

Abstract
The common oxidatively generated lesion, 8-oxo-7,8-dihydroguanine (8-oxoGua), is removed from DNA by base excision repair. The glycosylase primarily charged with recognition and removal of this lesion is 8-oxoGuaDNA glycosylase 1 (OGG1). When left unrepaired, 8-oxodG alters transcription and is mutagenic. Individuals homozygous for the less active OGG1 allele, Ser326Cys, have increased risk of several cancers. Here, small molecule enhancers of OGG1 were identified and tested for their ability to stimulate DNA repair and protect cells from the environmental hazard paraquat (PQ). PQ-induced mtDNA damage was inversely proportional to the levels of OGG1 expression whereas stimulation of OGG1, in some cases, entirely abolished its cellular effects. The PQ-mediated decline of mitochondrial membrane potential or nuclear condensation were prevented by the OGG1 activators. In addition, in Ogg1−/− mouse embryonic fibroblasts complemented with hOGG1S326C, there was increased cellular and mitochondrial reactive oxygen species compared to their wild type counterparts. Mitochondrial extracts from cells expressing hOGG1S326C were deficient in mitochondrial 8-oxodG incision activity, which was rescued by the OGG1 activators. These data demonstrate that small molecules can stimulate OGG1 activity with consequent cellular protection. Thus, OGG1-activating compounds may be useful in select humans to mitigate the deleterious effects of environmental oxidants and mutagens.
Keywords: 8-Oxoguanine DNA glycosylase-1, 8-oxo-7, 8-dihydroguanine, base excision repair, OGG1S326C, mitochondria, oxidative stress
Graphical abstract
INTRODUCTION
DNA in the nucleus and in the mitochondria is vulnerable to oxidative damage from endogenous and exogenous sources [1]. These non-bulky lesions are most often repaired by the base excision repair (BER) pathway [2]. The first step of this pathway, glycosylase incision, is an important point of specification, while other steps are common to any lesion repaired through BER [3]. A common and biologically important, mutagenic DNA lesion resulting from oxidative stress is 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxodG). The glycosylase primarily tasked with removal of 8-oxodG from the genome is 8-oxoGua DNA glycosylase (OGG1). OGG1 is present in both the nucleus and mitochondria, however, mitochondria accumulate 8-oxodG at rates 20-fold greater than the nucleus in mice deficient in Ogg1 [4]. In the absence of efficient glycosylase activity, oxidative stress can be highly mutagenic in both cellular compartments. In the mitochondria, DNA polymerase gamma (polγ), normally a high-fidelity replicative polymerase, inserts adenine opposite 8-oxodG greater than 25% of the time, resulting in a G-C to T-A transversion [5, 6]. Given that mtDNA is guanine-rich combined with its proximity to the primary cellular source of reactive oxygen species (ROS), it is particularly vulnerable to lesion formation and mutation. In the nucleus, the adduct is also mutagenic, as DNA polymerase delta (polδ) inserts A instead of C approximately 50% of the time opposite 8-oxodG, and may cooperate with translesion synthesis DNA polymerase eta (polη) to bypass this lesion [7]. In addition, RNA Polymerase II can readily bypass this lesion, but frequently misincorporates adenine opposite 8-oxodG lesions within coding regions, resulting in miscoded or prematurely terminated mRNAs [8]. Thus, fully active OGG1 is important for the clearance of 8-oxodG from the nuclear and mitochondrial genomes to prevent mutagenesis and transcriptional infidelity.
Although no disease has been associated with the loss of OGG1, several human OGG1 variants have been identified and correlated with disease risk. The most common disease-associated variant of hOGG1 is a single nucleotide substitution resulting in a serine to cysteine missense mutation at position 326 (hOGG1S326C). The Cys-326 allele occurs at frequencies of ~25–40% within Caucasian populations and ~40–60% in Asians [9]. Homozygotes of the hOGG1S326C allele are at elevated risk for developing several types of cancer [10–13] as well as the age-related degenerative disorder, macular degeneration [14]. A longevity study involving 7170 subjects, aged 55 to 70 at high risk for cardiovascular disease was recently reanalyzed for the S326C mutation and cancer, cardiovascular disease, and all-cause mortality [15]. Cys/Cys individuals had a hazard ratio of 1.69 over Ser carriers. The only dietary intervention that could be analyzed given the number of subjects in each group was vegetable intake as it related to variant vs. serine carriers. Cys/Cys individuals with low vegetable intake had significantly higher cardiovascular disease mortality (p<0.001). But Cys/Cys individuals that ate >314 g of vegetables per day were not at significantly higher risk than Ser/Ser (p=0.671). These data demonstrate that intervention can alter mortality risk in genetically predisposed individuals.
