

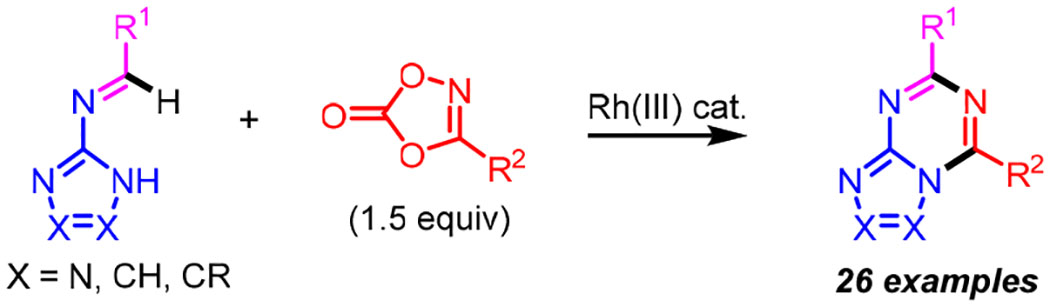
Abstract
A wide range of azolo[1,3,5]triazines were obtained by Rh(III)-catalyzed annulation of N-azolo imines and dioxazolones. The reaction proceeds by the first catalytic C-H amidation of an imidoyl C-H bond followed by cyclodehydration. Good yields were obtained for N-azolo imines derived from aminoazoles and aromatic and heteroaromatic aldehydes. A range of dioxazolone amidating reagents were employed to introduce aryl, heteroaryl, and alkyl substituents. The reaction was also performed with bench-top set up at 1 mmol scale using microwave heating.
Bridgehead N-fused [5,6]-bicyclic heterocycles are privileged pharmacophores in drug discovery and are present in numerous U.S. FDA approved drugs and many clinical candidates.1,2 We have recently reported the first examples of Rh(III)-catalyzed imidoyl C-H activation of N-azolo imines 1 enabling annulations with alkynes, diazoketones and sulfoxonium ylides to give azolopyrimidines 2 (Scheme 1A).3,4 The straightforward one-step preparation of diverse N-azolo imine starting materials 1 by simple condensation of readily available amino azoles with the enormous range of commercially available aromatic and heteroaromatic aldehydes is an important practical aspect of this method. Given that diverse N-azolo imines 1 can readily be accessed, we are extensively pursuing different types of transition metal-catalyzed imidoyl C-H functionalization of 1 for heterocycle synthesis, including for the preparation of other sub-classes having the privileged N-fused [5,6]-bicyclic heterocycle framework.
Scheme 1.
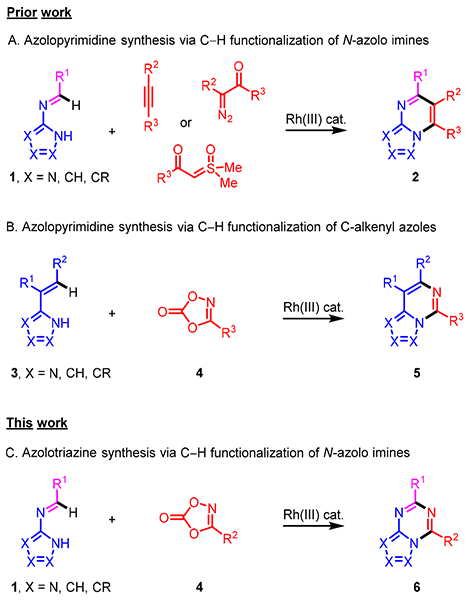
Preparation of Bridgehead N-Fused [5,6]-Bicyclic Heterocycles via Catalytic C-H Functionalization
The 1,4,2-dioxazol-5-one amidating reagents 4 were initially identified by Sauer and Mayer in the late 1960s as a safe alternative to acyl azides for N-acyl nitrene formation.5 This elegant finding was subsequently applied to direct C-N bond formation by Dubé6 and Bolm.7 Chang and co-workers later on introduced and developed 1,4,2-dioxazol-5-ones as extremely efficient amidating reagents in C-H functionalization reactions.8 These reagents have now been employed by a number of laboratories for C-H amidation.9 In work relevant to N-fused [5,6]-bicyclic heterocycle synthesis, we recently reported that C-alkenyl azoles 3 could be amidated with 1,4,2-dioxazol-5-ones 4 to afford azolopyrimidines 5 after cyclodehydration (Scheme 1B).10 Cheng and coworkers have also recently reported that tricyclic or higher order derivatives can be obtained by annulations with 2-arylimidazoles.11 Herein we describe Rh(III)-catalyzed imidoyl C-H amidation followed by cyclodehydration of readily available N-azolo imines 1 with 1,4,2-dioxazol-5-ones 4 having diverse electronic and steric properties to give azolo[1,3,5]triazines 6 (Scheme 1C), which have been recognized as privileged scaffolds in medicinal chemistry.12
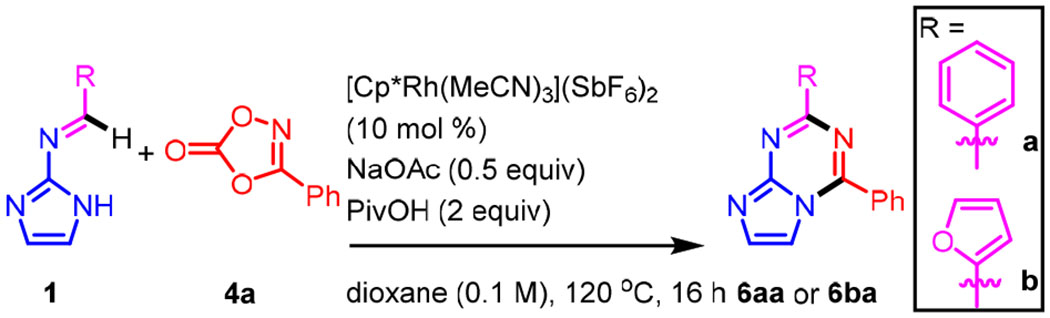
Annulation of N-azolo imine 1a and dioxazolone 4a provided azolotriazine 6aa in good yield using the cationic Rh(III) catalyst [Cp*Rh(MeCN)3](SbF6)2 with NaOAc and pivalic acid (PivOH) as additives in dioxane at 120 °C (entry 1, Table 1). Under these conditions, complete consumption of the starting imine 1a was observed and no significant by-products were detected. An active cationic catalyst could also be formed in situ from [Cp*RhCl2]2 and AgSbF6, but provided a slightly lower yield of product (entry 2). When the halide was not abstracted from [Cp*RhCl2]2, only trace amounts of product was observed (entry 3). As expected, product was not obtained when a Rh(III) catalyst was not added (entry 4). Both PivOH and NaOAc are essential for C-H amidation and cyclodehydration to azolotriazine 6aa. When either additive was excluded, only a small amount of 6aa was obtained, but with no remaining imine (entries 5 and 6).13 When stoichiometric NaOAc was used, a comparable yield of 6aa was obtained (entry 7); however, a sub-stoichiometric quantity of NaOAc was found to be more general (vide infra). Lowering the temperature to 100 °C led to a slight reduction in yield (entry 8). Doubling the concentration resulted in a significantly lower yield (entry 9) as did performing the reaction in toluene, DCE, or MeCN (entries 10–12). In comparison to Cp*Rh(III) catalysis, Cp*Ir(III) and Cp*Co(III) catalysts were ineffective and provided little to no product under the same reaction conditions (entries 13 and 14). The optimal conditions were also effective for furfural-derived imine 1b (entry 15). In contrast to imine 1a, furfural-derived imine 1b is more sensitive to the stoichiometry of the NaOAc additive. When 1 equiv rather than 0.5 equiv was used, a lower yield was observed (entry 16). Lowering the temperature to 100 °C also resulted in a lower yield (entry 17).
Table 1.
Reaction Parameters for Annulations to Azolotriazines 6a
| entry | imine | variation | Yield %b |
|---|---|---|---|
| 1 | 1a | none | 75 (73)c |
| 2 | 1a | [Cp*RhCl2]2, AgSbF6d | 60 |
| 3 | 1a | [Cp*RhCl2]2(5 mol %) | 8 |
| 4 | 1a | no Rh | 0 |
| 5 | 1a | no PivOH | 12 |
| 6 | 1a | no NaOAc | 16 |
| 7 | 1a | NaOAc (1 equiv) | 79 |
| 8 | 1a | 100 oC | 70 |
| 9 | 1a | 0.2 M | 46 |
| 10 | 1a | toluene as solvent | 11 |
| 11 | 1a | DCE as solvent | 9 |
| 12 | 1a | MeCN as solvent | 25 |
| 13 | 1a | [Cp*IrCl2]2, AgSbF6d | 10 |
| 14 | 1a | [Cp*Co(MeCN)3](SbF6)2 | 0 |
| 15 | 1b | none | 53 (55)e |
| 16 | 1b | NaOAc (1 equiv) | 37 |
| 17 | 1b | 100 °C | 43 |
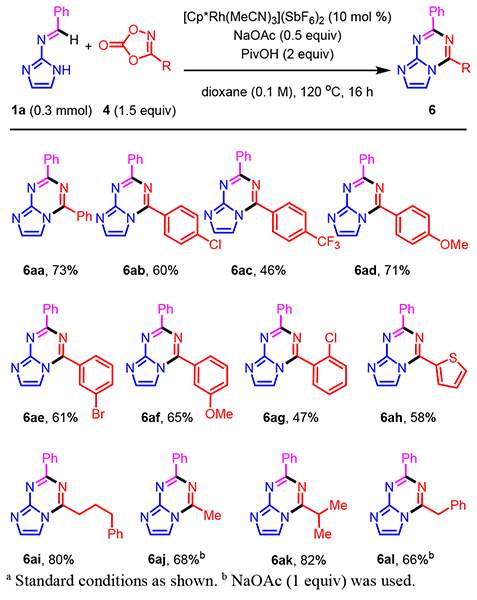
Using the optimal reaction conditions, we explored the scope of dioxazolone 4 for annulations with N-imidazo imine 1a (Scheme 2). A series of 3-aryl-substituted 1,4,2-dioxazol-5-ones 4a-g with different electronic properties were effective under the standard conditions giving bicyclic products 6aa-6ag in moderate to good yields (46–73%). Thiophene-containing dioxazolone (4h) was also a good coupling partner affording product 6ah in 58% yield. A variety of alkyl-substituted dioxazolones including n-alkyl (4i), methyl (4j), α-branched (4k), and benzyl (4l) served as effective inputs in the coupling reactions providing 6ai-6al in good to excellent yields (66–82%).
