

Abstract
We describe a unique Patient Derived Xenograft (PDX) and cell culture model of Succinate dehydrogenase-deficient gastrointestinal stromal tumor (SDH-deficient GIST), a rare mesenchymal tumor that can occur in association with paragangliomas in hereditary and non-hereditary syndromes This model is potentially important for what it might reveal specifically pertinent to this rare tumor type and, more broadly, to other types of SDH-deficient tumors. The primary tumor and xenografts show a very high proliferative fraction, and distinctive morphology characterized by tiny cells with marked autophagic activity. It is likely that these characteristics resulted from the combination of the germline SDHB mutation and a somatic KRAS G12D mutation. The most broadly relevant findings to date concern oxygen and oxidative stress. In paragangliomas harboring SDHx mutations, both hypoxic signaling and oxidative stress are putative drivers of tumor growth. However there are no models for SDH-deficient paragangliomas. This related model is the first from a SDHB-mutated human tumor that can be experimentally manipulated to study mechanisms of oxygen effects and novel treatment strategies. Our data suggest that tumor growth and survival require a balance between protective effects of hypoxic signaling versus deleterious effects of oxidative stress. While reduced oxygen concentration promotes tumor cell survival, a further survival benefit is achieved with antioxidants. This suggests potential use of drugs that increase oxidative stress as novel therapies. In addition, autophagy, which has not been reported as a major finding in any type of SDH-deficient tumor, is a potential target of agents that might trigger autophagic cell death.
Keywords: PGL syndrome, SDH-deficient GIST, SDHB, PDX model, xenograft, cell culture, hypoxia, metabolome
Introduction
SDH-deficient gastrointestinal stromal tumors (GISTs) are rare but sometimes lethal tumors that can occur in patients with paraganglioma (PGL) syndromes. These tumors do not harbor the KIT or PDGFRA mutations typical of conventional GISTs and are therefore not amenable to conventional GIST treatments with receptor tyrosine kinase inhibitors. They also metastasize to lymph nodes more frequently than conventional GISTs, and their clinical prognosis is not predictable by conventional risk stratification parameters (Boikos, et al. 2016; Mullassery and Weldon 2016; Ricci 2016). Similarly to PGLs, there is currently no cure for SDH-deficient GIST other than complete surgical excision, and there is a paucity of experimental models.
This paper describes a unique Patient Derived Xenograft (PDX) and cell culture model for basic and pre-clinical studies of SDH-deficient GIST. The model, which we have named “the Ian GIST model”, is derived from an aggressive gastric GIST that arose in a young man with a germline SDHB mutation and family history of multiple paragangliomas. Despite a remarkable multi-institutional translational research effort aimed at developing an effective treatment (“the Ian GIST Project”, https://www.forian.org/), the entire clinical course before the patient died was approximately 2 years. In addition to loss of SDHB protein expression and SDH activity, the tumor was found to harbor a somatic KRAS G12D mutation, possibly contributing to its aggressive behavior.
This GIST model is important for two reasons. First, it is a unique human-derived model for a rare type of tumor that occurs in patients with hereditary SDHB mutations. Although the basic metabolic defect in all SDH-deficient tumors is similar, different types of tumor such as GISTs and paragangliomas are likely to be affected by that defect in different ways because of intrinsic differences in their cells of origin. It is well established that different types of cancer as well as individually distinct cancers have varied genetic and epigenetic characteristics that dictate different approaches to treatment (Burgess, et al. 2017), and that even therapeutic outliers can lead to new markers of drug sensitivity (Burgess et al. 2017). Second, because SDH-deficient GIST does have similarities to other SDH-deficient tumors, information gained from studying this model might also lead to increased understanding of other tumor types and to improved treatments for them.
Materials and Methods
Origin of the model
The tumor of origin was a gastric GIST that arose in a 17 year-old patient who had a germline SDHB mutation (423+1G>A) and family history of paragangliomas in two male relatives, but was himself previously healthy. Approximately 3 months after resection of the primary tumor, he presented with a 15 cm recurrent tumor in the residual portion of stomach, with extensive local invasion and abdominal dissemination.
Our model was developed from a PDX established by a commercial service (Champions Oncology, Hackensack NJ) in Nude mice from a tumor deposit in the patient’s abdomen. With authorization from the deceased patient’s family we received cryopreserved passage 3 xenograft tissue and expanded through passage 6 in Nude and/or NSG mice. This project is approved by the Institutional Review Board at Tufts Medical Center.
Histology and Immunohistochemistry
Histologic sections of the primary tumor, the recurrence and xenografts were examined by a pathologist (AST) to assess phenotype drift in consecutive passages. Immunohistochemical stains were performed following previously reported protocols with antibodies shown in Supplementary Table1. All control tissues showed appropriate immunoreactivity.
Electron Microscopy
Fragments of xenograft tissue were fixed in commercially prepared fixative (Electron Microscopy Sciences, Hatfield PA) containing 2.5% glutaraldehyde and 2% formaldehyde in 0.1 M sodium cacodylate buffer. They were then postfixed in 2% OsO4, dehydrated in a graded ethanol series followed by propylene oxide, and embedded in Epon. Ultrathin (90 nm) sections on copper grids were stained with 0.2% lead citrate and 1% uranyl acetate and examined with a Philips 208S electron microscope at 80 kV.
Transcriptional profiling
RNA sequencing was performed by the Tufts genomics core with an Illumina HiSeq 2000. Paired-end RNA sequencing data was preprocessed using Trimmomatic (Bolger, et al. 2014) to cut or filter poor quality reads. The preprocessed data was then aligned using TopHat2 pipeline (Kim, et al. 2013). The featureCounts software (Liao, et al. 2014) was used to map reads to genes, and edgeR (Robinson, et al. 2010) was used to calculate RPKM (Reads Per Kilobase of transcript per Million mapped reads) values for genes.
Metabolomic analysis
In vivo metabolomic analyses were performed to test whether the major metabolites produced in the GIST xenografts are consistent with metabolic profiles reported in SDH-deficient paragangliomas and in an SDH-deficient line of fibroblast-like cells derived from mouse adrenal glands (Letouze, et al. 2013; Lussey-Lepoutre, et al. 2015). 1H NMR was used for unbiased metabolite profiling and 13C-NMR was used to trace the metabolic fate of glucose and determine the contribution of glycolysis and/or TCA cycle/anaplerotic pathways to the metabolite profile (Bruntz, et al. 2017). Four replicate subcutaneous tumor nodules (bilateral flank implants from two mice, each 1 cm in greatest dimension) were analyzed. A xenograft of a KIT-mutated, SDH-intact, GIST (GIST-TI) (Taguchi, et al. 2002) in NSG mouse was used as an SDH-intact control. Thirty minutes prior to sacrifice the mice were injected intraperitoneally with a 3 mmol/kg dose of 100% uniformly 13C6-labeled glucose (the natural abundance of 13C is 1%) (Eloqayli, et al. 2004). Tumor tissue was dissected cold and frozen in liquid nitrogen. Metabolites were extracted with chloroform/methanol. To each frozen tissue sample, 0.4 mL of ice-cold methanol and 0.085 mL water were added and the tissue was minced to a fine grain on ice. After sonication to homogeneity, 0.4 mL of cold chloroform was added, which was then processed as described by Wu et al (Wu, et al. 2008).
