

Abstract
Glutamatergic transmission in the nucleus accumbens shell (NAcSh) is a substrate for reward learning and motivation. Metabotropic glutamate (mGlu) receptors regulate NAcSh synaptic strength by inducing long-term depression (LTD). Inputs from prefrontal cortex (PFC) and medio-dorsal thalamus (MDT) drive opposing motivated behaviors yet mGlu receptor regulation of these synapses is unexplored. We examined Group I mGlu receptor regulation of PFC and MDT glutamatergic synapses onto specific populations of NAc medium spiny neurons (MSNs) using D1tdTom BAC transgenic mice and optogenetics. Synaptically evoked long-term depression (LTD) at MDT-NAcSh synapses required mGlu5 but not mGlu1 and was specific for D1(+) MSNs, whereas PFC LTD was expressed at both D1(+) and D1(−) MSNs and required mGlu1 but not mGlu5. Two weeks after five daily non-contingent cocaine exposures (15 mg/kg), LTD was attenuated at MDT-D1(+) synapses but was rescued by the mGlu5-positive allosteric modulator (PAM) VU0409551. These results highlight unique plasticity mechanisms regulating specific NAcSh synapses.
Introduction
The nucleus accumbens (NAc) integrates excitatory inputs encoding salient neuronal information, directing reward acquisition and shaping motivated behaviors. Strengthening discrete afferent-target glutamatergic synapses is thought to bias cellular computation of action selection by NAc circuitry and is widely regarded as a critical physiological process underlying reward learning and memory [1, 2]. Repeated exposure to salient stimuli restructures these synaptic connections, redirecting future responses to stimulus presentation. Maladaptive changes in synaptic strength and synaptic plasticity are implicated in the motivational deficits observed in depression, anxiety, and drug abuse [3, 4]. Understanding the molecular components underlying plasticity at specific NAc synapses may direct therapeutic interventions for treating motivational disorders.
NAc medium spiny neurons (MSNs) can be distinguished by their expression of either dopamine receptor type-1 [D1(+)], or type-2 [defined herein as D1(−)], and downstream projection targets [5–7]. Canonically, D1(+) MSNs promote reward seeking while D1(−) MSNs promote aversion [8, 9]. Excitatory inputs onto NAc MSNs are associated with unique characteristics of motivated behavior [10]. Glutamatergic inputs from the prefrontal cortex (PFC) and the medial dorsal thalamus (MDT) have contrasting effects on behavioral output. Animals will self-stimulate PFC-NAc afferents and activation promotes real-time place preference [5] whereas activation of MDT afferents inhibits acquisition of palatable rewards while inhibition alleviates negative affective behaviors associated with opiate withdrawal [11, 12].
Within the NAc, post-synaptic Group I mGlu receptors, comprised of mGlu1 and mGlu5, are known to trigger LTD of excitatory synaptic transmission. Activation of NAc Group I mGlu receptors induces a LTD of excitatory post-synaptic currents (EPSCs) via retrograde endocannabinoid signaling and/or post-synaptic internalization of AMPA receptors [13–19]. Notably NAc mGlu5 function is blunted/absent in mice exposed to cocaine via reduced surface expression [18–21]. Augmenting mGlu function using positive allosteric modulators (PAMs), agonists, or antagonists has been shown to inhibit drug-seeking behaviors and drug-induced physiological changes in mice [14, 22–24]. However, remodeling of NAc circuits following drug exposure differs by subregion, cell type, and afferent origin [5, 6, 25]. Elucidating how mGlu signaling modifies excitatory drive at discrete synapses is critical for understanding how they regulate the propagation of information through reward circuits.
Here, we utilize whole-cell patch clamp electrophysiology in D1tdTom BAC transgenic mice, viral-mediated gene transfer of channel rhodopsin (ChR2), and pharmacology to define modulation of PFC and MDT synapses onto NAcSh MSNs by mGlu receptors. We find low-frequency stimulation (LFS) of ChR2+ terminals elicits mGlu1 and mGlu5 dependent long-term depression (LTD) that is defined by afferent origin. Additionally, prior cocaine exposure inhibits mGlu5 dependent LTD at MDT-D1(+) synapses which is rescued by application of an mGlu5 PAM. Together, our results suggest that Group I mGlu receptor signaling selectively regulates distinct synaptic connections in the NAc of drug-naive animals and is impaired by cocaine history in a synapse-specific manner. These findings also suggest targeting of mGlu5 may be valuable in treating nuanced maladaptive synaptic remodeling following substance abuse.
Methods
Animals
All animals were bred and housed at Vanderbilt under the supervision of the Department of Animal Care. Transgenic BAC Drd1a-tdTomato mice were obtained from JAX laboratories and bred to C57BL/6J wild type females. Animals were housed on a 12-h light/dark cycle and fed ad lib. Breeding cages were given access to 5LOD chow (PicoLab®, 28.7% protein, 13.4% fat, 57.9% carbohydrate) to improve the viability of litters. Upon weaning at P21–28, experimental animals were switched to standard chow.
Stereotaxic surgery
All surgeries were performed in accordance to guidelines set by Vanderbilt IACUC. Briefly, 4–6 week male C57BL6 mice are anesthetized using a cocktail of ketamine (75 mg/kg) and dexdomitidor (0.5 mg/kg). Craniotomies were performed using a manual drill, AmScope microscope, and World Precision Instruments Aladdin Al-2000 syringe pump hydraulic system. Injection sites were based on coordinates listed in The Mouse Brain in Stereotaxic Coordinates [26]. PFC (AP 1.4, ML ± 0.5, DV −2.9 mm) and MDT (AP −1.2, ML 0.3, DV −3.00 mm) were located using Leica AngleTwo Stereotaxic software. AAV-CamKII-ChR2-EYFP (UNC Vector Core) was injected at 100 nL/min and allowed to permeate into the tissue for 10 min before removal of the syringe. Mice were revived using 0.5 mg/kg antisedan and treated with 5 mg/kg ketoprofen for 3 days following surgery.
