

SUMMARY
FANCA is a component of the Fanconi anemia (FA) core complex that activates DNA interstrand crosslink repair by monoubiquitination of FANCD2. Here, we report that purified FANCA protein catalyzes bidirectional single-strand annealing (SA) and strand exchange (SE) at a level comparable to RAD52, while a disease-causing FANCA mutant, F1263Δ, is defective in both activities. FANCG, which directly interacts with FANCA, dramatically stimulates its SA and SE activities. Alternatively, FANCB, which does not directly interact with FANCA, does not stimulate this activity. Importantly, five other patient-derived FANCA mutants also exhibit deficient SA and SE, suggesting that the biochemical activities of FANCA are relevant to the etiology of FA. A cell-based DNA double strand break (DSB) repair assay demonstrates that FANCA plays a direct role in the single-strand annealing sub-pathway (SSA) of DSB repair by catalyzing SA, and this role is independent of the canonical FA pathway and RAD52.
Keywords: FANCA, Fanconi anemia, single-strand annealing, strand exchange, DNA double strand break repair
In Brief
Benitez et al. report that FANCA biochemically catalyzes single-strand annealing and strand exchange. They find that the single-strand annealing activity of FANCA is relevant to the etiology of Fanconi anemia and responsible for its involvement in double strand break repair, which is independent of the canonical FA pathway and RAD52.
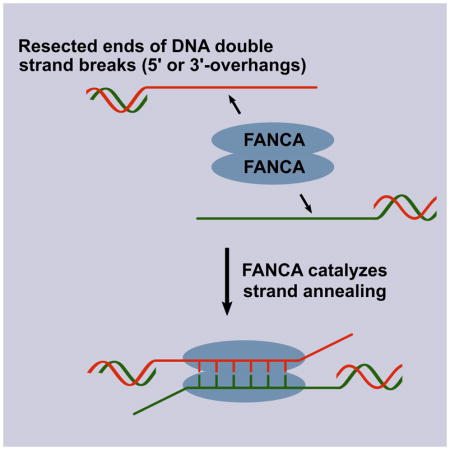
INTRODUCTION
Fanconi Anemia (FA) is a recessive hereditary disorder caused by biallelic mutations in at least one of 22 genes, and is clinically characterized by bone marrow failure, congenital abnormalities, and predisposition to cancer (Feeney et al., 2017; Inano et al., 2017; Knies et al., 2017; Moldovan and D’Andrea, 2009; Palovcak et al., 2017). Cells from FA patients show hypersensitivity to DNA crosslinking agents and accumulate chromosomal aberrations, supporting their important roles in the FA/BRCA pathway of DNA interstrand crosslink (ICL) repair (Kee and D’Andrea, 2012; Kottemann and Smogorzewska, 2013; Wang, 2007).
Eight FANC proteins assemble into the FA core complex including FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, FANCM, along with FA associated proteins (FAAPs) (Ceccaldi et al., 2016; Huang et al., 2014). Members of the FA core complex are required for successful activation of the FA/BRCA pathway through FANCD2 and FANCI monoubiquitination. FANC proteins, translesion synthesis polymerases, FAAPs, and structure-specific DNA endonucleases cooperate in the incision and bypass of ICLs and produce intermediate double-stranded DNA breaks (DSBs) (Benitez et al., 2014; Ceccaldi et al., 2016). These DSBs are repaired by homologous recombination (HR) proteins including BRCA2/FANCD1, BRCA1/FANCS, BRIP1/FANCJ, PALB2/FANCN, RAD51/FANCR, RAD51C/FANCO, and XRCC2/FANCU, which act downstream of the initial FANCD2 and FANCI monoubiquitination event (Crossan et al., 2011; Kee and D’Andrea, 2010; Stoepker et al., 2011; Wang, 2007). In the absence of these HR repair factors, DSBs can be repaired via non-homologous end joining (NHEJ), resulting in radial chromosomal structures that are commonly observed in FA patients (Moldovan and D’Andrea, 2009; Palovcak et al., 2017).
FANCA, a component of the FA core complex, is the most commonly affected complementation group in FA patients, accounting for ~64% of all mutations (Wang and Smogorzewska, 2015). FANCA plays a role in ICL repair and DSB repair through the canonical FA pathway via FANCD2 monoubiquitination (Howard et al., 2015; Nakanishi et al., 2005; Yang et al., 2005). Despite its importance in ICL repair, FANCA was found to be dispensable for FANCD2 monoubiquitination in an in vitro system (van Twest et al., 2017). Additionally, some FA disease-causing FANCA mutants have normal FANCD2 monoubiquitination and wild-type level of mitomycin C sensitivity (Adachi et al., 2002), suggesting that FANCA may have additional roles outside of the canonical FA pathway, but relevant to the etiology of FA disease. Here, we describe two biochemical activities of FANCA: single-strand annealing (SA) and strand exchange (SE). The SA and SE activities of FANCA directly explain its role in homology-directed DSB repair. Our cell-based DSB repair assay further demonstrates that FANCA contributes to the repair of DNA double strand breaks independently of the FA pathway and RAD52 in human cells.
RESULTS
FANCA Bidirectionally Anneals Single-Stranded DNA
The FA core complex has been previously reported to be involved in the SSA sub-pathway of DSB repair through the canonical FA pathway (Howard et al., 2015; Nakanishi et al., 2005; Yang et al., 2005). Nevertheless, double knockdowns of FANCA and FANCB, FANCA and FANCL, and FANCA and FANCD2 in a SSA reporter assay suggested that FANCA also plays an additional role in SSA repair independently of the FA core complex and FANCD2 (Figure S1A). Furthermore, the minimum effective processing segment of sequence homology required for the SSA repair was calculated to be ~29–33 bp (Sugawara et al., 2000), consistent with the minimum ssDNA binding requirement for FANCA (~30 bp) (Yuan et al., 2012). From this, we reasoned that FANCA might directly catalyze SA. Our biochemical analysis indicates that FANCA-WT (10–40 nM calculated as monomers) catalyzes annealing of fully complementary oligonucleotides in a concentration-dependent manner (Figure 1A, top panel lanes 3–6, and bottom panel). In contrast, the most prevalent Fanconi anemia disease-causing mutant, FANCA-F1263Δ (Fanconi Anemia Mutation Database), does not have any detectable activity (Figure 1A, top panel lanes 7–10, and bottom panel), indicating that the observed SA activity in WT protein is essential for FANCA function. This difference in SA efficiency between WT and F1263Δ is not due to impaired DNA binding since both proteins bind ssDNA and dsDNA with similar affinity (Figure S1B). Time-course analysis reveals that initial product formation occurs within 5 min of FANCA addition and no protein-independent formation of annealed DNA was observed throughout the entire experiment (Figure S2A). These results show that FANCA biochemically catalyzes SA.