The polar methanol/water layer was dried using a speed-vac and reconstituted in 50 mM phosphate D2O buffer pH 7.0 with 0.5 mM of NMR standard (DSS, 4,4-dimethyl-4-silapentane-1-sulfonic acid). 1H NMR spectra of each sample were collected at 25 °C on a Bruker Avance 600 spectrometer using 256 scans and a NOE1D pulse sequence. 13C NMR spectra were also measured using 10000 to 20000 scans. The 1H data were processed and analyzed using CHENOMX NMR Suite 8.0 to identify and quantify compounds present and measure their concentrations. Identity of compounds in 13C data were based on published reference spectra (Wishart, et al. 2009) (and on two-dimensional 1H-13C correlation spectra using HSQC (heteronuclear single-quantum coherence).
Immunoblots
Protocols for routine protein extraction and immunoblotting were as previously described (Powers, et al. 2000). Proteins were resolved on gradient 4–15% polyacrylamide gels to permit probing for a wide range of molecular weights. Blots were sequentially probed, stripped and re-probed with the same antibodies used for immunohistochemistry, at dilutions optimized for immunoblots.
For Redox immunoblots (Cox, et al. 2010) frozen GIST tissue samples were first placed in petri dishes on ice and soaked with 100 µL of 200 mM methyl methanethiolsulfonate (MMTS, purchased from Sigma) in PBS (1×). The samples were then sliced with razor blades and placed in a 1.5 mL tube along with an additional 900 µL of 200 mM MMTS in PBS. The tissue samples were incubated on ice for 30 minutes. After incubation, the tissue sample was centrifuged at 250 g for 5 minutes. The MMTS solution was removed from the tube and the cells were washed with 1 mL of PBS. The samples were then centrifuged and washed with PBS a second time. After centrifugation again, 100 µL RIPA buffer with HALT protease inhibitor cocktail (1×, Thermo Fisher) was added to the samples. The samples were then homogenized with a manual tissue homogenizer. The lysed cells were then re-pelleted at 12,000 g for 10 minutes. The supernatant of the solution was then collected and the protein content in the solution was assessed via the BCA assay. 20 µg of protein was loaded into a tris-tricine acrylamide gel and subjected to SDS-PAGE under non-reducing conditions (i.e. with no β-mercaptoethanol in the sample buffer). After the separated lysates were transferred to a PVDF membrane (Bio-Rad) (1 hour at 100V), the blot was blocked and incubated with either goat primary antibody for Prx2 (R&D Systems, Catalog # AF3489) (at a dilution of 1:800) or rabbit primary antibody for Prx3 (Abcam, Catalog #ab129206) (at a dilution of 1:10000) at 4 °C overnight and then with donkey anti-goat IR 688 (Licor) (at a dilution of 1:10000) or donkey anti-rabbit IR 800 (Licor) (at a dilution of 1:10000) for 1 hour at approximately 22 °C. β-tubulin was used as a loading control for the blots The tagged proteins were visualized using an Odyssey CLx Infrared Imaging System. In order to determine the fraction of oxidized protein for each tumor sample, the gel analyzer tool in ImageJ (https://imagej.nih.gov) was used. This tool calculates position-dependent intensities for the entirety of each band. The area under the intensity peak associated with the oxidized band was divided by the sum of the areas under the intensity peaks of both the oxidized and reduced bands..
Cell Cultures
Culture medium consisted of RPMI1640 with 15% fetal bovine serum, glutamine and penicillin/streptomycin, enriched with MEM non-essential amino acids (glycine, alanine, asparagine, aspartic acid, glutamic acid, proline, serine; each 0.1 mM, Life Technologies), Fatty Acid Supplement (Sigma), and uridine (50 ug/L, Sigma). Concentrations of aspartic acid and sodium pyruvate were further supplemented individually to final concentrations of 3mM and 5mM respectively (Supplementary Table 2).
Xenograft tissue at room temperature was minced into small fragments in complete culture medium, then gently triturated in a 1 mL polystyrene pipet followed by a 9” Pasteur pipet until approximately 80% of the tissue volume was dissociated. The resulting cell suspension was separated from the residual fragments, centrifuged at 500 g and resuspended in fresh medium.
In order to test the influence of ambient O2 concentration on cell growth and survival, we compared cultures routinely maintained in ~ 20% O2 (“normoxia”, 95% air/5% CO2) versus 10% or 5% O2 in Billups-Rothenberg modular incubator chambers. For the lowered O2 concentrations, the gas mixtures contained either 80% or 85% N2 plus 5% CO2. Cells were plated in black 96-well assay plates (Costar) at a density of 5×104 cells/well, and survival was quantitated at 3 days using a CyQuant NF Cell Proliferation Assay kit (Molecular Probes). Use of this DNA-based assay was chosen to avoid potential confounding effects of abnormal metabolism (Quent, et al. 2010). All cultures were maintained at 37°C in a water-saturated atmosphere.
Growth factors and inhibitors
Stem cell factor (SCF, also known as KIT ligand) was obtained from Cell Signaling Technologies. Recombinant human Insulin-like growth factor 1 (IGF1) was from Abcam. ERK inhibitors U0126 and PD98059 were from New England Biolabs and Promega, respectively.
Results
Histopathology
The primary tumor was a 4.5 cm ulcerated gastric mass. A few patchy areas of the tumor showed classic GIST patterns (Miettinen, et al. 2010) consisting of mildly discohesive, polygonal epithelioid cells with slightly eosinophilic cytoplasm and pleomorphic nuclei or hypercellular spindle cells with relatively bland nuclei. In contrast, at least 90% of cells in the tumor showed an unusual gradation from epithelioid to round shape and progressive decrease in cell size, with many cells becoming smaller than neutrophils (~12 um) and exhibiting almost no cytoplasm (Fig. 1A, Supplementary Fig. 1). Some areas showed prominent myxoid material between cells or in large pools, suggesting a possible collision tumor between GIST and adenocarcinoma (Supplementary Fig. 1B). This was ruled out by immunohistochemical stains for GIST markers KIT and DOG1 (Rammohan, et al. 2013) showing transition between cell types ((Fig. 1B, Supplementary Fig. 4), and by the absence of staining for cytokeratin markers indicative of carcinomas. Mitoses were very numerous in classic epithelioid areas (up to 5 mitoses per single high power field) (Supplementary Fig.2) and sparse or absent in the smallest cells. Staining for proliferation marker Ki67 showed a comparable pattern, ranging from approximately 80% to 20 % in different parts of the tumor (Fig. 1D). An abdominal tumor deposit sampled approximately one year after resection of the primary consisted almost entirely small cells with patches of myxoid material (Supplementary Fig. 3).
Figure 1.

Major characteristics of the primary gastric tumor (A–D) compared to passage 3 PDX (E–H). Bars = 50 um.
Tumor morphology (H&E stain, A and E).. Primary tumor (A) showing. relatively well differentiated spindle and epithelioid tumor cells with conspicuous eosinophilic cytoplasm (top left) transitioning to loosely cohesive, poorly differentiated tiny round cells, some smaller than 10 um, with almost undetectable cytoplasm (bottom right). PDX (E) consisting entirely of small to intermediate size cells, with prominent pools of extracellular myxoid material.
KIT (CD117) (B and F). Primary tumor (B) showing strong KIT expression in the largest cells with the most cytoplasm and reduced or absent expression in smaller cells. In the PDX (F) KIT expression shows a comparable association with the amount of cytoplasm.
SDHB (C and G), In both the primary tumor (C) and PDX (G) SDHB expression is lost in all tumor cells and retained in endothelial cells (arrows), which serve as positive controls. The PDX also contains scattered SDHB-positive mouse-derived inflammatory cells between tumor cells.
Ki67 (D and H). Proliferative fraction assessed by Ki67 expression is approximately twice as high in the xenograft as in the primary tumor.