Biochemical evidence demonstrates that hOGG1S326C is less active than the serine variant, particularly under oxidative conditions [16] where hOGG1S326C dimerizes through sulfide bonds of the cysteine residues [17]. The dimerized form of the enzyme cannot be stimulated by the downstream enzyme, apurinic/apyrimidinic endonuclease 1 (APE1). Stimulation by APE1 allows OGG1 to disassociate from the DNA more rapidly and repair more damaged substrate [17]. Consequently, the presence of the common variant adversely affects the efficiency of cellular DNA repair and may promote mutagenesis, especially within the mitochondrion, where oxidative stress is more severe.
Earlier work demonstrated that the bi-functional catalysis of OGG1, which includes a glycosylase step and an apurinic/apyrimidinic (AP) lyase reaction, could be stimulated by as much as 75-fold via a mechanism referred to as “product-assisted” catalysis [18]. In brief, the authors showed that nucleobase removal followed by the slower reaction controlled by the AP lyase could be improved by the presence of the guanine analogs, 8-bromoguanine or 8-aminoguanine. The question therefore arose as to whether small molecules with more “drug-like” properties could enhance Ogg1 activity to positively influence cellular function of either the serine or cysteine OGG1 variants. To explore the cellular benefits of enhancing OGG1 activity, we identified small molecule enhancers of OGG1 8-oxodG incision activity through an in vitro fluorescence-based screen and examined the cellular consequences of these molecules after oxidative stress. We identified compounds that lowered the cellular content of the 8-oxoG lesion, presumably through OGG1 activity.
The use of the toxic herbicide, paraquat is restricted in many countries, but is still widely used in Asia. PQ increases oxidative stress and has been shown to decrease function and increase damage in lung mitochondria of rodents [19]. Because of the prevalence of hOGG1S326C, particularly in Asian populations, and the importance of OGG1 in the cellular response to oxidative stress, we examined the role of OGG1 in PQ-induced oxidative damage and repair. We also investigated the potential of OGG1 stimulation to protect cells expressing hOGG1S326C. We utilized compounds that stimulated wild-type OGG1 or the S326C variant, or both. These tool compounds provide proof of concept that DNA repair can be stimulated and result in cellular improvement.
MATERIALS AND METHODS
Cell culture
Type II alveolar epithelial cells, A549 cells (ATCC; Manassas, VA), were grown to sub-confluence using complete Ham’s F12 medium containing 10% heat inactivated fetal bovine serum (FBS), and 1X GlutaMAX supplement (Life Technologies; Carlsbad, CA). The cells were maintained in a humidified atmosphere of 95% O2:5% CO2 at 37°C and the medium was replaced every 2–3 days. To sub-culture, A549 cells were treated with Trypsin/EDTA (Lonza Inc.; Walkersville, MD), counted using Trypan blue (Life Technologies; Carlsbad, CA) exclusion, and were seeded at a density of 100,000 cells/mL with 100 μL/well using 96-well black micro-clear bottom plates. Cells were incubated for 24 hours, medium was exchanged, and the cells were returned to the incubator until time of experimentation.
To generate cells devoid of functional mitochondria, i.e., rhoφ cells, A549 cells were treated with ethidium bromide (50 ng/mL; Life Technologies; Carlsbad, CA plus 100 μg/mL pyruvate, Life Technologies; Carlsbad, CA, and 50 μg/mL uridine, Sigma; St. Louis, MO) as noted previously (Brar et al., 2012; Holmuhamedov et al., 2002). The cells were sub-cultured through 14 population doublings (about 30 days) and A549 rhoφ were seeded into 96-well plates.
In some cases, the cells were exposed to paraquat (N,N′-dimethyl-4,4′-bipyridinium dichloride; Sigma; St. Louis, MO) at varying final concentrations (0.01 to 3 mM) and incubation periods (refer to figure legends). Paraquat was dissolved in Dulbecco’s phosphate buffered saline (PBS) and stored in aliquots (1M, −20°C) until use. It was thawed prior to use and diluted in cell culture medium. All measures were performed in triplicate.