Electrophysiology
Mice were anesthetized using isoflurane prior to sacrifice. Parasagittal sections (250 µm) containing the NAcSh were prepared from whole brain tissue using a Leica Vibratome. Slices were briefly placed in an N-methyl d-glucamine (NMDG) based recovery solution (2.5 KCL, 20 HEPES, 1.2 NaH2PO4, 25 Glucose, 93 NMDG, 30 NaHCO3, 5.0 sodium ascorbate, 3.0 sodium pyruvate, 10 MgCl2, and 0.5 mM CaCl2-2H2O) for 10–15 min at 32 °C before transfer to a chamber containing artificial cerebral spinal fluid (ACSF, 119 NaCl, 2.5 KCL, 1.3 MgCl2-6H2O, 2.5 CaCl2-2HO, 1.0 NaH2PO4-H2O, 26.2 NaHCO3, and 11 mM glucose) until use. All electrophysiology experiments were performed using a Scientifica Slicescope Pro System under a constant perfusion of 32°C ACSF at a rate of 2 mL/min. NAcSh MSNs were visualized with Scientifica PatchVision software and patched with 3–5 MΩ recording pipettes (P1000 Micropipette Puller) filled with a cesium-based internal solution (120 CsMeSO3, 15 CsCl, 8 NaCl, 10 HEPES, 0.2 EGTA, 10 TEA-Cl, 4.0 Mg2-ATP, 0.3 Na2-GTP, 0.1 spermine, and 5.0 mM QX 314 bromide). MSNs were identified based on visual appearance (size, morphology) as well as electrophysiological properties (membrane resistance, capacitance, and the presence of currents at +40 mV to exclude fast-spiking interneurons); D1(+) MSNs were identified by fluorescence of tdTomato. All experiments were performed in the presence of the GABAA channel blocker picrotoxin (50 µM). Experimental protocol execution, stimulation control, and data collection were performed using Molecular Devices pClamp 10 Analysis software. Control and monitoring of cell electrical properties were achieved using an Axopatch 500B Multiclamp amplifier and Axon Digidata 1550 low-noise data acquisition digitizer. Responses were filtered at 2 kHz and digitized at 10 kHz. Optical stimulation of ChR2-expressing terminals was achieved using a CoolLED pE-100 LED excitation system. 480 nm light was pulsed through the high-powered (×40) objective to excite ChR2+ terminals at 0.1 Hz for 0.5–1 msec. Light intensity was adjusted to evoke stable responses.
Behavior
Behavior was performed in MedAssociates Activity Test Chambers. Mice used in cocaine experiments were habituated to the behavior chambers and intraperitoneal (IP) saline injections in 15 min sessions over 2 days. The following 5 days, mice were given a single injection of vehicle (saline) or cocaine (15 mg/kg) IP immediately prior to being placed in the chamber. Mice were housed in home cages for the duration of the sessions and for at least 2 weeks following the final session before being sacrificed for electrophysiology recordings. Locomotor activity was tracked using Noldus Ethovision software.
Drugs
2-Methyl-6-(phenylethynyl)pyridine hydrochloride (MPEP, mGlu5 antagonist), LY367385 (mGlu1 antagonist), (RS)-3,5-dihydroxyphenylglycine (DHPG, Group I mGlu agonist), ketoprofen, dexdomitodor, and antisedan, were obtained from Tocris. Picrotoxin and cocaine hydrochloride were obtained from Sigma Aldrich. Ketamine was obtained from Patterson Veterinary Supply. VU0409551 was contributed by Jerri M. Rook, Craig W. Lindsley, and P. Jeffrey Conn.
Imaging
Widefield images were taken using an AZ-100 microscope housed in the Vanderbilt Cell Imaging Shared Resource Core facility. 250 µm brain slices originally prepared for electrophysiology experiments (see above) were fixed after recording with a 4% paraformaldehyde (PFA) solution for <24 h and stored in a 20% w/v sucrose solution until use. Images were processed using NIS Elements Viewer (Nikon) and ImageJ.
Data analysis
Electrophysiology experiments were analyzed using Clampfit 10.4 and Graphpad Prism v6.0. For LTD experiments, change in baseline and CV was calculated by averaging each value in the last 10 min of each recording and comparing to the average across baseline events. LTD was defined as a significant difference in amplitude of the last 10 min of the recording as measured using a one sample t-test vs. 100. Paired t-tests were performed to compare changes in CV over the course of experiments. Two-tailed t-tests were used to compare drug effects at specific synapses (defined by cell type and afferent origin). −70 mV decay was analyzed using a one way ANOVA and Tukey’s post-test. Behavior sensitization data was analyzed using two way repeated measures ANOVA with Sidak’s and Dunnet’s post-tests to examine individual days. For all analyses, alpha was set as 0.05.
Results
PFC and MDT innervate NAc MSNs
Both cortical (PFC) and thalamic (MDT) inputs into the NAc form excitatory synapses onto NAc MSNs [12, 27–31]. Whole-cell patch clamp electrophysiology was performed on acute parasagittal brain slices of d1tdTom BAC transgenic mice in order to isolate these NAcSh excitatory connections in a cell-type-specific manner. We used ChR2 to drive the activity of PFC and MDT projections in the NAcSh and uncover mechanisms mediating synaptic plasticity. AAV-CaMKII-CHR2-EYFP was stereotaxically injected into the infralimbic PFC and MDT (Fig. 1a). Three weeks post infection was sufficient for robust expression of ChR2-EYFP in both cell soma and afferents within NAcSh (Fig. 1b). A brief (0.5–1 ms) pulse of blue light was sufficient to activate ChR2 in EFYP+ neurons and evoke robust excitatory currents and action-potential firing with high fidelity (Fig. 1c). In the NAcSh, light-evoked EPSCs were observed at PFC and MDT synapses onto both MSN subtypes. EPSC amplitude varied with stimulation intensity, duration, and efficiency of viral infection at the injection site; however, values were not significantly different based on the synapse sampled (Fig. 1d, e). Interestingly, EPSC waveforms exhibited significant differences in decay kinetics of AMPA currents across cell types and inputs. (Fig. 1f). Specifically, −70 t1/2 was significantly greater at MDT synapses compared to those from the PFC of the same cell type (PFC-D1(+) vs. MDT-D1(+), p < 0.01; PFC-D1(−) vs. MDT-D1(−), p < 0.05, one way ANOVA, Tukey’s multiple comparisons test). These findings demonstrated both PFC and MDT form strong excitatory connections with NAcSh MSNs.
