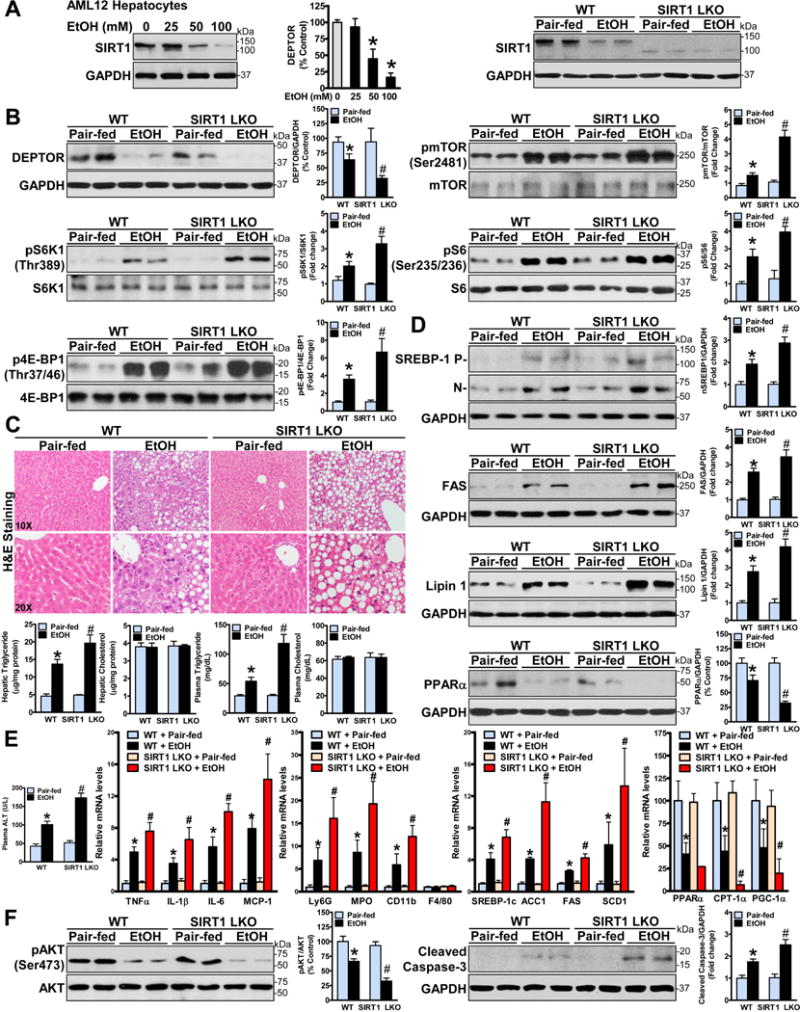
A. Protein levels of SIRT1 are measured in AML12 hepatocytes and in livers of wild-type (WT) mice and SIRT1 liver-specific knockout (SIRT1 LKO) mice under conditions of normal and ethanol exposure. B. Hepatocyte-specific deletion of SIRT1 downregulates DEPTOR and enhances mTORC1 activation in mice after chronic-binge ethanol feeding. C. Genetic ablation of SIRT1 in the liver increases the susceptibility to alcohol-induced fatty liver. D. Hepatic SIRT1 deficiency exacerbates alcohol-mediated abnormalities of lipid metabolism in mice. E–F. Hepatic SIRT1 loss aggravates alcohol-induced inflammation, apoptosis, and liver injury. The data are presented as the mean ± SEM, n = 4–8. *P < 0.05, vs. pair-fed WT mice; #P < 0.05, vs. ethanol-fed WT mice.
