

Abstract
The direct cyanomethylation of indoles at the 2- or 3-position was achieved via photoredox catalysis. The versatile nitrile synthon is introduced as a radical generated from bromoacetonitrile, a photocatalyst, and blue LEDs as a light source. The mechanism of the reaction is explored by determination of the Stern-Volmer quenching constants. By combining photo-physical data and mass spectrometry to follow the catalyst decomposition, the catalyst ligands were tuned to enable synthetically useful yields of radical coupling products. A range of indole substrates with alkyl, aryl, halogen, ester, and ether functional groups participate in the reaction affording products in 16–90% yields. The reaction allows the rapid construction of synthetically useful cyanomethyl indoles, products that otherwise require several synthetic steps.
Graphical Abstract
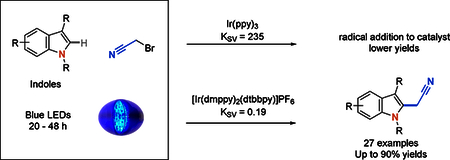
Introduction
The indole ring system is widely found in biologically active compounds. For example, indole-containing compounds derived from tryptophan, such as melatonin, serotonin, and psilocin, engage targets in the central nervous system.1-2 Chemical reactions designed to synthesize3 and modify indoles4 are therefore useful for the construction of pharmacologically active compounds with a variety of medicinal applications. Many reactions to functionalize indoles utilize the native nucleophilicity of the 3-position to generate 3-substituted analogs.5 We therefore sought to generate a synthetic method that preferentially functionalizes indoles at the 2-position in a single step. Additionally, we were most interested in developing a method to incorporate a synthetically versatile cyanomethyl group. Historically, this product was only accessible through lengthy reaction sequences, such as the elaboration of indole-2-carboxylic acid in five steps (Scheme 1a).6 A palladium catalyzed coupling reaction using a masked cyanomethyl group and indole stannane to rapidly afford 2-cyanomethyl-1-methylindole has been reported.7 Direct reaction of acetonitrile radical to indoles has been reported using a copper peroxide system but requires an excess of chemical oxidant and elevated reaction temperatures (Scheme 1b).8 We therefore sought to design a reaction that directly functionalizes indoles in a single step under mild reaction conditions and avoids the use of stoichiometric oxidants.
Scheme 1.
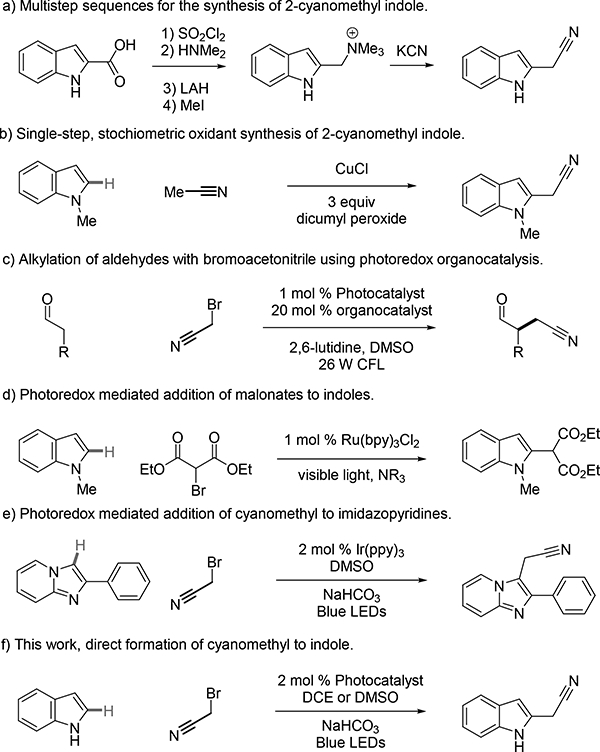
Synthesis of 2-cyanomethyl indoles.
Photoredox catalysis has developed into a highly useful platform for the incorporation of functional groups via singleelectron mechanisms.9–16 In addition to obtaining novel reactivity, photoredox catalysis avoids the use of stoichiometric redox reagents. For example, MacMillan has shown that by a dual photoredox chiral enamine catalysis mechanism, electron-deficient radicals add into catalytically generated enamines to afford enantioenriched α-alkylated aldehyde products.17
Subsequently, bromoacetonitrile was reported to be a competent radical precursor, affording chiral α-cyanomethylaldehydes via photoredox catalysis (Scheme 1c).18 Acetonitrile radical generated via photocatalysis with a heterogeneous Pd/TiO2 catalyst has also been shown to add into benzene.19 The Stephenson group have reported a new method to add, via photoredox, electron deficient radicals generated from bromomalonates into electron-rich heterocycles.20-21 Notably, they showed that a variety of indoles could be functionalized at the 2-position in this fashion (Scheme 1d). Sun and Lui have shown that photoredox mediated cyanomethylation of imidazopyridines adds selectively to the 3-position (Scheme 1e).22 Together, these examples illustrate the potential acetonitrile radical formation and its addition into an indole. Thus, we envisioned that direct alkylation of indoles at the 2-position with bromoacetonitrile could occur via photoredox catalysis (Scheme 1f).
The proposed reaction mechanism is shown in Scheme 2. A photo-excited catalyst reduces, via single-electron transfer, bromoacetonitrile (E1/2red = –0.69 V vs SCE in DMF).23 The radical anion decomposes to afford bromide anion and acetonitrile radical. The electron deficient radical then couples to electron-rich indole. The resulting benzyl radical is then oxidized by single-electron transfer to generate a benzyl cation, regenerating the photocatalyst and closing the catalytic cycle. Loss of a proton then restores aromaticity, affording 2-cyanomethyl indole.
Scheme 2.
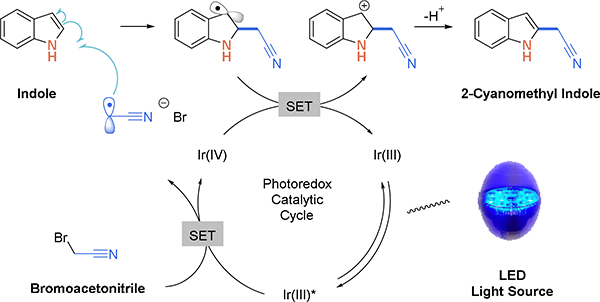
Proposed mechanism for photoredox catalyzed cyanoalkylation of indoles.
Results and Discussion
To test and optimize the reaction conditions, we chose N-methylindole (1), a substrate that lacks substituents that could bias the reaction (Table 1). Using DMSO as solvent and 2,6-lutidine as base, we initially tested a range of photocatalysts centered on Ru, Cu, or Ir, with excited state reduction potentials (E 1/2M+/M*: 4, –0.81 V24; 5, –1.43 V25; 6, –1.73 V26) above that of bromoacetonitrile (E1/2red = –0.69 V vs SCE in DMF).23 Photocatalysts 4 and 5 both resulted in 10% yield, while Ir(ppy)3 gave the best yield (catalyst 6, entry 3, 43%). We then tested commercially available iridium catalysts with altered ligands and redox potentials. Switching to a photocatalyst with a lower reduction potential [Ir(dtbbpy)(ppy)2]PF6 (E1/2II/III* = –0.96 V27, catalyst 7, entry 4) afforded a lower yield. For both catalysts 6 and 7, there is a dramatic change in the color of the reaction, likely indicating deactivation of the photocatalyst (Figure 1). Previously, it was reported that Ir(ppy)3 and other similar photocatalysts undergo deactivation through addition of malonate radical species into the phenylpyridine ligand.28
Table 1.
Catalyst optimization.
 | ||||
|---|---|---|---|---|
| Entry | Photocatalyst | mol % | Yield (%)a | |
| 1 | [Ru(bpy)3]CI2 | 4 | 1 | 10 |
| 2 | [Cu(dap)2]CI | 5 | 1 | 10 |
| 3 | fac-lr(ppy)3 | 6 | 2 | 43 |
| 4 | [lr(dtbbpy)(ppy)2]PF6 | 7 | 2 | 33 |
| 5 | [lr(dFCF3ppy)2(bpy)]PF6 | 8 | 2 | 54 |
| 6 | fac-lr(Fppy)3 | 9 | 2 | 53 |
| 7 | [lr(dtbbpy)(dtbppy)2]PF6 | 10 | 2 | 55 |
| 8 | [lr(dmppy)2(dtbbpy)]PF6 | 11 | 2 | 59 |
| 9b | fac-lr(ppy)3 | 6 | 2 | 0 |
| 10 | none | - | 0 | 0 |
Yield determined by HPLC calibrated assay, 1 M concentration, 0.5 mmol 2, 2 equiv 1.
No light.
Figure 1.
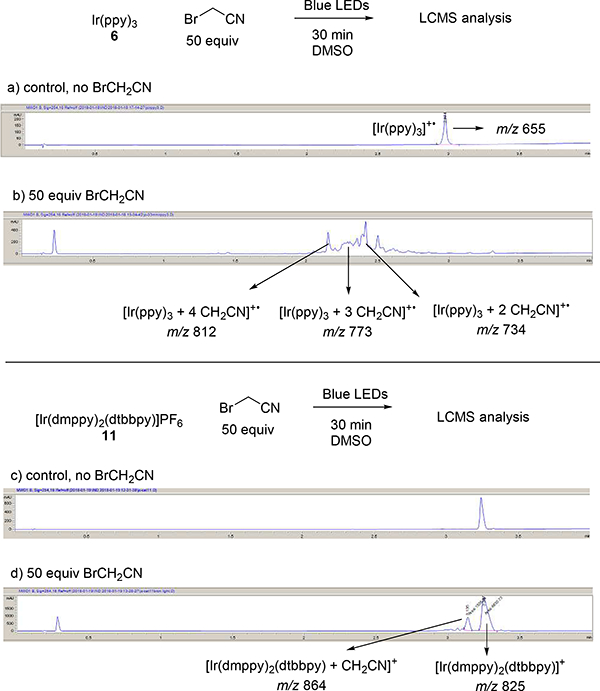
Observation of catalyst-cyanomethyl adducts by LCMS analysis after exposure to bromoacetonitrile and irradiation with blue LEDs using: a) Ir(ppy)3 (6), control no BrCH2CN; b) Ir(ppy)3, 50 equiv BrCH2CN ; c) [Ir(dmppy)2(dtbbpy)]PF6 (11), control no BrCH2CN; d) [Ir(dmppy)2(dtbbpy)]PF6 (11), 50 equiv BrCH2CN. Shown are the UV absorbance traces at 254 nm and the m/z ions detected for selected peaks.