Figure 1.

FANCA bidirectionally anneals ssDNA. Titration of the purified FANCA-WT and FANCA-F1263Δ mutant (10, 20, 30, and 40 nM calculated as monomers) on (A) fully complementary ssDNA oligos 74 nt in length (Bottom panel, quantification of the SA activity; Experiments were independently repeated 3 times; Error bars, standard deviation); (B) 3′-overhangs with a central homologous sequence stretch flanked by a dsDNA region (left panel) and a 3′-matched ssDNA without dsDNA (right panel); (C) 3′-overhangs with a central homologous sequence stretch flanked by a dsDNA region and a heterologous 3′ tail (left panel) and a center-matched ssDNA without dsDNA (right panel); (D) 5′-overhangs with a central homologous sequence stretch flanked by a dsDNA region (top panel) and a 5′-matched ssDNA without dsDNA (bottom panel); and (E) 5′-overhangs with a central homologous sequence stretch flanked by a dsDNA region and a heterologous 5′ tail (left panel) and a center-matched ssDNA without dsDNA (right panel). Quantification of SA activity is represented by % product below each gel. Red asterisks (*) indicate 32P labeling.
In cells, ssDNA derived from resected DSBs is quickly coated by ssDNA binding protein RPA (Rothenberg et al., 2008; Sugiyama et al., 1998). Our data indicates that FANCA is still able to catalyze SA with RPA-coated ssDNA, although with slightly lower activity (Figure S3A). To test whether FANCA is involved in the annealing of physiologically relevant substrates, we created two 3′-overhang substrates with a central homologous sequence stretch flanked by a dsDNA region with or without a heterologous 3′-tail. These structures were designed to mimic the 5′ to 3′ partially resected DSB intermediates that are substrates for the SSA pathway (Huertas, 2010). Importantly, FANCA-WT, but not FANCA-F1263Δ, effectively promotes annealing of the 3′-overhang structures, giving rise to dsDNA and 3′-flap products (Figures 1B and 1C, left panels lanes 4–7, and Figure S2B). The heterologous 3′-tail slightly reduces the annealing efficiency at the initial stage (compare the 3′-matched overhang (Figure 1B left) with the 3′-mismatched overhang substrate (Figure 1C left and Figure S2B)). Intriguingly, the flanking dsDNA region plays a critical role in promoting the SA activity of FANCA (Figures 1B and 1C, right panels, e.g. lane 5s and Figure S2B).
Double strand breaks can also be resected by 3′ to 5′ exonucleases such as MRE11 and EXD2 (Biehs et al., 2017), making it likely that 5′-overhangs could also be present during DSB repair. Our in vitro assays show that FANCA catalyzes annealing of both 5′-matched overhangs and mismatched overhangs with similar efficiency as their 3′-overhang counterparts (Compare Figures 1D and 1E with Figures 1B and 1C). Consistent with our 3′-overhang results, absence of a flanking dsDNA region reduces annealing (Figures 1D and 1E and Figure S2C).
RAD52 is a well-established factor in catalyzing SA in cells (Grimme et al., 2010; Mazina et al., 2017). A direct comparison of purified human RAD52 and FANCA under the same reaction conditions shows that the SA activities of FANCA are comparable to that of RAD52 (Figures S2D–S2F).
FANCA Promotes DNA Strand Exchange
In addition to SA, RAD52 also harbors strand exchange (SE) activity (Brown and Bishop, 2014; Huang et al., 2014; Lok and Powell, 2012). To examine whether FANCA catalyzes SE, we developed a SE assay with a splayed arm structure in combination with a third, unlabeled ssDNA oligonucleotide that is completely complementary to the strand of the fork with an exposed ssDNA 5′ flap. Our results show that FANCA-WT, but not FANCA-F1263Δ, strongly promotes formation of a 3′-overhang through SE. At 30–40 nM of FANCA-WT (calculated as monomers), about 70% of the splayed arm substrate has been converted to dsDNA with a 3′-overhang (Figure 2A, lanes 4–7).
Figure 2.

FANCA promotes strand exchange. Titration of the purified FANCA-WT and F1263Δ mutant (10, 20, 30, and 40 nM calculated as monomers) on (A) a pre-annealed splayed arm structure (Right panel, quantification of the SE activity; Experiments were independently repeated 3 times; Error bar, standard deviation); (B) a dsDNA structure with partial 5′-end resection of one strand and a short blunt-ended dsDNA with full homology to the uncovered flap of the invading DNA structure; and (C) a dsDNA structure with partial 3′-end resection of one strand and a short blunt-ended dsDNA with full homology to the uncovered flap of the invading DNA structure. Quantification of SE activity is represented by % product below each gel. (D) Titration of the purified FANCA-WT, FANCA-F1263Δ mutant, and RAD52 (10, 20, 30, 40, and 50 nM) in a D-loop formation assay. Red asterisks (*) indicate 32P labeling.
To further characterize the SE activity, we employed a different set of substrates (Masuda-Sasa et al., 2006) (Figure 2B), including a 3′-overhang DNA structure, which mimics partially resected dsDNA on the 5′-end, in combination with a short blunt-ended dsDNA with full homology to the uncovered flap of the invading 3′ overhang structure. This substrate is expected to be more difficult for SE because it requires the destabilization of a fully complementary blunt-ended dsDNA. Nevertheless, FANCA-WT, but not FANCA-F1263Δ (20–50 nM calculated as monomers), promotes SE of this substrate (Figure 2B, lanes 4–8), albeit the overall efficiency is several fold lower than that of the splayed arm substrate (compare Figure 2B, lanes 4–7 with Figure 2A, lanes 4–7). To determine the polarity of this activity we performed the same experiment using a 5′-overhang structure and we observed similar SE activity by FANCA-WT (Figure 2C, lanes 4–8).
A comparison of RAD52 and FANCA using the same substrates shows that the SE activity of FANCA is comparable to that of RAD52 (Figures S2G–S2I). Similar to RAD52, FANCA-WT, but not FANCA-F1263Δ, also promotes formation of joint molecules between ssDNA and a supercoiled, closed-circular dsDNA, in a D-loop formation assay (Kagawa et al., 2001) (Figure 2D).