The major morphological differences between xenografts and the patient’s tumors were a more compact architecture, increasingly monomorphous population of intermediate-to- small cells, loss or greatly decreased amounts of myxoid material, and uniform expression of Ki67 in 80–90% of the tumor cells. Immunohistochemical staining for KIT was maintained through at least in vivo passage 6, tending to be most intense in the largest cells and often undetectable in the smallest (Fig. 1). In contrast, staining for DOG1, which was sparse in the primary tumor, was almost lost by in vivo passage 4.
The most distinctive ultrastructural features were degenerating, malformed mitochondria and extensive autophagic vacuoles containing mitochondria and other cytoplasmic components. Many cells contained numerous cytoplasmic vacuoles and loss of most identifiable cytoplasmic structures (Fig. 2).
Figure. 2.

Ultrastructural features of the PDX cells. A. Electron micrograph of xenograft cell showing markedly abnormal mitochondria with swelling and loss of cristae. B. Xenograft cell with an autophagic vacuole containing a mitochondrion and amorphous material suggesting advanced degeneration of a mitochondrion or other cytoplasmic content. C. Xenograft cell containing numerous cytoplasmic vacuoles.
In both the primary tumor and xenografts, immunohistochemical staining for SDHB was lost in all tumor cells and retained in endothelial cells (Fig. 1 C and G) consistent with loss of the wild-type SDHB allele (Dahia, et al. 2005; van Nederveen, et al. 2009), while staining for SDHA (Korpershoek, et al. 2011; Oudijk, et al. 2013; Papathomas, et al. 2015) was retained.
Metabolite profile
Of 55 readily identifiable metabolites in the Ian GIST xenografts,, lactate comprised approximately 24% and succinate 11% of the total (Table 1). Fumarate was almost undetectable, with a succinate/fumarate ratio of 679 ± 151(n=4 tumors), consistent with loss of SDH activity. Four other metabolites that each comprised >5% of the total were taurine, glycine, myo-inositol and alanine. All others accounted for < 3%. The eight most abundant metabolites comprised >75% of the total (Table 1). In contrast, our analysis of a control xenograft of a KIT-mutated, SDHB-intact GIST model GIST-TI showed 0.54% succinate, consistent with reported values of less than 1% succinate in other non-SDH deficient tumors (Euceda, et al. 2017).
Table 1.
Most Abundant Metabolites in Xenograft Tissue
| Metabolite | % of Total Metabolites* (mean, n=4) |
Range |
|---|---|---|
| Lactate | 23.85 | 21.78–26.95 |
| Taurine | 11.49 | 10.82–12.54 |
| Succinate | 11.03 | 10.76–11.40 |
| Glycine | 8.63 | 8.18–9.30 |
| Myo-inositol | 8.04 | 6.65–8.63 |
| Alanine | 5.38 | 5.04–5.72 |
| O-Phosphoethanolamine | 4.10 | 3.78–4.37 |
| Glutamate | 3.3 | 2.15–4.32 |
| Sum | 75.82 |
The table lists the eight most abundant metabolites expressed as percentage of total metabolites, showing the average of percentages in 4 individual tumors.
The 13C-glucose label ended up as a small amount of glucose, and abundant lactate, alanine and glutamate, The labeled glutamate contained nearly equal amounts of [13C2-4,5]Glu and [13C2-2,3]Glu isoptomers (Supplementary Fig 5A), indicating nearly equal activity of pyruvate dehydrogenase (PDH) and the anaplerotic pathway catalyzed by pyruvate carboxylase (PC), respectively (Bruntz et al. 2017; Lussey-Lepoutre et al. 2015). There was also robust incorporation of glucose into succinate. In contrast, the ratio of [13C2-4,5]Glu and [13C2-2,3]Glu isoptomers in the GIST-T1 xenograft was approximately 6:1 (Supplementary Fig 5B). These findings are consistent with increased activity of PC previously reported in SDH-deficient cells (Lussey-Lepoutre et al. 2015).
Molecular indicators of elevated H2O2 in the mitochondria and cytosol
In light of recent studies that have linked complex II of the mitochondrial electron transport chain with increased levels of hydrogen peroxide (Kluckova, et al. 2015; Quinlan, et al. 2012), we hypothesized that SDH deficiency could result in increased steady-state fluxes of H2O2 in the mitochondria and perhaps in the cytosol. In order to measure relative H2O2 levels within the different GIST cells, we employed a previously described methodology (Cox et al. 2010) to measure the oxidation products of molecular targets of H2O2. Unlike free radical species, such as hydroxyl radical, which react readily with multiple targets inside cells, H2O2 reacts much more selectively within cells (Winterbourn 2008). In particular, studies have shown that virtually all of the H2O2 inside cells reacts with peroxiredoxins (Lim, et al. 2015; Winterbourn 2008), a family of cysteine-based antioxidant proteins that form disulfide-linked dimers upon reaction with H2O2, on the basis of the high second-order rate coefficient for their reaction with H2O2 and the intracellular abundance of the these proteins (on the order of 100 µM in many types of cells) (Peskin, et al. 2007; Selvaggio, et al. 2018).. Since these enzymes act as the primary sink for intracellular H2O2, several studies have used the oxidation status of peroxiredoxins (i.e. oxidized dimers versus reduced monomers) as proxies for the intracellular H2O2 level (Kumar, et al. 2009; Low, et al. 2007; Poynton and Hampton 2014; Sobotta, et al. 2013). This approach combines both the H2O2 specificity of peroxiredoxins as well as the H2O2 sensitivity of cells’ natural peroxide sensors.
In order to assay for oxidized peroxiredoxin dimers in both the mitochondria and cytosol in the two different types of GIST, lysates from GIST cells both with and without functional SDH were subjected to non-reducing SDS-PAGE (in which the proteins were denatured but the disulfide bonds were not reduced), transferred to a PVDF membrane, and stained for both peroxiredoxin 3 (Prx3; located in the mitochondria) and peroxiredoxin 2 (Prx2; located in the cytosol). The fraction of oxidized Prx3 and Prx2 dimers compared to reduced Prx3 and Prx2 monomers for each tumor indicated high levels of H2O2 in the mitochondria and cytosol of tumor cells in the xenograft tissue (Fig.3). Densitometry showed the ratio of oxidized PRX3 to total PRX3 protein to be consistently higher than the corresponding ratio for PRX2, (0.983 +/− 0.002 for PRX3 compared to 0.792 +/− 0.032 for PRX2, p=0.0095 two-tailed Student’s t-test), consistent with a greater degree of mitochondrial compromise. Comparison of the fraction of oxidized Prx3 and Prx2 in each of the Ian GIST xenografts to a SDH-intact primary GIST specimen also suggested that SDH-deficient tumors have slightly higher H2O2 levels in the mitochondria and cytosol, respectively, than the conventional GIST, but additional primary tumor specimens were not available to permit adequate statistical analysis.
Figure. 3.

Redox blot sequentially incubated with antibodies against human peroxiredoxin-2 (Prx2) and peroxiredoxin-3 (Prx3).
The bands at approximately 22 kDa represent the reduced monomer Prx2 or Prx-3 proteins, while bands at about 44 kDa represent the respective oxidized dimers. The amount of oxidized dimer relative to the total reduced monomer plus oxidized dimer in each lane is an indication of the relative amount of H2O2-induced stress (Cox et al. 2010). Prx2 acts as a sensor for H2O2 stress in the cytosol of the cells, while Prx3 acts in the mitochondria. Beta-tubulin is a loading control. Lanes 1–3 are from separate xenografts of SDH-deficient GIST. Lane 4, which shows the highest amounts of both oxidized dimers and reduced monomers,, is from a primary conventional GIST with intact SDHB.