Inspired by these findings, we tested photocatalysts that hinder addition of the electron-poor acetonitrile radical. Ligands with electron withdrawing groups, catalysts 8 and 9, reduce the rate of addition and afford better yields (entries 5–6: 53–54% yield). A consequence, however, of using the electrondeficient ligands is an increase in the oxidation potential (e.g. 8: E1/2III* /II = +0.89 V vs SCE in MeCN).27 This would limit the substrate scope to electron-poor indoles. We therefore tested catalysts that would block catalyst deactivation by incorporating alkyl groups on the ligands, [Ir(dtbbpy)(dtbppy)2]PF6 (10) and [Ir(dmppy)2(dtbbpy)]PF6 (11). Unlike the reactions using 6 or 7, the bright yellow color with 11 was maintained throughout the reaction and 59% yield was obtained. Electronically, catalyst 11 has a weaker reduction potential vs SCE, and the oxidation potential (E1/2III* /II +0.55 V)29 does not surpass that of methyl indole (+1.18 V),30 possibly limiting unwanted activity of the higher-potential catalysts.
Control reactions without photocatalyst or without light did not yield any product, indicating that this is a photocatalyzed reaction. To determine if the light source is required for the duration of the experiment, a control reaction was performed where the LED lamp was turned off throughout the reaction course (SI Figure S1). The reaction progress stops in the absence of light and restarts when light is reintroduced, indicating that a constant influx of photons is required, but a chain process is not excluded.31
Initially, using catalyst 6, a screen of solvent and bases commonly used in photoredox reactions was initiated (Table 2). Using 2,6-lutidine as a base with solvents DMF, NMP, or dioxane resulted in lower yields than DCE (69% yield). It is possible to use sodium bicarbonate in place of 2,6-lutidine (71% yield). This is synthetically convenient as 2,6-lutidine must be distilled before use and is often difficult to separate from the product during purification. Sodium bicarbonate is used as a solid without any special measures and removed post reaction by aqueous extraction. Additionally, for more polar substrates not soluble in DCE, it is possible to use DMSO (Table 2, entry 6, 65% yield). Using the optimal catalyst [Ir(dmppy)2(dtbbpy)]PF6 (11) with DCE and sodium bicarbonate resulted in 61% isolated yield. Increasing the reaction concentration to 2 M increased the yield to 75%. At higher concentrations (4 M), the reaction is heterogeneous, resulting in a lower yield (44%). Lower yields are obtained when a 1:1 or 1:2 ratio of indole to bromoacetonitrile is used (Table 2: 34 and 41% yield).
Table 2.
Optimization of reaction conditions.
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Base | [M] | Equiv 1 | Equiv 2 | Yield (%)a |
| fac-lr(ppy)3 6 | ||||||
| 1 | DMF | 2,6-lutidine | 1 | 2 | 1 | 52 |
| 2 | NMP | 2,6-lutidine | 1 | 2 | 1 | 55 |
| 3 | Dioxane | 2,6-lutidine | 1 | 2 | 1 | 56 |
| 4 | DCE | 2,6-lutidine | 1 | 2 | 1 | 69 |
| 5 | DCE | NaHC03 | 1 | 2 | 1 | 71 |
| 6 | DMSO | NaHC03 | 1 | 2 | 1 | 65 |
| [lr(dmppy)2(dtbbpy)]PF611 | ||||||
| 7 | DCE | NaHC03 | 1 | 2 | 1 | 58, 61b |
| 8 | DCE | NaHC03 | 2 | 2 | 1 | 72, 75b |
| 9 | DCE | NaHC03 | 4 | 2 | 1 | 44 |
| 10 | DCE | NaHC03 | 2 | 1 | 1 | 34 |
| 11 | DCE | NaHC03 | 2 | 1 | 1.5 | 46 |
| 12 | DCE | NaHC03 | 2 | 1 | 2 | 41b |
Yield determined by HPLC calibrated assay, concentration based on limiting reagent.
Isolated yield.
A set of LCMS experiments were undertaken to characterize the impact of ligand architecture on catalyst deactivation (Figure 1).32–33 The reaction of Ir(ppy)3 6 with bromoacetonitrile under photoredox conditions proceeds in under 30 minutes. At this time point, there is no detectable [Ir(ppy)3+• m/z 655 remaining, and signals that correlate to multiple cyanomethyl additions to the catalyst appear: [Ir(ppy)3 + 2 CH2CN]+• m/z 734, [Ir(ppy)3 + 3 CH2CN]+• m/z 773, [Ir(ppy)3 + 4 CH2CN]+• m/z 812 (Figure 1a). Control reactions without light show mostly remaining photocatalyst with 11% alkylation. In contrast, blue LED excitation of catalyst 11 with bromoacetonitrile is mostly intact after 30 minutes ([Ir(dmppy)2(dtbbpy)]+ m/z 825). The only additional peak to appear, 18% by relative peak area, is the mono-addition adduct [Ir(dmppy)2(dtbbpy) + CH2CN]+ m/z 864. In the absence of light, there is no catalyst modification. Two factors contribute to the longer lifetime of catalyst 11: 1) slower reaction with cyanomethyl radical from increased ligand sterics; 2) slower cyanomethyl radical generation from the decreased reducing power of the catalyst.
To further probe the mechanism of the reaction, Stern-Volmer quenching experiments were performed using bromoacetonitrile, bromomalonate, or indole with catalyst 6 or 11 (Figure 2). In contrast to the electrochemical reduction potentials, the Stern-Volmer quenching constant using catalyst 6 is twofold higher for bromomalonate (−1.69 V, KSV 489) vs bromoacetonitrile (–0.69 V, KSV 235). Indole is not found to quench the fluorescence of catalyst 6. This indicates for either bromomalonate or bromoacetonitrile the photoredox catalytic cycle most likely follows the mechanism outlined in Scheme 2 with reductive quenching of the excited state photocatalyst. This study also demonstrates that despite the lower electrochemical reduction potential, bromoacetonitrile is a more difficult photoredox substrate than bromomalonate. To observe quenching of catalyst 11, a 10-fold higher concentration of substrate was necessary resulting in a bromomalonate KSV of 2 vs 489 for catalyst 6. For catalyst 11, the lower reducing potential (E1/2IV/III* vs SCE: 6, –0.87 V; 11, –1.73 V) and increased steric bulk contribute to the lower quenching constants. The KSV values for 11 with bromoacetonitrile and indole are 0.18 and 0.52 respectively. Data for indole could not be accurately collected above 0.1 M as the signal from indole overlapped with the photocatalyst emission peak. This indicates that the reaction for bromoacetonitrile and indole could proceed via either an oxidative or reductive pathway. The low quenching constants measured using catalyst 11 correlate with the prolonged reaction times and high concentrations that were found necessary to achieve higher product yields.
Figure 2.
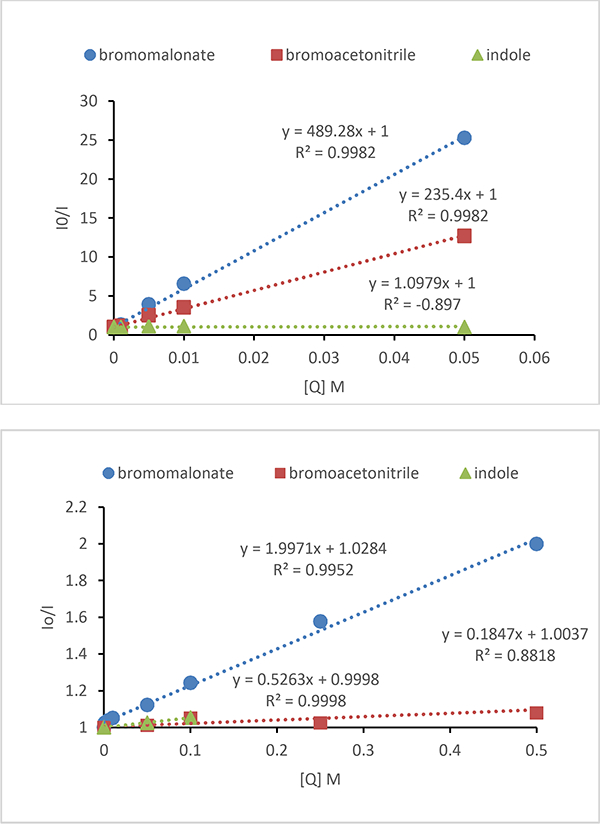
Stern-Volmer plots for bromomalonate, bromoacetonitrile, and indole using photocatalysts: a) Ir(ppy)3 (6) and b) [Ir(dmppy)2(dtbbpy)]PF6 (11).
A time-course following the generation of product is shown in Figure S2. Catalyst 6 rapidly generates product but then plateaus at 40% assay yield. Catalyst 11 also achieves 40% conversion at 7h but continues on to reach slightly higher assay yield (43%) at 24h. Since catalyst 11 is less prone to radical addition, consuming less bromoacetonitrile, we chose to explore the substrate scope with catalyst 11 (Figure 3).
Since the indole coupling partner is the more valuable reaction component, we first explored the substrate scope with 1.5 equiv of bromoacetonitrile (Scheme 3. For low yielding substrates, results using excess indole are also reported. Direct reaction with indole (12a) gave a modest 34% yield of cy-anomethylated indole (13a). Importantly, this showed that radical alkylation using an unprotected indole is possible. Next, we tested the scope of unprotected indoles functionalized at the 3-position and observed increased yields for methyl (13b, 61%) and phenyl (13c, 72%) analogs. Substituents at the 3-position likely increase yield by stabilizing the tertiary vs secondary benzyl radical intermediate enabling useful synthetic yields with just 1 equiv of indole and 1.5 equiv bromoacetonitrile. Changing the electronics of the 3-aryl substituent had little impact on reaction yield: fluoro (13d, 70%), chloro (13e, 61%), methoxy (13f, 50%), and thiophene (13g, 62%) were all well tolerated. For the more challenging substrates indole 12a or 3-methyl indole 12b, the yield is increased by using 2 equiv of indole and bromoacetonitrile as the limiting reagent (13a, 73%; 13b, 81%).
Scheme 3.
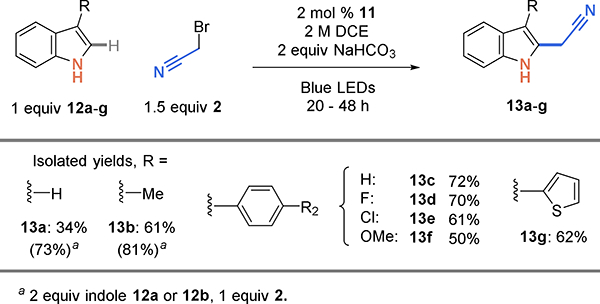
Cyanoalkylation scope of indoles substituted at the 3-position.
To probe the effect of electronic changes and functional group tolerance on the indole core, a set of 5-substituted indoles were tested (Scheme 4). Halogens F, Cl, and Br were well tolerated 15a-15c (64–78% yield). Although methyl ester 15d was obtained in high yield (73%), nitrile-containing 15e (33%) and trifluoromethyl 15f (40%) were not. Electron donating methyl 14g and methoxy 14h substituents resulted in lower yields of the desired cyanomethyl product (15g, 44%; 15h, 36%). During the reactions with electron-rich indoles, there is a marked color change from the initial yellow color of the catalyst to dark brown, which may indicate catalyst decomposition.