FANCA Forms a Homodimer that is Critical for Its SA and SE Activities
During purification, native FANCA-WT behaves like a homodimer. Gel filtration analysis (using Superdex-200) shows that the peak of purified FANCA-WT corresponds to a molecular weight of 320 kDa, which is twice its calculated weight of 163 kDa (Figure S3B, FANCA-WT fractions #24-25). We have further identified that a FANCA variant L1069A/L1076A, which contains mutations within its leucine zipper motif (Garcia-Higuera et al., 2000; Yuan et al., 2012), elutes within peak fractions corresponding to the predicted molecular weight of 163 kDa, indicating that this mutant is not able to form a homodimer (Figure S3B, L1069A/L1076A fractions #26-27). Intriguingly, this FANCA L1069A/1076A mutant does not exhibit impaired DNA binding activity (Figure S1B), but is catalytically inactive for SA and SE (Figure S3C), suggesting that the ability of FANCA to form a homodimer is a prerequisite for the catalysis of strand annealing and exchange.
FANCG, but not FANCB, Stimulates FANCA-Mediated SE and SA
FANCG has been shown to physically interact with FANCA (Garcia-Higuera et al., 2000; Kruyt et al., 1999; Waisfisz et al., 1999). To test whether FANCG has an impact on the observed FANCA-mediated SA and SE, we purified human FANCG expressed in insect cells. As a control, we also purified FANCB, a member of the FA core complex that does not directly interact with FANCA (Figure S4A). In the absence of FANCA, FANCG does not have any SA and SE activities (Figures S4B–S4I, lanes 9–12). In the presence of suboptimal concentration of FANCA, FANCG dramatically stimulates FANCA-mediated SA and SE in a concentration-dependent manner (Figures S4B–S4I, compare lanes 4 with FANCA only and lanes 5–8 with increasing amount of FANCG, 5–30 nM), while FANCB has no detectable effect on the SA and SE activities of FANCA (Figures S4J–S4K).
Patient-Derived FANCA Mutants are Defective in SA and SE
Patient derived FANCA truncation mutant Q772X does not bind DNA whereas its complementing C-terminal fragment C772-1455 does (Figure S1B) (Yuan et al., 2012). Our results show that the SA and SE activities of FANCA require both the N-terminal and C-terminal portions of the protein (Figures 3A and 3B, lanes 4 and 5) and that the DNA binding activity of FANCA alone is insufficient to catalyze SA or SE.
Figure 3.

FANCA mutants are defective in SA and SE. Genetic variants of FANCA: C-terminal truncation (Q772X), N-terminal truncation (C772-1455), D598N, R951W, R1117G, Q1128E, and F1263Δ were analyzed for (A) SA and (B) SE activities. Red asterisks (*) indicate 32P labeling. Quantification of each activity is represented by the bar graphs on the bottom. Experiments were independently repeated 3 times. Statistical significance is represented by the black asterisks (** p ≤ 0.01, * p ≤ 0.05).
To determine whether the SA and SE activities of FANCA are defective in other patient-derived FANCA mutations we tested D598N, R951W, R1117G, and Q1128E (Qian et al., 2013). As shown in Figure 3, all purified FANCA variants exhibit significantly decreased SA and SE activities while retaining WT level of DNA binding affinity (Figure S1B). Notably, D598N and Q1128E are proficient in FANCD2 monoubiquitination and mitomycin C resistance (Adachi et al., 2002), suggesting that the SA and SE activities of FANCA may not be related to activation of DNA interstrand crosslink repair. These data suggest that the SA and SE activities of FANCA are relevant to the etiology of Fanconi anemia.
The SA and SE Activities of FANCA Play an Important Role in DSB Repair in Cells
To assess the physiological role of the biochemical activities of FANCA, we employed the I-SceI DSB repair GFP reporter assay which can monitor repair products from all four unique DSB repair sub-pathways, including homologous recombination (HR), single-strand annealing (SSA), microhomology-mediated end joining (MMEJ), and non-homologous end joining (NHEJ) (Gunn and Stark, 2012).
A FANCA siRNA knockdown screening analysis in U2OS cells showed that the SSA repair sub-pathway is the most significantly impacted by FANCA deficiency (Figure 4A), while little difference is seen for the three remaining pathways. Next, we tested whether overexpression of FANCA affects DSB repair efficiency in U2OS WT cells (Figure 4B, left panel). Intriguingly, we found that overexpression of FANCA-WT did not perturb DSB repair. However, overexpression of the F1263Δ mutant significantly reduced the efficiency of both SSA and HR repair sub-pathways (Figure 4B, right panel).
Figure 4.

FANCA participates in the SSA pathway of DSB repair by catalyzing SA in cells. (A) In vivo repair efficiency of four DSB repair sub-pathways, MMEJ, NHEJ, HR, and SSA in FANCA siRNA knockdown U2OS cells as measured by the GFP reporter assay. Left panel, Western blots. Right panel, relative repair efficiency normalized to siRNA control (siCtrl) (**p ≤ 0.01). (B) DSB repair analysis in cells with FANCA-WT and F1263Δ overexpression. Left panel, Western blot. Right panel, relative repair efficiency normalized to FANCA-WT (**p ≤ 0.01). (C) SSA repair analysis in CRISPR FANCA knockout, and FANCA-WT, FANCA-F1263Δ, FANCA-D598N mutant complemented U2OS cells. Top left panel, Western blot. Bottom panel, representative flow graphs that were statistically analyzed and summarized in the bar graph (top right panel, **p ≤ 0.01, *p≤ 0.05). A positive control for FANCD2 monoubiquitination was obtained by treating cells with 1μM MMC for 24h. Products were resolved in a 6% SDS-PAGE gel until the targeted FANCD2 was at ~2/3 of the gel. No MMC treatment was included in cells for the SSA assay. (D) SSA repair analysis of FANCA and RAD52 double knockdown in U2OS cells. Top panel, Western blot. Bottom panel, relative SSA repair efficiency normalized to siCtrl (*p ≤ 0.05 when compared to either single knockdown). All experiments were independently repeated 3 times. Error bars, standard deviation.
Importantly, a CRISPR/Cas9 knockout of FANCA showed ~4.5-fold reduction of SSA repair (Figure 4C). Complementation of the knockout cells with FANCA-WT fully restores the SSA efficiency (Figure 4C, top right panel). As expected, the F1263Δ mutant, that is deficient in both SA and FANCD2 monoubiquitination (Adachi et al., 2002), is not able to complement the repair defect (Fig. 4C, top right panel). FANCA-D598N, which is defective in SA (Figure 3) but proficient in FANCD2 monoubiquitination (Figure 4C, top left panel), is also incapable of fully complementing the SSA repair defect, suggesting that the role of FANCA in the SSA repair pathway depends on its SA activity, but not FANCD2 monoubiquitination.