Scheme 2.
Dioxazolone Scope for Rh(III)-Catalyzed Annulation to Give Azolotriazines 6a
We next investigated the scope for the N-azolo imines 1 derived from several different amino azoles and various aromatic and heteroaromatic aldehydes (Scheme 3). As indicated in the optimization studies, furfural-derived imine 1b coupled with 3-phenyl-substituted 1,4,2-dioxazol-5-one 4a to generate heterocycle 6ba (55%). The same imine coupled equally well with alkyl-substituted dioxazolone 4i to afford 6bi in 58% yield. In addition to 1a, a series of imines derived from 2-amino-imidazole and benzaldehydes bearing electron-deficient (1c, 1d, and 1f) and electron-rich (1e) substituents at the para- and meta-positions efficiently coupled with both aryl-and alkyl-substituted dioxazolones under standard conditions to give bicyclic heterocycles 6ca-6fi (59–77%). Ortho-substituted benzaldimine 1g also coupled to give azolotriazine 6gi, albeit with a slight reduction in yield (46%). Significantly, the bromo- (1f) and chloro- (1g) substituted imines afforded products that are amenable to subsequent cross-coupling chemistry. Annulations of N-azolo imines 1 from enolizable aldehydes were not investigated because this type of imine was difficult to prepare.
Scheme 3.
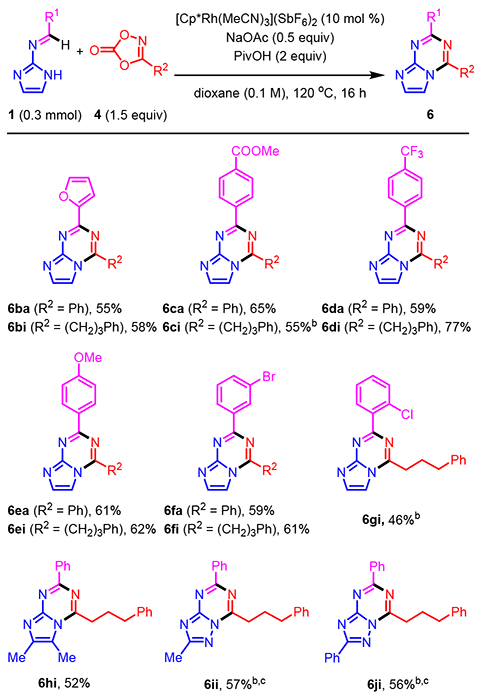
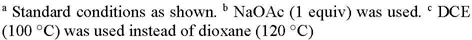
N-Azolo Imine Scope for Rh(III)-Catalyzed Annulations to Give Azolotriazines 6a
Imine 1h with dimethyl substitution on the imidazole ring was also an effective coupling partner to give imidazotriazine 6hi. Although imines derived from 3-aminopyrazoles were not effective coupling partners (data not shown), N-triazolo imines 1i and 1j afforded triazolotriazines 6ii and 6ji in reasonable yields (56–57%). It is notable that imine 1j with multiple potential sites for directed C-H functionalization was still selective for imidoyl C-H activation to give 6ji.
Lastly, 6ai was prepared on the bench-top at 1 mmol scale (Scheme 4). For practical purposes, the reaction was performed using a microwave reactor with a 2 h reaction time. Bicyclic product 6ai was isolated in reasonable yield (65%).
Scheme 4.
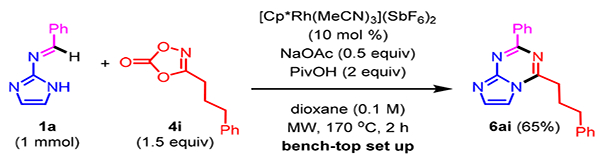
Bench-top Reaction Set Up on 1 mmol Scale
A possible mechanism for the annulation is depicted in Scheme 5. Based upon our previous study on annulations of N-azolo imines with alkynes, diazoketones and sulfoxonium ylides,3 we propose that imine 1 undergoes concerted metalation-deprotonation to give rhodacycle A. In our previously published report,3 a rhodacycle obtained by imidoyl C-H activation was rigorously characterized by X-ray crystallography and was shown to be competent in the catalytic cycle. In accord with Chang’s detailed mechanistic studies on dioxazolone-mediated C-H amidation,8a insertion of the N-acyl nitrene with release of CO2 then generates the six-membered rhodacycle B. Proto-demetalation affords amide C to regenerate the active Rh(III) catalyst. Under the reaction conditions, amide C undergoes cyclodehydration to provide the bicyclic heterocycle product 6.
Scheme 5.
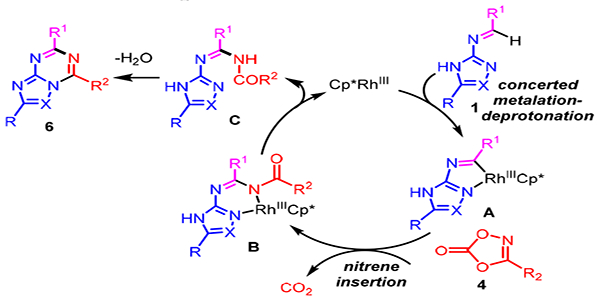
Proposed Mechanism for Annulation
In conclusion, Rh(III)-catalyzed imidoyl C-H amidation of imines 1 followed by cyclodehydration affords azolotriazines 6. The reaction proceeds in good yields for a range of aryl, heteroaryl, benzyl, and alkyl dioxazolones 4. N-Azolo imines 1 derived from amino imidazoles and triazoles and a variety of different aromatic and heteroaromatic aldehydes are also effective inputs. Moreover, the reaction is applicable to straightforward bench-top set up at 1 mmol scale using microwave heating.
EXPERIMENTAL SECTION
General Information.
Unless otherwise noted, all commercially available reagents were purchased and used as received. Solvents including 1,4-dioxane, toluene, 1,2-dichloroethane (DCE), and acetonitrile (MeCN) were deoxygenated by sparging with argon and stored over activated 3Å molecular sieves in a nitrogen filled glove box. The microwave reaction was performed using a microwave reactor with an external IR sensor and in a closed reaction vessel. Commercial AgSbF6 was stored in a nitrogen filled glove box. 1H-, 13C-, and 19F-NMR spectra were recorded on 400 MHz, 500 MHz or 600 MHz spectrometers. The chemical shift [δ (ppm)], coupling constants [J (Hz)], multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, pent = pentet, m = multiplet, br = broad), and integration are reported. Chemical shifts for 1H- and 13C-NMR are reported relative to residual undeuterated solvent in CDCl3 (7.24 ppm for 1H-NMR and 76.99 ppm for 13C-NMR) and (CD3)2SO (2.47 ppm for 1H-NMR and 39.94 ppm for 13C-NMR). Flash chromatography was carried out with slica gel with 40–63 μm particle size and with 230400 mesh. Partial data are provided for IR spectra. Melting points are reported uncorrected. High-resolution mass spectra (HRMS) were obtained using electrospray ionization (ESI) on a time of flight (TOF) mass spectrometer (Yale University) or electron ionization (EI+) obtained by University of Illinois SCS Mass Spectrometry Laboratory.
Preparation of catalysts and reactants.