To further investigate a causal relationship between SIRT1 and DEPTOR in the setting of ALD, liver-specific SIRT1 knockout mice (SIRT1 LKO) were used to investigate the role of genetic SIRT1 ablation in hepatocytes on alcoholic activation of mTORC1. SIRT1 LKO mice displayed a slight decrease in DEPTOR and a mild increase in the basal mTORC1 activity when compared with pair-fed WT mice. Intriguingly, DEPTOR levels were reduced by chronic-binge ethanol feeding and further attenuated by hepatic SIRT1 deficiency. Consequently, phosphorylation of mTOR and its substrates, S6K1 and 4E-BP1, was enhanced by ethanol feeding, and such an induction was further potentiated by hepatic SIRT1 deficiency (Fig. 7B).
To determine the in vivo functional consequence of hepatocyte-specific SIRT1 loss on ALD, H&E staining revealed that SIRT1 LKO mice exhibited a higher degree of steatosis when compared to the WT mice that underwent the same ethanol treatment, although SIRT1 LKO mice were phenotypically normal under a chow diet, similarly to previous findings (20, 21). Consistent with more severe hepatic steatosis, ethanol-fed SIRT1 LKO mice exhibited much higher hepatic and plasma triglyceride levels than WT mice without affecting hepatic and plasma cholesterol levels (Fig. 7C). Thus, hepatic ablation of SIRT1 disrupts DEPTOR function and exacerbates the development of alcoholic fatty liver.
To further understand the mechanisms by which SIRT1 LKO mice are more susceptible to developing alcoholic fatty liver, expression or activity of the major regulators controlling lipid metabolic pathways were examined. As shown in Fig. 7D and fig. S7C, the accumulation of the active, nuclear form of SREBP-1 and expression of lipin 1 were significantly higher in ethanol-fed SIRT1 LKO mice than those in WT controls. Accordingly, chronic ethanol feeding significantly elevated expression of key lipogenic enzymes including ACC1, FAS and SCD1 in WT mice, and these increases were more pronounced in ethanol-fed SIRT1 LKO mice. Moreover, SIRT1 loss further reinforced alcohol-mediated reduction of PPARα and PGC-1α. More severe steatosis phenotype in SIRT1 LKO mice likely results from elevated fatty acid synthesis and diminished fatty acid oxidation.
As shown in Fig. 7E–F, SIRT1 LKO mice also displayed higher ALT levels than WT mice after chronic-binge feeding. Hepatic expression of TNFα and IL-1β, two major pro-inflammatory cytokines that are associated with metabolic disease (22), were significantly increased in SIRT1 LKO mice. Notably, ethanol-fed SIRT1 LKO mice had increased neutrophil markers, while hepatic expression of the macrophage marker F4/80 was comparable between WT and SIRT1 LKO mice in both normal and ethanol feeding conditions. More severe liver injury seen in ethanol-fed SIRT1 LKO mice was further characterized by reduced Akt activity and increased apoptosis. Collectively, the argument of alcoholic liver injury by SIRT1 loss is characterized by abnormal hepatocellular fat accumulation, reduced cell survival, and increased inflammatory and apoptotic responses.
Disrupting SIRT1 and DEPTOR signaling is a molecular signature in patients with ALD
To gain clinical evidence for the relationship among SIRT1, DEPTOR, and lipogenesis, liver samples from healthy subjects (n = 6) and patients with ALD (n =8) were used as described previously (10, 23). As shown in Fig. 8A–B, protein levels of SIRT1 and DEPTOR were reduced by ~50% in liver tissues from patients with ALD compared to normal livers. In opposite to DEPTOR inhibition, mTORC1 autophosphorylation at Ser2481 was increased ~2.6-fold in patients with ALD. An over 4-fold increase in S6 phosphorylation, as a marker of S6K1 activity, was also observed. Likewise, hepatic phosphorylation of 4E-BP1, the downstream target of mTORC1, was increased ~3-fold in patients with ALD. While emerging evidence shows much lower SIRT1 levels in patients with ALD (20), our animal and human studies provide additional evidence that aberrant inhibition of DEPTOR is associated with defective SIRT1 in the progression of ALD.