To test the epistatic relationship between FANCA and RAD52 in SSA repair, we performed a double knockdown of FANCA and RAD52. The significant reduction in SSA repair in the double knockdown cells indicates that either FANCA or RAD52 plays a compensatory role in SSA when the other is absent, suggesting that the role of FANCA in SSA is independent of RAD52 (Figure 4D).
Deficient FANCA may result in chromatin accumulation of 53BP1 that can lead to a decrease in SSA (Ochs et al., 2016; Renaud et al., 2016). We determined the level of chromatin-bound 53BP1 in FANCA knockdown cells (Figures S1C and S1D) and as expected found a slight increase (Figure S1C, compare lane 3 with 1). To test whether an increase in chromatin-bound 53BP1 has an effect on SSA we overexpressed 53BP1. Multi-fold chromatin-bound 53BP1 increase resulted in only a ~30% reduction in SSA (Figure S1C), suggesting a slight increase in chromatin 53BP1 is likely not responsible for the significant reduction in SSA seen in FANCA knockdowns. Furthermore, 53BP1 overexpression with FANCA knockdown does not additively increase the level of chromatin-bound 53BP1, but leads to a significant reduction in SSA when compared with either 53BP1 overexpression or FANCA knockdown (Figure S1C), suggesting that FANCA and 53BP1 have independent roles in SSA.
DISCUSSION
In this study, we showed that FANCA has intrinsic SA and SE activities, and further demonstrated that these activities play a direct role in the SSA sub-pathway of DSB repair. These results shed light into the molecular mechanism of the previously reported role of FANCA (Nakanishi et al., 2005; Yang et al., 2005).
Importantly, we show that five patient-derived mutations in FANCA with various levels of FANCD2 monoubiquitination and ICL repair capacity (Adachi et al., 2002) are deficient in both activities, providing etiological insight into FA. Some FANCA mutants, such as D598N and Q1128E, that are proficient in FANCD2 monoubiquitination (Figure 4C and (Adachi et al., 2002)) and have wild-type level of mitomycin C sensitivity (Adachi et al., 2002), have reduced SA and SE (Figure 3), suggesting that defects in SA and SE, but not FANCD2 monoubiquitination, are directly linked to the disease presentation in these FANCA patients. Meanwhile, our data with D598N and 1128E also suggest SA or SE is not a critical requirement for the canonical FA pathway in ICL resistance. Altogether, our data suggests that FANCA plays a role in DSB repair by catalyzing SA and/or SE outside of FANCD2 monoubiquitination.
Furthermore, FANCA drives SA and SE in the absence of ATP (No ATP is present in any of our biochemical assays), in a similar manner to RAD52 (Mortensen et al., 1996). Based on our primary sequence analysis, FANCA does not contain a known ATPase or conserved ATP-binding domain. It is well established that RAD52, a protein that biochemically promotes SA in vitro, is required for SSA (Lok et al., 2013; Singleton et al., 2002; Stark et al., 2004; Wu et al., 2008). Cells that lack functional RAD52 present with a relatively weak SSA phenotype, raising the possibility that additional factors are involved in catalyzing the annealing steps in mammalian DSB repair (Liu and Heyer, 2011). Our double knockdown experiment with RAD52 and FANCA indeed supports that FANCA has a role in SSA that is independent of RAD52 (Figure 4D).
FANCA has high affinity for ssDNA and relatively low affinity for dsDNA (Yuan et al., 2012). It is likely that the homodimeric FANCA brings two complimentary ssDNAs into close proximity for dsDNA formation, through its high affinity for ssDNA. Once dsDNA is formed, FANCA turns over by releasing the dsDNA product through its low dsDNA affinity, initiating the next round of catalysis. It is important to note that FANCA mutants with single residue changes are defective in strand annealing, but proficient in DNA binding (Figure S1B). This suggests that affinity to DNA per se is insufficient to anneal ssDNA, and that FANCA-catalyzed strand annealing requires a dynamic interaction between FANCA and DNA.
It has been previously shown that restoration of FANCA protein in FANCA-null cells also increased HR (Howard et al., 2015; Nakanishi et al., 2005; Yang et al., 2005). Our knockdown experiment does not fully recapitulate its role in HR (Figure 4A), likely due to differences in the assay system or siRNA selection. However, we show that overexpression of the F1263Δ mutant results in HR and SSA repair deficiency when cells are challenged with DSB formation. It is possible that the unaffected DNA binding activity of this mutant, combined with its defective SA and SE activities, makes it a dominant-negative candidate when overexpressed in a wild-type background (Figure 4B). Nevertheless, we are not able to distinguish whether it is the SA or SE activity that contributes to this defect in HR. Furthermore; FANCA directly interacts with BRCA1 (Folias et al., 2002), which also promotes HR and SSA, yet the functional relevance of this interaction has not been determined. It would be interesting to study the interplay of FANCA’s biochemical activities with BRCA1 and other HR factors.
Previous biochemical work from our lab showed that FANCA is involved in stimulating the incision of ICLs on the leading strand via MUS81/EME1 endonuclease (Benitez et al., 2014). Incision of ICLs in a replication fork on the leading strand produces intermediate DSB structures with 5′-3′ overhangs. In untreated cells, FANCA depletion from Xenopus egg extracts results in accumulation of DSBs (Sobeck et al., 2006), suggesting a role for FANCA in DSB repair. FANCA may also participate in the reestablishment of collapsed replication forks through its bidirectional SA and SE activities.
In summary, our data suggests that the SA and SE activities of FANCA are physiologically relevant and directly involved in DSB repair in cells. Additionally, the SA catalytic role of FANCA in the SSA repair sub-pathway is independent of the FA core complex, FANCD2, and RAD52. As a next step, it will be of considerable interest to investigate whether and how FANCA physically and functionally interacts with other repair proteins to exert its role in DSB repair and achieve successful maintenance of genome stability.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yanbin Zhang (yzhang4@med.miami.edu)
METHOD DETAILS
Protein expression and purification
cDNAs for FANCA, FANCG, and FANCB were obtained from Dr. Weidong Wang at the National Institute on Aging, NIH. All genes were cloned into pFastBac1 (for native proteins) and pFastBacHT (for HIS6-tagged proteins) vectors and subsequently sequenced. Suspected mutations were screened against the human single nucleotide polymorphism (SNP) collection at NCBI (http://www.ncbi.nlm.nih.gov/sites/entrez). True mutations were corrected by PCR-mediated site-specific mutagenesis and verified by re-sequencing. Baculoviruses were subsequently prepared according to the manufacturer’s protocol (Invitrogen).