Scheme 4.
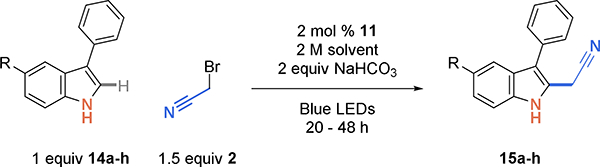
Cyanoalkylation scope of indoles substituted at the 5-position.
Reactions with 3-phenylindole and N-alkyl substituents such as methyl or isopropyl, shown in Scheme 5, afforded the cyanomethyl products in modest yields (17a, 67%; 17b, 47%). Using the standard conditions, 3-(4-fluoro)phenyl-N-isopropyl indole afforded 56% product (17c). Using 2 equiv of indole increased the yield to 90%. When cyclopropyl indole 16d was used, no formation of a ring-opened product was observed; however, the yield was restricted to 19% (17d).
Scheme 5.
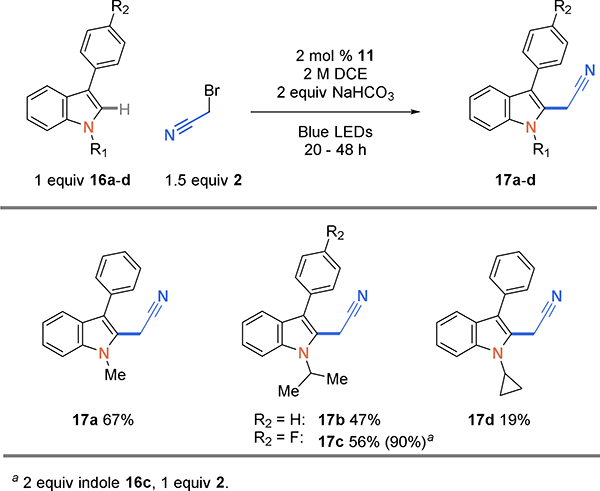
Cyanoalkylation scope of N-substituted indoles.
The addition of carbon-centered radicals to the 2-position of indoles is well precedented.34 However, when the 2-position is blocked, radical addition still proceeds at the electron-rich 3-position. For example, use of 2-methyl indole (Scheme 6, 18a) affords 2-methyl-3-cyanomethyl indole 19a in 16% yield. Using 3 equiv of indole increases the yield to 41%. An indole functionalized with an ethyl ester at the 2-position affords the 3-cyanomethylated product 19b in 51% yield. Aryl substituents at the 2-position are tolerated, and products with phenyl, 4-fluoro-phenyl, and 4-bromo-phenyl are obtained in 71–81% yield (19c-e).
Scheme 6.
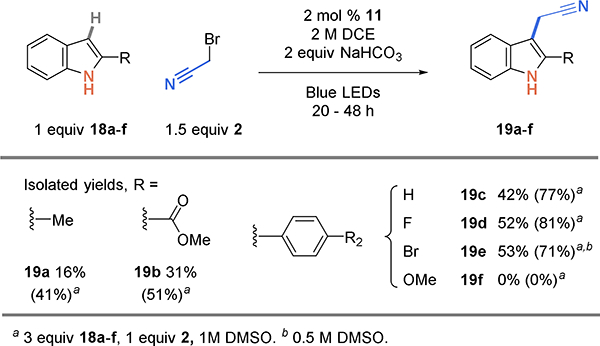
Cyanoalkylation scope of indoles substituted at the 2-position forcing alkylation at the 3-position.
Reactions with 5-azaindole (20) and 3-pyridoindole (22) afford undesired pyridinium-cyanoalkylated by-products in high yields (Scheme 7, 21 and 23). Further control reactions showed that the iridium photocatalyst is not necessary for product formation. Reaction of pyridine and chloroacetontirile to yield similar pyridinium-cyanoalkylated species are reported.35 This reactivity shows that although the photoredoxmediated cyanomethylation of indole provides high yields of product without N-alkylation, pyridines are readily alkylated and pose a limitation to the method.
Scheme 7.
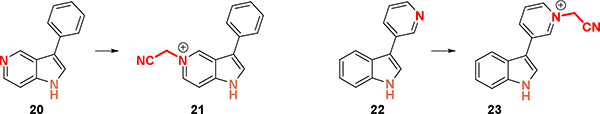
Failed reactions with pyridine substrates.
Conclusion
In summary, the direct cyanoalkylation of indoles via photoredox catalysis is demonstrated. Indole substrates with alkyl, aryl, halogen, ester, and ether functional groups underwent addition in 16–90% yields. The reaction provides rapid access to functionalized indoles without the use of high heat, high catalyst loading, or protecting groups, and greatly decreases the number of synthetic steps required for the formation of these biologically useful products.
Experimental Section
General Information.
Unless otherwise stated, all commercial reagents were purchased from Combi-Blocks. Iridium catalysts 6-11 were purchased from Aspira Chemical. All anhydrous solvents were purchased from EMD Millipore. Bromoacetonitrile was purchased from Acros and distilled prior to use. All other reagents and solvents were used without additional purification. Reactions were monitored by LCMS (Agilent Technologies G6100 Series LC/MSD Single Quad). Flash chromatography was carried out on a CombiFlash Rf + purification system using RediSep Rf Gold silica gel (20–40 μm) purchased from Teledyne Isco, Inc. Organic solutions were concentrated under reduced pressure on a Heidolph rotary evaporator. 1H and 13C{1H} NMR spectra were recorded on a Bruker Avance (400 MHz) spectrometer and are internally referenced to residual protio solvent signals (note: DMSO referenced at δ 2.50 ppm for 1H and δ 39.52 for 13C). Data for 1H NMR are reported as follows: chemical shift (δ ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, p =pentet, dd = doublet of doublets, ddd = doublet of doublet of doublets, m = multiplet, br = broad), integration, coupling constant (Hz) and assignment. HRMS data was collected at the University of California, Irvine Mass Spectrometry facility using a Waters Micromass LCT Premier Mass Spectrometer and analyzed by time-of-flight (TOF) measurements. The light source for photochemical reactions is a Westinghouse 15W, 120V Blue LED PAR38 reflector indoor outdoor lightbulb, model 03151.
Characterization of catalyst-cyanomethyl adducts by LCMS shown in Figure 1.
Catalyst (10 μmol, 6.5 mg 6 or 9.7 mg 11) was added to a flame-dried 2 dram vial. The vial was then capped, evacuated, and backfilled with argon 3 times. Then under positive pressure of argon, DMSO (0.25 mL) and bromoacetonitrile (35 μL, 0.5 mmol, 50 equiv) were added successively. The reaction mixture was then degassed with argon five minutes. The vial was subsequently placed directly in front of blue LEDs (at a distance of 0.75 cm from the front of the vial to the front of the light) and stirred for 30 min at room temperature. A 0.1 mL sample was then removed and diluted with 1 mL acetonitrile/water 0.5% TFA and analyzed by LCMS.
Luminescence quenching experiments shown in Figure 2.
Luminescence quenching experiments were carried out using a Varian Eclipse fluorometer. Catalyst 6 was excited at 385 nM, and emission was measured at 520 nM. Catalyst 11 was excited at 385 nM, and emission was measured at 592 nM. Emission intensities were recorded using 0.5 μM Ir(ppy)3 6 or 5 μM [Ir(dmppy)2(dtbbpy)]PF611 and the appropriate amount of quencher. Prior to analysis, solutions were degassed with argon for 15 min.
Synthesis of Substrates
3-(4-Methoxyphenyl)-1H-indole (12f).
Step 1: 4-methoxyphenethyl alcohol (826 mg, 5.42 mmol, 1.0 equiv) was added to a flame-dried round bottom flask and dissolved in dry dichloromethane (5 mL). The reaction mixture was cooled to 0 °C. Dess-Martin periodinane (DMP) (2.41 g, 5.69 mmol, 1.05 equiv) was slowly added over a 5 minute period. The reaction mixture was allowed to stir for 10 minutes at 0 °C before being warmed to room temperature and allowed to stir for an additional hour. After being determined complete by TLC, the reaction mixture was filtered. The filtrate was diluted in dichloromethane. The organic layer was quenched with saturated sodium thiosulfate (20 mL) and extracted. The organic layer was then washed with saturated aqueous sodium bicarbonate (20 mL) and brine (20 mL). The organic layer was extracted, dried over anhydrous magnesium sulfate, and filtered. The filtrate was concentrated in vacuo to give (4-methoxylphenyl)acetaldehyde (0.776 g, 95% yield) as a colorless oil.
Step 2: (4-methoxylphenyl)acetaldehyde (700 mg, 4.66 mmol, 1.0 equiv) and phenylhydrazine (503 mg, 4.66 mmol, 1.0 equiv) were added to a flame-dried vial. The vial was then fitted with a reflux condenser and stirred at room temperature for 30 minutes, followed by stirring at 100 °C for one hour. After cooling to room temperature, a solution of zinc chloride (1.14 g, 8.38 mmol, 2.0 equiv) in ethanol (10 mL) was added and allowed to stir at reflux for 24 hours. The reaction was cooled to room temperature and determined complete by TLC.
To dry load the product on silica for purification, silica gel and dichloromethane were added successively to the reaction mixture. The reaction mixture was then concentrated in vacuo before being purified by silica gel column chromatography, eluting with a gradient ramp of heptanes:ethyl acetate 2:1 to give 3-(4-methoxyphenyl)-1H-indole (12f) (0.911 g, 87% yield) as a light brown solid. Analytical data are consistent with reported values.36 1H NMR (400 MHz, DMSO-d6) δ 11.25 (br s, 1H), 7.81 (d, J = 8.07 Hz, 1H), 7.53 – 7.64 (m, 3H), 7.43 (d, J = 7.83 Hz, 1H), 7.14 (t, J = 7.09 Hz, 1H), 7.07 (t, J = 7.09 Hz, 1H), 6.96 – 7.04 (m, 2H), 3.79 (s, 3H). 13C NMR (101 MHz, DMSO-d6) d 157.7, 137.3, 128.8, 128.1, 125.6,123.0, 121.7, 119.8, 119.4, 116.0, 114.7, 112.3, 55.5.
3-Phenyl-1H-indole (12c).
Synthesized according to the procedure described for substrate 12f. Analytical data are consistent with reported values (854 mg, 88% yield).36 1H NMR (400 MHz, DMSO-d6) δ 11.36 (br. s., 1H), 7.87 (d, J = 8.07 Hz, 1H), 7.65 – 7.73 (m, 3H), 7.39 – 7.50 (m, 3H), 7.20 – 7.28 (m, 1H), 7.13 −7.19 (m, 1H), 7.07 – 7.12 (m, 1H). 13C NMR (101 MHz, DMSO-d6) d 137.4, 136.3, 129.2, 127.0, 125.7, 125.4, 123.9, 121.9, 120.1, 119.5, 116.1, 112.4.