Cell culture studies
Cell culture experiments were undertaken with two complementary objectives. The first was to study short term primary cultures in order to identify critical factors regulating tumor cell survival, growth and differentiation independently of potentially confounding changes that might occur in cell lines. The second was to use data gained from those studies to guide efforts to establish cell lines, which would be needed for more extensive mechanistic investigations.
A striking and almost immediate finding was that the cells are averse to oxygen. Primary cultures were initially plated in a routinely employed atmosphere of 95% air / 5% CO2. This resulted in rapid cell death, often obvious within hours and leading to loss of almost all tumor cells within two weeks. In light of the high levels of oxidative stress already present in vivo, as implied by the Prx blots, we therefore tested the effects of decreased O2 concentrations, both alone and in combination with antioxidants. Over a 3-day period in culture, there was a marked, concentration-dependent increase in tumor cell survival in the presence of 10% or 5% O2, and survival was further enhanced by addition of either N-acetylcysteine or a proprietary antioxidant mix. However, despite this beneficial effect, there was a sharp decline in Ki67 expression compared to the tumor cells in vivo (Fig. 4). By two weeks in culture almost all tumor cells had died or been overgrown by mouse cells, which grew vigorously under hypoxic conditions. The rapid switch between populations of human and mouse cells was confirmed by immunoblots that showed almost undetectable SDHB during the initial 3-day culture period, consistent with the overwhelming predominance of SDH-deficient human tumor cells, and its re-emergence by 2 weeks. In addition, immunoreactivity for Ki67 using a human-selective antibody disappeared over the same time course.
Figure. 4.

Effects of O2 concentration on GIST cell survival in culture.
Cells were maintained at the indicated O2 concentrations for 3 days in complete medium in the presence or absence of N-acetylcysteine (NAC, 5uM) or a proprietary antioxidant (SAO, Sigma antioxidant; 1uL/mL). A. Enhanced tumor cell survival in reduced O2 (representative experiment, each value mean ± SEM of quadruplicate wells, p<.03, for 5 or 10% vs 20%, difference between 5 and 10% is not significant (Mann-Whitney U test). B. Representative immunoblot showing effects of reduced O2 in cultures maintained concurrently with those in Panel A. A single immunoblot from a 4–15% gradient polyacrylamide gel was consecutively probed, stripped and re-probed for each of the indicated proteins. Increased survival and proliferative activity of GIST cells in both 10% and 5% versus 20% O2 are reflected by greater amounts of KIT and Ki67, and by decreased amounts of cleaved poly ADP-ribose polymerase (PARP), a marker of apoptosis (Kaufmann, et al. 1993). C and D. Phase contrast photomicrographs of GIST cells in 10% and 20% O2, respectively. E. Immunocytochemical staining for Ki67 of GIST cells in a 10% O2 well represented in Panel A (arrows indicate stained nuclei). Note only a few scattered cells labeled, compared to nearly all cells in xenograft tissue.
In order to get clues to understand the basis for loss of tumor cell proliferation in culture, RNA-Sequencing analysis was performed on the xenograft and the cultured cells to identify changes in mRNA expression. We found 226 genes upregulated by >10 fold and 120 genes down regulated by the same amount in cells cultured in 10% O2 as compared to the xenograft. The complete list is given in Supplementary Table 3. Analysis of pathways with Ingenuity Pathway Analysis software indicated that genes and pathways most affected in upregulated genes are matrix metalloproteases, cytokines, and wnt5a and wnt5b. The cytokine induction could be an indicator of a senescence associated secretory phenotype (Lasry and Ben-Neriah 2015). The downregulated genes were most involved in connective tissue and growth regulation pathways. Figure 5 shows the changes in expression of genes of known interest to GIST. While there was little change in the GIST marker DOG1, there was a > 30 fold decrease in IGF1 expression when cells were put into culture. However, addition of IGF-1 to the culture of these cells was not sufficient to rescue the growth defect (Data not shown). There was an almost 10-fold increase in HIF2 expression consistent with an increase in hypoxic stress in culture. There was a 4-fold increase in KIT ligand expression, but a 2 fold decrease in KIT receptor expression. Consistent with this, there was nearly a 3 fold increase in CBL expression which can downregulate the KIT receptor through the ubiquitin pathway (Zeng, et al. 2005). Thus, below we examined the expression of KIT protein in the cultures.
Figure 5.

Fold change in expression of selected genes in cell culture compared to the xenograft.
RNA sequencing analysis was preformed from the xenograft and from cells cultures in 10% oxygen for 3 days and analysis was performed as described in Methods.
In addition to its SDHB mutation and consequent loss of SDH activity, this GIST was unusual in also harboring a somatic KRAS mutation. This suggested that the unusual morphology, extremely high proliferative fraction, partial loss of the characteristic marker KIT, and extreme fragility of the tumor cells in vitro might result from aberrant RAS/MAPK signaling. To test the effects of inhibiting this signaling cascade we used two structurally different inhibitors of MAP kinase signaling, U0126 and PD98059 (Dokladda, et al. 2005). Low concentrations of U0126, sufficient to cause approximately a 50% decrease in ERK phosphorylation, caused a 2–2.5-fold increase in KIT expression (n=4 independent experiments). This effect was not seen with PD98059. Paradoxically, this modest inhibition of ERK also maintained short-term proliferative activity, as indicated by Ki67 expression, at a higher level than in control cultures (Fig.6). However, the percentage of labeled cells in culture remained markedly lower than in xenograft tissue. Long term maintenance with either inhibitor did not result in sustained proliferation or establishment of cell lines.
Figure 6.

Representative immunoblot showing effects of ERK inhibitor U0126 on the GIST cells in culture.
Cells were maintained in 10% O2 for 3 days in complete medium with the indicated concentrations of U0126. A single immunoblot from a 4–15% gradient polyacrylamide gel was consecutively probed, stripped and re-probed for each of the indicated proteins. (Bands are arranged to highlight effects on KIT and Ki67, not by molecular weight. Anomalous migration accounts for artifactually low Ki67 in lane 3).
Discussion
The Ian GIST model is a unique human-derived model for SDH-deficient GIST, a rare mesenchymal tumor that can occur in patients with SDHB mutations.
GISTs comprise <1% of all gastrointestinal tumors(Oudijk et al. 2013). SDH-deficient GISTs, accountfor ~3% of all GISTs and ~5% of gastric GISTs in an unselected adult population (Gill, et al. 2011). Approximately 50% arise in the context of Carney Triad, a non-hereditary syndrome caused by post-zygotic promoter hypermethylation of SDHC and consisting of SDH-deficient PGL, SDH-deficient GIST, and pulmonary chondroma. Approximately 30% are associated with germline mutations of SDHA (Oudijk et al. 2013). In a 2016 NIH study, 34 of 63 SDH-mutant GISTs harbored mutations in SDHA, 16 in SDHB, 12 in SDHC, and 1 in SDHD (Boikos et al. 2016). All developed in the stomach, consistent with earlier reports.
SDH-deficient GISTs may occur in patients with or without a history of PGLs, and the lifetime risk of developing SDH-deficient GIST in patients with SDHB mutations is uncertain. It is also unclear whether SDHB confers a worse prognosis than other mutated SDHx genes, as is the case with paraganglioma (Gimenez-Roqueplo, et al. 2008). However, most SDH-deficient GISTs follow an indolent course even after they disseminate (Boikos et al. 2016). The tumor represented by the Ian GIST model was unusual for its morphology, high proliferative fraction, decreased expression of the GIST markers KIT and DOG1, and rapid clinical course. It is likely that these characteristics resulted from the combination of germline SDHB and somatic KRAS mutations.