Fig. 8. Integrated downregulation of SIRT1 and DEPTOR contributes to the pathogenesis in patients with ALD.
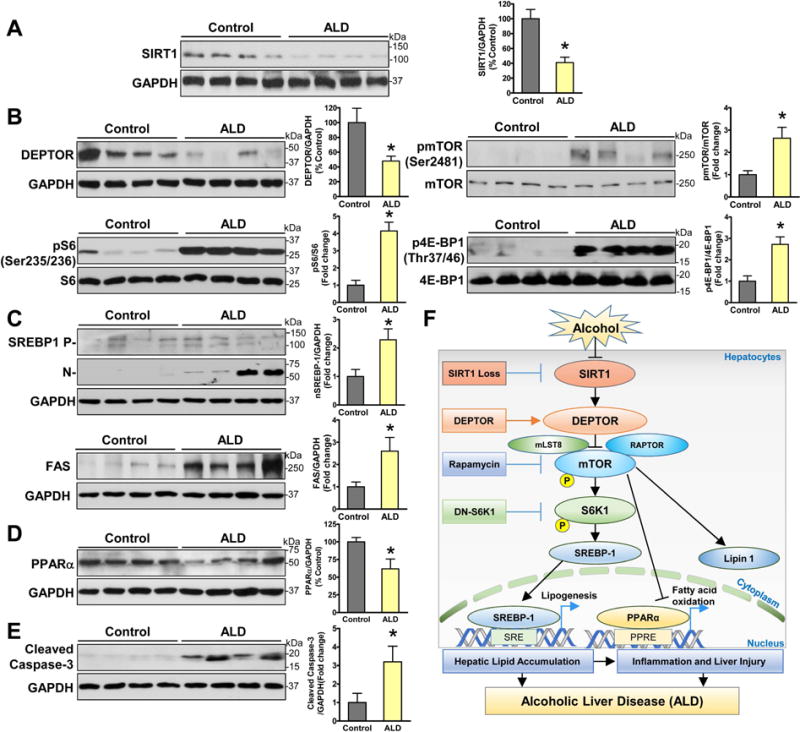
A–E. Representative immunoblots and densitometric quantification for SIRT1, mTORC1 signaling, lipid metabolic regulators, and apoptosis in normal liver tissues (n= 6) and liver tissues from patients with ALD (n =8). The data are presented as the mean ± SEM. *P < 0.05 vs. normal liver tissue. F. The proposed model for the deregulation of the SIRT1-DEPTOR-mTORC1 axis in the pathogenesis of ALD in mice and humans. Chronic alcohol consumption causes SIRT1 suppression in hepatocytes, which is coupled to the downregulation of DEPTOR and activation of mTORC1 and S6K1. Aberrant activation of mTORC1 by alcohol stimulates the proteolytic processing, nuclear translocation, and transcriptional activity of SREBP-1, promotes the cytoplasmic translocation of lipin 1, and inhibits the transcriptional activity of PPARα, which in turn increases fatty acid synthesis and downregulates fatty acid oxidation. Alcohol-induces hepatic lipogenic process acts through parallel S6K1-dependent and independent pathway. Alcohol feeding acts largely via SIRT1 inhibition and mTORC1 activation to induce excess fat accumulation and apoptosis in hepatocytes. Hepatic lipotoxicity and inflammation likely contribute to the development of acute-on-chronic alcoholic liver injury. Hepatic loss of SIRT1 impairs DEPTOR function, stimulates mTORC1 and lipogenesis, and promotes triglyceride overproduction, thereby leading to inflammation and liver injury in ALD. DEPTOR-dependent inhibition of mTORC1 provides a potential druggable target for treating ALD in humans.