3-(4-Fluorophenyl)-1H-indole (12d).
Synthesized according to the procedure described for substrate 12f. Analytical data are consistent with reported values (305 mg, 76% yield).37 1H NMR (400 MHz, DMSO-d6) δ 11.37 (br s, 1H), 7.83 (d, J = 7.58 Hz, 1H), 7.65 – 7.76 (m, 3H), 7.45 (d, J = 8.07 Hz, 1H), 7.22 – 7.30 (m, 2H), 7.13 – 7.19 (m, 1H), 7.06 – 7.12 (m, 1H). 13C NMR (101 MHz, DMSO-d6) d 162.0, 159.6, 137.3, 132.8, 132.7, 128.7, 128.6, 125.3, 123.9, 121.9, 120.1, 119.2, 116.1, 115.9, 115.1, 112.4.
3-(4-Chlorophenyl)-1H-indole (12e).
Synthesized according to the procedure described for substrate 12f. Analytical data are consistent with reported values (260 mg, 65% yield).36 1H NMR (400 MHz, DMSO-d6) δ 11.45 (br s, 1H), 7.86 (d, J = 8.7 Hz, 1H), 7.69 – 7.80 (m, 3H), 7.41 – 7.51 (m, 3H), 7.06 – 7.21 (m, 2H). 13C NMR (101 MHz, DMSO-d6) d 137.4, 135.3, 130.0, 129.2, 128.4, 125.2, 124.4, 122.0, 120.3, 119.3, 114.8, 112.5.
3-(Thiophen-2-yl)-1H-indole (12g).
Synthesized according to the procedure described for substrate 12f. Analytical data are consistent with reported values (248 mg, 61% yield).38 1H NMR (400 MHz, DMSO-d6) δ 11.42 (br s, 1H), 7.88 (d, J =7.82 Hz, 1H), 7.72 (d, J = 2.69 Hz, 1H), 7.45 (d, J = 7.58 Hz, 1H), 7.39 (dd, J = 1.10, 5.01 Hz, 1H), 7.33 (dd, J = 0.98, 3.67 Hz, 1H), 7.05 – 7.23 (m, 4H). 13C NMR (101 MHz, DMSO-d6) d 138.4, 137.0, 128.2, 125.1, 123.7, 122.6, 122.2, 122.0, 119.5, 112.5, 110.2.
5-Bromo-3-phenyl-1H-indole (14a).
Synthesized according to the procedure described for substrate 12f. Analytical data are consistent with reported values (449 mg, 75% yield).37,1H NMR (400 MHz, DMSO-d6) δ 11.60 (br s, 1H), 7.97 (d, J = 1.96 Hz, 1H), 7.77 (d, J = 2.69 Hz, 1H), 7.62 – 7.69 (m, 2H), 7.41 – 7.49 (m, 3H), 7.22 – 7.31 (m, 2H). 13C NMR (101 MHz, DMSO-d6) d 135.8, 135.5, 129.4, 127.0, 126.5, 126.1, 125.7, 124.8, 121.9, 118.6, 116.0, 114.0.
5-Chloro-3-phenyl-1H-indole (14b).
Synthesized according to the procedure described for substrate 12f. Analytical data are consistent with reported values (461 mg, 78% yield).37 1H NMR (400 MHz, DMSO-d6) δ 11.59 (br s, 1H), 7.84 (d, J = 1.96 Hz, 1H), 7.79 (d, J = 2.69 Hz, 1H), 7.62 – 7.72 (m, 2H), 7.39 – 7.52 (m, 3H), 7.21 – 7.29 (m, 1H), 7.17 (dd, J = 1.96, 8.56 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) d 136.0, 135.5, 127.2, 127.1, 126.1, 125.5, 124.4, 121.5, 115.9, 114.5, 112.8.
5-Fluoro-3-phenyl-1H-indole (14c).
Synthesized according to the procedure described for substrate 12f. Analytical data are consistent with reported values (332 mg, 52% yield).37,1H NMR (400 MHz, DMSO-d6) δ 11.48 (br s, 1H), 7.79 (d, J =2.69 Hz, 1H), 7.67 (d, J = 7.09 Hz, 2H), 7.57 (dd, J = 2.45, 10.51 Hz, 1H), 7.40 – 7.49 (m, 3H), 7.20 – 7.28 (m, 1H), 7.01 (dt, J = 2.57, 9.11 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) d 159.1, 156.8, 135.8, 134.1, 129.3, 126.8, 125.9, 125.9, 125.6, 125.4, 116.4, 116.4, 113.5, 113.4, 110.2, 110.0, 104.3, 104.1.
Methyl-3-phenyl-1H-indole-5-carboxylate (14d).
Synthesized according to the procedure described for substrate 12f 407 mg, 71% yield). 1H NMR (400 MHz, DMSO-d6) δ 11.78 (br s, 1H), 8.46 – 8.56 (m, 1H), 7.78 – 7.84 (m, 2H), 7.64 – 7.70 (m, 2H), 7.54 – 7.62 (m, 1H), 7.49 (t, J =7.83 Hz, 2H), 7.24 – 7.34 (m, 1H), 3.86 (s, 3H). 13C NMR (101 MHz, DMSO-d6) d167.7, 139.9, 135.4, 129.4, 127.3, 126.4, 125.7, 125.1, 122.9, 121.9, 121.5, 117.6, 112.5, 52.3.
3-Phenyl-1H-indole-5-carbonitrile (14e).
Synthesized according to the procedure described for substrate 12f. Analytical data are consistent with reported values (268 mg, 49% yield).39 1H NMR (400 MHz, DMSO-d6) δ 11.95 (br s, 1H), 8.34 (s, 1H), 7.92 (d, J = 2.45 Hz, 1H), 7.68 – 7.75 (m, 2H), 7.60 – 7.65 (m, 1H), 7.41 – 7.54 (m, 3H), 7.29 (t, J = 7.34 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) d 139.0, 134.8, 129.4, 127.3, 126.5, 126.5, 125.2, 125.2, 124.7, 121.2, 117.2, 113.7, 102.2.
3-Phenyl-5-(trifluoromethyl)-1H-indole (14f).
Synthesized according to the procedure described for substrate 12f (287 mg, 56% yield). 1H NMR (400 MHz, DMSO-d6) δ 11.84 (br s, 1H), 8.13 (s, 1H), 7.89 (s, 1H), 7.62 – 7.74 (m, 3H), 7.48 (t, J = 7.70 Hz, 3H), 7.29 (t, J = 7.34 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) d 138.8, 135.2, 129.5, 127.3, 126.4, 126.2, 124.8, 121.0, 120.7, 118.4, 118.3, 117.3, 116.8, 116.7, 113.3.
5-Methyl-3-phenyl-1H-indole (14g).
Synthesized according to the procedure described for substrate 12f. Analytical data are consistent with reported values (516 mg, 78% yield).37 1H NMR (400 MHz, DMSO-d6) δ 11.23 (br. s., 1H), 7.65 – 7.74 (m, 3H), 7.63 (d, J = 2.69 Hz, 1H), 7.42 (t, J = 7.70 Hz, 2H), 7.33 (d, J = 8.07 Hz, 1H), 7.18 – 7.26 (m, 1H), 6.93 – 7.04 (m, 1H), 2.42 (s, 3H). 13C NMR (101 MHz, DMSO-d6) d 136.6, 135.8, 129.2, 128.6, 126.9, 125.7, 125.6, 123.9, 123.5, 119.1, 115.7, 112.1,21.9.
5-Methoxy-3-phenyl-1H-indole (14h).
Synthesized according to the procedure described for substrate 12f. Analytical data are consistent with reported values (354 mg, 62% yield).36 1H NMR (400 MHz, DMSO-d6) δ 11.23 (br. s., 1H), 7.58 – 7.72 (m, 3H), 7.39 – 7.46 (m, 2H), 7.36 (d, J = 8.56 Hz, 1H), 7.31 (d, J = 2.20 Hz, 1H), 7.19 – 7.26 (m, 1H), 6.82 (dd, J = 2.45, 8.80 Hz, 1H), 3.80 (s, 3H). 13C NMR (101 MHz, DMSO-d6) d 154.4, 136.5, 132.5, 129.3, 126.8, 125.7, 125.6, 124.5, 116.0, 113.1, 112.0, 101.2, 55.8.
1-Methyl-3-phenyl-1H-indole (16a).
3-phenyl-1H-indole (200 mg, 1.03 mmol, 1.0 equiv) was added to a flame-dried vial and dissolved in dry N,N-dimethylformamide. The reaction mixture was cooled to 0 °C, and sodium hydride (41.1 mg, 1.03 mmol, 1.0 equiv) was slowly added and allowed to stir at 0 °C for ten minutes before being warmed to room temperature and stirred for 1 hour. The reaction was then cooled to 0 °C, and iodomethane (174 mg, 1.23 mmol, 1.2 equiv) was added and allowed to stir at 0 °C for ten minutes before being warmed to room temperature and stirred overnight. The reaction was determined complete by TLC. The reaction mixture was then diluted in dichloromethane and the organic layer was washed with saturated aqueous sodium bicarbonate (15 mL) before being extracted, washed with brine (15 mL), dried over anhydrous magnesium sulfate, and filtered. To dry load the product on silica for purification, silica gel and dichloromethane were added successively to the resulting filtrate. The reaction mixture was then concentrated in vacuo before being purified by silica gel column chromatography eluting with a gradient ramp of heptanes:ethyl acetate 3:1 to give 1-methyl-3-phenyl-1H-indole (16a, 0.186 g, 87% yield) as a pale yellow solid. Analytical data are consistent with reported values.37 1H NMR (400 MHz, DMSO-d6) δ 7.90 (d, J = 8.07 Hz, 1H), 7.64 – 7.71 (m, 3H), 7.50 (d, J = 8.07 Hz, 1H), 7.41 – 7.48 (m, 2H), 7.21 – 7.30 (m, 2H), 7.11 – 7.20 (m, 1H), 3.83 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 137.7, 136.0, 129.3, 128.1, 126.9, 125.8, 122.0, 120.3, 119.7, 115.3, 110.7, 33.0.
1-Isopropyl-3-phenyl-1H-indole (16b).
Synthesized according to the procedure described for 16a. Analytical data are consistent with reported values (161 mg, 89% yield).37,1H NMR (400 MHz, DMSO-d6) δ 7.80 – 7.95 (m, 2H), 7.66 – 7.74 (m, 2H), 7.58 (d, J = 8.07 Hz, 1H), 7.40 – 7.48 (m, 2H), 7.18 – 7.28 (m, 2H), 7.08– 7.17 (m, 1H), 4.81 (spt, J = 6.68 Hz, 1H), 1.52 (d, J = 6.60 Hz, 6H). 13C NMR (101 MHz, DMSO-d6) d 136.6, 136.1, 129.2, 127.0, 125.8, 125.8, 123.4, 121.9, 120.3, 119.8, 115.8, 110.8, 47.0, 22.9.