[Cp*Rh(MeCN)3(SbF6)2] was synthesized according to literature procedures.8a N-Azolo imine substrates 1a-g were synthesized according to literature procedures.3,14 Dioxazolones 4a-h and 4j-l were synthesized according to literature procedures.8a,11,15 Imine 1h was prepared via literature procedures14 with slight modification. Imines 1i and 1j were synthesized as described below. Dioxazolone 4i was prepared according to a literature procedure with slight modification.8a
(E)-N-(4,5-Dimethyl-1H-imidazol-2-yl)-1-phenylmethanimine (1h):
Imine 1h was prepared via literature procedures14 with slight modification. To a flame-dried 50-mL round bottom flask was added 2-amino-4,5-dimethylimidazole ethyl sulfate16 (1.9 g, 8.0 mmol, 1.0 equiv). The flask was degassed and filled with nitrogen. To the flask was added CH2Cl2 (10 mL) then benzaldehyde (0.81 mL, 8.0 mmol, 1.0 equiv), Ti(OiPr)4 (3.79 mL, 12.8 mmol, 1.60 equiv), and Et3N (4.46 mL, 32.0 mmol, 4.00 equiv) were sequentially added dropwise. The resultant mixture was stirred overnight at rt, and the reaction was quenched with water (20 mL). The resulting mixture was immediately filtered and washed with CH2Cl2. The filtrate was transferred to a separatory funnel. The organic layer was separated, and the aqueous layer was washed with CH2Cl2 (x2). The combined organic extracts were dried (anhyd. N2SO4) and concentrated under reduced pressure. Purification by silica gel column chromatography (20–40% ethyl acetate/hexanes) afforded 1h (814 mg, 51%) as a yellow solid. mp 199–201 °C. FTIR (neat) 2918, 1600, 1572, 1450, 1431, 1246, 872, 755, 685, 561, 494 cm−1. 1H-NMR (600 MHz, CDCl3) δ 10.07 (s, 1H), 9.22 (s, 1H), 7.85 (d, J = 6.7 Hz, 2H), 7.47–7.34 (m, 3H), 2.17 (s, 3H), 2.03 (s, 3H). 13C-NMR (151 MHz, CDCl3) δ 158.6, 148.1, 135.7, 132.9, 131.5, 129.0, 128.8, 121.7, 12.5, 9.5. HRMS (EI+): m/z [M-H]+ calcd for C12H12N3, 198.1031; found 198.1029.
(E)-N-(3-Methyl-1H-1,2,4-triazol-5-yl)-1-phenylmethanimine (1i):
Inside a glove box, to a flame-dried 10–20-mL Biotage microwave vial (#354833) was added 3-methyl-1H-1,2,4-triazol-5-amine (441 mg, 4.50 mmol, 1.00 equiv), benzaldehyde (0.460 mL, 4.50 mmol, 1.00 equiv), 3Å molecular sieves (approximately 7.5 g), and THF (20.0 mL). The vial was capped with a Teflon-lined cap, removed from the glove box, and the mixture was stirred with a pre-heated stem block filled with oil at 100 °C overnight. The resulting mixture was filtered through a pad of celite, washed with ethyl acetate, and concentrated under reduced pressure. The crude residue was recrystallized with hot ethyl acetate to afford 1i (621 mg, 74%) as a white solid. mp 139–141 °C. FTIR (neat) 1622, 1558, 1450, 1314, 1220, 1149, 1063, 878, 763, 687, 544, 496 cm−1. 1H-NMR (600 MHz, CDCI3) δ 13.49 (s, 1H), 9.30 (s, 1H), 7.95 (d, J = 8.0 Hz, 2H), 7.50 (t, J = 7.2 Hz, 1H), 7.45 (t, J = 7.4 Hz, 2H), 2.59 (s, 3H). 13C-NMR (151 MHz, CDCI3) δ 165.0, 164.4, 155.6, 135.3, 132.4, 129.5, 128.9, 13.0. HRMS (EI+): m/z [M-H]+ calcd for C10H9N4, 185.0827; found 185.0824.
(E)-1-Phenyl-N-(3-phenyl-1H-1,2,4-triazol-5-yl)methanimine (1j):
Inside a glove box, to a flame-dried 2–5-mL Biotage microwave vial (#351521) was added 3-phenyl-1H-1,2,4-triazol-5-amine (801 mg, 5.00 mmol, 1.00 equiv), benzaldehyde (0.510 mL, 5.00 mmol, 1.00 equiv), 3Å molecular sieves (approximately 200 mg), and toluene (2.00 mL). The vial was capped with a Teflon-lined cap, removed from the glove box, and the mixture was stirred with a pre-heated stem block filled with oil at 100 °C overnight (Note: a white solid precipitated out of the reaction mixture after about 2 h). The solid was scrapped off, transferred to a filtered frit, and washed thoroughly with ethyl acetate. The resulting solid (mixed with molecular sieves) was dissolved with hot chloroform and filtered through a pad of celite. The filtrate was then concentrated under reduced pressure to afford 1j (992 mg, 80%) as a white solid. mp 169–172 °C. FTIR (neat) 1618, 1564, 1472, 1381, 1219, 1158, 987, 770, 694, 572, 509 cm−1. 1H-NMR (600 MHz, (CD3)2SO) δ 9.30 (s, 1H), 8.07–8.00 (m, 4H), 7.60–7.42 (m, 6H). 13C-NMR (151 MHz, (CD3)2SO) δ 164.9, 163.3, 157.7, 135.6, 133.0, 130.1, 129.8, 129.5, 129.3, 126.3. HRMS (EI+): m/z [M-H]+ calcd for C15H11N4, 247.0984; found 247.0984.
3-(3-Phenylpropyl)-1,4,2-dioxazol-5-one (2i):
The starting hydroxamic acid (N-hydroxy-4-phenylbutanamide) was prepared according to a literature procedure for a related compound.17 To a flame-dried 100-mL round bottom flask was added 4-phenylbutanoic acid (4.43 g, 27.0 mmol, 1.00 equiv) and THF (45.0 mL). After adding carbonyldiimidazole (CDI) (6.57 g, 40.5 mmol, 1.50 equiv), the reaction mixture was stirred under nitrogen at rt for 1 h, and then hydroxylamine chloride (3.75 g, 54.0 mmol, 2.00 equiv) was added. After an overnight-stir at rt, the resultant mixture was transferred to a separatory funnel, diluted with aqueous KHSO4 (300 mL), and extracted with ethyl acetate (3 × 100 mL). The combined organic extracts were washed with brine, dried (anhyd. Na2S04) and concentrated under reduced pressure to afford crude N-hydroxy-4-phenylbutanamide (4.9 g, quantitative) as a white solid, which was used in the next step without further purification.
Dioxazolone 4i was prepared via a literature procedure for a related compound.8a The above crude N-hydroxy-4-phenylbutanamide (4.85 g, 27.0 mmol, 1.00 equiv) was redissolved in CH2C12 (300 mL). After adding CDI (4.38 g, 27.0 mmol, 1.00 equiv), the reaction mixture was stirred under nitrogen at rt for 30 mins and quenched with 1N HCl (150 mL). The resulting mixture was transferred to a separatory funnel and the organic layer was separated. The aqueous layer was extracted with CH2C12 (x2). The combined organic extracts were dried (anhyd. Na2SO4) and concentrated under reduced pressure. Purification by silica gel column chromatography (10% ethyl acetate/hexanes) afforded 4i (5.03 g, 91%) as a colorless liquid. FTIR (neat) 1871, 1824, 1636, 1148, 981, 752, 699 cm−1. 1H-NMR (500 MHz, CDCl3) δ 7.32 (t, J = 7.5 Hz, 2H), 7.23 (t, J = 7.4 Hz, 1H), 7.18 (d, J = 6.9 Hz, 2H), 2.75 (t, J = 7.4 Hz, 2H), 2.62 (t, J = 7.5 Hz, 2H), 2.07 (p, J = 7.4 Hz, 2H). 13C-NMR (126 MHz, CDCl3) δ 166.4, 154.1, 139.7, 128.7, 128.4, 126.5, 34.5, 25.9, 24.0. HRMS (EI+): m/z [M]+ calcd for C11H11O3N, 205.0739; found 205.0742.
General Procedures for C-H Functionalization of N-Azolo Imines with 3-Substituted-1,4,2-dioxazol-5-ones (0.3 mmol scale).
To a flame-dried 2–5 mL Biotage microwave vial (#351521) charged with a stir bar in a glove box was added N-azolo imine (0.300 mmol, 1.00 equiv), 3-substituted-1,4,2-dioxazol-5-one (0.450 mmol, 1.50 equiv), [Cp*Rh(MeCN)3](SbF6)2 (10 mol %, 0.0300 mmol, 25.0 mg), PivOH (0.600 mmol, 2.00 equiv, 61.2 mg), NaOAc (0.150 mmol, 0.500 equiv, 12.3 mg), and dioxane (0.100 M, 3.00 mL). The vial was capped with a Teflon-lined cap, removed from the glove box, and the mixture was stirred with a preheated stem block filled with oil at 120 °C for 16 h. The resultant mixture was then cooled to rt, filtered through a pad of celite, washed thoroughly with acetone, and concentrated under reduced pressure. The product was purified by silica gel column chromatography.
2,4-Diphenylimidazo[1,2-a][1,3,5]triazine (6aa):
The reaction was performed according to the general procedure employing 51.4 mg of N-imidazo imine 1a and 73.4 mg of 3-phenyl-1,4,2-dioxazol-5-one 4a. Purification by silica gel column chromatography (5–30% acetone/hexanes) afforded 6aa (59.9 mg, 73%) as a yellow solid. mp 156–158 °C. FTIR (neat) 1593, 1572, 1477, 1391, 1329, 1257, 1132, 759, 737, 687, 607, 479 cm−1. 1H-NMR (400 MHz, CDCI3) δ 8.72–8.55 (m, 2H), 8.09 (d, J = 6.6 Hz, 2H), 7.84–7.76 (m, 2H), 7.72–7.60 (m, 3H), 7.53–7.46 (m, 3H). 13C-NMR (101 MHz, CDCl3) δ 159.6, 155.1, 150.7, 136.6, 135.8, 132.6, 131.8, 131.6, 129.2, 128.8, 128.7, 128.5, 109.1. HRMS (ESI): m/z [M + H]+ calcd for C17H13N4+, 273.1135; found 273.1121.