The most distinctive morphological feature of this tumor model is the presence of tiny cells with marked autophagic activity. Comparable findings have been reported in a model of nutrient starvation caused by growth factor deprivation (Lum, et al. 2005). In that model, decreased cell size was caused by autophagic consumption of cytoplasmic constituents, with progressive loss of ribosomes and the ER/Golgi network. The latter would also result in reduced expression of markers associated with differentiated function. We hypothesized that in the present model conditions comparable to exogenous nutrient starvation might be caused by superimposing metabolic demands caused by KRAS mutation (Eser, et al. 2014) on loss of SDH activity, which alone results in decreased function of TCA cycle anaplerotic pathways (Lussey-Lepoutre et al. 2015) and loss of ATP production by oxidative phosphorylation.
As a preliminary test of this hypothesis we used two MEK inhibitors, U0126 and PD98059, which have previously been employed to study effects of kinome remodeling on cellular function and differentiation (Duncan, et al. 2012). Consistent with this hypothesis, concentrations of U0126 causing approximately a 50% decrease in ERK phosphorylation in cell culture produced a 2–2.5-fold increase in KIT expression and also maintained short-term proliferative activity as indicated by Ki67 expression. PD9059 did not affect KIT but had a comparable effect on Ki67 (data not shown). While neither inhibitor is entirely specific for MEK and both might have targets outside the RAS/MAPK cascade (Dang and Lowik 2004; Dokladda et al. 2005), these results suggest that use of the Ian GIST model to study kinome remodeling (Duncan et al. 2012) might be a fruitful area of investigation. They are of particular interest in view of recent studies showing that treatment of cancer patients with kinase inhibitors can select for survival of subsets of tumor cells known as drug tolerant persister (DTP) populations (Terai, et al. 2018)
Considering its unusual features of, it might be asked how relevant the Ian GIST model is likely to be either to other SDH-deficient GISTS or to paragangliomas and other SDH-deficient tumors. Our most relevant findings to date concern oxygen and antioxidants. Paragangliomas are known to occur with increased frequency at high altitudes, and hypoxic signaling is a putative phenotypic modifier and driver of tumor growth in patients harboring SDHx mutations (Her, et al. 2015). Based largely on murine knockdown models, hyperoxia has been proposed as a potential treatment (Her et al. 2015). In addition, SDHx-mutated paragangliomas are reported to show high levels of markers associated with oxidative stress (Fliedner, et al. 2012), which has been proposed as both a driver and inhibitor of tumor progression in other tumor types (Pani, et al. 2004). Because of the latter role, superoxide dismutase has been proposed as a therapeutic target (Pani et al. 2004). The Ian GIST model is the first from an actual human tumor of a type known to be associated with SDHB mutations that can be experimentally manipulated and utilized to study mechanisms of oxygen effects and novel treatment strategies. The evidence thus far suggests that tumor growth and survival require a balance between protective effects of hypoxic signaling versus deleterious effects of oxidative stress. While reduced oxygen concentration clearly promotes tumor cell survival, a further survival benefit for tumor cells is achieved with N-acetylcysteine or other antioxidants. This suggests use of drugs that increase rather than decrease oxidative stress as novel therapies (Huang, et al. 2016; Raj, et al. 2011). Another major finding is the role of autophagy in maintaining tumor cell survival in xenografts. Autophagy is largely uninvestigated in any type of SDH-deficient tumor. However, autophagy pathways are potential targets of agents that may cooperate with kinase inhibitors and other drugs as triggers of apoptotic cell death (Terai et al. 2018). It is of interest that taurine, a sulfur containing amino acid present at very high levels in our xenografts, is both an antioxidant (Schuller-Levis and Park 2003) and a negative regulator of autophagy (Zhang, et al. 2017)
The deleterious effects of oxygen observed in cell culture are not entirely unexpected because it has been recognized for some time that routine culture conditions in 95% air/5% CO2 are not physiological either for normal tissues or tumors (Carreau, et al. 2011). While the pO2 in inspired air (~20% O2) is 150 mm Hg, the value drops dramatically in normal solid tissues. For example, in liver and kidney pO2 has been measured at ~ 41 and 72 mm Hg (equivalent to ~ 5% and 10% O2) respectively. In tumors of different histologic types, pO2 ranged from 0–54 mm Hg (Carreau et al. 2011). The most important consequence of the present study might therefore be the way the dramatic response to O2 may inform our approaches to other SDH-deficient tumors, both in terms of model development and pre-clinical drug testing. For more than 40 years attempts to establish human paraganglioma and pheochromocytoma cell lines in cultures maintained at 95% air/5% CO2 have been unsuccessful. Most pre-clinical drug testing of substitute cell lines has also been conducted under these conditions, which can exaggerate the efficacy of many chemotherapeutic agents (Carreau et al. 2011). Revisiting these studies may yield interesting results.
In summary, the Ian GIST model is potentially important both for what it might reveal specifically pertinent to this tumor type and more broadly to other types of SDH-deficient tumors, especially regarding metabolic and signaling characteristics shared across the spectrum of SDH-deficient tumor types. The presence of the KRAS mutation could make the model challenging as a tool for development of therapies
Supplementary Material
Supplementary Figure. 1 Histologic sections of the primary gastric tumor. A. Relatively well differentiated area of tumor showing loosely cohesive epithelioid cells and a minor component of spindle cells (at center). B. Poorly differentiated area of tumor showing numerous rounded tumor cells in a myxoid matrix. There is a gradient of cell size and appearance, ranging from epithelioid cells with conspicuous cytoplasm as in panel A to tiny round cells, some smaller than 10 um, with almost undetectable cytoplasm. Bar = 50 um
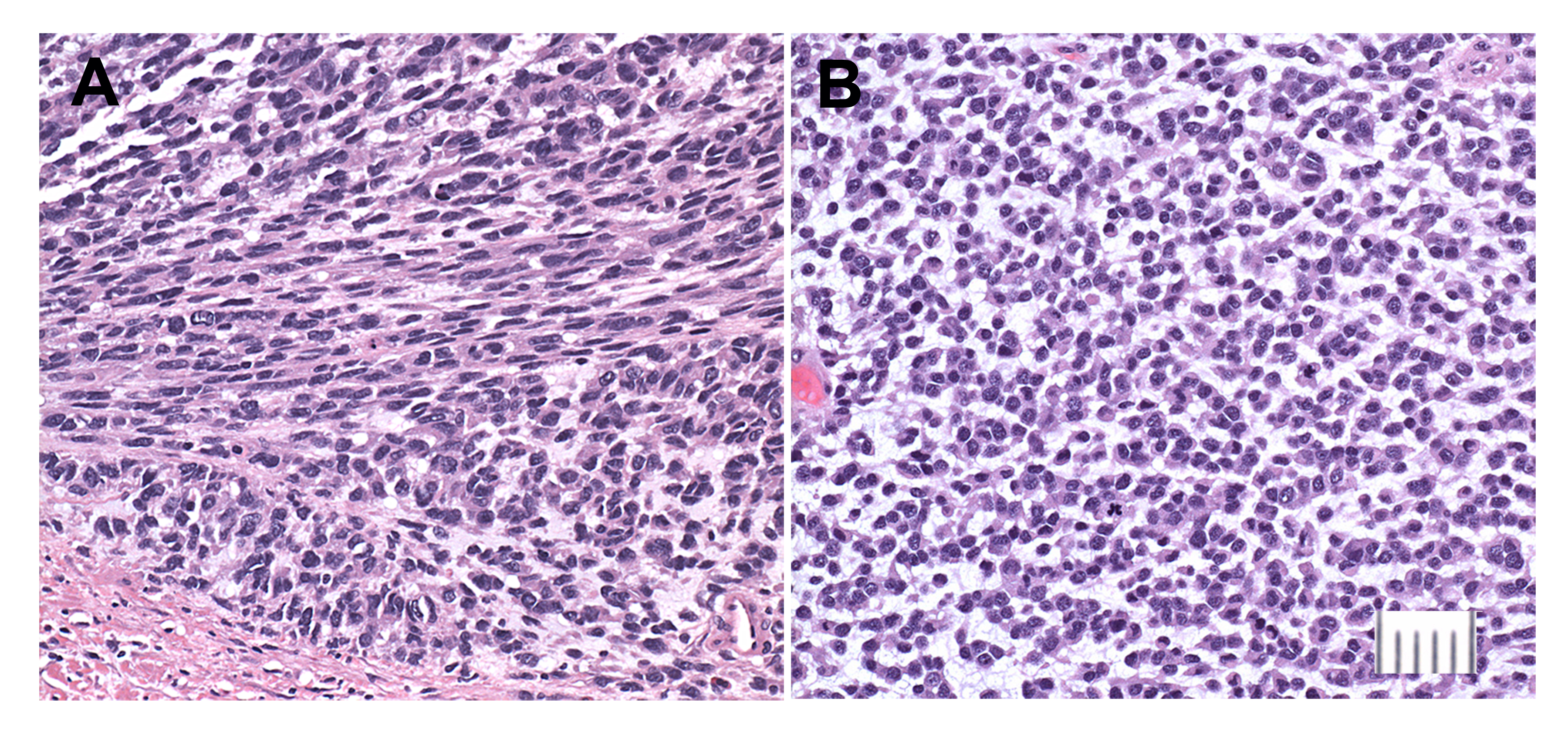{kind=link}
Supplementary Figure. 2 High magnification of epithelioid tumor cells showing marked pleomorphism and numerous mitoses (arrows). Bar = 50 um
Supplementary Figure. 3 Abdominal tumor deposit, consisting almost entirely of small rounded cells (A) and prominent pools of myxoid material (B). Bar = 50 um.