1-Cyclopropyl-3-phenyl-1H-indole (16d).
3-phenyl-1H-indole (300 mg, 1.55 mmol, 1.0 equiv), cyclopropylboronic acid (266 mg, 3.10 mmol, 2.0 equiv), copper(II) acetate (281 mg, 1.55 mmol, 1.0 equiv), 2,2’-bipyridine (1, 242 mg, 1.55 mmol 1.0 equiv), and sodium carbonate (328 mg, 3.10 mmol, 2.0 equiv) were added to a vial and dissolved in 1,2-dichloroethane (8 mL). The reaction mixture was allowed to stir at room temperature, open to the atmosphere, overnight. The reaction was determined complete by TLC. To dry load the product on silica for purification, silica gel and dichloromethane were added successively to the reaction mixture. The reaction mixture was then concentrated in vacuo before being purified by column chromatography eluting with a gradient ramp of heptanes:ethyl acetate 2:1 to give 1-cyclopropyl-3-phenyl-1H-indole (16d, 0.303 g, 84% yield) as a colorless oil. 1H NMR (400 MHz, DMSO-d6) δ 7.87 (d, J = 7.83 Hz, 1H), 7.60 – 7.73 (m, 4H), 7.43 (t, J = 7.70 Hz, 2H), 7.22 – 7.32 (m, 2H), 7.13 – 7.20 (m, 1H), 3.50 (tt, J = 3.73, 7.03 Hz, 1H), 0.94 – 1.17 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ 138.4, 135.8, 129.2, 127.1, 126.8, 126.2, 125.9, 122.2, 120.8, 119.9, 115.6, 111.2, 27.4, 6.5.
3-phenyl-1H-pyrrolo[2,3-c]pyridine (20).
Step 1: tert-butyl-3-bromo-1H-pyrrolo[2,3-c]pyridine-1-carboxylate (300 mg, 1.00 mmol, 1.0 equiv), phenylboronic acid (182 mg, 1.50 mmol, 1.5 equiv), [1,1’Bis(diphenylphosphino)ferrocene]palladium(II) dichloride (36.3 mg, 50.0 μmol, 0.05 equiv), and cesium carbonate (651 mg, 2.00 mmol, 2.0 equiv) were added to a flame-dried vial and dissolved in dioxane (4 mL). The solution was then sparged with argon for five minutes. Then, under positive pressure, water (0.4 mL) was added to the reaction mixture. The reaction vial was sealed and stirred for 16 hours at 100 °C. The reaction was cooled to room temperature and determined complete by TLC. To dry load the product on silica for purification, silica gel and dichloromethane were added successively to the reaction mixture. The reaction mixture was then concentrated in vacuo before being purified by silica gel column chromatography eluting with a gradient ramp of dichloromethane:methanol 9:1 to give tert-butyl 3-phenyl-1H-pyrrolo[2,3-c]pyridine-1-carboxylate (0.212 g, 71% yield) as a white solid.
Step 2: tert-butyl-3-phenyl-1H-pyrrolo[2,3-c]pyridine-1-carboxylate (200 mg, 679 μmol, 1.0 equiv) was added to a vial and dissolved in methanol (3mL). 4M HCl in dioxane (3.37mL) was added to the reaction mixture and allowed to stir at room temperature for one hour, then at 50 °C for two hours. The crude mixture was then cooled to room temperature and concentrated in vacuo. The resulting solid was dissolved in dichloromethane and washed with saturated sodium bicarbonate (15 mL) solution. The organic layer was extracted, dried over anhydrous sodium sulfate, and concentrated in vacuo to give 3-phenyl-1H-pyrrolo[2,3-c]pyridine (20, 0.122 g, 93% yield) as a colorless oil. 1H NMR (400 MHz, DMSO-d6) δ 11.77 (br s, 1H), 9.17 (s, 1H), 8.24 (d, J = 5.62 Hz, 1H), 7.82 (s, 1H), 7.73 – 7.79 (m, 2H), 7.40 – 7.50 (m, 3H), 7.21 – 7.32 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 142.6, 140.8, 140.8, 135.1, 129.4, 127.2, 126.4, 124.9, 122.6, 116.1, 107.7.
3-(pyridin-3-yl)-1H-indole (22).
Step 1: tert-butyl 3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-indole-1 -carboxylate (415 mg, 1.21 mmol, 1.1 equiv), 1,1’Bis(diphenylphosphino)ferrocene] palladium(II) dichloride (40.0 mg, 55.0 μmol, 0.05 equiv), and cesium carbonate (716 mg, 2.20 mmol, 2.0 equiv) were added to a flame-dried vial. The vial was capped, and under positive pressure of argon, 1,4-dioxane (5 mL) and 3-bromopyridine (175 mg, 1.10 mmol, 1.0 equiv) were added successively. The reaction mixture was then sparged with argon for five minutes. Then, under positive pressure of argon, water (0.5 mL) was added. The vial was then heated to 90 °C for twelve hours. The reaction was cooled to room temperature and determined complete by TLC. To dry load the product on silica for purification, silica gel and dichloromethane were added successively to the reaction mixture. The reaction mixture was then concentrated in vacuo before being purified by silica gel column chromatography eluting with a gradient ramp of dichloromethane:methanol 19:1 to give tert-butyl 3-(pyridin-3-yl)-1H-indole-1-carboxylate (0.319 g, 90% yield) as a pale yellow solid.
Step 2: tert-butyl 3-(pyridin-3-yl)-1H-indole-1-carboxylate was added to a vial and dissolved in methanol (3mL). 4M HCl in dioxane (3.37mL) was added to the reaction mixture and allowed to stir at room temperature for one hour, then at 50 °C for two hours. The crude mixture was then cooled to room temperature and concentrated in vacuo. The resulting solid was dissolved in dichloromethane and washed with saturated sodium bicarbonate solution (15 mL). The organic layer was extracted, dried over anhydrous sodium sulfate, and concentrated in vacuo to give 3-(pyridin-3-yl)-1H-indole 22 (0.195 g, 92% yield) as a colorless oil. Analytical data are consistent with reported values.40 ‘H NMR (400 MHz, DMSO-d6) δ 11.85 (br s, 1H), 9.16 (s, 1H), 8.60 – 8.76 (m, 2H), 8.10 (d, J = 2.93 Hz, 1H), 7.98 (d, J = 7.83 Hz, 1H), 7.90 (dd, J = 5.50, 7.95 Hz, 1H), 7.53 (d, J = 7.83 Hz, 1H), 7.12 – 7.30 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 141.3, 140.6, 139.5, 137.5, 134.9, 126.8, 126.7, 124.8, 122.7, 121.1, 119.2, 112.9, 110.4.
Synthesis of Cyanomethyl-indole Products
2-(3-phenyl-1H-indol-2-yl)acetonitrile (13c).
3-phenyl-1H-indole (12c, 67.6 mg, 350 μmol, 1.0 equiv), sodium bicarbonate (58.8 mg, 700 μmol, 2.0 equiv), and the [4,4’-Bis(tert-butyl)-2,2’-bipyridine]bis[5-methyl-2-(4-methyl-2-pyridinyl) phenyl]iridium(IH) hexafluorophosphate (11, 6.8 mg, 7 μmol, 2.0 mol %) were added to a flame-dried 2 dram vial. The vial was then capped, evacuated, and backfilled with argon 3 times. Then under positive pressure of argon, 1,2-dichloroethane (175 μL) and bromoacetonitrile (36.5 μL, 525 μmol, 1.5 equiv) were added successively. The reaction mixture was then degassed with argon for five minutes. The vial was subsequently placed directly in front of blue LEDs (at a distance of 0.75 cm from the front of the vial to the front of the light) and stirred for 24 hours at room temperature. The reaction was then dry loaded onto silica by adding silica gel, dichloromethane, and evaporating to dryness. The product was then purified by silica gel column chromatography using a 40g silica gel column eluting with a gradient of 0–25% ethyl acetate in heptanes. 2-(3-phenyl-1H-indol-2-yl)acetonitrile (13c, yellow solid, 58 mg, 72% yield). Melting point 109–111 °C. ‘H NMR (400 MHz, DMSO-d6) δ 11.61 (s, 1H), 7.41 – 7.65 (m, 7H), 7.33 – 7.40 (m, 1H), 7.15 – 7.23 (m, 1H), 7.04 – 7.12 (m, 1H), 4.18 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 136.1, 134.3, 129.4, 129.4, 126.9, 126.8, 124.7, 122.6, 120.4,119.0, 118.3, 115.2, 112.0, 15.9. HRMS (ESI): [M-H]-, C16H11N2, calculated: 231.0922; found: 231.0928.
2-(1-methyl-1H-indol-2-yl)acetonitrile (3).
Synthesized according to the procedure described for 13c. Light brown solid, 32 mg, 75% yield. Melting point 87–89 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.53 (d, J = 7.83 Hz, 1H), 7.44 (d, J = 8.31 Hz, 1H), 7.13 – 7.22 (m, 1H), 7.01 – 7.08 (m, 1H), 6.47 (s, 1H), 4.33 (s, 2H), 3.71 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 137.9, 130.5, 127.2, 121.9, 120.5, 119.9, 118.2, 110.1,101.0, 30.0, 16.1. HRMS (ESI): [M]+• C11H10N2, calculated: 170.0844; found: 170.0844.
2-(1H-indol-2-yl)acetonitrile (13a).
Synthesized according to the procedure described for 13c using 2 equiv of indole 12a and 1 equiv bromoacetonitrile 2. White solid, 29 mg, 73% yield. Analytical data are consistent with reported values.41 1H NMR (400 MHz, DMSO-d06) δ 11.29 (br s, 1H), 7.49 (d, J =7.82 Hz, 1H), 7.35 (dd, J = 0.86, 8.19 Hz, 1H), 7.08 (dt, J = 1.10, 7.52 Hz, 1H), 6.94 – 7.03 (m, 1H), 6.35 – 6.44 (m, 1H), 4.19 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 136.9, 128.9, 128.1, 121.8, 120.3, 119.7, 118.5, 111.6, 101.0, 17.1. HRMS (ESI): [M-H]-, C10H7N2, calculated: 155.0609; found: 155.0611.
2-(3-Methyl-1H-indol-2-yl)acetonitrile (13b).
Synthesized according to the procedure described for 13c using 2 equiv of indole 12b and 1 equiv bromoacetonitrile 2. Pale yellow solid, 34 mg, 81% yield. Analytical data are consistent with reported values.42 1H NMR (400 MHz, DMSO-d6) δ 11.02 (br. s., 1H), 7.45 (d, J = 7.83 Hz, 1H), 7.32 (d, J = 8.07 Hz, 1H), 7.05 – 7.12 (m, 1H), 6.97 – 7.03 (m, 1H), 4.12 (s, 2H), 2.23 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 136.0, 128.7, 124.1, 121.9, 119.0, 118.7, 118.4, 111.5, 108.3, 14.9, 8.5. HRMS (ESI): [M-H]-, C11H9N2, calculated: 169.0766; found: 169.0764.