4-(4-Chlorophenyl)-2-phenylimidazo[1,2-a] [1,3,5]triazine (6ab):
The reaction was performed according to the general procedure employing 51.4 mg of N-imidazo imine 1a and 88.9 mg of 3-(4-chlorophenyl)-1,4,2-dioxazol-5-one 4b. Purification by silica gel column chromatography (10–30% acetone/hexanes) afforded 6ab (55.1 mg, 60%) as a yellow solid. mp >200 °C. FTIR (neat) 3171, 3099, 1605, 1589, 1565, 1471, 1367, 1330, 1254, 1131, 1064, 731, 685, 485 cm−1. 1H-NMR (400 MHz, CDCl3) δ 8.70–8.53 (m, 2H), 8.06 (d, J = 8.6 Hz, 2H), 7.84–7.74 (m, 2H), 7.63 (d, J = 8.5 Hz, 2H), 7.54–7.44 (m, 3H). 13C-NMR (101 MHz, CDCl3) δ 159.5, 154.0, 150.7, 139.1, 136.9, 135.6, 131.7, 130.1, 130.1, 129.6, 128.7, 128.6, 108.8. HRMS (ESI): m/z [M + H]+ calcd for C17H12ClN4+, 307.0745; found 307.0736.
2-Phenyl-4-(4-(trifluoromethyl)phenyl)imidazo[1,2-a] [1,3,5]triazine (6ac):
The reaction was performed according to the general procedure employing 51.4 mg of N-imidazo imine 1a and 104 mg of 3-(4-(trifluoromethyl)phenyl)-1,4,2-dioxazol-5-one 4c. Purification by silica gel column chromatography (10–30% acetone/hexanes) afforded 6ac (47.1 mg, 46%) as a yellow solid. mp 158–161 °C. FTIR (neat) 1593, 1571, 1479, 1390, 1324, 1254, 1168, 1110, 1064, 1014, 848, 768, 716, 687, 440 cm−1. 1H-NMR (400 MHz, CDCl3) δ 8.688.52 (m, 2H), 8.22 (d, J = 8.1 Hz, 2H), 7.92 (d, J = 8.2 Hz, 2H), 7.82 (d, J = 1.7 Hz, 1H), 7.75 (d, J = 1.6 Hz, 1H), 7.54–7.44 (m, 3H). 13C-NMR (101 MHz, CDCl3) δ 159.5, 153.7, 150.5, 137.1, 135.5, 135.1, 134.3 (q, J = 33.1 Hz), 131.8, 129.2, 128.8, 128.6, 126.3 (q, J = 3.7 Hz), 123.4 (q, J = 272.9 Hz), 108.7. 19F-NMR (376 MHz, CDCl3) δ −63.19. HRMS (ESI): m/z [M + H]+ calcd for C18H12N4+, 341.1009; found 341.1010.
4-(4-Methoxyphenyl)-2-phenylimidazo[1,2-a] [1,3,5]triazine (6ad):
The reaction was performed according to the general procedure employing 51.4 mg of N-imidazo imine 1a and 86.9 mg of 3-(4-methoxyphenyl)-1,4,2-dioxazol-5-one 4d. Purification by silica gel column chromatography (5–30% acetone/hexanes) afforded 6ad (64.6 mg, 71%) as a yellow foam. FTIR (neat) 1574, 1501, 1473, 1391, 1329, 1260, 1246, 1132, 1038, 766, 713, 687, 611 cm−1. 1H-NMR (400 MHz, CDCl3) δ 8.65–8.56 (m, 2H), 8.08 (d, J = 8.8 Hz, 2H), 7.81 (d, J = 1.7 Hz, 1H), 7.75 (d, J = 1.7 Hz, 1H), 7.52–7.43 (m, 3H), 7.10 (d, J= 8.9 Hz, 2H), 3.91 (s, 3H). 13C-NMR (101 MHz, CDCl3) δ 163.1, 159.5, 154.7, 150.9, 136.4, 136.0, 131.4, 130.7, 128.7, 128.5, 123.9, 114.5, 109.0, 55.6. HRMS (ESI): m/z [M + H]+ calcd for C18H15N4O+, 303.1240; found 303.1242.
4-(3-Bromophenyl)-2-phenylimidazo[1,62-a] [1,3,5]triazine (6ae):
The reaction was performed according to the general procedure employing 51.4 mg of N-imidazo imine 1a and 109 mg of 3-(3-bromophenyl)-1,4,2-dioxazol-5-one 4e. Purification by silica gel column chromatography (10–30% acetone/hexanes) afforded 6ae (64.2 mg, 61%) as a yellow solid. mp 147–150 °C. FTIR (neat) 3167, 3133, 3104, 3063, 1588, 1562, 1464, 1389, 1327, 1253, 1131, 901, 869, 765, 731, 688, 609 cm−1. 1H-NMR (400 MHz, CDCl3) δ 8.69–8.55 (m, 2H), 8.22 (t, J = 1.8 Hz, 1H), 8.01 (d, J = 7.8 Hz, 1H), 7.84–7.78 (m, 2H), 7.77 (d, J = 1.6 Hz, 1H), 7.56–7.46 (m, 4H). 13C-NMR (101 MHz, CDCl3) δ 159.5, 153.6, 150.5, 136.9, 135.6, 135.5, 133.6, 131.8, 131.7, 130.7, 128.8, 128.6, 127.1, 123.4, 108.9. HRMS (ESI): m/z [M + H]+ calcd for C17H12BrN4+, 351.0240; found 351.0238.
4-(3-Methoxyphenyl)-2-phenylimidazo[1,2-a] [1,3,5] triazine (3af):
The reaction was performed according to the general procedure employing 51.4 mg of N-imidazo imine 1a and 86.9 mg of 3-(3-methoxyphenyl)-1,4,2-dioxazol-5-one 4f. Purification by silica gel column chromatography (10–30% acetone/hexanes) afforded 6af (59.2 mg, 65%) as a yellow foam. FTIR (neat) 1574, 1501, 1473, 1391, 1329, 1289, 1260, 1246, 1132, 1038, 766, 713, 687, 611 cm−1. 1H-NMR (400 MHz, CDCl3) δ 8.67–8.57 (m, 2H), 7.83–7.75 (m, 2H), 7.66–7.44 (m, 6H), 7.19 (dd, J = 8.3, 1.9 Hz, 1H), 3.91 (s, 3H). 13C-NMR (101 MHz, CDCl3) δ 160.1, 159.6, 155.0, 150.6, 136.5, 135.8, 132.9, 131.6, 130.3, 128.8, 128.5, 120.7, 118.3, 114.4, 109.2, 55.6. HRMS (ESI): m/z [M + H]+ calcd for C18H15N4O+, 303.1240; found 303.1242.
4-(2-Chlorophenyl)-2-phenylimidazo[1,2-a] [1,3,5]triazine (6ag):
The reaction was performed according to the general procedure employing 51.4 mg of N-imidazo imine 1a and 88.9 mg of 3-(2-chlorophenyl)-1,4,2-dioxazol-5-one 4g. Purification by silica gel column chromatography (10–30% acetone/hexanes) afforded 6ag (43.1 mg, 47%) as a tan solid. mp 156–158 °C. FTIR (neat) 1603, 1584, 1500, 1462, 1399, 1329, 1251, 1135, 1026, 763, 712, 690, 607, 438 cm−1. 1H-NMR (400 MHz, CDCl3) δ 8.67–8.52 (m, 2H), 7.76 (d, J = 1.6 Hz, 1H), 7.68 (d, J = 8.4 Hz, 1H), 7.65–7.57 (m, 2H), 7.56–7.44 (m, 4H), 7.23 (d, J = 1.7 Hz, 1H). 13C-NMR (101 MHz, CDCl3) δ 159.3, 153.7, 149.6, 136.6, 135.7, 132.7, 132.7, 131.6, 130.9, 130.7, 130.6, 128.9, 128.6, 127.6, 109.5 HRMS (ESI): m/z [M + H]+ calcd for C17H12ClN4+, 307.0745; found 307.0754.
2-Phenyl-4-(thiophen-2-yl)imidazo[1,2-a] [1,3,5]triazine (6ah):
The reaction was performed according to the general procedure employing 51.4 mg of N-imidazo imine 1a and 76.1 mg of 3-(2-thiophenyl)-1,4,2-dioxazol-5-one 4h. Purification by silica gel column chromatography (10–30% acetone/hexanes) afforded 6ah (48.3 mg, 58%) as a yellow solid. mp 141–144 °C. FTIR (neat) 3143, 3093, 1580, 1531, 1468, 1425, 1329, 1254, 1131, 862, 761, 706, 683, 480 cm−1. 1H-NMR (600 MHz, CDCl3) δ 8.65–8.58 (m, 2H), 8.15 (d, J = 3.8 Hz, 1H), 8.01 (d, J = 1.6 Hz, 1H), 7.86 (d, J= 1.6 Hz, 1H), 7.78 (d, J = 4.9 Hz, 1H), 7.54–7.45 (m, 3H), 7.31 (t, J = 4.4 Hz, 1H). 13C-NMR (151 MHz, CDCl3) δ 159.0, 151.1, 148.9, 137.3, 135.7, 135.1, 133.6, 132.0, 131.5, 128.8, 128.7, 128.5, 108.9. HRMS (ESI): m/z [M + H]+ calcd for C15H11N4S+, 279.0699; found 279.0698.