Fig. 1.

AAV-CamKII-ChR2-EYFP infection in the PFC and MDT results in robust expression of ChR2 at injection site and NAc terminals. a Experimental timeline for recording optically evoked EPSCs in the NAc. b Widefield fluorescence imaging of EYFP expression in the PFC, MDT, and resulting expression in NAc afferent fibers. c Representative optically evoked action-potential firing (top) and positive current (bottom) via 10 Hz stimulation of ChR2 in an EYFP-expressing neuron. d Representative optically evoked EPSCs elicited by light stimulation of PFC (top) and MDT (bottom) synapses onto NAc D1(+) (red) and D1(−) (black) MSNs. Scale bars: 100 pA/10 ms. e Summary plot of optical EPSC amplitudes recorded from NAc MSNs in all naive LTD experiments prior to LFS. f Decay kinetics for optical EPSCs evoked from PFC and MDT synapses in the NAcSh.
mGlu receptor-dependent LTD is differentially expressed at PFC and MDT synapses
Recent work has demonstrated that inputs into the NAcSh are susceptible to differing types of synaptic modulation based on afferent origin [32]. Several studies have demonstrated that 10–13 Hz stimulation of NAc core glutamatergic afferents induces an mGlu5-dependent LTD [17–19]. Therefore, we sought to determine whether this mGlu-dependent LTD was similarly induced in the NAcSh adding both cell type and input specificity. Following a 5 min 10 Hz stimulation, we observed a robust light-evoked LTD at PFC synapses at both D1(+) and D1(−) MSNs (Fig. 2a–d PFC-D1(+), 69.16 ± 5.65, n = 10, p < 0.01; PFC-D1(−), 72.05 ± 4.327, n = 10, p < 0.001 one sample t-test). However, LFS-induced LTD at MDT synapses onto D1(+) but not D1(−) MSNs (Fig. 2f–i MDT-D1(+), 75.76 ± 6.221, n = 10, p < 0.01; MDT-D1(−), 104.3 ± 7.487, n = 7, p = 0.584, one sample t-test). Surprisingly, we were not able to reliably sample the paired pulse ratio (PPR) at PFC and MDT synapses, as paired stimulation resulted in run-down and near failures following repeated exposure (data not shown). In lieu of this, 1/CV2 was analyzed. Light-evoked LTD did not induce a change in 1/CV2 at PFC or MDT synapses (Fig. 2e, j PFC: D1(+) base vs. LTD, p = 0.271; D1(−) base vs. LTD, p = 0.39, paired t-test. MDT: D1(+) base vs. LTD, p = 0.893; D1(−) base vs. LTD, p = 0.149, paired t-test). These results suggested that this mechanism likely does not involve presynaptic changes. We concluded that LTD is present at PFC synapses onto both D1(+) and D1(−) NAcSh MSNs while LTD is specific for MDT-D1(+) synapses and likely occurs via a post-synaptic mechanism.
Fig. 2.

LFS of PFC and MDT inputs to NAcSh elicits robust LTD. a, b Representative LTD induction at PFC D1(+) [red] and D1(−) [gray] synapses. c Representative traces averaged from baseline and the last 10 min following LTD induction (30-40). Scale bars 100 pA/10 ms. d Averaged LTD experiments at PFC synapses. e 1/CV2 during baseline and following LTD induction. f, g Representative LTD induction at MDT D1(+) and D1(−) synapses. h Representative traces averaged from baseline and the last 10 min following LTD induction (min 30–40). Scale bars 100 pA/10 ms. i Averaged LTD experiments sampled at MDT synapses. LTD was not evoked at MDT D1(−) synapses but was present at D1(+). j 1/CV2 during baseline and following LTD induction.
To determine the molecular mediators of 10 Hz LTD at PFC and MDT synapses in the NAcSh, we utilized pharmacological antagonists of Group I mGlu receptors. We found that 10 Hz LTD was intact in the presence of the mGlu5 antagonist MPEP at both D1(+) and D1(−) PFC synapses (Fig. 3a, c D1(+) +MPEP, 69.07 ± 4.962, n = 6, p > 0.05; D1(−) + MPEP, 58.0 ± 12.6, n = 5, p > 0.05, one sample t-test vs. 100; D1(+) naive vs. MPEP, p > 0.05; D1(−) naive vs. MPEP, p > 0.05, t-test). As LTD was not observed at MDT D1(−) synapses we focused on MDT-D1(+). LTD was blocked by MPEP at MDT-D1(+) synapses (Fig. 3b, c D1(+) 103.7 ± 7.651, p = 0.652, n = 6, one sample t-test vs. 100; naive vs. MPEP, p < 0.01, t-test), suggesting mGlu5 regulation of glutamatergic transmission is synapse specific. mGlu1 has been shown to regulate PFC afferents in the NAcSh but only in rats withdrawn from cocaine self-administration [15, 28, 33]. Nevertheless, we examined whether 10 Hz stimulation of PFC-NAc synapses was dependent on mGlu1 activation. Surprisingly, we found that the mGlu1 antagonist LY367385 (50 µM) was indeed able to completely block LTD at PFC-NAcSh synapses (Fig. 3d, f D1(+) + LY, 106.9 ± 10.78, n = 7, p = 0.548; D1(−) + LY, 98.84 ± 13.4, n = 5, p = 0.934, one sample t-test vs. 100; D1(+) naive vs. LY, p < 0.01, D1(−) naive vs. LY, p < 0.05, t-test). The presence of 50 µM LY367385 did not block but enhanced LTD at MDT-D1(+) synapses (Fig. 3e, f D1(+) + LY, 24.8 ± 2.404, n = 6, p < 0.001, one sample t-test vs. 100; D1(+) naive vs. LY, p < 0.01, t-test). These results demonstrated that mGlu1-mediated LTD is present at PFC-NAcSh synapses while LTD at MDT-D1(+) MSNs requires mGlu5 in naive mice.