2-(3(4-Fluorophenyl)-1H-indol-2-yl)acetonitrik (13d).
Synthesized according to the procedure described for 13c. Pale yellow solid, 44 mg, 70% yield. Melting point 129–131 °C. ‘H NMR (400 MHz, DMSO-d6) δ 11.61 (s, 1H), 7.43 – 7.56 (m, 4H), 7.29 – 7.41 (m, 2H), 7.18 (t, J = 7.09 Hz, 1H), 7.02 – 7.11 (m, 1H), 4.18 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ162.6, 160.2, 136.0, 131.3, 131.2, 130.6, 126.9, 124.7, 122.7,120.4, 118.8, 118.2, 116.3, 116.1, 114.2, 112.0, 15.8. HRMS ESI): [M]+·, C16H11FN2, calculated: 250.0906; found: 250.0901.
2-(3-(4-Chlorophenyl)-1H-indol-2-yl)acetonitrile (13e).
Synthesized according to the procedure described for 13c. Yellow solid, 41 mg, 61% yield. Melting point 139–142 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.68 (s, 1H), 7.38 – 7.64 (m, 6H), 7.19 (dt, J = 0.98, 7.58 Hz, 1H), 7.05 – 7.13 (m, 1H). 13C NMR (101 MHz, DMSO-d6) δ 136.1, 133.2, 131.4, 131.1, 129.4, 126.7, 125.1, 122.8, 120.5, 118.8, 118.1, 113.9, 112.1, 15.9. HRMS (ESI): [M-H]-, C16H10ClN2, calculated: 265.0533; found: 265.0528.
2-(3-(4-Methoxyphenyl)-1H-indol-2-yl)acetonitrile (13f).
Synthesized according to the procedure described for 13c. Yellow solid, 33 mg, 50% yield. Melting point 143–144 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.52 (s, 1H), 7.52 (d, J = 8.07 Hz, 1H), 7.45 (d, J = 8.31 Hz, 1H), 7.37 – 7.42 (m, 2H), 7.14 – 7.22 (m, 1H), 7.02– 7.13 (m, 3H), 4.14 (s, 2H), 3.82 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 158.4, 136.1, 130.5, 127.1, 126.4, 124.2, 122.5, 120.2, 119.0, 118.3, 115.0, 114.9, 112.0, 55.6, 15.9. HRMS (ESI): [M-H]-, C17H13N2O, calculated: 261.1028; found: 261.1027.
2-(3-(Thiophen-2-yl)-1H-indol-2-yl)acetonitrile (13g).
Synthesized according to the procedure described for 13c. Yellow solid, 37 mg, 62% yield. Melting point 162–164 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.73 (s, 1H), 7.73 (d, J = 8.07 Hz, 1H), 7.61 (dd, J = 0.98, 5.14 Hz, 1H), 7.47 (d, J = 8.07 Hz, 1H), 7.17 – 7.27 (m, 3H), 7.06 – 7.17 (m, 1H), 4.27 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 135.9, 135.6, 128.3, 126.7,125.5, 125.4, 125.3, 123.0, 120.7, 119.2, 117.9, 112.2, 108.4, 16.2. HRMS (ESI): [M-H]-, C14H9N2S, calculated: 237.0486; found: 237.0488.
2-(5-Bromo-3-phenyl-1H-indol-2-yl)acetonitrile (15a).
Synthesized according to the procedure described for 13c. White solid, 57 mg, 74% yield. Melting point 180–182 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.87 (s, 1H), 7.63 (d, J = 1.71 Hz, 1H), 7.50 – 7.59 (m, 2H), 7.43 – 7.49 (m, 3H), 7.35 – 7.42 (m, 1H), 7.31 (dd, J = 1.83, 8.68 Hz, 1H), 4.21 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 134.8, 133.5, 129.5, 129.4, 128.7,127.1, 126.4, 125.2, 121.1, 118.0, 114.9, 114.2, 112.9, 15.9. HRMS (ESI): [M-H]-, C16H10BrN2, calculated: 309.0027; found: 309.0025.
2-(5-Chloro-3-phenyl-1H-indol-2-yl)acetonitrile (15b).
Synthesized according to the procedure described for 13c. White solid, 52 mg, 78% yield. Melting point 178–179 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.84 (s, 1H), 7.44 – 7.61 (m, 7H), 7.36 – 7.42 (m, 1H), 7.15 – 7.23 (m, 1H), 4.21 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 134.6, 133.5, 129.5, 129.4,128.0, 127.1, 126.6, 125.0, 122.6, 118.1, 118.0, 115.0, 113.7,16.0 HRMS (ESI): [M-H]-, C16H10ClN2, calculated: 265.0538; found: 265.0538.
2-(5-Flouro-3-phenyl-1H-indol-2-yl)acetonitrile (15c).
Synthesized according to the procedure described for 13c. White solid, 40 mg, 64% yield. Melting point 153–155 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.73 (s, 1H), 7.44 – 7.58 (m, 6H),7.33 – 7.41 (m, 1H), 7.24 (dd, J = 2.32, 9.90 Hz, 1H), 7.04 (dt, J = 2.57, 9.11 Hz, 1H), 4.20 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 159.2, 156.8, 133.8, 132.7, 129.5, 129.2, 127.1,127.0, 127.0, 126.8, 118.1, 115.4, 115.3, 113.3, 113.2, 111.0,110.7, 103.8, 103.6, 16.0. HRMS (ESI): [M]+• C16H11FN2, calculated: 250.0906; found: 250.0907.
Methyl 2-(Cyanomethyl)-3-phenyl-1H-indole-5-carboxylate (15d).
Synthesized according to the procedure described for 13c using DMSO instead of DCE. Yellow solid, 53 mg, 73% yield. Melting point 235–238 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.04 (s, 1H), 8.19 (s, 1H), 7.82 (dd, J = 1.47, 8.56 Hz, 1H), 7.53 – 7.61 (m, 3H), 7.46 – 7.51 (m, 2H), 7.37 – 7.45 (m, 1H), 4.23 (s, 2H), 3.83 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 167.5, 138.7, 133.4, 129.6, 129.5, 127.3, 126.7, 126.6,123.6, 121.8, 121.5, 118.0, 116.5, 112.2, 52.3, 15.9. HRMS (ESI): [M-H]-, C18H13N2O2, calculated: 289.0977; found: 289.0984.
2-(Cyanomethyl)-3-phenyl-1H-indole-carbonitrile (15e).
Synthesized according to the procedure described for 13c using DMSO instead of DCE. Light brown solid, 21 mg, 33% yield. Melting point 172–174 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.23 (s, 1H), 7.99 (s, 1H), 7.62 – 7.66 (m, 1H), 7.49 – 7.58 (m, 6H), 7.38 – 7.44 (m, 1H), 4.26 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 137.9, 132.9, 129.6, 129.5, 127.6, 127.4,126.7, 125.4, 124.7, 120.9, 117.8, 116.0, 113.4, 102.6, 16.0. HRMS (ESI): [M-H]-, C17H10N3, calculated: 256.0875; found: 256.0876.
2-(3-phenyl-5-(trifluoromethyl)-1H-mdol-2-yl)acetonitrile (15f).
Synthesized according to the procedure described for 13c using DMSO instead of DCE. White solid, 31 mg, 41% yield. Melting point 156–158 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.12 (s, 1H), 7.81 (s, 1H), 7.68 (d, J = 8.56 Hz, 1H), 7.53 – 7.60 (m, 2H), 7.47 – 7.52 (m, 3H), 7.38 – 7.44 (m, 1H),4.25 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 137.7, 133.2,129.6, 129.5, 127.4, 127.3, 127.2, 126.3, 124.5, 121.4, 121.1,119.1 119.1, 117.9, 116.4, 116.3, 116.1, 113.0, 15.9. HRMS (ESI): [M-H]-, C17H10F3N2, calculated: 299.0796; found: 299.0797.
2-(5-Methyl-3-phenyl-1H-indol-2-yl)acetonitrile (15g).
Synthesized according to the procedure described for 13c using DMSO instead of DCE. Brown solid, 27 mg, 44% yield. Melting point 149–151 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.46 (s, 1H), 7.41 – 7.56 (m, 4H), 7.27 – 7.39 (m, 3H), 6.95 – 7.06 (m, 1H), 4.14 (s, 2H), 2.37 (s, 3H). 13C NMR (101 MHz, DMSO-d6) d 134.5, 129.4, 128.9, 127.1, 126.7, 124.6, 124.2, 118.5, 118.3, 114.8, 111.7, 21.7, 15.9. HRMS (ESI): [M-H]-, C17H13N2, calculated: 245.1079; found: 245.1079.
2-(5-Methoxy-3-phenyl-1H-indol-2-yl)acetonitrile (15h).
Synthesized according to the procedure described for 13c using DMSO instead of DCE. White solid, 24 mg, 36% yield. Melting point 132–134 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.45 (s, 1H), 7.45 – 7.55 (m, 4H), 7.30 – 7.40 (m, 2H), 7.00 (d, J = 2.20Hz, 1H), 6.83 (dd, J = 2.45, 8.80 Hz, 1H), 4.14 (s, 2H), 3.73 (s, 3H). 13C NMR (101 MHz, DMSO-d6) d 4.5, 134.5,131.1, 129.4, 129.2, 127.2, 126.7, 125.2, 118.3, 115.0, 112.8, 100.5, 55.8, 16.0. HRMS (ESI): [M-H]-, C17H13N2O, calculated: 261.1028; found: 261.1035.
2-(1-Methyl-3-phenyl-1H-indol-2-yl)acetonitrile (17a).
Synthesized according to the procedure described for 13c. Yellow oil, 41 mg, 67% yield. Melting point 64–66 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.49 – 7.60 (m, 4H), 7.45 (d, J = 6.85 Hz, 2H), 7.34 – 7.42 (m, 1H), 7.24 – 7.30 (m, 1H), 7.07 – 7.17 (m, 1H), 4.29 (s, 2H), 3.88 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 137.2, 134.1, 129.6, 129.5, 127.0, 126.2, 126.1, 122.9, 119.2, 117.9, 115.7, 110.6, 30.4, 14.6. HRMS (ESI): [M]+\ C17H14N2, calculated: 246.1157; found: 246.1162.
2-(1-Isopropyl-3-phenyl-1H-indol-2-yl)acetonitrile (17b).
Synthesized according to the procedure described for 13c. Yellow solid, 32 mg, 47% yield. Melting point 67–69 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.74 (d, J = 8.31 Hz, 1H), 7.49–7.60 (m, 3H), 7.35 – 7.47 (m, 3H), 7.21 (t, J = 7.58 Hz, 1H), 7.09 (t, J = 7.46 Hz, 1H), 4.80 – 4.97 (m, 1H), 4.25 (s, 2H), 1.67 (d, J = 6.85 Hz, 6H). 13C NMR (101 MHz, DMSO-d6) δ134.7, 134.2, 129.9, 129.5, 127.4, 127.1, 125.7, 122.6, 120.3, 118.2, 115.8, 112.8, 48.1, 21.4, 15.0. HRMS (ESI): [M]+• C19H18N2, calculated: 274.1470; found: 274.1477.