To test whether mTORC1 hyperactivation is clinically relevant to the lipogenic and apoptotic processes in ALD, we took advantage of our observation that the master lipogenic regulator SREBP-1 directly binds the FAS promoter and upregulates its transcription, thereby promoting triglyceride synthesis in human hepatocytes (13). Hepatic nuclear SREBP-1 levels and FAS expression were increased in patients with ALD (Fig. 8C), aligned with elevated lipogenesis in patients with NAFLD (24). Excess fat accumulation was also accompanied by the downregulation of PPARα (Fig. 8D). Consistent with our finding that mTORC1 inhibition ameliorates liver injury in ethanol-fed mice (Fig. 6), mTORC1 activation was associated with the excessive hepatocyte apoptosis in human ALD, as evidenced by increased cleaved caspase-3 (Fig. 8E). Collectively, the deregulation of SIRT1-DEPTOR-mTORC1 signaling and the imbalance of lipogenesis and fatty acid oxidation may represent major pathological features of human ALD.
DISCUSSION
We have discovered that mTORC1 activity is enhanced in experimental animals and patients of ALD, characterized by an increase in mTOR-mediated phosphorylation and activity of S6K1. Sustained mTORC1 activation by alcohol is likely attributed to the defects of SIRT1 and DEPTOR. Mechanistically, mTORC1 is necessary for alcohol-induced lipogenesis, as reflected by the proteolytic activation, nuclear translocation and transcriptional activity of the critical lipogenic transcriptional factor SREBP-1 and by the cytoplasmic accumulation of the key lipogenic regulator lipin 1. Pharmacological intervention with rapamycin protects against alcohol-induced lipogenesis and ameliorates steatosis, inflammation and acute-on-chronic liver injury. Importantly, the concomitant reduction of SIRT1 and DEPTOR signaling is linked to elevated lipogenesis and decreased fatty acid oxidation in human liver specimens with ALD. As depicted in Fig. 8F, these translational studies, for the first time to our knowledge, uncover that the dysregulation of the SIRT1-DEPTOR-mTORC1 axis is a critical determinant of ALD pathology.
DEPTOR converges upstream of mTORC1 signaling and attenuates alcoholic hepatic steatosis and liver injury
mTORC1 has been appreciated to be implicated in NAFLD (7, 25), but whether and how the mTORC1 pathway is activated by alcohol feeding have yet to be investigated. Here using the murine model of approximating human ALD, several lines of evidence suggest a novel function of DEPTOR in hepatocytes. First, chronic-binge ethanol feeding leads to a robust increase in phosphorylation and activity of mTOR and its downstream kinase S6K1 in vivo. Alcohol-induced mTORC1 signaling mainly occurs in hepatocytes, consistent with the hepatocyte being a major cell type for ethanol metabolism. Second, DEPTOR is highly expressed in hepatocytes of normal mice and is markedly suppressed in mice after chronic binge ethanol feeding, indicating a pathological role of DEPTOR impairment in ALD. This notion is further supported by the fact that gain-of-function of DEPTOR confers insensitivity of mTORC1 signaling to ethanol exposure in hepatocytes and in vivo. The clinical relevance of mTORC1 induction to alcoholic liver disease is evidenced by decreased DEPTOR and increased S6K1 signaling in patients with ALD. Third, it is being increasingly recognized that excess lipid accumulation in the liver promotes inflammatory cell infiltration, thereby stimulating local inflammation and eventually liver injury (22). Since decreased fat accumulation by mTORC1 inhibition enables us to examine its impact on hepatic inflammation, we find that pharmacological inhibition of mTORC1 not only protects against hepatosteatosis but also ameliorates alcoholic liver injury, possibly through lowered hepatocellular lipids, promoted hepatocyte survival, and reduced hepatic neutrophil infiltration and inflammation. Therefore, dysregulation of DEPTOR-mTORC1 signaling may represent a previously unrecognized mechanism for the pathogenesis of ALD.