Purification of FANCA-WT and mutants were carried out as described previously (Yuan et al., 2012). In brief, upon expression of the recombinant FANCA proteins in insect cells, the cells were homogenized using a Dounce homogenizer to prepare extracts. FANCA were purified by using HiTrap Q Sepharose Fast Flow, 5-mL HiTrap Blue, Mono S, Mono Q, and/or Superdex 200 gel filtration columns (GE Healthcare, Piscataway, NJ), and/or a 2-mL high-resolution hydroxylapatite column (Calbiochem, La Jolla, CA) and by tracing FANCA protein through SDS-PAGE and Western blot.
To express and purify recombinant human FANCB protein, 15 T-175 flasks of High-Five insect cells (90% confluency) were infected with pFastBac-HTb-FANCB baculovirus. Cells were harvested after 48 hours of infection. Cell pellet was resuspended in 25 ml of low salt buffer (20 mM Hepes.KOH pH 7.5, 0.5 mM MgCl2, 5 mM KCl, 5 mM β-Mercaptoethanol, and protease inhibitors (0.5 mM PMSF, 0.3 mg/ml benzamidine hydrochloride, 0.5 μg/ml pepstatin A, 0.5 μg/ml leupeptin, 0.5 μg/ml antipain)). The suspended cells were lysed by 10 strokes on ice in a Dounce homogenizer. The homogenized sample was centrifuged at 4200 rpm 4°C for 6 min. The nuclei pellet was resuspended in 30-ml cold extraction buffer (50 mM Hepes.KOH pH 7.5, 10% sucrose, 160 mM NaCl, protease inhibitors, and 5 mM β-Mercaptoethanol) and incubated on ice for 10 min. After centrifugation at 16000 rpm for 1 hr, the supernatant containing FANCB (by Western blot) was diluted 1.5X in nickel A buffer (50 mM Sodium Phosphate buffer PH 8.0, 15% glycerol, 300 mM NaCl, 5 mM β-Mercaptoethanol, and protease inhibitors), and resolved on a nickel-charged HiTrap Chelating column. FANCB peak was eluted around 200 mM of imidazole in Nickel A buffer. The pooled FANCB was desalted to 100 mM KCl in buffer Q (50mM KPO4 pH 7.4, 0.5 mM EDTA, 0.01% NP-40, 10% glycerol, 2 mM DTT, protease inhibitors) and resolved on a Mono-Q column. The purified FANCB was eluted at 320 mM KCl in buffer Q.
For FANCG expression and purification, 15 T-175 flasks of High-Five insect cells (90% confluency) were infected with pFastBac-HTb-FANCG baculovirus. Cells were harvested after 60 hours of infection. Cell pellet was resuspended in 25 ml of low salt buffer (20 mM Hepes.KOH pH 7.5, 0.5 mM MgCl2, 5 mM KCl, 5 mM β-Mercaptoethanol, and protease inhibitors). The suspended cells were lysed by 10 strokes on ice in a Dounce homogenizer. The homogenized sample was centrifuged at 15000 rpm for 1 hr, the supernatant containing FANCG (by Western blot) was diluted 2X by nickel A buffer (50 mM NaPO4 buffer pH 8.0, 15% glycerol, 300 mM NaCl, 5 mM β-Mercaptoethanol, and protease inhibitors), and resolved on a nickel-charged HiTrap Chelating column. FANCG peak was eluted around 70 mM of imidazole in Nickel A buffer. The pooled FANCG was further purified on a Superdex-200 gel filtration column in buffer Q with 220 mM KCl.
Human RPA heterotrimeric complex was expressed in E. coli BL21 (DE3) cells and purified to homogeneity as previously described (Henricksen et al., 1994). In brief, the homogenized supernatant from 2 liters of an induced culture was loaded to a 10-ml Affi-Gel blue column equilibrated with HI buffer (30 mM Hepes.KOH pH 7.5, 1 mM DTT, 0.25 mM EDTA, 0.75% inositol, 0.01% NP-40, and protease inhibitors) containing 50 mM KCl. The column was washed sequentially with 50 ml each of HI buffer containing 50 mM KCl, 0.8 M KCl, 0.5 M NaSCN, or 1.5 M NaSCN. RPA was eluted in the 1.5 M NaSCN wash. After desalting by a hydroxylapatite column, the pooled RPA sample was loaded onto a Mono-Q column equilibrated with HI buffer containing 100 mM KCl. The column was washed with 10 ml of HI buffer containing 100 mM KCl and then developed with a 30-ml linear salt gradient of 100–500 mM KCl. The peak of RPA was eluted at ~300 mM KCl.
Human RAD52 was purified as described previously (Kagawa et al., 2001). In brief, RAD52 was purified sequentially by Ni-NTA agarose beads, a Heparin-Sepharose column, and a Mono-S column. Protein concentration was determined by the Coomassie (Bradford) Protein Assay Reagent (Pierce, Rockford, CA). The purified proteins were stored in −80°C in aliquots.
DNA Substrates and chromatin fractionation
Sequences and substrate designs are available in the Supplemental Information. Oligonucleotides used in the strand exchange assay were adopted from Masuda-Sasa et al. 2006 (Masuda-Sasa et al., 2006). The 5′-end of oligos was labeled with γ-32P-ATP and T4 polynucleotide kinase enzyme (New England Biolabs) using the protocol specified by the manufacturer. For assays in which dsDNA structures were required as substrates the oligonucleotides involved were pre-annealed at 1:1 molar ratios. Annealing was carried out in a water bath within ~5 h by slowly cooling from 85°C to 20 °C. The quality of annealing was monitored by native gel electrophoresis. Proper annealing was verified by the mobility of a corresponding substrate. Chromatin fractionation of U2OS cells was carried out as described previously (Mendez and Stillman, 2000). In brief, 5 × 106 cells were used to produce appropriate amount of the chromatin fraction. Buffer A (10 mM HEPES pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM DTT, and supplementary protease inhibitors) with 0.1% Triton X-100 was used first to disrupt cell membrane and extract the soluble cytoplasmic fraction. Pellets from centrifugation were incubated with Buffer B (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT, and supplementary protease inhibitors) for the extraction of nuclear soluble fraction. Insoluble fraction, which is enriched with chromatin, was reconstituted in SDS sample buffer with boiling. Hsp90, PCNA, and Histone H3 were used to monitor the quality of fractionation.