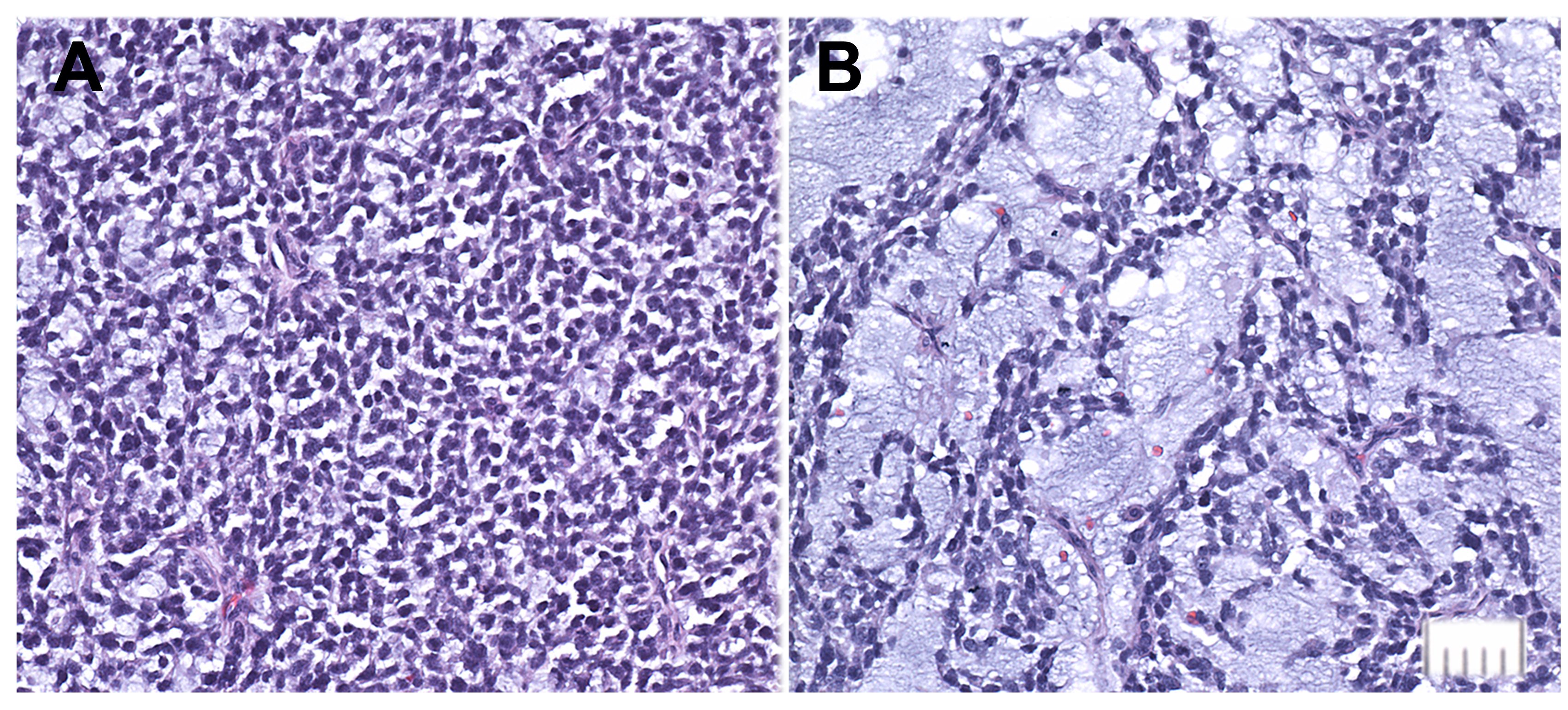{kind=link}
Supplementary Figure. 4 DOG1 expression in the primary tumor. Staining is less overall than for KIT (cf. Fig. 1), and is absent in most of the small cells. Bar = 50 um
Supplementary Figure 5 representative 13C NMR spectra of Ian GIST (Panel A) compared to control SDH-intact GIST-T1 (Panel B) xenografts.. Select resonance multiplets are labeled with the corresponding molecule and carbon using IUPAC numbering.
Acknowledgments
The authors thank Dr. Andrey Gritzman for providing pathology slides of the patient’s primary tumor and recurrence, and Dr. Andrea Califano for sharing transcriptome data not shown in this paper.