2-Phenyl-4-(3-phenylpropyl)imidazo[1,2-a][1,3,5]triazine (6ai):
The reaction was performed according to the general procedure employing 51.4 mg of N-imidazo imine 1a and 92.3 mg of 3-(3-phenylpropyl)-1,4,2-dioxazol-5-one 4i. Purification by silica gel column chromatography (5–35% acetone/hexanes) afforded 6ai (75.6 mg, 80%) as an off-white solid. mp 121–123 °C. FTIR (neat) 3104, 2930, 1592, 1501, 1400, 1259, 1141, 747, 705, 697, 621, 501 cm−1. 1H-NMR (400 MHz, CDCl3) δ 8.68–8.49 (m, 2H), 7.72 (apparent s, 1H), 7.53–7.44 (m, 3H), 7.34 (d, J = 1.6 Hz, 1H), 7.33–7.18 (m, 5H) 3.07 (t, J = 7.4 Hz, 2H), 2.85 (t, J = 7.4 Hz, 2H), 2.38 (p, J = 7.4 Hz, 2H). 13C-NMR (101 MHz, CDCl3) δ 159.1, 158.0, 149.7, 140.7, 136.3, 135.9, 131.5, 128.74, 128.6, 128.5, 128.5, 126.3, 107.2, 35.0, 32.3, 26.1. HRMS (ESI): m/z [M + H]+ calcd for C20H19N4+, 315.1604; found 315.1605.
4-Methyl-2-phenylimidazo[1,2-a][1,3,5]triazine (6aj):
The reaction was performed according to the general procedure with slight modification (1 equiv (24.6 mg) of NaOAc was used) employing 51.4 mg of N-imidazo imine 1a and 45.5 mg of 3-methyl-1,4,2-dioxazol-5-one 4j. Purification by silica gel column chromatography (10–35% acetone/hexanes) afforded 6aj (43.1 mg, 68%) as a yellow solid. mp >200 °C. FTIR (neat) 1605, 1592, 1503, 1432, 1261, 1145, 768, 717, 707, 685, 542 cm−1. 1H-NMR (400 MHz, CDCl3) δ 8.658.47 (m, 2H), 7.75 (d, J = 1.6 Hz, 1H), 7.52–7.45 (m, 3H), 7.44 (d, J = 1.6 Hz, 1H), 2.87 (s, 3H). 13C-NMR (101 MHz, CDCl3) δ 159.3, 155.4, 149.6, 136.3, 135.8, 131.5, 128.7, 128.5, 107.6, 20.8. HRMS (ESI): m/z [M + H]+ calcd for C12H11N4+, 211.0978; found 211.0976.
4-Isopropyl-2-phenylimidazo[1,2-a] [1,3,5] triazine (6ak):
The reaction was performed according to the general procedure employing 51.4 mg of N-imidazo imine 1a and 58.1 mg of 3-isopropyl-1,4,2-dioxazol-5-one 4k. Purification by silica gel column chromatography (5–35% acetone/hexanes) afforded 6ak (58.4 mg, 82%) as a white solid. mp 102–104 °C. FTIR (neat) 3120, 3097, 2971, 2934, 1606, 1593, 1501, 1402, 1332, 1252, 1126, 771, 739, 692, 619, 525, 483 cm−1. 1H-NMR (400 MHz, CDCl3) δ 8.69–8.49 (m, 2H), 7.73 (d, J = 1.7 Hz, 1H), 7.51 (d, J = 1.7 Hz, 1H), 7.49–7.42 (m, 3H) 3.40 (hept, J = 6.8 Hz, 1H), 1.51 (d, J = 6.8 Hz, 6H). 13C-NMR (101 MHz, CDCl3) δ 162.6, 159.3, 149.9, 136.2, 136.0, 131.4, 128.7, 128.5, 107.3, 32.3, 19.1. HRMS (ESI): m/z [M + H]+ calcd for C14H15N4+, 239.1291; found 239.1292.
4-Benzyl-2-phenylimidazo[1,2-a][1,3,5]triazine (6al):
The reaction was performed according to the general procedure with slight modification (1 equiv (24.6 mg) of NaOAc was used) employing 51.4 mg of N-imidazo imine 1a and 79.7 mg of 3-benzyl-1,4,2-dioxazol-5-one 4l. Purification by silica gel column chromatography (5–30% acetone/hexanes) afforded 6al (56.5 mg, 66%) as a yellow solid. mp 161–163 °C. FTIR (neat) 1608, 1592, 1498, 1402, 1330, 1133, 909, 732, 703, 686, 623, 563, 537 cm−1. 1H-NMR (400 MHz, CDCl3) δ 8.64–8.51 (m, 2H), 7.68 (d, J = 1.7 Hz, 1H), 7.56–7.45 (m, 3H), 7.42 (d, J = 1.7 Hz, 1H), 7.38–7.25 (m, 5H) 4.48 (s, 2H). 13C-NMR (101 MHz, cdcl3) δ 159.3, 156.5, 149.9, 136.4, 135.8, 132.7, 131.5, 129.1, 128.9, 128.8, 128.5, 127.9, 107.8, 40.9. HRMS (ESI): m/z [M + H]+ calcd for C18H15N4+, 287.1291; found 287.1293.
2-(Furan-2-yl)-4-phenylimidazo[1,2-a] [1,3,5]triazine (6ba):
The reaction was performed according to the general procedure employing 48.3 mg of N-imidazo imine 1b and 73.4 mg of 3-phenyl-1,4,2-dioxazol-5-one 4a. Purification by silica gel column chromatography (10–35% acetone/hexanes) afforded 6ba (43.2 mg, 55%) as a yellow solid. mp 130–132 °C. FTIR (neat) 1567, 1503, 1476, 1446, 1325, 1259, 1132, 759, 740, 694, 631, 594, 465 cm−1. 1H-NMR (400 MHz, CDCl3) δ 8.06–7.98 (m, 2H), 7.77–7.74 (m, 2H), 7.70–7.56 (m, 4H), 7.49 (d, J = 3.5 Hz, 1H), 6.59 (dd, J = 3.5, 1.7 Hz, 1H). 13C-NMR (101 MHz, CDCl3) δ 155.7, 152.3, 150.8, 149.9, 146.1, 136.7, 132.7, 131.4, 129.3, 128.7, 115.9, 112.5, 109.3. HRMS (ESI): m/z [M + H]+ calcd for C15H11N40+, 263.0927; found 263.0929.
2-(Furan-2-yl)-4-(3-phenylpropyl)imidazo[1,2-a][1,3,5]triazine (6bi):
The reaction was performed according to the general procedure employing 48.3 mg of N-imidazo imine 1b and 92.3 mg of 3-phenyl-1,4,2-dioxazol-5-one 4i. Purification by silica gel column chromatography (10–35% acetone/hexanes) afforded 6bi (53.2 mg, 58%) as a tan solid. mp 117119 °C. FTIR (neat) 3108, 2925, 2668, 1603, 1583, 1498, 1455, 1414, 1327, 1264, 1166, 1110, 1005, 750, 701, 594, 497 cm−1. 1H-NMR (400 MHz, CDCl3) δ 7.70 (d, J = 1.7 Hz, 1H), 7.65 (dd, J = 1.7, 0.9 Hz, 1H), 7.44 (dd, J = 3.5, 0.9 Hz, 1H), 7.357.13 (m, 6H), 6.57 (dd, J = 3.5, 1.8 Hz, 1H), 3.05 (t, J = 7.4 Hz, 2H), 2.82 (t, J = 7.4 Hz, 2H), 2.31 (p, J = 7.5 Hz, 2H). 13C-NMR (101 MHz, CDCl3) δ 158.7, 151.9, 150.9, 149.0, 146.1, 140.6, 136.4, 128.6, 128.5, 126.4, 115.8, 112.5, 107.5, 35.0, 32.5, 26.4. HRMS (ESI): m/z [M + H]+ calcd for C18H17N4O+, 305.1397; found 305.1396.