OGG1 over expression
To alter the content of OGG1 in A549 cells, BacMam virus constructs were generated using expression vector, pDONRhumanOGG1v1a_FL (GeneCopoeia; Rockville, MD). This was a Gateway PLUS shuttle clone containing the full-length human OGG1 transcript variant 1a (8-oxoGua DNA glycosylase, OGG1, nuclear gene encoding mitochondrial protein, transcript variant 1a, reference sequence NM_002542.5). The cDNA of OGG1v1a fl was sub-cloned into a pFNCMV-DEST vector using a LR clonase reaction. The resulting expression OGG1v1a construct was transfected into SF9 insect cells for BacMam virus production (Life Technologies Bac-to-Bac Baculovirus Expression Systems version 1.2, Carlsbad, CA, Kost et al., 2008). After A549 cells were seeded into 96-well plates and incubated overnight, the culture medium was replaced with complete medium containing (v:v) full length OGG1 BacMam virus, MnSOD-OGG1-GFP, or null virus. Successful transduction was completed within 48 hours and cells were exposed to paraquat.
OGG1 siRNA
To lower the OGG1 content, A549 cells were subjected to OGG1 SMARTpool siRNA (Thermo Scientific; Waltham, MA) according to the manufacturer’s instructions. The siRNA (20 μM) was diluted 1:20 in Opti-MEM® I buffer (Life Technologies; Carlsbad, CA) and incubated at room temperature for 5 min. Dharmafect® 1 (Thermo Scientific; Rockford, IL) that was diluted (1:75 v:v) with Opti-MEM® I buffer was mixed with an equal volume of the diluted siRNA, incubated for 20 min at room temperature and further diluted (1:5, v:v) in Ham’s F12 medium. The latter replaced the cell medium and contained 100 nM human OGG1 SMARTpool siRNA. Controls were included on each cell plate with cells treated with Dharmafect® 1 transfection reagent or media and siRNA buffer. A scrambled non-targeting siRNA and a non-affected gene control (GAPDH) were included to assess non-specific effects of the siRNA treatment. The cells were incubated for 48 hours followed by exchange of the serum-free medium for complete culture medium. Under these conditions OGG1 gene expression was down-regulated as measured by PCR (data not shown).
Activators of OGG1 (Table 1) were identified from a high-throughput screen of the GlaxoSmithKline compound collection using a fluorescent OGG1 enzymatic assay. Briefly, a hairpin oligonucleotide containing an 8-oxoG base pair was used as a fluorescence labeled substrate. The assay measured an increase in fluorescence in response to dissociation of the 5′FAM (fluorescein) from the FRET (fluorescence resonance energy transfer) quench pair coupled to the 3′ DAB (Dabcyl) fluorescent quencher upon cleavage of the DNA substrate at the nucleobase 8-oxoG by purified hOGG1. In the presence of an activator, the very low basal catalytic activity of OGG1 was accelerated by 1–2 orders of magnitude, enabling this assay to detect activators present in the compound collection. 8-Bromoguanine was used as a positive control on each plate along with buffer only controls (Lukina et al., 2013). All the OGG1 activators used in the present study were shown to positively bind purified OGG1 through surface plasmon resonance assays (data not shown). The OGG1 activators (Table 1) were dissolved in 100% dimethyl sulfoxide (DMSO, final concentration = 0.1%, Thermo Scientific; Rockford, IL) and were added 4 hours prior to paraquat exposure. Control samples in the absence of OGG1 activators also contained 0.1% DMSO.
Table 1.