DNA binding assay
DNA binding EMSA analysis was performed as described previously (Yuan et al., 2012) in a 10 μl reaction containing 25 mM Tris-HCl pH 7.5, 100 mM NaCl, 5 mM EDTA, 1 mM DTT, 6% glycerol, 1 nM 5′-32P-labeled oligonucleotide substrates, and the indicated amounts of protein. The reactions were incubated at room temperature for 45 min, followed by the addition of 4 μl of 50% (w/v) sucrose buffered by 10 mM Tris-HCl pH 7.5. The reaction mixtures were resolved by electrophoresis through a 4% non-denaturing polyacrylamide gel in 40 mM Tris acetate (pH 7.6) and 10 mM EDTA with 6% glycerol using the Owl P9DS electrophoresis system (Thermo Scientific) at 100 V (~1.5 watts/gel) for 40 or 90 min as indicated. DNA substrates and shifted bands were visualized by autoradiography.
Assays for strand annealing, strand exchange, and D-loop formation
Strand annealing and strand exchange activities were carried out as previously described (Mir et al., 2013). In brief, a total of 0.5 nM 5′-32P-labeled DNA substrate and the indicated amounts of protein were incubated in a 10 μl reaction (25 mM Tris-HCl pH 8.0, 100 mM NaCl, 1 mM EDTA). The reaction mixture was incubated at room temperature for 40 min or as indicated in the figure legend and stopped with 1 μl of 10x stop solution (200 mM EDTA, 32% Glycerol, 1% SDS, 0.024% Bromophenol Blue), 3 μg protease K, and 10 min incubation at room temperature. Products were separated on a 6 or 10% native PAGE gel at 100V for 1.5 or 4 hr depending on substrate size. Substrate and product bands were visualized by autoradiography. D-loop formation was carried out essentially as described previously (Kagawa et al., 2001) with modifications. In brief, a total of 1.5 nM of 32P-labeled ssDNA A18 (Table S1 for sequence) was pre-incubated with indicated amounts of either FANCA or RAD52 protein in a 10 μl reaction of 50 mM Hepes.KOH pH 7.5, 5 mM MgCl2, 50 mM NaCl, 0.5 mg/ml BSA, and 5 mM DTT at RT for 10 min. Afterwards, 7.5 nM of pBR322 supercoiled DNA was added to the reaction mixtures, incubated at 37 °C for 10 min and the reaction was terminated with 1 μl of 10X stop solution (200 mM EDTA, 32% Glycerol, 1% SDS, 0.024% Bromophenol Blue), 3 μg protease K, and 10 min incubation at room temperature. The reaction mixtures were resolved on a 1.5% agarose gel in 1X TBE buffer at 100V for 2hrs. After gel drying, substrate and product bands were visualized by autoradiography.
Cell-based DSB repair assay
The I-SceI construct and four U2OS cell lines carrying MMEJ, NHEJ, HR, and SSA reporter constructs were generous gifts from Dr. Jeremy Stark at the City of Hope Medical Center. Cells were cultured in DMEM complemented with 10% FBS, antibiotics and glutamine. Knockdown of FANCA, FANCB, FANCL, RAD52, and FANCD2 was carried out using FANCA siRNA (5.6 nM), FANCB siRNA (24 nM), FANCL siRNA (0.8 nM, custom siRNA: GACAAGAGCUGUAUGCACUUU), RAD52 siRNA (2 nM), FANCD2 siRNA (5.6 nM), and Control siRNA, and verified by Western blot. FANCA CRISPR knockout constructs in Cas9-GFP backbone with a sgRNA sequence of CCAAGGCCATGTCCGACTCG were purchased from Genscript. Knockout lines were generated by electroporation of CRISPR constructs followed by FACS sorting and Western blot confirmation. I-SceI based fluorescent reporter assay was carried out as previously described with some modifications (Gunn and Stark, 2012). U2OS cells were seeded in 6-well plates at a density of 250K/well. On the following day, cells were subjected to transfection of 1μg per well of I-SceI plasmid in the presence or absence of siRNA as indicated by using lipofectamine 2000 to initiate double strand break production. 48 hours after transfection, cells were washed once with PBS, trypsinized with 300 μL trypsin, and neutralized with 400 μL media. FACS analysis was carried out within 1 hour with PE channel to assist the exclusion of auto-fluorescence.
QUANTIFICATION AND STATISTICAL ANALYSIS
ImageJ was used for quantification for all strand annealing and exchange assays. FlowJo was used for flow cytometry analysis. Microsoft excel software was used to perform all statistical analyses. Statistical differences between groups were determined by two-tailed Student t-test. Significance is denoted as * for p < 05, and ** for p < 01.
Supplementary Material
Highlights.
FANCA catalyzes bidirectional single-strand annealing and strand exchange
FANCG stimulates FANCA-mediated strand annealing and strand exchange
Fanconi anemia patient-derived FANCA mutants are deficient in both activities
The single-strand annealing activity of FANCA plays a direct role in DSB repair
Acknowledgments
This work was supported by National Institutes of Health Grants HL131013 and ES027058 to Y.Z., CA188347 to A.V.M., a Flight Attendant Medical Research Institute grant to Y.Z., and a Drexel Coulter Program Award to A.V.M. Cells and constructs for DSB repair reporter assays were generously provided by Jeremy Stark at the City of Hope National Medical Center.
Footnotes
AUTHOR CONTRIBUTIONS
A.B., W.L., A.P., Y.Z, X.H.P., A.V.M., F.Y., and Y.Z. designed the experiments. A.B., W.L., A.P., G.W., J.M., K.A., A.K, K.Z., Y.Z., F.B., F.Y., and Y.Z collected data. A.B., W.L, A.P., F.Y., and Y.Z wrote the manuscript. All authors reviewed the manuscript and approved the conclusions.
DECLARATION OF INTERESTS
The authors declare no competing interests.