Methyl 4-(4-phenylimidazo[1,2-a][1,3,5]triazin-2-yl)benzoate (6ca):
The reaction was performed according to the general procedure employing 68.8 mg of N-imidazo imine 1c and 73.4 mg of 3-phenyl-1,4,2-dioxazol-5-one 4a. Purification by silica gel column chromatography (5–30% acetone/hexanes) afforded 6ca (64.3 mg, 65%) as a yellow solid. mp >200 °C. FTIR (neat) 3160, 3104, 1719, 1612, 1576, 1477, 1283, 1254, 1107, 761, 735, 692 cm−1. 1H-NMR (400 MHz, CDCl3) δ 8.70 (d, J = 8.6 Hz, 2H), 8.15 (d, J = 8.5 Hz, 2H), 8.10 (d, J = 6.7 Hz, 2H), 7.89–7.81 (m, 2H), 7.76–7.61 (m, 3H), 3.94 (s, 3H). 13C-NMR (101 MHz, CDCl3) δ 166.7, 158.5, 155.3, 150.4, 139.9, 137.1, 132.8, 132.5, 131.6, 129.7, 129.3, 128.7, 128.6, 109.3, 52.3. HRMS (ESI): m/z [M + H]+ calcd for C19H15N4O2+, 331.1190; found 331.1191.
Methyl 4-(4-(3-phenylpropyl)imidazo[1,2-a][1,3,5]triazin-2-yl)benzoate (6ci):
The reaction was performed according to the general procedure with slight modification (1 equiv (24.6 mg) of NaOAc was used) employing 68.8 mg of N-imidazo imine 1c and 92.3 mg of 3-phenyl-1,4,2-dioxazol-5-one 4i. Purification by silica gel column chromatography (5–30% acetone/hexanes) afforded 6ci (61.1 mg, 55%) as a white solid. mp 108–110 °C. FTIR (neat) 1721, 1610, 1593, 1504, 1495, 1410, 1273, 1252, 1016, 870, 770, 731, 718, 699 cm−1. 1H-NMR (400 MHz, CDCl3) δ 8.64 (d, J = 8.5 Hz, 2H), 8.14 (d, J = 8.5 Hz, 2H), 7.77 (d, J= 1.6 Hz, 1H), 7.38 (d, J = 1.6 Hz, 1H), 7.34–7.15 (m, 5H), 3.94 (s, 3H), 3.10 (t, J = 7.4 Hz, 2H), 2.86 (t, J = 7.4 Hz, 2H), 2.40 (p, J = 7.4 Hz, 2H). 13C-NMR (101 MHz, cdcl3) δ 166.7, 158.3, 158.0, 149.4, 140.6, 139.9, 136.7, 132.4, 129.7, 128.6, 128.5, 126.4, 107.5, 52.3, 35.0, 32.4, 26.1. HRMS (ESI): m/z [M + H]+ calcd for C22H211N4O2+, 373.1659; found 373.1657.
4-Phenyl-2-(4-(trifluoromethyl)phenyl)imidazo[1,2-a] [1,3,5] triazine (6da):
The reaction was performed according to the general procedure employing 71.8 mg of N-imidazo imine 1d and 73.4 mg of 3-phenyl-1,4,2-dioxazol-5-one 4a. Purification by silica gel column chromatography (5–30% acetone/hexanes) afforded 6da (59.8 mg, 59%) as a yellow solid. mp 143–146 °C. FTIR (neat) 1619, 1595, 1573, 1478, 1318, 1257, 1169, 1104, 1064, 1015, 858, 774, 694, 590, 440 cm−1. 1H-NMR (400 MHz, CDCl3) δ 8.74 (d, J = 8.1 Hz, 2H), 8.09 (d, J = 7.4 Hz, 2H), 7.84 (d, J = 8.8 Hz, 2H), 7.79–7.58 (m, 5H). 13C-NMR (101 MHz, CDCl3) δ 158.1, 155.4, 150.3, 139.1, 137.1, 132.9 (q, J = 32.6 Hz), 132.9, 131.5, 129.3, 129.0, 128.7, 125.5 (q, J = 3.7 Hz), 124.0 (q, J = 272.4 Hz), 109.4. 19F-NMR (376 MHz, CDCl3) δ −62.86. HRMS (ESI): m/z [M + H]+ calcd for C18H12F3N4+, 341.1009; found 341.1013.
4-(3-Phenylpropyl)-2-(4-(trifluoromethyl)phenyl)imidazo[1,2-a][1,3,5]triazine (6di):
The reaction was performed according to the general procedure employing 71.8 mg of N-imidazo imine 1d and 92.3 mg of 3-phenyl-1,4,2-dioxazol-5-one 4i. Purification by silica gel column chromatography (5–30% acetone/hexanes) afforded 6di (87.8 mg, 77%) as a white solid. mp 96–99 °C. FTIR (neat) 1617, 1595, 1496, 1415, 1319, 1253, 1164, 1107, 1063, 1015, 857, 784, 739, 697, 592, 489 cm−1. 1H-NMR (400 MHz, CDCl3) δ 8.68 (d, J = 8.2 Hz, 2H), 7.78 (d, J = 1.6 Hz, 1H), 7.73 (d, J = 8.3 Hz, 2H), 7.39 (d, J= 1.6 Hz, 1H), 7.34–7.26 (m, 2H), 7.26–7.17 (m, 3H), 3.11 (t, J = 7.4 Hz, 2H), 2.87 (t, J = 7.4 Hz, 2H), 2.40 (p, J = 7.4 Hz, 2H). 13C-NMR (101 MHz, CDCl3) δ 158.4, 157.6, 149.3, 140.5, 139.2, 136.8, 132.8 (q, J = 32.5 Hz), 128.9, 128.6, 128.4, 126.4, 125.5 (q, J = 3.7 Hz), 124.0 (q, J = 272.4 Hz), 107.5, 35.0, 32.4, 26.1. 19F-NMR (376 MHz, CDCl3) δ–62.86. HRMS (ESI): m/z [M + H]+ calcd for C21H18F3N4+, 383.1478; found 383.1480.
2-(4-Methoxyphenyl)-4-phenylimidazo[1,2-a] [1,3,5]triazine (6ea):
The reaction was performed according to the general procedure employing 60.4 mg of N-imidazo imine 1e and 73.4 mg of 3-phenyl-1,4,2-dioxazol-5-one 4a. Purification by silica gel column chromatography (5–30% acetone/hexanes) afforded 6ea (55.1 mg, 61%) as a yellow solid. mp 173–176 °C. FTIR (neat) 1591, 1573, 1500, 1471, 1448, 1391, 1310, 1249, 1165, 1138, 1023, 851, 777, 704, 691, 577, 516 cm−1. 1H-NMR (400 MHz, CDCl3) δ 8.57 (d, J = 8.9 Hz, 2H), 8.07 (d, J = 6.8 Hz, 2H), 7.78–7.71 (m, 2H), 7.70–7.59 (m, 3H), 6.98 (d, J = 8.9 Hz, 2H), 3.87 (s, 3H). 13C-NMR (101 MHz, CDCl3) δ 162.6, 159.5, 154.9, 150.8, 136.2, 132.5, 131.9, 130.6, 129.1, 128.7, 128.4, 113.9, 108.9, 55.4. HRMS (ESI): m/z [M + H]+ calcd for C18H15N4O+, 303.1240; found 303.1255.
2-(4-Methoxyphenyl)-4-(3-phenylpropyl)imidazo[1,2-a][1,3,5]triazine (6ei):
The reaction was performed according to the general procedure employing 60.4 mg of N-imidazo imine 1e and 92.3 mg of 3-phenyl-1,4,2-dioxazol-5-one 4i. Purification by silica gel column chromatography (10–30% acetone/hexanes) afforded 6ei (64.1 mg, 62%) as a yellow solid. mp 101–104 °C. FTIR (neat) 1593, 1508, 1493, 1399, 1307, 1249, 1165, 1028, 842, 784, 744, 698, 582, 503 cm−1. 1H-NMR (400 MHz, CDCl3) δ 8.53 (d, J = 9.0 Hz, 2H), 7.67 (d, J = 1.6 Hz, 1H), 7.34–7.18 (m, 6H), 6.98 (d, J = 8.9 Hz, 2H), 3.87 (s, 3H), 3.05 (t, J = 7.4 Hz, 2H), 2.85 (t, J = 7.4 Hz, 2H), 2.37 (p, J = 7.4 Hz, 2H). 13C-NMR (101 MHz, CDCl3) δ 162.5, 159.0, 157.7, 149.9, 140.8, 135.9, 130.6, 128.6, 128.5, 128.5, 126.3, 113.9, 106.9, 55.4, 35.0, 32.3, 26.1. HRMS (ESI): m/z [M + H]+ calcd for C21H21N4O+ 345.1710; found 345.1707.
2-(3-Bromophenyl)-4-phenylimidazo[1,2-a] [1,3,5] triazine (6fa):
The reaction was performed according to the general procedure employing 75.0 mg of N-imidazo imine 1f and 73.4 mg of 3-phenyl-1,4,2-dioxazol-5-one 4a. Purification by silica gel column chromatography (10–30% acetone/hexanes) afforded 6fa (62.2 mg, 59%) as a yellow solid. mp 179–182 °C. FTIR (neat) 1597, 1572, 1504, 1479, 1380, 1324, 1248, 1227, 1131, 1069, 764, 735, 690, 674, 661, 608 cm−1. 1H-NMR (400 MHz, CDCl3) δ 8.78 (t, J = 1.9 Hz, 1H), 8.56 (d, J = 7.9 Hz, 1H), 8.14–8.05 (m, 2H), 7.87–7.79 (m, 2H), 7.73–7.59 (m, 4H), 7.36 (t, J = 7.9 Hz, 1H). 13C-NMR (101 MHz, CDCl3) δ 158.1, 155.3, 150.3, 137.9, 136.9, 134.4, 132.8, 131.7, 131.5, 130.1, 129.3, 128.8, 127.3, 122.8, 109.3. HRMS (ESI): m/z [M + H]+ calcd for C17H12BrN4+, 351.0240; found 351.0245.