Inhibition of DEPTOR may represent a molecular mechanism responsible for alcohol-induced metabolic reprograming
One of the most important findings is that mTORC1 is responsible for alcohol-mediated metabolic switch from fatty acid oxidation toward fatty acid synthesis. The current study has utilized in vivo molecular and pharmacological approaches to manipulate DEPTOR-mTORC1 signaling: (1) Gain-of-function of DEPTOR in livers of chronic-binge ethanol-fed mice; (2) Pharmacological inhibition of mTORC1 with rapamycin; (3) Loss-of-function of hepatic S6K1; and (4) Liver-specific SIRT1 knockout mice. These combined studies suggest that alcohol-induced de novo lipogenesis is, at least partially, mediated through the disruption of DEPTOR and activation of mTORC1 and S6K1. Our in vivo and in vitro data is the first to indicate that inhibition of mTORC1 by DEPTOR counteracts the ability of ethanol to stimulate SREBP-1-dependent lipogenesis. The present study identifies a novel functional connection between DEPTOR and SREBP-1 in hepatocytes at multiple levels. First, our critical finding with high clinical significance is that hepatic DEPTOR levels inversely correlate with SREBP-1 activity in patients with ALD, suggesting that individuals with lower levels of DEPTOR may be predisposed to the deleterious consequence of excess alcohol consumption. Conversely, DEPTOR-dependent inhibition of mTORC1 is sufficient to eliminate the proteolytic processing, nuclear translocation, and transcriptional activity of SREBP-1 in chronic-binge ethanol-fed mice, the mouse model that nearly resembles the pathological features of human ALD (10). Second, recent studies show that mTORC1 directly phosphorylates lipin 1, the key enzyme for diacylglycerol synthesis, and promotes nuclear exclusion and cytoplasmic accumulation of lipin 1, and mTORC1 inhibition decreases SREBP-1 processing in a lipin 1-dependent fashion (26). As demonstrated in animals treated with rapamycin, mTORC1 is necessary for the cytoplasmic accumulation of lipin 1 and nuclear translocation of SREBP-1 in response to alcohol feeding. The regulation of SREBP-1 by DEPTOR is further supported by our findings that exogenous expression of DEPTOR reduces the cytoplasmic abundance of lipin 1 and renders mice insensitive to alcohol-induced SREBP-1 processing. Third, it has been shown that constitutive activation of mTORC1 by TSC1/2 loss promotes SREBP-1 processing in cultured cells, and this effect is inhibited by S6K1 knockdown (27). The data presented here show that the beneficial effects of hepatic overexpression of DN-S6K1 on fatty liver could be explained by the downregulation of SREBP-1 processing, suggesting that DEPTOR reduces SREBP-1 activity possibly by repressing S6K1. Interestingly, alcohol-mediated stimulation of lipin 1 acts through a S6K1-independent mechanism. Finally, because the effect of mTORC1 inhibition on SREBP-1 in ethanol-fed mice share some features seen in mice with liver-specific knockout of SCAP, the major lipid sensor that regulates SREBP-1 processing (14), we suspect that SCAP may be involved in the effect of DEPTOR on SREBP-1. Therefore, the molecular mechanisms by which DEPTOR downregulates SREBP-1 activity are more complex than originally envisioned. It is likely that DEPTOR cooperates with mTORC1 and S6K1 to control SREBP-1 processing by modulating distinct regulators.