Funding
This research was supported principally by grants to AST from the SDHB Pheo Para Coalition, the Pheo Para Alliance, and the Paradifference Foundation. The work utilized NMR instrumentation that was purchased with funding from a National Institutes of Health SIG grant (S10OD020073). KTS acknowledges support from the NSF GRFP
Footnotes
Declaration of Interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
- Boikos SA, Pappo AS, Killian JK, LaQuaglia MP, Weldon CB, George S, Trent JC, von Mehren M, Wright JA, Schiffman JD, et al. Molecular Subtypes of KIT/PDGFRA Wild-Type Gastrointestinal Stromal Tumors: A Report From the National Institutes of Health Gastrointestinal Stromal Tumor Clinic. JAMA Oncol. 2016;2:922–928. doi: 10.1001/jamaoncol.2016.0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruntz RC, Lane AN, Higashi RM, Fan TW. Exploring cancer metabolism using stable isotope-resolved metabolomics (SIRM) J Biol Chem. 2017;292:11601–11609. doi: 10.1074/jbc.R117.776054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess MR, Hwang E, Mroue R, Bielski CM, Wandler AM, Huang BJ, Firestone AJ, Young A, Lacap JA, Crocker L, et al. KRAS Allelic Imbalance Enhances Fitness and Modulates MAP Kinase Dependence in Cancer. Cell. 2017;168:817–829. e815. doi: 10.1016/j.cell.2017.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreau A, El Hafny-Rahbi B, Matejuk A, Grillon C, Kieda C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J Cell Mol Med. 2011;15:1239–1253. doi: 10.1111/j.1582-4934.2011.01258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox AG, Winterbourn CC, Hampton MB. Measuring the redox state of cellular peroxiredoxins by immunoblotting. Methods Enzymol. 2010;474:51–66. doi: 10.1016/S0076-6879(10)74004-0. [DOI] [PubMed] [Google Scholar]
- Dahia PL, Ross KN, Wright ME, Hayashida CY, Santagata S, Barontini M, Kung AL, Sanso G, Powers JF, Tischler AS, et al. A HIF1alpha Regulatory Loop Links Hypoxia and Mitochondrial Signals in Pheochromocytomas. PLoS Genet. 2005;1:e8. doi: 10.1371/journal.pgen.0010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang ZC, Lowik CW. Differential effects of PD98059 and U0126 on osteogenesis and adipogenesis. J Cell Biochem. 2004;92:525–533. doi: 10.1002/jcb.20087. [DOI] [PubMed] [Google Scholar]
- Dokladda K, Green KA, Pan DA, Hardie DG. PD98059 and U0126 activate AMP-activated protein kinase by increasing the cellular AMP:ATP ratio and not via inhibition of the MAP kinase pathway. FEBS Lett. 2005;579:236–240. doi: 10.1016/j.febslet.2004.11.084. [DOI] [PubMed] [Google Scholar]
- Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS, Johnson NL, Granger DA, Jordan NV, Darr DB, et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell. 2012;149:307–321. doi: 10.1016/j.cell.2012.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eloqayli H, Dahl CB, Gotestam KG, Unsgard G, Sonnewald U. Changes of glial-neuronal interaction and metabolism after a subconvulsive dose of pentylenetetrazole. Neurochem Int. 2004;45:739–745. doi: 10.1016/j.neuint.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Eser S, Schnieke A, Schneider G, Saur D. Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer. 2014;111:817–822. doi: 10.1038/bjc.2014.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Euceda LR, Hill DK, Stokke E, Hatem R, El Botty R, Bieche I, Marangoni E, Bathen TF, Moestue SA. Metabolic Response to Everolimus in Patient-Derived Triple-Negative Breast Cancer Xenografts. J Proteome Res. 2017;16:1868–1879. doi: 10.1021/acs.jproteome.6b00918. [DOI] [PubMed] [Google Scholar]
- Fliedner SM, Kaludercic N, Jiang XS, Hansikova H, Hajkova Z, Sladkova J, Limpuangthip A, Backlund PS, Wesley R, Martiniova L, et al. Warburg effect's manifestation in aggressive pheochromocytomas and paragangliomas: insights from a mouse cell model applied to human tumor tissue. PLoS One. 2012;7:e40949. doi: 10.1371/journal.pone.0040949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill AJ, Chou A, Vilain R, Clarkson A, Lui M, Jin R, Tobias V, Samra J, Goldstein D, Smith C, et al. Immunohistochemistry for SDHB divides gastrointestinal stromal tumors (GISTs) into 2 distinct types. Am J Surg Pathol. 2011;34:636–644. doi: 10.1097/PAS.0b013e3181d6150d. [DOI] [PubMed] [Google Scholar]
- Gimenez-Roqueplo AP, Burnichon N, Amar L, Favier J, Jeunemaitre X, Plouin PF. Recent advances in the genetics of phaeochromocytoma and functional paraganglioma. Clin Exp Pharmacol Physiol. 2008;35:376–379. doi: 10.1111/j.1440-1681.2008.04881.x. [DOI] [PubMed] [Google Scholar]
- Her YF, Nelson-Holte M, Maher LJ., 3rd Oxygen concentration controls epigenetic effects in models of familial paraganglioma. PLoS One. 2015;10:e0127471. doi: 10.1371/journal.pone.0127471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang BK, Langford TF, Sikes HD. Using Sensors and Generators of H2O2 to Elucidate the Toxicity Mechanism of Piperlongumine and Phenethyl Isothiocyanate. Antioxid Redox Signal. 2016;24:924–938. doi: 10.1089/ars.2015.6482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann SH, Desnoyers S, Ottaviano Y, Davidson NE, Poirier GG. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: an early marker of chemotherapy-induced apoptosis. Cancer Res. 1993;53:3976–3985. [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluckova K, Sticha M, Cerny J, Mracek T, Dong L, Drahota Z, Gottlieb E, Neuzil J, Rohlena J. Ubiquinone-binding site mutagenesis reveals the role of mitochondrial complex II in cell death initiation. Cell Death Dis. 2015;6:e1749. doi: 10.1038/cddis.2015.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpershoek E, Favier J, Gaal J, Burnichon N, van Gessel B, Oudijk L, Badoual C, Gadessaud N, Venisse A, Bayley JP, et al. SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. J Clin Endocrinol Metab. 2011;96:E1472–1476. doi: 10.1210/jc.2011-1043. [DOI] [PubMed] [Google Scholar]
- Kumar V, Kitaeff N, Hampton MB, Cannell MB, Winterbourn CC. Reversible oxidation of mitochondrial peroxiredoxin 3 in mouse heart subjected to ischemia and reperfusion. FEBS Lett. 2009;583:997–1000. doi: 10.1016/j.febslet.2009.02.018. [DOI] [PubMed] [Google Scholar]
- Lasry A, Ben-Neriah Y. Senescence-associated inflammatory responses: aging and cancer perspectives. Trends Immunol. 2015;36:217–228. doi: 10.1016/j.it.2015.02.009. [DOI] [PubMed] [Google Scholar]
- Letouze E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, Janin M, Menara M, Nguyen AT, Benit P, et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23:739–752. doi: 10.1016/j.ccr.2013.04.018. [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Lim JB, Huang BK, Deen WM, Sikes HD. Analysis of the lifetime and spatial localization of hydrogen peroxide generated in the cytosol using a reduced kinetic model. Free Radic Biol Med. 2015;89:47–53. doi: 10.1016/j.freeradbiomed.2015.07.009. [DOI] [PubMed] [Google Scholar]
- Low FM, Hampton MB, Peskin AV, Winterbourn CC. Peroxiredoxin 2 functions as a noncatalytic scavenger of low-level hydrogen peroxide in the erythrocyte. Blood. 2007;109:2611–2617. doi: 10.1182/blood-2006-09-048728. [DOI] [PubMed] [Google Scholar]
- Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–248. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- Lussey-Lepoutre C, Hollinshead KE, Ludwig C, Menara M, Morin A, Castro-Vega LJ, Parker SJ, Janin M, Martinelli C, Ottolenghi C, et al. Loss of succinate dehydrogenase activity results in dependency on pyruvate carboxylation for cellular anabolism. Nat Commun. 2015;6:8784. doi: 10.1038/ncomms9784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miettinen M, Fletcher CDM, Kindblom L-G, Tsui WMS. Mesenchymal tumours of the tomach. In: Bosman FT, Carneiro F, Hruban RH, Theise ND, editors. WHO Classification of Tumours of the Digestive System. 4. Lyon: IARC; 2010. pp. 74–76. [Google Scholar]