2-(3-Bromophenyl)-4-(3-phenylpropyl)imidazo[1,2-a] [1,3,5]triazine (6fi):
The reaction was performed according to the general procedure employing 75.0 mg of N-imidazo imine 1f and 92.3 mg of 3-phenyl-1,4,2-dioxazol-5-one 4i. Purification by silica gel column chromatography (10–30% acetone/hexanes) afforded 6fi (72.1 mg, 61%) as a white solid. mp 100–102 °C. FTIR (neat) 1600, 1495, 1455, 1319, 1253, 1131, 807, 779, 739, 719, 696, 673, 436 cm−1. 1H-NMR (400 MHz, CDCl3) δ 8.74 (t, J = 1.8 Hz, 1H), 8.50 (d, J = 7.9 Hz, 1H), 7.74 (d, J = 1.6 Hz, 1H), 7.61 (ddd, J = 7.9, 2.1, 1.1 Hz, 1H), 7.43–7.12 (m, 8H), 3.09 (t, J = 7.4 Hz, 2H), 2.86 (t, J = 7.4 Hz, 2H), 2.38 (p, J = 7.4 Hz, 2H). 13C-NMR (101 MHz, CDCl3) δ 158.3, 157.6, 149.4, 140.6, 138.0, 136.6, 134.3, 131.7, 130.1, 128.6, 128.5, 127.2, 126.4, 122.8, 107.4, 35.0, 32.4, 26.1. HRMS (ESI): m/z [M + H]+ calcd for C20H18BrN4+, 393.0709; found 393.0708.
2-(2-Chlorophenyl)-4-(3-phenylpropyl)imidazo[1,2-a][1,3,5]triazine (6gi):
The reaction was performed according to the general procedure with slight modification (1 equiv (24.6 mg) of NaOAc was used) employing 61.7 mg of N-imidazo imine 1g and 92.3 mg of 3-phenyl-1,4,2-dioxazol-5-one 4i. Purification by silica gel column chromatography (5–30% acetone/hexanes) afforded 6gi (48.3 mg, 46%) as a clear oil. FTIR (neat) 1587, 1495, 1329, 1261, 1247, 1139, 1107, 1049, 910, 761, 735, 698 cm−1. 1H-NMR (600 MHz, CDCl3) δ 7.97–7.92 (m, 1H), 7.81 (d, J = 1.6 Hz, 1H), 7.53–7.48 (m, 1H), 7.41 (d, J = 1.6 Hz, 1H), 7.41–7.36 (m, 2H), 7.31–7.27 (m, 2H), 7.24–7.17 (m, 3H), 3.11 (t, J = 7.4 Hz, 2H), 2.85 (t, J = 7.4 Hz, 2H), 2.36 (p, J = 7.4 Hz, 2H). 13C-NMR (151 MHz, CDCl3) δ 159.8, 158.1, 149.0, 140.7, 136.6, 136.0, 133.3, 132.1, 131.0, 130.9, 128.6, 128.5, 126.8, 126.3, 107.3, 34.9, 32.5, 26.2. HRMS (ESI): m/z [M + H]+ calcd for C20H18ClN4+, 349.1215; found 349.1214.
6,7-Dimethyl-2-phenyl-4-(3-phenylpropyl)imidazo[1,2-a][1,3,5]triazine (6hi):
The reaction was performed according to the general procedure employing 59.8 mg of N-imidazo imine 1h and 92.3 mg of 3-phenyl-1,4,2-dioxazol-5-one 4i. Purification by silica gel column chromatography (10–35% acetone/hexanes) afforded 6hi (53.5 mg, 52%) as a yellow solid. mp 149–152 °C. FTIR (neat) 1606, 1595, 1495, 1487, 1405, 1264, 1229, 1133, 1022, 756, 737, 697,565, 487 cm−1. 1H-NMR (400 MHz, CDCl3) δ 8.58–8.45 (m, 2H), 7.52–7.43 (m, 3H), 7.34–7.17 (m, 5H), 3.26 (t, J = 7.5 Hz, 2H), 2.84 (t, J = 7.4 Hz, 2H), 2.49 (s, 3H), 2.35 (s, 3H), 2.30 (p, J = 7.5 Hz, 2H). 13C-NMR (101 MHz, CDCl3) δ 158.1, 157.2, 148.6, 143.5, 141.0, 136.1, 131.0, 128.5, 128.4, 128.4, 126.2, 114.0, 34.9, 33.0, 27.9, 13.6, 11.9. HRMS (ESI): m/z [M + H]+ calcd for C22H23N4+, 343.1917; found 343.1918.
2-Methyl-5-phenyl-7-(3-phenylpropyl)-[1,2,4]triazolo[1,5-a] [1,3,5]triazine (6ii):
The reaction was performed according to the general procedure with slight modification (1 equiv (24.6 mg) of NaOAc was used; DCE was used instead of dioxane; temp = 100 °C) employing 55.9 mg of N-triazolo imine 1i and 92.3 mg of 3-phenyl-1,4,2-dioxazol-5-one 4i. Purification by silica gel column chromatography (10–30% ethyl acetate/hexanes) afforded 6ii (56.4 mg, 57%) as a white solid. mp 117–120 °C. FTIR (neat) 1610, 1593, 1529, 1478, 1445, 1407, 1376, 1312, 768, 746, 687, 677, 587, 570, 491 cm−1. 1H-NMR (400 MHz, CDCl3) δ 8.58 (d, J = 6.8 Hz, 2H), 7.57–7.46 (m, 3H), 7.29–7.15 (m, 5H), 3.35 (t, J = 7.5 Hz, 2H), 2.84 (t, J = 7.6 Hz, 2H), 2.62 (s, 3H), 2.39 (p, J = 7.6 Hz, 2H). 13C-NMR (101 MHz, CDCl3) δ 167.6, 163.3, 160.1, 157.4, 140.8, 135.2, 132.3, 129.3, 128.7, 128.5, 128.4, 126.2, 35.2, 31.4, 26.5, 15.2. HRMS (ESI): m/z [M + H]+ calcd for C20H20N5+, 330.1713; found 330.1721.
2,5-Diphenyl-7-(3-phenylpropyl)-[1,2,4]triazolo[1,5-a] [1,3,5]triazine (6ji):
The reaction was performed according to the general procedure with slight modification (1 equiv (24.6 mg) of NaOAc was used; DCE was used instead of dioxane; temp = 100 °C) employing 74.5 mg of N-triazolo imine 1j and 92.3 mg of 3-phenyl-1,4,2-dioxazol-5-one 4i. Purification by silica gel column chromatography (5–10% ethyl acetate/hexanes) afforded 6ji (65.7 mg, 56%) as a white solid. mp 136–138 °C. FTIR (neat) 1607, 1595, 1527, 1511, 1449, 1410, 1375, 1276, 767, 703, 697, 688, 607, 501 cm−1. 1H-NMR (400 MHz, CDCl3) δ 8.62 (d, J = 6.7 Hz, 2H), 8.40–8.32 (m, 2H), 7.60–7.47 (m, 6H), 7.31–7.15 (m, 5H), 3.43 (t, J = 7.4 Hz, 2H), 2.88 (t, J = 7.5 Hz, 2H), 2.44 (p, J = 7.5 Hz, 2H). 13C-NMR (101 MHz, CDCl3) δ 166.8, 163.3, 160.5, 157.7, 140.9, 135.2, 132.4, 131.1, 129.8, 129.3, 128.7, 128.7, 128.5, 128.4, 127.8, 126.2, 35.2, 31.4, 26.5. HRMS (ESI): m/z [M + H]+ calcd for C25H22N5+ 392.1870; found 392.1875.
Procedure for C-H Functionalization of N-Imidazo Imine 1a with 3-(3-Phenylpropyl)-1,4,2-dioxazol-5-one 4i Using a Microwave Reactor (1 mmol scale).
With bench-top set up, to a flame-dried 10–20-mL Biotage microwave vial (#354833) charged with a stir bar was added N-imidazo imine 1a (171 mg, 1.00 mmol, 1.00 equiv), [Cp*Rh(MeCN)3](SbF6)2 (10 mol %, 0.100 mmol, 83.3 mg), PivOH (204 mg, 2.00 mmol, 2 equiv), NaOAc (41.0 mg, 0.500 mmol, 0.500 equiv). The vial was capped with a Teflon-lined cap and flushed with N2 for ca. 5 min, and then dioxane (10.0 mL, 0.100 M) was added. To the resulting mixture was added 3-(3-phenylpropyl)-1,4,2-dioxazol-5-one 4i (0.260 mL, 306 mg, 0.450 mmol, 1.50 equiv) dropwise via syringe. The reaction vial was flushed with N2 for further ca. 5 min before heating with a Biotage Initiator+ (#356007), which employs an external IR sensor and a closed reaction vessel. The resultant mixture was stirred in the microwave reactor for 2 h at 170 °C using the following settings (absorption level: low, vial type: 10–20 mL, prestirring: 0, initial power: 0, dynamic deflector optimization: ON, pressure: OFF, power: OFF, fixed hold time: ON, stir rate: 600). After cooling to rt, the resultant mixture was filtered through a pad of celite, washed thoroughly with acetone, and concentrated under reduced pressure. Purification by silica gel column chromatography (5–35% acetone/hexanes) afforded 6ai (205.3 mg, 65%) as an off-white solid. 1H- and 13C-NMR spectra matched with 6ai obtained from small scale (0.3 mmol).