Defective SIRT1 is functionally linked to DEPTOR inhibition in mice and humans with ALD
An intriguing observation in the present study is that defective SIRT1 is coupled to the dysregulation of DEPTOR-mTORC1-S6K1 signaling in hepatocytes exposed to ethanol and in patients with ALD. We have previously identified that SIRT1 stimulates AMP-activated protein kinase and lowers triglyceride accumulation in hepatocytes in insulin resistant states (28). We have generated mice with overexpression of hepatic SIRT1 and liver-specific deletion of SIRT1 (SIRT1 LKO mice) using the Cre/LoxP system. These mice show opposite phenotypes with regards to lipid homeostasis. Hepatic overexpression of SIRT1 attenuates fatty liver and glucose intolerance in obese, diabetic mice (7). In contrast, SIRT1 knockout in hepatocytes of mice reduces the hepatocyte-derived hormone FGF21 and accelerates the development of fatty liver caused by fasting (21). Because recent studies show that SIRT1 expression is reduced in patients with ALD (20), the current study further expands to explore whether SIRT1 loss promotes lipid accumulation associated with ALD. SIRT1 LKO mice develop more severe hepatic steatosis phenotype than control mice upon alcohol feeding, accompanied by diminished DEPTOR and activated mTORC1-S6K1 signaling. The convergence of these defects contribute to increased lipogenesis and impaired fatty acid oxidation, as evidenced by aberrant activation of SREBP-1 and lipin 1 as well as repression of PPARα and PGC-1α. In support of these findings, the overall triglyceride content of the liver, a well-established indicator of lipotoxicity, is profoundly increased in SIRT1 LKO mice; alterations in lipid metabolism could contribute to pathological features of ALD including hepatocyte death, neutrophil infiltration and inflammation. Although recent studies by the You Group indicate that ethanol-mediated impairment of hepatic SIRT1 signaling via lipin 1 contributes to fatty liver (26), our studies provide additional evidence that loss of SIRT1 may further decrease alcohol-mediated reduction of DEPTOR levels to accelerate the development of ALD. While this regulation appears to contribute to alcohol-induced lipid metabolic disturbance, the precise mechanism for the link between SIRT1 and DEPTOR is not understood. The regulation of DEPTOR stability involves its phosphorylation, ubiquitination, and degradation (6). Given the fact that mTOR kinase regulates the phosphorylation and ubiquitin-proteasome degradation of DEPTOR (6), future studies are needed to elucidate the mechanistic link between SIRT1 and DEPTOR by determining whether SIRT1 directly binds to DEPTOR itself or whether SIRT1 affects DEPTOR function through these modulations or through a cofactor that functions cooperatively with DEPTOR.
In conclusion, the integrated defects of SIRT1 and DEPTOR may represent a previously unrecognized mechanism responsible for the pathogenesis of ALD in mice and humans. The disruption of SIRT1 and DEPTOR plays a causal role in alcohol-induced mTORC1 activation, hepatocyte lipogenesis and apoptosis. Therapeutically, pharmacological and molecular inhibition of mTORC1 with rapamycin and DEPTOR overexpression ameliorates alcoholic steatosis, inflammation, and acute-on-chronic liver injury. Therefore, targeting SIRT1 and DEPTOR could be of therapeutic potential for treating ALD in humans.
Materials and Methods
Animal models
The Bin-binge mouse model of early stage ALD was recently developed as described (9, 10). Hepatocyte-specific SIRT1 knockout (Sirt1 LKO) mice were achieved by crossing albumin-Cre recombinase transgenic mice with floxed SIRT1Δex4 mice containing the deleted SIRT1 allele with floxed exon 4 (21).
All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Texas Health Science Center at San Antonio (UTHSCSA).
Human Liver Tissue Samples
Normal human liver samples and alcoholic liver disease tissues were obtained from donor livers or recipient livers during liver transplantation from the Liver Tissue Procurement and Distribution System (LTPDS) at University of Minnesota as described previously (10, 23).
Statistical analysis
Data are presented as means ± standard error (S.E.M). Using GraphPad Prism 5.0 software, results were analyzed by one-way ANOVA between multiple groups, when appropriate, and results were analyzed by a two-tailed Student’s t-test between two groups. P< 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We sincerely thank Dr. Jianping Ye (Louisiana State University) for the adenovirus encoding a dominant negative mutant of S6K1 (S6K1 K100R). We greatly appreciate Dr. Richard A. Cohen and Dr. Pranoti Mandrekar for insightful discussion. We would like to thank members of Zang laboratory for helpful discussion.