Supplemental information includes one table and four supplemental figures and figure legends.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adachi D, Oda T, Yagasaki H, Nakasato K, Taniguchi T, D’Andrea AD, Asano S, Yamashita T. Heterogeneous activation of the Fanconi anemia pathway by patient-derived FANCA mutants. Hum Mol Genet. 2002;11:3125–3134. doi: 10.1093/hmg/11.25.3125. [DOI] [PubMed] [Google Scholar]
- Benitez A, Yuan F, Nakajima S, Wei L, Qian L, Myers R, Hu JJ, Lan L, Zhang Y. Damage-dependent regulation of MUS81-EME1 by Fanconi anemia complementation group A protein. Nucleic acids research. 2014;42:1671–1683. doi: 10.1093/nar/gkt975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biehs R, Steinlage M, Barton O, Juhasz S, Kunzel J, Spies J, Shibata A, Jeggo PA, Lobrich M. DNA Double-Strand Break Resection Occurs during Non-homologous End Joining in G1 but Is Distinct from Resection during Homologous Recombination. Mol Cell. 2017;65:671–684 e675. doi: 10.1016/j.molcel.2016.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Bishop DK. DNA Strand Exchange and RecA Homologs in Meiosis. Cold Spring Harb Perspect Biol. 2014:7. doi: 10.1101/cshperspect.a016659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R, Sarangi P, D’Andrea AD. The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol. 2016;17:337–349. doi: 10.1038/nrm.2016.48. [DOI] [PubMed] [Google Scholar]
- Crossan GP, van der Weyden L, Rosado IV, Langevin F, Gaillard PH, McIntyre RE, Gallagher F, Kettunen MI, Lewis DY, Brindle K, et al. Disruption of mouse Slx4, a regulator of structure-specific nucleases, phenocopies Fanconi anemia. Nat Genet. 2011;43:147–152. doi: 10.1038/ng.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feeney L, Munoz IM, Lachaud C, Toth R, Appleton PL, Schindler D, Rouse J. RPA-Mediated Recruitment of the E3 Ligase RFWD3 Is Vital for Interstrand Crosslink Repair and Human Health. Mol Cell. 2017;66:610–621 e614. doi: 10.1016/j.molcel.2017.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folias A, Matkovic M, Bruun D, Reid S, Hejna J, Grompe M, D’Andrea A, Moses R. BRCA1 interacts directly with the Fanconi anemia protein FANCA. Human molecular genetics. 2002;11:2591–2597. doi: 10.1093/hmg/11.21.2591. [DOI] [PubMed] [Google Scholar]
- Garcia-Higuera I, Kuang Y, Denham J, D’Andrea AD. The fanconi anemia proteins FANCA and FANCG stabilize each other and promote the nuclear accumulation of the Fanconi anemia complex. Blood. 2000;96:3224–3230. [PubMed] [Google Scholar]
- Grimme JM, Honda M, Wright R, Okuno Y, Rothenberg E, Mazin AV, Ha T, Spies M. Human Rad52 binds and wraps single-stranded DNA and mediates annealing via two hRad52-ssDNA complexes. Nucleic Acids Res. 2010;38:2917–2930. doi: 10.1093/nar/gkp1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn A, Stark JM. I-SceI-based assays to examine distinct repair outcomes of mammalian chromosomal double strand breaks. Methods Mol Biol. 2012;920:379–391. doi: 10.1007/978-1-61779-998-3_27. [DOI] [PubMed] [Google Scholar]
- Henricksen LA, Umbricht CB, Wold MS. Recombinant replication protein A: expression, complex formation, and functional characterization. J Biol Chem. 1994;269:11121–11132. [PubMed] [Google Scholar]
- Howard SM, Yanez DA, Stark JM. DNA damage response factors from diverse pathways, including DNA crosslink repair, mediate alternative end joining. PLoS Genet. 2015;11:e1004943. doi: 10.1371/journal.pgen.1004943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Leung JW, Lowery M, Matsushita N, Wang Y, Shen X, Huong D, Takata M, Chen J, Li L. Modularized functions of the Fanconi anemia core complex. Cell Rep. 2014;7:1849–1857. doi: 10.1016/j.celrep.2014.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huertas P. DNA resection in eukaryotes: deciding how to fix the break. Nat Struct Mol Biol. 2010;17:11–16. doi: 10.1038/nsmb.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inano S, Sato K, Katsuki Y, Kobayashi W, Tanaka H, Nakajima K, Nakada S, Miyoshi H, Knies K, Takaori-Kondo A, et al. RFWD3-Mediated Ubiquitination Promotes Timely Removal of Both RPA and RAD51 from DNA Damage Sites to Facilitate Homologous Recombination. Mol Cell. 2017;66:622–634 e628. doi: 10.1016/j.molcel.2017.04.022. [DOI] [PubMed] [Google Scholar]
- Kagawa W, Kurumizaka H, Ikawa S, Yokoyama S, Shibata T. Homologous pairing promoted by the human Rad52 protein. J Biol Chem. 2001;276:35201–35208. doi: 10.1074/jbc.M104938200. [DOI] [PubMed] [Google Scholar]
- Kee Y, D’Andrea AD. Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev. 2010;24:1680–1694. doi: 10.1101/gad.1955310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee Y, D’Andrea AD. Molecular pathogenesis and clinical management of Fanconi anemia. The Journal of clinical investigation. 2012;122:3799–3806. doi: 10.1172/JCI58321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knies K, Inano S, Ramirez MJ, Ishiai M, Surralles J, Takata M, Schindler D. Biallelic mutations in the ubiquitin ligase RFWD3 cause Fanconi anemia. J Clin Invest. 2017;127:3013–3027. doi: 10.1172/JCI92069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottemann MC, Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature. 2013;493:356–363. doi: 10.1038/nature11863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruyt FA, Abou-Zahr F, Mok H, Youssoufian H. Resistance to mitomycin C requires direct interaction between the Fanconi anemia proteins FANCA and FANCG in the nucleus through an arginine-rich domain. The Journal of biological chemistry. 1999;274:34212–34218. doi: 10.1074/jbc.274.48.34212. [DOI] [PubMed] [Google Scholar]
- Liu J, Heyer WD. Who’s who in human recombination: BRCA2 and RAD52. Proc Natl Acad Sci U S A. 2011;108:441–442. doi: 10.1073/pnas.1016614108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lok BH, Carley AC, Tchang B, Powell SN. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene. 2013;32:3552–3558. doi: 10.1038/onc.2012.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lok BH, Powell SN. Molecular pathways: understanding the role of Rad52 in homologous recombination for therapeutic advancement. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18:6400–6406. doi: 10.1158/1078-0432.CCR-11-3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda-Sasa T, Polaczek P, Campbell JL. Single strand annealing and ATP-independent strand exchange activities of yeast and human DNA2: possible role in Okazaki fragment maturation. The Journal of biological chemistry. 2006;281:38555–38564. doi: 10.1074/jbc.M604925200. [DOI] [PubMed] [Google Scholar]