- Mullassery D, Weldon CB. Pediatric/"Wildtype" gastrointestinal stromal tumors. Semin Pediatr Surg. 2016;25:305–310. doi: 10.1053/j.sempedsurg.2016.09.004. [DOI] [PubMed] [Google Scholar]
- Oudijk L, Gaal J, Korpershoek E, van Nederveen FH, Kelly L, Schiavon G, Verweij J, Mathijssen RH, den Bakker MA, Oldenburg RA, et al. SDHA mutations in adult and pediatric wild-type gastrointestinal stromal tumors. Mod Pathol. 2013;26:456–463. doi: 10.1038/modpathol.2012.186. [DOI] [PubMed] [Google Scholar]
- Pani G, Colavitti R, Bedogni B, Fusco S, Ferraro D, Borrello S, Galeotti T. Mitochondrial superoxide dismutase: a promising target for new anticancer therapies. Curr Med Chem. 2004;11:1299–1308. doi: 10.2174/0929867043365297. [DOI] [PubMed] [Google Scholar]
- Papathomas TG, Oudijk L, Persu A, Gill AJ, van Nederveen F, Tischler AS, Tissier F, Volante M, Matias-Guiu X, Smid M, et al. SDHB/SDHA immunohistochemistry in pheochromocytomas and paragangliomas: a multicenter interobserver variation analysis using virtual microscopy: a Multinational Study of the European Network for the Study of Adrenal Tumors (ENS@T) Mod Pathol. 2015;28:807–821. doi: 10.1038/modpathol.2015.41. [DOI] [PubMed] [Google Scholar]
- Peskin AV, Low FM, Paton LN, Maghzal GJ, Hampton MB, Winterbourn CC. The high reactivity of peroxiredoxin 2 with H(2)O(2) is not reflected in its reaction with other oxidants and thiol reagents. J Biol Chem. 2007;282:11885–11892. doi: 10.1074/jbc.M700339200. [DOI] [PubMed] [Google Scholar]
- Powers JF, Evinger MJ, Tsokas P, Bedri S, Alroy J, Shahsavari M, Tischler AS. Pheochromocytoma cell lines from heterozygous neurofibromatosis knockout mice. Cell Tissue Res. 2000;302:309–320. doi: 10.1007/s004410000290. [DOI] [PubMed] [Google Scholar]
- Poynton RA, Hampton MB. Peroxiredoxins as biomarkers of oxidative stress. Biochim Biophys Acta. 2014;1840:906–912. doi: 10.1016/j.bbagen.2013.08.001. [DOI] [PubMed] [Google Scholar]
- Quent VM, Loessner D, Friis T, Reichert JC, Hutmacher DW. Discrepancies between metabolic activity and DNA content as tool to assess cell proliferation in cancer research. J Cell Mol Med. 2010;14:1003–1013. doi: 10.1111/j.1582-4934.2010.01013.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan CL, Orr AL, Perevoshchikova IV, Treberg JR, Ackrell BA, Brand MD. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J Biol Chem. 2012;287:27255–27264. doi: 10.1074/jbc.M112.374629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj L, Ide T, Gurkar AU, Foley M, Schenone M, Li X, Tolliday NJ, Golub TR, Carr SA, Shamji AF, et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature. 2011;475:231–234. doi: 10.1038/nature10167. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Rammohan A, Sathyanesan J, Rajendran K, Pitchaimuthu A, Perumal SK, Srinivasan U, Ramasamy R, Palaniappan R, Govindan M. A gist of gastrointestinal stromal tumors: A review. World J Gastrointest Oncol. 2013;5:102–112. doi: 10.4251/wjgo.v5.i6.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci R. Syndromic gastrointestinal stromal tumors. Hered Cancer Clin Pract. 2016;14:15. doi: 10.1186/s13053-016-0055-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuller-Levis GB, Park E. Taurine: new implications for an old amino acid. FEMS Microbiology Letters. 2003;226:195–202. doi: 10.1016/S0378-1097(03)00611-6. [DOI] [PubMed] [Google Scholar]
- Selvaggio G, Coelho P, Salvador A. Mapping the phenotypic repertoire of the cytoplasmic 2-Cys peroxiredoxin - Thioredoxin system. 1. Understanding commonalities and differences among cell types. Redox Biol. 2018;15:297–315. doi: 10.1016/j.redox.2017.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobotta MC, Barata AG, Schmidt U, Mueller S, Millonig G, Dick TP. Exposing cells to H2O2: a quantitative comparison between continuous low-dose and one-time high-dose treatments. Free Radic Biol Med. 2013;60:325–335. doi: 10.1016/j.freeradbiomed.2013.02.017. [DOI] [PubMed] [Google Scholar]
- Taguchi T, Sonobe H, Toyonaga S, Yamasaki I, Shuin T, Takano A, Araki K, Akimaru K, Yuri K. Conventional and molecular cytogenetic characterization of a new human cell line, GIST-T1, established from gastrointestinal stromal tumor. Lab Invest. 2002;82:663–665. doi: 10.1038/labinvest.3780461. [DOI] [PubMed] [Google Scholar]
- Terai H, Kitajima S, Potter DS, Matsui Y, Quiceno LG, Chen T, Kim TJ, Rusan M, Thai TC, Piccioni F, et al. ER Stress Signaling Promotes the Survival of Cancer "Persister Cells" Tolerant to EGFR Tyrosine Kinase Inhibitors. Cancer Res. 2018;78:1044–1057. doi: 10.1158/0008-5472.CAN-17-1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Nederveen FH, Gaal J, Favier J, Korpershoek E, Oldenburg RA, de Bruyn EM, Sleddens HF, Derkx P, Riviere J, Dannenberg H, et al. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis. Lancet Oncol. 2009;10:764–771. doi: 10.1016/S1470-2045(09)70164-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol. 2008;4:278–286. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- Wishart DS, Knox C, Guo AC, Eisner R, Young N, Gautam B, Hau DD, Psychogios N, Dong E, Bouatra S, et al. HMDB: a knowledgebase for the human metabolome. Nucleic Acids Res. 2009;37:D603–610. doi: 10.1093/nar/gkn810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Southam AD, Hines A, Viant MR. High-throughput tissue extraction protocol for NMR- and MS-based metabolomics. Anal Biochem. 2008;372:204–212. doi: 10.1016/j.ab.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Zeng S, Xu Z, Lipkowitz S, Longley JB. Regulation of stem cell factor receptor signaling by Cbl family proteins (Cbl-b/c-Cbl) Blood. 2005;105:226–232. doi: 10.1182/blood-2004-05-1768. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Ren S, Liu Y, Gao K, Liu Z, Zhang Z. Inhibition of Starvation-Triggered Endoplasmic Reticulum Stress, Autophagy, and Apoptosis in ARPE-19 Cells by Taurine through Modulating the Expression of Calpain-1 and Calpain-2. Int J Mol Sci. 2017;18 doi: 10.3390/ijms18102146. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure. 1 Histologic sections of the primary gastric tumor. A. Relatively well differentiated area of tumor showing loosely cohesive epithelioid cells and a minor component of spindle cells (at center). B. Poorly differentiated area of tumor showing numerous rounded tumor cells in a myxoid matrix. There is a gradient of cell size and appearance, ranging from epithelioid cells with conspicuous cytoplasm as in panel A to tiny round cells, some smaller than 10 um, with almost undetectable cytoplasm. Bar = 50 um
Supplementary Figure. 2 High magnification of epithelioid tumor cells showing marked pleomorphism and numerous mitoses (arrows). Bar = 50 um
Supplementary Figure. 3 Abdominal tumor deposit, consisting almost entirely of small rounded cells (A) and prominent pools of myxoid material (B). Bar = 50 um.
Supplementary Figure. 4 DOG1 expression in the primary tumor. Staining is less overall than for KIT (cf. Fig. 1), and is absent in most of the small cells. Bar = 50 um
Supplementary Figure 5 representative 13C NMR spectra of Ian GIST (Panel A) compared to control SDH-intact GIST-T1 (Panel B) xenografts.. Select resonance multiplets are labeled with the corresponding molecule and carbon using IUPAC numbering.