Fig. 3.

LTD of PFC and MDT synapses is differentially controlled by Group I mGlu receptor subtype. a Bath application of 10 µM MPEP does not impair LTD of PFC-NAcSh synapses. b Bath application of MPEP completely blocks induction of LTD at MDT-D1(+) NAcSh synapses. c Summary of experiments performed in the presence of 10 µM MPEP. Values shown as percent baseline. d Application of 50 µM LY367385 completely blocks induction of LTD at PFC-NAcSh synapses. e LY367385 did not impair LTD at MDT-D1(+) synapses. f Summary of experiments performed in the presence of 50 µM LY367385. Values shown as percent baseline. g, h 5 min bath application of the Group I mGlu agonist R,S-DHPG (100 µM) induces a robust LTD at both PFC and MDT-D1(+)synapses in the NAcSh. i Summary of DHPG experiments at PFC and MDT synapses. Values shown as percent baseline.
In order to verify that Group I mGlu receptor activation was sufficient to induce LTD we applied the Group I mGlu agonist DHPG. 100 µM DHPG-induced a robust depression of synaptic transmission at PFC and MDT synapses onto NAC MSNs (Fig. 3g–i PFC: D1(+) + DHPG, 63.51 ± 9.523, n = 7, p < 0.01; D1(−) + DHPG, 66.66 ± 11.5, n = 6, p < 0.05, one sample t-test vs. 100. MDT: D1(+) + DHPG, 74.52 ± 6.97, n = 7, p < 0.05, one sample t-test vs. 100). We concluded that mGlu receptors function differentially at PFC and MDT NAc synapses to regulate synaptic strength.
Cocaine selectively impairs mGlu-LTD at D1(+) MSNs
Impairments in mGlu function in the NAc have been correlated to both acute and repeated exposure to cocaine. Diminished pharmacological mGlu LTD has been observed in the NAcSh 14-21 day drug-free period, which we refer to herein as “abstinence” from repeated cocaine exposure [20]. Therefore, we chose to examine whether a repeated drug exposure and abstinence paradigm would impair mGlu LTD at PFC and MDT synapses. Following stereotaxic surgery, mice were subjected to a 7-day cocaine sensitization task (Fig. 4a) before being returned to the home-cage for a 14-day abstinence. Across repeated cocaine injections, mice exhibited a robust increase in locomotor activity relative to saline treated controls and relative to the first day of cocaine exposure, consistent with drug-induced neurological adaptations (Fig. 4a—two way ANOVA of saline vs. cocaine, interaction F(6, 66) = 18.1. ***p < 0.001, ****p < 0.0001, Cocaine compared to day 1 of drug, Dunnet’s multiple comparisons. #p < 0.05, ###p < 0.01, ####p < 0.001, saline vs. cocaine (daily), Sidak’s multiple comparisons). Following cocaine abstinence, we observed a robust LTD at PFC-D1(−) synapses in both saline and cocaine treated mice (Fig. 4c Saline, 67.45 ± 7.658, n = 5, p < 0.05; cocaine, 66.93 ± 5.329, n = 7, p < 0.05, one sample t-test). Similarly, LFS-LTD at PFC-D1(+) synapses was not significantly inhibited in mice exposed to cocaine when compared to saline controls (Fig. 4b, c PFC-D1(+), saline, 72.83 ± 6.60, n = 11, p < 0.05; cocaine, 87.83 ± 3.581, n = 9, p < 0.05, one sample t-test. sal vs. coc, p = 0.07, t-test). However, we observed a trend towards a reduction in LTD magnitude, indicating LTD may be impaired but not absent. Interestingly, LFS-LTD of MDT synapses onto D1(+) synapses was absent in mice treated with cocaine (Fig. 4e, g saline, 68.1 ± 6.38, n = 5, p < 0.01; cocaine, 95.32 ± 6.24, n = 6, p > 0.05, one sample t-test. Sal vs. Coc, p < 0.05, t-test). LFS-LTD was still absent at MDT-D1(−) synapses in cocaine treated mice (MDT-D1(−) + Coc, 108.6 ± 19.9, n = 3, data not shown). Taken together, these results suggested that mGlu5 dependent LFS-LTD is selectively impaired MDT-D1(+) synapses in mice exposed to cocaine.
Fig. 4.

LTD at D1(+) synapses in the NAc is impaired following cocaine exposure. a Timeline of experimental procedures leading up to electrophysiological recordings. Mice exposed to cocaine (15 mg/kg) exhibited robust locomotor sensitization following repeated exposures. b LTD at PFC D1(+) MSNs was not significantly impaired in mice exposed to cocaine compared to saline controls and EPSC amplitude was significantly different from baseline in cocaine-LTD experiments. c LTD at PFC D1(−) MSNs was not affected by cocaine exposure. d Summary of LTD experiments from sal/coc exposed mice at PFC-NAc synapses. Values shown as percent baseline. e LTD at MDT D1(+) synapses was absent in mice exposed to cocaine and significantly different from saline controls. f Bath application of the mGlu5 PAM VU551 (10 µM) was sufficient to rescue LTD deficits seen at MDT-D1(+) synapses in mice exposed to cocaine. g Summary of LTD experiments from sal/coc exposed mice at MDT D1(+) synapses. Values shown as percent baseline.