- Mazina OM, Keskin H, Hanamshet K, Storici F, Mazin AV. Rad52 Inverse Strand Exchange Drives RNA-Templated DNA Double-Strand Break Repair. Mol Cell. 2017;67:19–29 e13. doi: 10.1016/j.molcel.2017.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez J, Stillman B. Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol Cell Biol. 2000;20:8602–8612. doi: 10.1128/mcb.20.22.8602-8612.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mir T, Huang SH, Kobryn K. The telomere resolvase of the Lyme disease spirochete, Borrelia burgdorferi, promotes DNA single-strand annealing and strand exchange. Nucleic Acids Res. 2013;41:10438–10448. doi: 10.1093/nar/gkt832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldovan GL, D’Andrea AD. How the fanconi anemia pathway guards the genome. Annu Rev Genet. 2009;43:223–249. doi: 10.1146/annurev-genet-102108-134222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen UH, Bendixen C, Sunjevaric I, Rothstein R. DNA strand annealing is promoted by the yeast Rad52 protein. Proc Natl Acad Sci U S A. 1996;93:10729–10734. doi: 10.1073/pnas.93.20.10729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi K, Yang YG, Pierce AJ, Taniguchi T, Digweed M, D’Andrea AD, Wang ZQ, Jasin M. Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair. Proc Natl Acad Sci U S A. 2005;102:1110–1115. doi: 10.1073/pnas.0407796102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochs F, Somyajit K, Altmeyer M, Rask MB, Lukas J, Lukas C. 53BP1 fosters fidelity of homology-directed DNA repair. Nat Struct Mol Biol. 2016;23:714–721. doi: 10.1038/nsmb.3251. [DOI] [PubMed] [Google Scholar]
- Palovcak A, Liu W, Yuan F, Zhang Y. Maintenance of genome stability by Fanconi anemia proteins. Cell Biosci. 2017;7:8. doi: 10.1186/s13578-016-0134-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian L, Yuan F, Rodriguez-Tello P, Padgaonkar S, Zhang Y. Human Fanconi anemia complementation group a protein stimulates the 5′ flap endonuclease activity of FEN1. PLoS One. 2013;8:e82666. doi: 10.1371/journal.pone.0082666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renaud E, Barascu A, Rosselli F. Impaired TIP60-mediated H4K16 acetylation accounts for the aberrant chromatin accumulation of 53BP1 and RAP80 in Fanconi anemia pathway-deficient cells. Nucleic Acids Res. 2016;44:648–656. doi: 10.1093/nar/gkv1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenberg E, Grimme JM, Spies M, Ha T. Human Rad52-mediated homology search and annealing occurs by continuous interactions between overlapping nucleoprotein complexes. Proc Natl Acad Sci U S A. 2008;105:20274–20279. doi: 10.1073/pnas.0810317106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton MR, Wentzell LM, Liu Y, West SC, Wigley DB. Structure of the single-strand annealing domain of human RAD52 protein. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:13492–13497. doi: 10.1073/pnas.212449899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobeck A, Stone S, Costanzo V, de Graaf B, Reuter T, de Winter J, Wallisch M, Akkari Y, Olson S, Wang W, et al. Fanconi anemia proteins are required to prevent accumulation of replication-associated DNA double-strand breaks. Mol Cell Biol. 2006;26:425–437. doi: 10.1128/MCB.26.2.425-437.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark JM, Pierce AJ, Oh J, Pastink A, Jasin M. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Molecular and cellular biology. 2004;24:9305–9316. doi: 10.1128/MCB.24.21.9305-9316.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoepker C, Hain K, Schuster B, Hilhorst-Hofstee Y, Rooimans MA, Steltenpool J, Oostra AB, Eirich K, Korthof ET, Nieuwint AW, et al. SLX4, a coordinator of structure-specific endonucleases, is mutated in a new Fanconi anemia subtype. Nat Genet. 2011;43:138–141. doi: 10.1038/ng.751. [DOI] [PubMed] [Google Scholar]
- Sugawara N, Ira G, Haber JE. DNA length dependence of the single-strand annealing pathway and the role of Saccharomyces cerevisiae RAD59 in double-strand break repair. Molecular and cellular biology. 2000;20:5300–5309. doi: 10.1128/mcb.20.14.5300-5309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama T, New JH, Kowalczykowski SC. DNA annealing by RAD52 protein is stimulated by specific interaction with the complex of replication protein A and single-stranded DNA. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:6049–6054. doi: 10.1073/pnas.95.11.6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Twest S, Murphy VJ, Hodson C, Tan W, Swuec P, O’Rourke JJ, Heierhorst J, Crismani W, Deans AJ. Mechanism of Ubiquitination and Deubiquitination in the Fanconi Anemia Pathway. Mol Cell. 2017;65:247–259. doi: 10.1016/j.molcel.2016.11.005. [DOI] [PubMed] [Google Scholar]
- Waisfisz Q, de Winter JP, Kruyt FA, de Groot J, van der Weel L, Dijkmans LM, Zhi Y, Arwert F, Scheper RJ, Youssoufian H, et al. A physical complex of the Fanconi anemia proteins FANCG/XRCC9 and FANCA. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:10320–10325. doi: 10.1073/pnas.96.18.10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AT, Smogorzewska A. SnapShot: Fanconi Anemia and Associated Proteins. Cell. 2015;160:354–354 e351. doi: 10.1016/j.cell.2014.12.031. [DOI] [PubMed] [Google Scholar]
- Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet. 2007;8:735–748. doi: 10.1038/nrg2159. [DOI] [PubMed] [Google Scholar]
- Wu Y, Kantake N, Sugiyama T, Kowalczykowski SC. Rad51 protein controls Rad52-mediated DNA annealing. The Journal of biological chemistry. 2008;283:14883–14892. doi: 10.1074/jbc.M801097200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YG, Herceg Z, Nakanishi K, Demuth I, Piccoli C, Michelon J, Hildebrand G, Jasin M, Digweed M, Wang ZQ. The Fanconi anemia group A protein modulates homologous repair of DNA double-strand breaks in mammalian cells. Carcinogenesis. 2005;26:1731–1740. doi: 10.1093/carcin/bgi134. [DOI] [PubMed] [Google Scholar]
- Yuan F, Qian L, Zhao X, Liu JY, Song L, D’Urso G, Jain C, Zhang Y. Fanconi anemia complementation group A (FANCA) protein has intrinsic affinity for nucleic acids with preference for single-stranded forms. The Journal of biological chemistry. 2012;287:4800–4807. doi: 10.1074/jbc.M111.315366. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.