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the NIH (R35GM122473 to J.A.E). The Villum Foundation (VKR023371) is also acknowledged for postdoctoral support to K.S.H.
Footnotes
ASSOCIATED CONTENT
Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
NMR spectra (PDF)
The authors declare no competing financial interest
REFERENCES
- (1).For selected approved drugs incorporating a [5,6]-bicyclic heterocycle core with a ring junction nitrogen, see: anagliptin, olprinone, minodronic acid, vardenafil, zalepton, acalabrutinib, zolpidem, and ponatinip. The compound structure, bioactivity, list of literature, and access to ongoing clinical trials, applications, and usage can be obtained by searching the compound name in PubChem.
- (2).For select phase II and III clinical candidates incorporating a [5,6]-bicyclic heterocycle core with a ring junction nitrogen, see: LY2090314, dipraglurant, AMG-337, irbinitinib, dinaciclib, empesertib, fligotinib, entospletinib, andvolitinib. The compound structure, bioactivity, list of literature, and access to ongoing clinical trials, applications, and usage can be obtained by searching the compound name in PubChem.
- (3).Halskov KS; Witten MR; Hoang GL; Mercado BQ; Ellman JA Rhodium(III)-Catalyzed Imidoyl C-H Activation for Annulations to Azolopyrimidines. Org. Lett 2018, 20, 2464–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) For selected reviews on transition metal-catalyzed C-H functionalization that include applications to nitrogen heterocycle synthesis, see: Satoh T; Miura M Oxidative Coupling of Aromatic Substrates with Alkynes and Alkenes under Rhodium Catalysis. Chem. Eur. J 2010, 16, 11212–11222. [DOI] [PubMed] [Google Scholar]; (b) Song G; Wang F; Li X C-C, C-O and C-N Bond Formation via Rhodium(III)-Catalyzed Oxidative C-H Activation. Chem. Soc. Rev 2012, 41, 3651–3678. [DOI] [PubMed] [Google Scholar]; (c) Yamaguchi J; Yamaguchi AD; Itami K C-H Bond Functionalization: Emerging Synthetic Tools for Natural Products and Pharmaceuticals. Angew. Chem. Int. Ed 2012, 51, 8960–9009. [DOI] [PubMed] [Google Scholar]; (d) Mesganaw T; Ellman JA Convergent Synthesis of Diverse Tetrahydropyridines via Rh(I)-Catalyzed C-H Functionalization Sequences. Org. Process Res. Dev 2014, 18, 1097–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Gulías M; Mascareñas JL Metal-Catalyzed Annulations through Activation and Cleavage of C-H Bond. Angew. Chem. Int. Ed 2016, 55, 11000–11019. [DOI] [PubMed] [Google Scholar]; (f) Newton CG; Wang S-G; Oliveira CC; Cramer N Catalytic Enantioselective Transformations Involving C-H Bond Cleavage by Transition-Metal Complexes. Chem. Rev 2017, 117, 8908–8976. [DOI] [PubMed] [Google Scholar]; (g) Hummel JR; Boerth JA; Ellman JA Transition-Metal-Catalyzed C-H Bond Addition to Carbonyls, Imines, and Related Polarized π Bonds. Chem. Rev 2017, 117, 91639227. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Park Y; Kim Y; Chang S Transition Metal-Catalyzed C-H Amination: Scope, Mechanism, and Applications. Chem. Rev 2017, 117, 9247–9301. [DOI] [PubMed] [Google Scholar]; (i) Dong Z; Ren Z; Thompson SJ; Xu Y; Dong G Transition-Metal-Catalyzed C-H Alkylation Using Alkenes. Chem. Rev 2017, 117, 9333–9403. [DOI] [PubMed] [Google Scholar]
- (5).Sauer J; Mayer KK Thermolyse und Photolyse von 3.4-Diphenyl-Δ2-1.2.4-oxdiazolinon-(5) und 2.4-Diphenyl-Δ2-1.3.4-oxdiazolinon-(5). Tetrahedron Lett. 1968, 9, 319–324. [Google Scholar]
- (6).Dubé P; Nathel NFF; Vetelino M; Couturier M; Aboussafy CL; Pichette S; Jorgensen ML; Hardink M Carbonyldiimidazole-Mediated Lossen Rearrangement. Org. Lett 2009, 11, 5622–5625. [DOI] [PubMed] [Google Scholar]
- (7).Bizet V; Buglioni L; Bolm C Light-Induced Ruthenium-Catalyzed Nitrene Transfer Reactions: A Photochemical Approach towards N-Acyl Sulfimides and Sulfoximines. Angew. Chem. Int. Ed 2014, 53, 5639–5642. [DOI] [PubMed] [Google Scholar]
- (8).(a) Park Y; Park KT; Kim JG; Chang S Mechanistic Studies on the Rh(III)-Mediated Amido Transfer Process Leading to Robust C-H Amination with a New Type of Amidating Reagent. J. Am. Chem. Soc 2015, 137, 4534–4542. [DOI] [PubMed] [Google Scholar]; (b) Park Y; Jee S; Kim JG; Chang S Study of Sustainability and Scalability in the Cp*Rh(III)-Catalyzed Direct C-H Amidation with 1,4,2-Dioxazol-5-ones. Org. Process Res. Dev 2015, 19, 1024–1029. [Google Scholar]
- (9).For a recent review including 1,4,2-dioxazol-5-one as an efficient amidating reagent in C-H functionalization, see reference 4h and references cited therein.
- (10).Halskov KS; Roth HS; Ellman JA Synthesis of [5,6]-Bicyclic Heterocycles with a Ring-Junction Nitrogen Atom: Rhodium(III)-Catalyzed C-H Functionalization of Alkenyl Azoles. Angew. Chem. Int. Ed 2017, 56, 9183–9187, DOI: 10.1002/anie.201703967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Wu X; Sun S; Xu S; Cheng J Rh-Catalyzed Annulation of ortho-C-H Bonds of 2-Arylimidazoles with 1,4,2-Dioxazol-5-ones toward 5-Arylimidazo[1,2-c]quinazolines. Adv. Synth. Catal 2018, 360, 1111–1115, DOI: 10.1002/adsc.201701331. [DOI] [Google Scholar]
- (12).Lim FPL; Dolzhenko AV 1,3,5-Triazine-Based Analogues of Purine: From Isosteres to Privileged Scaffolds in Medicinal Chemistry. Eur. J. Med. Chem, 2014, 85, 371–390. [DOI] [PubMed] [Google Scholar]
- (13).Li reported that Zn(OAc)2 in conjunction with a cobalt catalyst provide efficient system for annulations to enamides and pyrimidones (see Liu, Y.; Xie, F.; Jia, A.-Q.; Li, X. Cp*Co(III)-Catalyzed Amidation of Olefinic and Aryl C-H Bonds: Highly Selective Synthesis of Enamides and Pyrimidones. Chem. Commun. 2018, 54, 4345–4348). Co(III) and Rh(III) catalysts employed with Zn(OAc)2 instead of NaOAc/PivOH were tried but proved unsuccessful: no desired bicyclic product was detected by crude 1H-NMR.
- (14).Li H; Zhao J; Zeng L; Hu W Organocatalytic Asymmetric Domino Aza-Michael-Mannich Reaction: Synthesis of Tetrahydroimidazopyrimidine Derivatives. J. Org. Chem 2011, 76, 8064–8069. [DOI] [PubMed] [Google Scholar]
- (15).(a) Mishra A; Mukherjee U; Vats TK; Deb I Ir(III)/MPAA-Catalyzed Mild and Selective C-H Amidation of N-Sulfonyl Ketimines: Access to Benzosultam-Fused Quinazolines/Quinazolinones. J. Org. Chem 2018, 83, 3756–3767. [DOI] [PubMed] [Google Scholar]; (b) Chen Y; Zhang R; Peng Q; Xu L; Pan X Rhodium(III)-Catalyzed Directed C-H Amidation of N-Nitrosoanilines and Subsequent Formation of 1,2-Disubstituted Benzimidazoles. Chem. Asian J 2017, 12, 2804–2808. [DOI] [PubMed] [Google Scholar]; (c) Bizet V; Bolm C Sulfur Imidations by Light-Induced Ruthenium-Catalyzed Nitrene Transfer Reactions. Eur. J. Org. Chem 2015, 2854–2860. [Google Scholar]
- (16).Little TL; Webber SE A Simple and Practical Synthesis of 2-Aminoimidazoles. J. Org. Chem 1994, 59, 7299–7305. [Google Scholar]
- (17).Usachova N; Leitis G; Jirgensons A; Kalvinsh I Synthesis of Hydroxamic Acids by Activation of Carboxylic Acids with N,N’-Carbonyldiimidazole: Exploring the Efficiency of the Method. Synth. Commun 2010, 40, 927–935. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.