The mGlu5-positive allosteric modulator VU0409551 rescues MDT LTD impaired by cocaine experience
Modulation of Group I mGlu receptors has been proposed as a valuable therapeutic strategy for treating a range of psychiatric disorders. Recent findings have demonstrated that mGlu5 PAMs have strong efficacy in ameliorating behavioral deficits in a schizophrenia mouse model [34]. Additionally, mGlu1 PAMs have been efficacious in reducing cue-induced drug seeking in rats and reverse some aspects of physiological adaptations in the NAc [15]. Therefore, having demonstrated that an mGlu5-dependent LTD at MDT-D1(+) synapses is impaired following cocaine, we chose to examine whether a selective mGlu5 PAM, VU0409551 (VU551) was sufficient to rescue LTD in cocaine treated mice. To assess this, 10 µM VU551 (PAM) was included in the ACSF during baseline and LTD induction. LTD at MDT-D1(+) synapses was rescued in the presence of VU551 to levels near saline controls (Fig. 4f, g D1(+) + Pam, 55.83 ± 3.58, n = 5, p < 0.01, one sample t-test; coc vs. coc + PAM, p < 0.05, t-test). These results demonstrated that potentiating mGlu5 can restore plasticity at MDT-D1(+) NAcSh synapses.
Discussion
NAc mGlu receptors are potent regulators of glutamatergic synaptic strength [17–19, 35] and are impacted by cocaine history [23, 36]. However, how these receptors control excitatory transmission from discrete inputs is unknown. Here, we demonstrated an mGlu1 LFS-LTD is present at PFC synapses onto both cell types, but plasticity from MDT inputs is specific for D1(+) MSNs and requires mGlu5. Following cocaine exposure, this plasticity is attenuated selectively at MDT D1(+) synapses (Fig. 5). These results are consistent with recent studies demonstrating a heterogeneity of molecular regulatory mechanisms functioning at specific NAc synapses and input and cell-type-specific changes induced by drug exposure [12, 27–29, 32]. These findings broaden the known role of mGlu receptors in shaping reward circuit function.
Fig. 5.
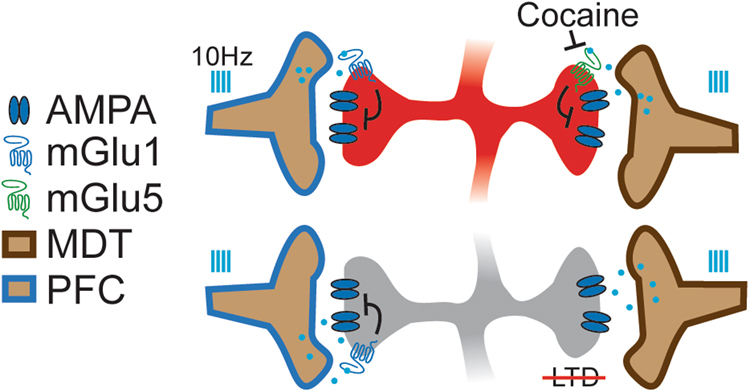
Model of mGlu regulation of NAcSh PFC and MDT synapses. 10 Hz light-evoked Group I mGlu LTD is differentially expressed at PFC and MDT synapses, with cortical synapses recruiting mGlu1 while thalamic inputs onto D1(+) MSNs recruits mGlu5. LTD at all synapses likely occurs via a post-synaptic mechanism as indicated by absence of changes in 1/CV2. Prior cocaine exposure blocks the expression of LTD at thalamic—D1(+) synapses but does not impair mGlu1 LTD at PFC synapses.
Distinct glutamatergic NAcSh inputs are hypothesized to drive nuanced aspects of motivated behavior. Specifically, the PFC is thought to direct reward seeking and enhance positive environment-associated valence [5, 10]. Conversely, inputs from the MDT have been shown to drive conditioned place aversion and inhibit palatable reward seeking [11]. Using region-specific expression of ChR2 (Fig. 1a, b) and D1-tdTom marker mice, we determined that, despite these differences in behavioral affect, light-evoked EPSCs did not vary in amplitude across inputs or between cell types. However, we observed significantly greater decay kinetics at MDT-NAc synapses compared to PFC (Fig. 1e). While differences in kinetics have been observed due to AMPA subunit composition [37], these results are inconsistent with recent findings showing the presence of GluA2-lacking AMPA receptors in naive and saline treated rats at MDT-NAc synapses [29]. However, these differences could also be explained by synaptic morphology, dendritic locus, or AMPA auxiliary protein association [38, 39] and may serve as the basis for future studies.
We next determined whether mGlu receptor-dependent LTD occurs at PFC and MDT synapses. We found that optical LFS is able to induce a robust LTD at PFC synapses on both MSN subtypes (Fig. 2). However, LTD at MDT synapses was specific for D1(+) MSNs. Additionally, light-evoked LTD was not accompanied by a change in 1/CV2 (Fig. 2h, i) suggesting it may occur via a post-synaptic mechanism. We next confirmed the necessity of Group I mGlu receptor activation for light-evoked LTD at PFC and MDT NAcSh synapses. Using specific Group I mGlu receptor antagonists, we found that LTD at PFC-NAcSh synapses was independent of mGlu5 but required mGlu1 (Fig. 3). This result was surprising as previous publications have almost exclusively implicated mGlu5 as the triggering mechanism for this plasticity within the NAc. Specifically, Ma et al. demonstrated an mGlu1-dependent plasticity at PFC-NAc synapses using a 1 Hz stimulation protocol that also required NMDA receptors in rats trained to self-administer cocaine [28]. Additionally, others have shown a switch from mGlu5-dependent eCB signaling in drug-naive animals to an mGlu1-dependent internalization of AMPA receptors in animals that had self-administered cocaine [15, 28]. However, these studies did not differentiate MSN subtype, were performed in rats, and evoked Group I mGlu LTD using a different stimulation protocol or the Group I mGlu agonist DHPG. Thus, our findings suggest that mGlu1 activation may favor PFC inputs and require more substantial glutamate release for recruitment in naive animals.