2-(3-(4-Fluorophenyl)-1-isoropyl-1H-indol-2-yl)acetonitrile (17c).
Synthesized according to the procedure described for 13c using 2 equiv of indole 16c and 1 equiv bromoacetonitrile 2. White solid, 66 mg, 90% yield. Melting point 127–129 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.74 (d, J = 8.56 Hz, 1H), 7.41 – 7.53 (m, 3H), 7.33 – 7.40 (m, 2H), 7.21 (dt, J = 1.22,7.70, Hz, 1H), 7.06 – 7.12 (m, 1H), 4.87 (quin, J = 6.91 Hz, 1H), 4.25 (s, 2H), 1.66 (d, J = 6.85 Hz, 6H). 13C NMR (101 MHz, DMSO-d6) δ 162.8, 160.4, 134.6, 131.8, 131.8, 130.5, 130.4, 127.4, 125.8, 122.6, 120.3, 119.5, 118.1, 116.4, 116.2,114.8, 112.8, 48.1, 21.4, 15.0. HRMS (ESI): [M]+• C19H17FN2, calculated: 292.1376; found: 292.1386.
2-(1-Cyclopropyl-3-phenyl-1H-indol-2-yl)acetonitrile (17d).
Synthesized according to the procedure described for 13c. Yellow solid, 13 mg, 19% yield. Melting point 68–71 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.66 (d, J = 8.31 Hz, 1H), 7.50–7.58 (m, 3H), 7.45 (d, J = 7.09 Hz, 2H), 7.37 – 7.43 (m, 1H), 7.28 (t, J = 7.21 Hz, 1H), 7.09 – 7.17 (m, 1H), 4.27 (s, 2H),3.35– 3.40 (m, 1H), 1.27 – 1.35 (m, 2H), 1.11 – 1.19 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 137.8, 133.9, 129.7, 129.5,127.6, 127.2, 126.3, 123.1, 120.9, 119.4, 118.1, 116.4, 111.6,25.8, 15.3, 7.5. HRMS (ESI): [M]+• C19H16N2, calculated: 272.1313; found: 272.1309.
2-(2-methyl-1H-indol-3-yl)acetonitrile (19a).
Synthesized according to the procedure described for 13c using DMSO instead of DCE with 3 equiv of indole 18a and 1 equiv of bromoacetonitrile 2. Yellow solid, 17 mg, 41% yield. Melting point 83–85 °C. Analytical data are consistent with reported values.43 1H NMR (400 MHz, DMSO-d6) δ 11.12 (br s, 1H),7.26 (d, J = 8.07 Hz, 1H), 7.00 (t, J = 7.58 Hz, 1H), 6.93 (d, J = 7.09 Hz, 1H), 6.20 – 6.28 (m, 1H), 4.11 (s, 2H), 2.41 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 136.5, 136.4, 127.8, 121.1,120.6, 119.7, 118.6, 110.8, 97.7, 21.1, 13.9. HRMS (ESI): [M-H]-, C11H9N2, calculated: 169.0766; found: 169.0763.
Methyl-3-(cyanomethyl)-1H-indole-2-carboxylate (19b).
Synthesized according to the procedure described for 13c using DMSO instead of DCE with 3 equiv of indole 18b and 1 equiv of bromoacetonitrile 2. Colorless oil, 27 mg, 51% yield. Melting point 150–152 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.08 (br s, 1H), 7.84 (d, J = 8.07 Hz, 1H), 7.49 (d, J = 8.31 Hz, 1H), 7.34 (t, J = 7.70 Hz, 1H), 7.17 (t, J = 7.58 Hz, 1H), 4.38 (s, 2H), 3.94 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 162.0,136.6, 126.5, 126.0, 124.2, 120.9, 120.4, 119.1, 113.3, 111.2, 52.4, 13.4. HRMS (ESI): [M-H]-, C12H9N2O2, calculated: 213.0664; found: 213.0659.
2-(2-phenyl-1H-indol-3-yl)acetonitrile (19c).
Synthesized according to the procedure described for 13c using DMSO instead of DCE with 3 equiv of indole 18c and 1 equiv of bromoacetonitrile 2. Yellow solid, 45 mg, 77% yield. Melting point 87–89 °C. Analytical data are consistent with reported values.44 1H NMR (400 MHz, DMSO-d6) δ 11.59 (s, 1H),7.65– 7.78 (m, 3H), 7.56 – 7.64 (m, 2H), 7.43 – 7.50 (m, 2H), (t, J = 7.46 Hz, 1H), 7.13 (t, J = 7.34 Hz, 1H), 4.16 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 136.2, 136.2, 132.0,129.5, 128.6, 128.4, 128.1, 122.7, 120.0, 119.7, 118.7, 112.0,100.8, 13.6. HRMS (ESI): [M-H]-, C16H11N2, calculated: 231.0922; found: 231.0925.
2-(2-(4-fluorophenyl)-1H-mdol-3-yl)acetomtrile (19d).
Synthesized according to the procedure described for 13c using DMSO instead of DCE with 3 equiv of indole 18d and 1 equiv of bromoacetonitrile 2. Pale yellow solid, 51 mg, 81% yield. Melting point 138–141 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.59 (s, 1H), 7.64 – 7.76 (m, 3H), 7.35 – 7.47 (m, 3H), 7.16 – 7.22 (m, 1H), 7.08 – 7.16 (m, 1H), 4.15 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 163.6, 161.1, 136.2, 135.2, 130.6, 130.6,128.6, 128.5, 128.0, 122.7, 120.0, 119.7, 118.7, 116.6, 116.4,112.0, 100.8, 13.5. HRMS (ESI): [M]+• C16H11FN2, calculated: 250.0906; found: 250.0907.
2-(2-(4-bromophenyl)-1H-indol-3-yl)acetonitrile (19e).
Synthesized according to the procedure described for 13c using DMSO (0.5 M) instead of DCE with 3 equiv of indole 18e and 1 equiv of bromoacetonitrile 2. White solid, 55 mg, 71% yield. Melting point 184–187 °C. ‘H NMR (400 MHz, DMSO-d6) δ11.65 (s, 1H), 7.76 – 7.81 (m, J = 8.56 Hz, 2H), 7.73 (d, J =7.83 Hz, 1H), 7.59 – 7.65 (m, J = 8.31 Hz, 2H), 7.45 (d, J =8.7 Hz, 1H), 7.18 – 7.27 (m, 1H), 7.09 – 7.16 (m, 1H), 4.18 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 136.3, 134.9, 132.4,131.1, 130.4, 128.0, 123.0, 121.9, 120.1, 119.6, 118.8, 112.1,101.3, 13.5. HRMS (ESI): [M-H]-, C16H10BrN2, calculated: 309.0027; found: 309.0024.
5-(cyanomethyl)-3-phenyl-1H-pyrrolo[3,2-c]pyridin-5-ium (21).
Synthesized according to the procedure described for 13c using DMSO instead of DCE. Yellow solid, 49 mg, 84% yield. Melting point 213–215 °C. Analytical data are consistent with reported values.35 1H NMR (400 MHz, DMSO-d6) δ 13.30 (br s, 1H), 9.80 (s, 1H), 8.69 (d, J = 7.09 Hz, 1H), 8.41 (s, 1H), 8.17 (d, J = 7.09 Hz, 1H), 7.83 (d, J = 7.34 Hz, 2H), 7.56 (t, J = 7.58 Hz, 2H), 7.40 – 7.48 (m, 1H), 5.97 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 142.4, 140.8, 134.9, 132.1, 131.3,129.6, 128.1, 127.8, 122.5, 119.9, 115.8, 111.3, 46.9.
1-(cyanomethyl)-3-(1H-indol-3-yl)pyridin-1-ium (23).
Synthesized according to the procedure described for 13c using DMSO instead of DCE. Yellow solid, 51 mg, 87% yield. Melting point 226–229 °C. 1H NMR (400 MHz, DMSO-d6) δ12.7 (br s, 1H), 9.48 (s, 1H), 8.94 – 9.05 (m, 2H), 8.18 – 8.27 (m, 2H), 8.12 (d, J = 7.58 Hz, 1H), 7.57 (d, J = 7.58 Hz, 1H),7.27 (dd, J = 7.21, 8.68 Hz, 2H), 6.02 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 142.8, 141.7, 141.1 137.6, 137.3, 129.0,128.3, 124.2, 123.2, 121.5, 119.5, 115.0, 113.1, 108.8, 48.3. HRMS (ESI): [M]+, C15H12N3, calculated: 234.1026; observed: 234.1031.
Supplementary Material
Figure 3.
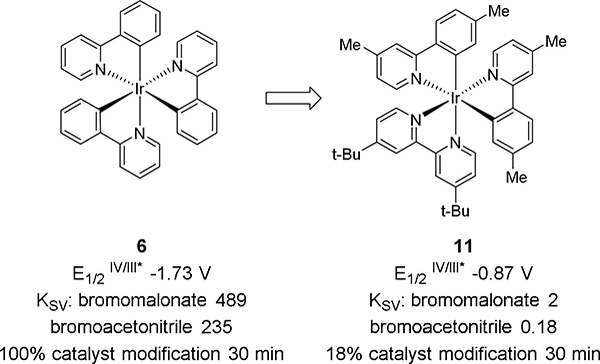
Summary of catalyst properties for Ir(ppy)3 (6) and [Ir(dmppy)2(dtbbpy)]PF6 (11).
ACKNOWLEDGMENT
We thank Sandeep Raikar and Jean-Marc Grandjean for useful discussions. We also thank Lionel Cheruzel and Laura Miller Conrad at SJSU for experimental assistance. We thank Stanley B. Prusiner for providing support. This work was supported by grants from the National Institutes of Health (AG002132 and AG031220) and by support from Daiichi Sankyo (Tokyo, Japan).
Footnotes
ASSOCIATED CONTENT
Supporting Information
1H and 13C NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The Institute for Neurodegenerative Diseases has a research collaboration with Daiichi Sankyo (Tokyo, Japan).