Tool OGG1 small molecule activators
| Number | Name |
|---|---|
| Compound A | (S)-2-((6-methyl-2-(phenylamino)pyrimidin-4-yl)amino)-N-phenylpropanamide |
| Compound B | 2-(4-fluorophenyl)-N,1,7-trimethyl-1H-pyrrolo[2,3-d]pyridazin-4-amine |
| Compound C | N-cyclohexyl-2-cyclopropylquinazolin-4-amine |
| Compound D | 2-(4-(4-(1H-imidazol-1-yl)phenoxy)-5-methylpyrimidin-2-yl)octahydropyrrolo[1,2-a]pyrazine |
| Compound E | 1-(4-(4-ethylphenyl)-6,7-dihydro-1H-imidazo[4,5-c]pyridin-5(4H)-yl)-2-(1-methyl-1H-indol-3-yl)ethanone |
| Compound F | 1-cyclohexyl-1-(2,4-dichlorophenyl)-2-(1H-imidazol-1-yl)ethanol |
8-OxoGua Immunofluorescence
For determination of 8-oxoGua, cell culture medium from A549 cells was aspirated and initially replaced with 250 nM MitoTracker® orange in serum-free medium, and incubated for 30 min (37°C, 5% CO2). Fixation of the cells was accomplished by the addition of an equal volume of 8% paraformaldehyde (4% final concentration, Electron Microscopy Sciences; Hatfield, PA). The cell plates were incubated for 20 min at room temperature in the dark, rinsed with media and stained for 8-oxoGua using methods adapted from previous methods (Marella et al., 2007; Moiseeva et al., 2009; Richardson et al., 2009). The fixative was aspirated, and the cells were washed once with phosphate buffered saline (PBS). RNase A (100 μg/mL in PBS, Qiagen; Valencia, CA) was added to the cells and incubated for 1 hour at 37°C, and the cells were subjected to an acidic wash of hydrochloric acid (2M) and then pH was rebalanced with 0.1 M sodium borate, pH 8.5. A549 cells were washed and blocked in PBS containing 3% bovine serum albumin (BSA, Sigma; St. Louis, MO), 5% donkey serum (Millipore; Billerica, MA), and 0.1% Triton X-100 (Sigma; St. Louis MO). The blocking buffer was then exchanged with additional blocking buffer containing diluted mouse anti-8-oxoGua antibody (1:100, Millipore; Billerica, MA). Wells containing mouse IgM were included as an isotype control on each cell plate. Following incubation overnight, the cell plates were brought out to room temperature, washed and diluted labeled goat-anti-mouse IgG (H &L)-Alexa Fluor® 647 (1:500, Life Technologies; Carlsbad, CA) and 5 μg/mL Hoechst 33342 in PBS containing 3% BSA was added for 1 hour at RT. Afterward, the cells were washed again, and the plates were imaged and analyzed using the Operetta Imaging system (Perkin Elmer, Waltham, MA). Segmentation was performed using the Hoechst 33342 and MitoTracker® orange dyes, and the fluorescence intensity of 8-oxoGua was quantified in the cytoplasmic region. Single cell analysis was performed, and the cells were classified and selected based on their initial un-corrected mean cytoplasmic region intensity of 8-oxoGua. Cells with 8-oxoGua intensity above the peak mean distribution from the untreated control were characterized as a “high responding” by using several experiments to establish threshold settings. The percentage of cells in this group was calculated based on the total number of cells imaged in each well. The shape and size of both the nuclear and the cellular regions were measured for area, width/length, and roundness as part of the morphological assessment. Finally, the mean fluorescence intensity for the isotype control was subtracted from each treatment group, correcting the value based on non-specific binding.
Mitochondrial Membrane Potential (A549 cells)
The cationic carbocyanine, ratiometric dye JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolycarbocyanine iodide; Life Technologies; Carlsbad, CA) was used to measure mitochondrial membrane potential [20]. Cells were stained using the manufacturer’s suggested protocol for JC-1 (2 μg/mL, Life Technologies; Carlsbad, CA). The dye was incubated for 30 min (37°C, 5% CO2) and then 5 μg/mL Hoechst 33342 and 5 μg/mL CellMask™ deep red were added to the cells. Following an additional 30 min (37°C, 5% CO2) incubation, the medium was exchanged for live cell imaging solution. Image analysis was performed with Hoechst 33342 and CellMask™ deep red stain for nuclear and cytoplasmic region identification, respectively. Mean fluorescence intensity of the JC-1 aggregate and the JC-1 monomer were each quantified within the cellular region and the data was reported as a ratio of the average fluorescence intensity of the monomer relative to the aggregate.
Image acquisition
A549 cells in black, micro-clear bottom 96-well plates were imaged using an Operetta Imaging platform (Perkin Elmer; Waltham, MA) with the following settings: four fields per well, 20X, long-working distance objective, and in wide-field mode. The cell plates were exposed to varying excitation spectra depending on the dyes in each well, and the emission spectrum for each dye was detected. Each dye was added to untreated cells (controls) to adjust for background staining and to assess dye interference from each color overlay. Cells intersecting the border of each image were excluded from the data analysis. Each image contains a reference bar which describes the level of spatial resolution.