Unlike the PFC, LFS-LTD of MDT inputs was specific for D1(+) MSNs. The presence of this LTD specifically at D1(+) MSNs may contribute to the aversive effects of 10–30 Hz stimulation of MDT synapses in the NAc in vivo [11]. LTD of D1(+) but not D1(−) would bias MDT-driven NAc function towards D1(−) MSNs output, a phenomenon shown to oppose appetitive behavior [40]. Additionally, this plasticity was blocked by the mGlu5 antagonist MPEP and insensitive to the mGlu1 antagonist LY367385 (Fig. 5d,e). Bath application of DHPG was similarly sufficient to induce LTD at MDT-D1(+) synapses. Taken together, these findings demonstrate an input and cell-type-specific regulation of NAc synapses by Group I mGlu receptors, with pan Group I activation favoring decreased PFC influence and enhanced MDT-D1(−) excitatory drive. While this demonstrated differential regulation of these inputs by Group I mGlu receptors, both mGlu1 and mGlu5 are expressed at PFC and MDT NAc terminals [41]. Thus, both mGlu1 and mGlu5 are likely functional but are recruited by different conditions.
Alterations in glutamatergic transmission in the NAcSh occur following abstinence/withdrawal from cocaine, a physiological correlate of the incubation of drug craving [18, 36]. These changes broadly include an increase in glutamatergic quantal size, changes in MSN excitability, generation of silent synapses, increases in presynaptic release, and disruptions in mGlu-dependent plasticity [6]. Thus, we investigated whether mGlu-dependent LTD was affected following abstinence from cocaine at PFC and MDT synapses. We observed no significant difference in LFS-LTD at PFC synapses in mice exposed to cocaine compared to saline controls (Fig. 4c, d). However, LFS-LTD at MDT-D1(+) synapses was absent in mice exposed to cocaine. This is consistent with multiple reports demonstrating cocaine-induced adaptations in NAc circuitry is specific for D1(+) MSNs [31, 32, 42] as well as observed deficits in mGlu-dependent plasticity in rodents following cocaine [15, 18, 19]. These findings are supported by decreased DHPG-induced LTD in the NAcSh following abstinence from experimenter-delivered cocaine [20, 43]. While others have demonstrated enhanced LFS-LTD at PFC-D1(+) synapses following withdrawal from contingent cocaine self-administration [32], multiple reports have failed to see differences in glutamatergic synaptic strength of PFC synapses in the NAcSh following non-contingent drug delivery [5, 27]. Taken together, these results are congruent with reports highlighting differential effects of contingent and experimenter-delivered drug regimens on NAc circuitry function [33] and attenuation of mGlu-dependent plasticity in withdrawal from non-contingent cocaine exposure.
Resetting synaptic signaling via in vivo optogenetics is sufficient to ameliorate drug-induced behavioral adaptations. By “normalizing” the connectivity of the PFC with the NAcSh using in vivo optogenetic LFS mimicking mGlu plasticity ex vivo, cue-induced cocaine seeking is reduced [32]. Additionally, dampening MDT input in to the NAc using hM4Di (inhibitory) designer receptors exclusively activated by designer drugs or optogenetic silencing was sufficient to reduce morphine withdrawal-induced aversion [12]. While mGlu1 agonists and PAMs have been shown efficacious in rescuing cocaine-induced physiological and behavioral effects [15], it is unknown whether mGlu5 PAMs are also able to ameliorate drug-induced changes in the NAc. Thus, we utilized VU551, an mGlu5 PAM shown to potentiate mGlu5 signaling independent of NMDA receptor activation [44] and reduce psycho-mimetic behavior in rodents. VU551 was able to rescue the induction of mGlu5 LTD at MDT-D1(+) synapses in cocaine treated mice. Notably, mGlu5 in the NAc had been shown to promote resilience to chronic stress [45], and mGlu5 antagonists are capable of reducing lever pressing for cocaine [46]. We posit that the multimodal effects of targeting mGlu5 may be due in part to regulation of specific afferent-MSN connections. While agonizing mGlu5 appears to run counter to preventing drug seeking, targeting mGlu5 may alternatively be useful for treating anhedonia, anxiety, and dysphoria following the cessation of drug intake.
Our findings demonstrate that synaptic recruitment of Group I mGlu receptors occurs differentially at discrete NAc afferents, highlighting unique roles for mGlu1 and mGlu5 in regulating PFC-NAc and MDT-NAc synapses, respectively. While it is unclear whether these findings are specific to PFC and MDT synapses, these results highlight the necessity for synapse-specific approaches in future studies aimed at deconstructing molecular mediators of synaptic connectivity within the reward circuitry. Expanding our understanding of synapse-specific plasticity mechanisms serves to clarify how unique synaptic profiles allow for integration of neuronal reward-encoding information and opens the door for targeted pharmacological approaches to remodel reward circuit function. Our results broaden the understanding of mGlu regulation of NAc reward circuitry, bolstering the potential for targeting mGlu receptors in motivational disorders.
Acknowledgements
We would like to thank Kevin Manz and Drs Max Joffe and Carrie Grueter for their comments and critiques of this manuscript.
Funding
This was supported by NIDA R00 DA031699 (for BAG). CWL is an inventor on multiple composition of matter patents protecting allosteric modulators of GPCRs. JMR’s work has been funded by NIH, Alzheimer’s Drug Discovery Foundation, Harrington Discovery Institute, Thome Alzheimer’s Disease Drug Discovery Foundation, and Brain & Behavior Research Foundation. She is an inventor on patents that protect different classes of muscarinic receptor allosteric modulators. PJC has been funded by NIH, Michael J. Fox Foundation, Dystonia Medical Research Foundation, CHDI Foundation and others.