REFERENCES
- (1).Chadha N; Silakari O Indoles as Therapeutics of Interest in Medicinal Chemistry: Bird’s Eye View. Eur. J. Med. Chem 2017, 134, 159. [DOI] [PubMed] [Google Scholar]
- (2).Sravanthi TV; Manju SL Indoles — a Promising Scaffold for Drug Development. Eur. J. Pharm. Sci 2016, 91, 1. [DOI] [PubMed] [Google Scholar]
- (3).Humphrey GR; Kuethe JT Practical Methodologies for the Synthesis of Indoles. Chem. Rev 2006, 106, 2875. [DOI] [PubMed] [Google Scholar]
- (4).Bandini M; Eichholzer A Catalytic Functionalization of Indoles in a New Dimension. Angew. Chem. Int. Ed 2009, 48, 9608. [DOI] [PubMed] [Google Scholar]
- (5).Reaction at the 2-position can be activated with a BF3K group, see for example: Lee S; MacMillan DWC Organocatalytic Vinyl and Friedel-Crafts Alkylations with Trifluoroborate Salts. J. Am. Chem. Soc 2007, 129, 15438. [DOI] [PubMed] [Google Scholar]
- (6).Rajagopal S; Brooks ME; Nguyen T-M; Novak M Synthesis and Characterization of the Aqueous Solution Chemistry of the Food-Derived Carcinogen Model N-Acetoxy-N-(1-Methyl-5h-Pyrido[4,5-b]Indol-3-yl)Acetamide and Its N-Pivaloyloxy Analogue. Tetrahedron 2003, 59, 8003. [Google Scholar]
- (7).Lindsay-Scott PJ; Clarke A; Richardson J Two-Step Cyanomethylation Protocol: Convenient Access to Functionalized Aryl- and Heteroarylacetonitriles. Org. Lett 2015, 17, 476. [DOI] [PubMed] [Google Scholar]
- (8).Liu Z-Q; Li Z Radical-Promoted Site-Specific Cross Dehydrogenative Coupling of Heterocycles with Nitriles. Chem. Commun 2016, 52, 14278. [DOI] [PubMed] [Google Scholar]
- (9).Douglas JJ; Sevrin MJ; Stephenson CRJ Visible Light Photocatalysis: Applications and New Disconnections in the Synthesis of Pharmaceutical Agents. Org. Process Res. Dev 2016, 20, 1134. [Google Scholar]
- (10).Romero NA; Nicewicz DA Organic Photoredox Catalysis. Chem. Rev 2016, 116, 10075. [DOI] [PubMed] [Google Scholar]
- (11).Stephenson C; Yoon T Enabling Chemical Synthesis with Visible Light. Acc. Chem. Res 2016, 49, 2059. [DOI] [PubMed] [Google Scholar]
- (12).Shaw MH; Twilton J; MacMillan DWC Photoredox Catalysis in Organic Chemistry. J. Org. Chem 2016, 81, 6898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kozlowski M; Yoon T Editorial for the Special Issue on Photocatalysis. J. Org. Chem 2016, 81, 6895. [DOI] [PubMed] [Google Scholar]
- (14).Prier CK; Rankic DA; MacMillan DWC Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev 2013, 113, 5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Staveness D; Bosque I; Stephenson CRJ Free Radical Chemistry Enabled by Visible Light-Induced Electron Transfer. Acc. Chem. Res 2016, 49, 2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Silvi M; Melchiorre P Enhancing the Potential of Enantioselective Organocatalysis with Light. Nature 2018, 554, 41. [DOI] [PubMed] [Google Scholar]
- (17).Nicewicz DA; MacMillan DWC Merging Photoredox Catalysis with Organocatalysis: The Direct Asymmetric Alkylation of Aldehydes. Science 2008, 322, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Welin ER; Warkentin AA; Conrad JC; MacMillan DWC Enantioselective α-Alkylation of Aldehydes by Photoredox Organocatalysis: Rapid Access to Pharmacophore Fragments from β-Cyanoaldehydes. Angew. Chem. Int. Ed 2015, 54, 9668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Yoshida H; Fujimura Y; Yuzawa H; Kumagai J; Yoshida T A Heterogeneous Palladium Catalyst Hybridised with a Titanium Dioxide Photocatalyst for Direct C-C Bond Formation between an Aromatic Ring and Acetonitrile. Chem. Commun 2013, 49, 3793. [DOI] [PubMed] [Google Scholar]
- (20).Swift EC; Williams TM; Stephenson CRJ Intermolecular Photocatalytic C-H Functionalization of Electron-Rich Heterocycles with Tertiary Alkyl Halides. Synlett 2016, 27, 754. [Google Scholar]
- (21).Furst L; Matsuura BS; Narayanam JMR; Tucker JW; Stephenson CRJ Visible Light-Mediated Intermolecular C-H Functionalization of Electron-Rich Heterocycles with Malonates. Org. Lett 2010, 12, 3104. [DOI] [PubMed] [Google Scholar]
- (22).Chang Q; Liu Z; Liu P; Yu L; Sun P Visible-Light-Induced Regioselective Cyanomethylation of Imidazopyridines and Its Application in Drug Synthesis. J. Org. Chem 2017, 82, 5391. [DOI] [PubMed] [Google Scholar]
- (23).Isse AA; Gennaro A Homogeneous Reduction of Haloacetonitriles by Electrogenerated Aromatic Radical Anions: Determination of the Reduction Potential of •CH2CN. J. Phys. Chem. A 2004, 108, 4180. [Google Scholar]
- (24).Bock CR; Connor JA; Gutierrez AR; Meyer TJ; Whitten DG; Sullivan BP; Nagle JK Estimation of Excited-State Redox Potentials by Electron-Transfer Quenching. Application of Electron-Transfer Theory to Excited-State Redox Processes. J. Am. Chem. Soc 1979, 101, 4815. [Google Scholar]
- (25).Pirtsch M; Paria S; Matsuno T; Isobe H; Reiser O [Cu(dap)2Cl] as an Efficient Visible-Light-Driven Photoredox Catalyst in Carbon-Carbon Bond-Forming Reactions. Chem. Eur. J 2012, 18, 7336. [DOI] [PubMed] [Google Scholar]
- (26).Flamigni L; Barbieri A; Sabatini C; Ventura B;Barigelletti F, Photochemistry and Photophysics of Coordination Compounds:Iridium In Photochemistry and Photophysics of Coordination Compounds Ii, Balzani V; Campagna S, Eds. Springer Berlin Heidelberg: Berlin, Heidelberg, 2007; pp 143. [Google Scholar]
- (27).Lowry MS; Goldsmith JI; Slinker JD; Rohl R; Pascal RA; Malliaras GG; Bernhard S Single-Layer Electroluminescent Devices and Photoinduced Hydrogen Production from an Ionic Iridium(III) Complex. Chem. Mater 2005, 17, 5712. [Google Scholar]
- (28).Devery III JJ; Douglas JJ; Nguyen JD; Cole KP; Flowers II RA; Stephenson CRJ Ligand Functionalization as a Deactivation Pathway in a fac-Ir(ppy)3-Mediated Radical Addition. Chem. Sci 2015, 6, 537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Terrett JA; Clift MD; MacMillan DWC Direct β-Alkylation of Aldehydes Via Photoredox Organocatalysis. J. Am. Chem. Soc 2014, 136, 6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Merenyi G; Lind J; Shen X Electron Transfer from Indoles, Phenol, and Sulfite (SO32-) to Chlorine Dioxide (ClO2.). J. Phys. Chem. A 1988, 92, 134. [Google Scholar]
- (31).Cismesia MA; Yoon TP Characterizing Chain Processes in Visible Light Photoredox Catalysis. Chem. Sci 2015, 6, 5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Cai Y; Wang J; Zhang Y; Li Z; Hu D; Zheng N; Chen H Detection of Fleeting Amine Radical Cations and Elucidation of Chain Processes in Visible-Light-Mediated [3 + 2] Annulation by Online Mass Spectrometric Techniques. J. Am. Chem. Soc 2017, 139, 12259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Triandafillidi I; Kokotou MG; Kokotos CG Photocatalytic Synthesis of γ-Lactones from Alkenes: High-Resolution Mass Spectrometry as a Tool to Study Photoredox Reactions. Org. Lett 2018, 20, 36. [DOI] [PubMed] [Google Scholar]
- (34).Baciocchi E; Muraglia E Manganese(III)-Promoted Homolytic Methylmalonylation of Pyrrole and Indole Derivatives. A Simple Route to α-Methyl-2-Pyrrole and α-Methyl-3-Indoleacetic Acids. J. Org. Chem 1993, 58, 7610. [Google Scholar]
- (35).Voskressensky LG; Storozhenko OA; Festa AA; Novikov RA; Varlamov AV Synthesis of Chromenoimidazoles, Annulated with an Azaindole Moiety, through a Base-Promoted Domino Reaction of Cyano-Methyl Quaternary Salts. Synthesis 2017, 49, 2753. [Google Scholar]
- (36).Joucla L; Batail N; Djakovitch L “On Water” Direct and SiteDSelective Pd Catalysed C-H Arylation of (NH) Indoles. Advanced Synthesis & Catalysis 2010, 352, 2929. [Google Scholar]
- (37).Yang R; Yu J-T; Sun S; Zheng Q; Cheng J Copper-Mediated Intramolecular Aza-Wacker-Type Cyclization of 2-Alkenylanilines toward 3-Aryl Indoles. Tetrahedron Letters 2017, 58, 445. [Google Scholar]
- (38).Tsuchimoto T; Iwabuchi M; Nagase Y; Oki K; Takahashi H Indium Catalyzed Heteroaryl-Heteroaryl Bond Formation through Nucleophilic Aromatic Substitution. Angew. Chem. Int. Ed 2011, 50, 1375. [DOI] [PubMed] [Google Scholar]
- (39).Chen S; Liao Y; Zhao F; Qi H; Liu S; Deng G-J Palladium-Catalyzed Direct Arylation of Indoles with Cyclohexanones. Org. Lett 2014, 16, 1618. [DOI] [PubMed] [Google Scholar]
- (40).Okada M; Sugita T; Wong CP; Wakimoto T; Abe I Identification of Pyridinium with Three Indole Moieties as an Antimicrobial Agent. J. Nat. Prod 2017, 80, 1205. [DOI] [PubMed] [Google Scholar]
- (41).Blechert S Hetero Cope Umlagerungen. 4. Mitteilung. Regiokontrollierte Indolsynthesen.Helv. Chim. Acta 1985, 68, 1835. [Google Scholar]
- (42).Ikeda M; Matsugashita S; Tamura Y Photochemistry of Ethyl 2-Cyano-1,2-Dihydroquinoline-1-Carboxylates (Reissert Compounds): Synthesis of 2-Cyanomethylinodole-1-Carboxylates. J. Chem. Soc., Perkin Trans 1 1976, 2587. [Google Scholar]
- (43).Merour JY; Coadou JY; Tatibouet F Syntheses of 2(5)-Substituted 1-Acetyl-3-Oxo-2,3-Dihydroindoles, 3-Acetoxy-1-Acetylindoles, and of 2-Methyl-5-Methoxyindole-3-Acetic Acid. Synthesis 1982, 1982, 1053. [Google Scholar]
- (44).Tobisu M; Fujihara H; Koh K; Chatani N Synthesis of 2-Boryl- and Silylindoles by Copper-Catalyzed Borylative and Silylative Cyclization of 2-Alkenylaryl Isocyanides. J. Org. Chem 2010, 75, 4841. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.