Fund: This work was supported by the National Institutes of Health Grants R21 AA021181, RO1 DK100603, and RO1 DK076942 (to M.Z.). M.Z. also received the Distinguished Chair Endowment Fund from the Ewing Halsell Foundation at the University of Texas Health Science Center at San Antonio.
Footnotes
Author Contributions: M.Z., B.G., and H.C. contributed to designed experiments. H.C., F.S., A.S., A.N., Y.L., H.W., M.J., and X.L performed the experiments and analyzed research data. B.J., J.H., D.L. N.L., and F.N. provided technical assistance and L.G., B.J. and J.H. provided reagents and material support. B.G., P.P., and J.F. provided human liver samples and insightful discussion. M.Z. and H.C. wrote the manuscript. B.G., B.J., L.G., F.L., A.K., N.M., and P.P. reviewed the manuscript. M.Z. obtained the funding.
The authors disclose no conflicts of interest.
References
- 1.Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology. 2011;141:1572–1585. doi: 10.1053/j.gastro.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Louvet A, Mathurin P. Alcoholic liver disease: mechanisms of injury and targeted treatment. Nat Rev Gastroenterol Hepatol. 2015;12:231–242. doi: 10.1038/nrgastro.2015.35. [DOI] [PubMed] [Google Scholar]
- 3.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thedieck K, Holzwarth B, Prentzell MT, Boehlke C, Klasener K, Ruf S, Sonntag AG, et al. Inhibition of mTORC1 by astrin and stress granules prevents apoptosis in cancer cells. Cell. 2013;154:859–874. doi: 10.1016/j.cell.2013.07.031. [DOI] [PubMed] [Google Scholar]
- 5.Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, Gray NS, et al. DEPTOR Is an mTOR Inhibitor Frequently Overexpressed in Multiple Myeloma Cells and Required for Their Survival. Cell. 2009;137:873–886. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao DM, Inuzuka H, Tan MKM, Fukushima H, Locasale JW, Liu PD, Wan LX, et al. mTOR Drives Its Own Activation via SCF(beta TrCP)-Dependent Degradation of the mTOR Inhibitor DEPTOR. Molecular Cell. 2011;44:290–303. doi: 10.1016/j.molcel.2011.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Y, Xu S, Giles A, Nakamura K, Lee JW, Hou X, Donmez G, et al. Hepatic overexpression of SIRT1 in mice attenuates endoplasmic reticulum stress and insulin resistance in the liver. FASEB J. 2011;25:1664–1679. doi: 10.1096/fj.10-173492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu M-J, Cai Y, Wang H, Altamirano J, Chang B, Bertola A, Odena G, et al. Fat-Specific Protein 27/CIDEC Promotes Development of Alcoholic Steatohepatitis in Mice and Humans. Gastroenterology. 2015;149:1030–1041.e1036. doi: 10.1053/j.gastro.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model) Nat Protocols. 2013;8:627–637. doi: 10.1038/nprot.2013.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bertola A, Park O, Gao B. Chronic plus binge ethanol feeding synergistically induces neutrophil infiltration and liver injury in mice: a critical role for E-selectin. Hepatology. 2013;58:1814–1823. doi: 10.1002/hep.26419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schalm SS, Blenis J. Identification of a conserved motif required for mTOR signaling. Curr Biol. 2002;12:632–639. doi: 10.1016/s0960-9822(02)00762-5. [DOI] [PubMed] [Google Scholar]