Conflict of interest
Over the past 3 years PJC has served on the Scientific Advisory Boards for Michael J. Fox Foundation, Stanley Center for Psychiatric Research Broad Institute (MIT/Harvard), Karuna Pharmaceuticals, Lieber Institute for Brain Development, Clinical Mechanism (POCM) and Proof of Concept (POC) Consortium, and Neurobiology Foundation for Schizophrenia and Bipolar Disorder. He is an inventor on patents that protect different classes of metabotropic glutamate and muscarinic receptor allosteric modulators. The remaining authors declare that they have no conflict of interest.
References
- 1.Koob GF. Neurobiology of addiction. Focus. 2011;9:55–65. doi: 10.1176/foc.9.1.foc55. [DOI] [Google Scholar]
- 2.Luscher C, Malenka RC. Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron. 2011;69:650–63. doi: 10.1016/j.neuron.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Joffe ME, Grueter CA, Grueter BA. Biological substrates of addiction. Wiley Interdiscip Rev Cogn Sci. 2014;5:151–71. doi: 10.1002/wcs.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Russo SJ, Nestler EJ. The brain reward circuitry in mood disorders. Nat Rev Neurosci. 2013;14:609–25. doi: 10.1038/nrn3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Britt JP, Benaliouad F, McDevitt RA, Stuber GD, Wise RA, Bonci A. Synaptic and behavioral profile of multiple glutamatergic inputs to the nucleus accumbens. Neuron. 2012;76:790–803. doi: 10.1016/j.neuron.2012.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grueter BA, Rothwell PE, Malenka RC. Integrating synaptic plasticity and striatal circuit function in addiction. Curr Opin Neurobiol. 2012;22:545–51. doi: 10.1016/j.conb.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kreitzer AC, Malenka RC. Striatal plasticity and basal ganglia circuit function. Neuron. 2008;60:543–54. doi: 10.1016/j.neuron.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calipari ES, Bagot RC, Purushothaman I, Davidson TJ, Yorgason JT, Pena CJ, et al. In vivo imaging identifies temporal signature of D1 and D2 medium spiny neurons in cocaine reward. Proc Natl Acad Sci USA. 2016;113:2726–31. doi: 10.1073/pnas.1521238113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith RJ, Lobo MK, Spencer S, Kalivas PW. Cocaine-induced adaptations in D1 and D2 accumbens projection neurons (a dichotomy not necessarily synonymous with direct and indirect pathways) Curr Opin Neurobiol. 2013;23:546–52. doi: 10.1016/j.conb.2013.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Everitt BJ, Robbins TW. Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci. 2005;8:1481–9. doi: 10.1038/nn1579. [DOI] [PubMed] [Google Scholar]
- 11.Do-Monte FH, Minier-Toribio A, Quinones-Laracuente K, Medina-Colon EM, Quirk GJ. Thalamic regulation of sucrose seeking during unexpected reward omission. Neuron. 2017;94:388–400.e4. doi: 10.1016/j.neuron.2017.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu Y, Wienecke CF, Nachtrab G, Chen X. A thalamic input to the nucleus accumbens mediates opiate dependence. Nature. 2016;530:219–22. doi: 10.1038/nature16954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loweth JA, Scheyer AF, Milovanovic M, LaCrosse AL, Flores-Barrera E, Werner CT, et al. Synaptic depression via mGluR1 positive allosteric modulation suppresses cue-induced cocaine craving. Nat Neurosci. 2014;17:73–80. doi: 10.1038/nn.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loweth JA, Tseng KY, Wolf ME. Adaptations in AMPA receptor transmission in the nucleus accumbens contributing to incubation of cocaine craving. Neuropharmacology. 2014;76(Pt B):287–300. doi: 10.1016/j.neuropharm.2013.04.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCutcheon JE, Loweth JA, Ford KA, Marinelli M, Wolf ME, Tseng KY. Group I mGluR activation reverses cocaine-induced accumulation of calcium-permeable AMPA receptors in nucleus accumbens synapses via a protein kinase C-dependent mechanism. J Neurosci. 2011;31:14536–41. doi: 10.1523/JNEUROSCI.3625-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohno-Shosaku T, Kano M. Endocannabinoid-mediated retrograde modulation of synaptic transmission. Curr Opin Neurobiol. 2014;29:1–8. doi: 10.1016/j.conb.2014.03.017. [DOI] [PubMed] [Google Scholar]
- 17.Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ. Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc Natl Acad Sci USA. 2002;99:8384–8. doi: 10.1073/pnas.122149199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fourgeaud L, Mato S, Bouchet D, Hemar A, Worley PF, Manzoni OJ. A single in vivo exposure to cocaine abolishes endocannabinoid-mediated long-term depression in the nucleus accumbens. J Neurosci. 2004;24:6939–45. doi: 10.1523/JNEUROSCI.0671-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grueter BA, Brasnjo G, Malenka RC. Postsynaptic TRPV1 triggers cell type-specific long-term depression in the nucleus accumbens. Nat Neurosci. 2010;13:1519–25. doi: 10.1038/nn.2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang CC, Yeh CM, Wu MY, Chang AY, Chan JY, Chan SH, et al. Cocaine withdrawal impairs metabotropic glutamate receptor-dependent long-term depression in the nucleus accumbens. J Neurosci. 2011;31:4194–203. doi: 10.1523/JNEUROSCI.5239-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szumlinski KK, Abernathy KE, Oleson EB, Klugmann M, Lominac KD, He DY, et al. Homer isoforms differentially regulate cocaine-induced neuroplasticity. Neuropsychopharmacology. 2006;31:768–77. doi: 10.1038/sj.npp.1300890. [DOI] [PubMed] [Google Scholar]
- 22.Grueter BA, McElligott ZA, Robison AJ, Mathews GC, Winder DG. In vivo metabotropic glutamate receptor 5 (mGluR5) antagonism prevents cocaine-induced disruption of postsynaptically maintained mGluR5-dependent long-term depression. J Neurosci. 2008;28:9261–70. doi: 10.1523/JNEUROSCI.2886-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grueter BA, McElligott ZA, Winder DG. Group I mGluRs and long-term depression: potential roles in addiction? Mol Neurobiol. 2007;36:232–44. doi: 10.1007/s12035-007-0037-7. [DOI] [PubMed] [Google Scholar]
- 24.Loweth JA, Tseng KY, Wolf ME. Using metabotropic glutamate receptors to modulate cocaine’s synaptic and behavioral effects: mGluR1 finds a niche. Curr Opin Neurobiol. 2013;23:500–6. doi: 10.1016/j.conb.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stuber GD, Britt JP, Bonci A. Optogenetic modulation of neural circuits that underlie reward seeking. Biol Psychiatry. 2012;71:1061–7. doi: 10.1016/j.biopsych.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. Cambridge, MA: Academic Press; 2008.
- 27.Joffe ME, Grueter BA. Cocaine experience enhances thalamo-accumbens N-methyl-d-aspartate receptor function. Biol Psychiatry. 2016;80:671–81. doi: 10.1016/j.biopsych.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma YY, Lee BR, Wang X, Guo C, Liu L, Cui R, et al. Bidirectional modulation of incubation of cocaine craving by silent synapse-based remodeling of prefrontal cortex to accumbens projections. Neuron. 2014;83:1453–67. doi: 10.1016/j.neuron.2014.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neumann PA, Wang Y, Yan Y, Wang Y, Ishikawa M, Cui R, et al. Cocaine-induced synaptic alterations in thalamus to nucleus accumbens projection. Neuropsychopharmacology. 2016;41:2399–410. doi: 10.1038/npp.2016.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pascoli V, Turiault M, Luscher C. Reversal of cocaine-evoked synaptic potentiation resets drug-induced adaptive behaviour. Nature. 2011;481:71–75. doi: 10.1038/nature10709. [DOI] [PubMed] [Google Scholar]
- 31.Terrier J, Luscher C, Pascoli V. Cell-type specific insertion of GluA2-lacking AMPARs with cocaine exposure leading to sensitization, cue-induced seeking, and incubation of craving. Neuropsychopharmacology. 2016;41:1779–89. doi: 10.1038/npp.2015.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pascoli V, Terrier J, Espallergues J, Valjent E, O’Connor EC, Luscher C. Contrasting forms of cocaine-evoked plasticity control components of relapse. Nature. 2014;509:459–64. doi: 10.1038/nature13257. [DOI] [PubMed] [Google Scholar]
- 33.McCutcheon JE, Wang X, Tseng KY, Wolf ME, Marinelli M. Calcium-permeable AMPA receptors are present in nucleus accumbens synapses after prolonged withdrawal from cocaine self-administration but not experimenter-administered cocaine. J Neurosci. 2011;31:5737–43. doi: 10.1523/JNEUROSCI.0350-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Foster DJ, Conn PJ. Allosteric modulation of GPCRs: new insights and potential utility for treatment of schizophrenia and other CNS disorders. Neuron. 2017;94:431–46. doi: 10.1016/j.neuron.2017.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Turner BD, Kashima DT, Manz KM, Grueter CA, Grueter BA. Synaptic plasticity in the nucleus accumbens: lessons learned from experience. ACS Chem Neurosci. 2018. [DOI] [PMC free article] [PubMed]
- 36.Wolf ME. Synaptic mechanisms underlying persistent cocaine craving. Nat Rev Neurosci. 2016;17:351–65. doi: 10.1038/nrn.2016.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu W, Shi Y, Jackson AC, Bjorgan K, During MJ, Sprengel R, et al. Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron. 2009;62:254–68. doi: 10.1016/j.neuron.2009.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Herring BE, Shi Y, Suh YH, Zheng CY, Blankenship SM, Roche KW, et al. Cornichon proteins determine the subunit composition of synaptic AMPA receptors. Neuron. 2013;77:1083–96. doi: 10.1016/j.neuron.2013.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Greger IH, Watson JF, Cull-Candy SG. Structural and functional architecture of AMPA-type glutamate receptors and their auxiliary proteins. Neuron. 2017;94:713–30. doi: 10.1016/j.neuron.2017.04.009. [DOI] [PubMed] [Google Scholar]
- 40.Lobo MK, Covington HE, 3rd, Chaudhury D, Friedman AK, Sun H, Damez-Werno D, et al. Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science. 2010;330:385–90. doi: 10.1126/science.1188472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mitrano DA, Pare JF, Smith Y. Ultrastructural relationships between cortical, thalamic, and amygdala glutamatergic inputs and group I metabotropic glutamate receptors in the rat accumbens. J Comp Neurol. 2010;518:1315–29. doi: 10.1002/cne.22277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.MacAskill AF, Cassel JM, Carter AG. Cocaine exposure reorganizes cell type- and input-specific connectivity in the nucleus accumbens. Nat Neurosci. 2014;17:1198–207. doi: 10.1038/nn.3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang CC, Liang YC, Lee CC, Hsu KS. Cocaine withdrawal impairs mGluR5-dependent long-term depression in nucleus accumbens shell neurons of both direct and indirect pathways. Mol Neurobiol. 2015;52:1223–33. doi: 10.1007/s12035-014-8926-z. [DOI] [PubMed] [Google Scholar]
- 44.Rook JM, Xiang Z, Lv X, Ghoshal A, Dickerson JW, Bridges TM, et al. Biased mGlu5-positive allosteric modulators provide in vivo efficacy without potentiating mGlu5 modulation of NMDAR currents. Neuron. 2015;86:1029–40. doi: 10.1016/j.neuron.2015.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shin S, Kwon O, Kang JI, Kwon S, Oh S, Choi J, et al. mGluR5 in the nucleus accumbens is critical for promoting resilience to chronic stress. Nat Neurosci. 2015;18:1017–24. doi: 10.1038/nn.4028. [DOI] [PubMed] [Google Scholar]
- 46.Moussawi K, Pacchioni A, Moran M, Olive MF, Gass JT, Lavin A, et al. N-Acetylcysteine reverses cocaine-induced metaplasticity. Nat Neurosci. 2009;12:182–9. doi: 10.1038/nn.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]