- 12.Holz MK, Ballif BA, Gygi SP, Blenis J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell. 2005;123:569–580. doi: 10.1016/j.cell.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 13.Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, Park O, et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011;13:376–388. doi: 10.1016/j.cmet.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moon Y-A, Liang G, Xie X, Frank-Kamenetsky M, Fitzgerald K, Koteliansky V, Brown Michael S, et al. The Scap/SREBP Pathway Is Essential for Developing Diabetic Fatty Liver and Carbohydrate-Induced Hypertriglyceridemia in Animals. Cell Metabolism. 2012;15:240–246. doi: 10.1016/j.cmet.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ji C, Chan C, Kaplowitz N. Predominant role of sterol response element binding proteins (SREBP) lipogenic pathways in hepatic steatosis in the murine intragastric ethanol feeding model. J Hepatol. 2006;45:717–724. doi: 10.1016/j.jhep.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 16.Yang Z, Tsuchiya H, Zhang Y, Lee S, Liu C, Huang Y, Vargas GM, et al. REV-ERBalpha Activates C/EBP Homologous Protein to Control Small Heterodimer Partner-Mediated Oscillation of Alcoholic Fatty Liver. Am J Pathol. 2016;186:2909–2920. doi: 10.1016/j.ajpath.2016.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takeuchi K, Reue K. Biochemistry, physiology, and genetics of GPAT, AGPAT, and lipin enzymes in triglyceride synthesis. Am J Physiol Endocrinol Metab. 2009;296:E1195–1209. doi: 10.1152/ajpendo.90958.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu M, Wang F, Li X, Rogers CQ, Liang X, Finck BN, Mitra MS, et al. Regulation of hepatic lipin-1 by ethanol: role of AMP-activated protein kinase/sterol regulatory element-binding protein 1 signaling in mice. Hepatology. 2012;55:437–446. doi: 10.1002/hep.24708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang J, Gao Z, Yin J, Quon MJ, Ye J. S6K directly phosphorylates IRS-1 on Ser-270 to promote insulin resistance in response to TNF-(alpha) signaling through IKK2. J Biol Chem. 2008;283:35375–35382. doi: 10.1074/jbc.M806480200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yin H, Hu M, Liang X, Ajmo JM, Li X, Bataller R, Odena G, et al. Deletion of SIRT1 from hepatocytes in mice disrupts lipin-1 signaling and aggravates alcoholic fatty liver. Gastroenterology. 2014;146:801–811. doi: 10.1053/j.gastro.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y, Wong K, Giles A, Jiang J, Lee JW, Adams AC, Kharitonenkov A, et al. Hepatic SIRT1 attenuates hepatic steatosis and controls energy balance in mice by inducing fibroblast growth factor 21. Gastroenterology. 2014;146:539–549 e537. doi: 10.1053/j.gastro.2013.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ertunc ME, Hotamisligil GS. Lipid signaling and lipotoxicity in metaflammation: indications for metabolic disease pathogenesis and treatment. J Lipid Res. 2016;57:2099–2114. doi: 10.1194/jlr.R066514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mukhopadhyay P, Horvath B, Rajesh M, Varga ZV, Gariani K, Ryu D, Cao Z, et al. PARP inhibition protects against alcoholic and non-alcoholic steatohepatitis. J Hepatol. 2017;66:589–600. doi: 10.1016/j.jhep.2016.10.023. [DOI] [PubMed] [Google Scholar]
- 24.Min HK, Kapoor A, Fuchs M, Mirshahi F, Zhou H, Maher J, Kellum J, et al. Increased hepatic synthesis and dysregulation of cholesterol metabolism is associated with the severity of nonalcoholic fatty liver disease. Cell Metab. 2012;15:665–674. doi: 10.1016/j.cmet.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gong Q, Hu Z, Zhang F, Cui A, Chen X, Jiang H, Gao J, et al. Fibroblast growth factor 21 improves hepatic insulin sensitivity by inhibiting mammalian target of rapamycin complex 1 in mice. Hepatology. 2016 doi: 10.1002/hep.28523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E, Guertin DA, et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146:408–420. doi: 10.1016/j.cell.2011.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39:171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hou X, Xu S, Maitland-Toolan KA, Sato K, Jiang B, Ido Y, Lan F, et al. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J Biol Chem. 2008;283:20015–20026. doi: 10.1074/jbc.M802187200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.