Purification of hOGG1 and hOGG1S326C
A plasmid including the open reading frame (ORF) encoding human OGG1, 1a transcript variant, was purchased from GeneCopoeia (catalog number GC-S0001). The ORF was PCR-amplified and subcloned into pENTR/TEV/D-TOPO to insert a TEV-protease cleavage site at the N-terminus, and then subcloned via GATEWAY cloning (Invitrogen) into pDEST-T7FLAGHis6-ST. The S326C mutant was generated by site directed mutagenesis of the wild type ORF in the pENTR/TEV/D-TOPO vector using PCR with Pfu UltraHF polymerase (Agilent Technologies) and forward primer: 5′-GCCGACCTGCGCCAATgCCGCCATGCTCAGGAG-3′ and reverse primer: 5′-CTCCTGAGCATGGCGGcATTGGCGCAGGTCGGC-3′. The mutant ORF was transferred to the pDest-T7FLAGHis6-ST expression vector using GATEWAY cloning as above.
The expression plasmid was co-transformed into BL21(DE3) * Escherichia coli cells along with pRR692, a plasmid encoding rare tRNA species to aid recombinant protein expression. One ml of the expression strain was inoculated into 1 L of LB broth supplemented with 1% glucose, 100 mg/ml carbenicillin, and 25 mg/ml chloramphenicol, and grown overnight at 37°C with shaking. For production, the seed culture was inoculated into an Applikon 343 bioreactor containing 19 L 2xYT broth supplemented with Vogel-Bonner salts (8.4 mM sodium citrate, KH2PO4, 25mM NH4NO3, 0.81mM MgSO4, 0.68mM CaCl2, 23.8mM citric acid, 17.4mM ZnSO4, 2.55mM Fe(NH4)2(SO4)2, 1mM CuSO4, 0.27mM MnSO4, 0.81mM H3BO3, 0.21mM Na2MoO4, 20.5nM biotin), 1% glucose, 100 mg/ml carbenecillin, and 25 mg/ml chloramphenicol. The culture grown in the reactor at 37 °C to an OD600 ~ 5, at which point the reactor temperature was reduced to 15°C and protein expression was induced by the addition of 0.2 mM IPTG. The cells were harvested 20 h post-induction by centrifugation at 10,000 × g in a PSAngelus Powerfuge.
For purification, 100 g w/w of cell paste was resuspended in 1L of buffer A (50 mM Tris HCl, pH7.5, 250 mM NaCl, 0.1 mM TCEP) and lysed with two passes through an Avestin Emulsiflex homogenizer at 10,000 psi. Twenty microliters of Benzonase® nuclease (Sigma) were added to the homogenate and the mixture was clarified by centrifugation at 38,000 × g at 4°C for 30min. The protein was affinity purified by batch capture from the supernate on 20 ml of NiNTA agarose resin for 4h at 4°C. The resin was packed into an XK26 column (GE Healthcare) and equilibrated with buffer A. The protein was stepwise eluted from the column using 100 mM imidazole and 250 mM imidazole in buffer A. Fractions containing the protein of interest were identified by analysis on SDS-PAGE, pooled, and the tags removed by overnight cleavage with 20 mg TEV protease. LC/MS analysis confirmed that the tags were 100% removed from the Ogg1 protein. The pooled and cleaved OGG1 protein was desalted on a G25 column in buffer B (50 mM MES, pH 6.0, 1 mM EDTA, 20% glycerol, and 10 mM β-mercaptoethanol), and loaded onto a 10/100 GL Mono S column (GE Healthcare) equilibrated in buffer B. The protein was eluted with a 0–1 M gradient of NaCl in buffer B. The protein peak fractions were collected, pooled and stored in buffer B + 400 mM NaCl at −80°C until further use [21].
In vitro incision assays, purified hOGG1
The indicated concentrations of hOGG1 and hOGG1S326C were incubated with 48 fmol of 5′ γ-32P-ATP labeled substrate (Figure 1A). The reaction buffer consisted of: 70 mM HEPES-KOH, pH 7.6, 5 mM EDTA, 1 mM DTT, 75 mM NaCl, and 10% glycerol in a 20 μL total volume [22]. Reactions were incubated at 37°C for 1 hour. Reactions were terminated with 10 μL formamide gel loading dye (96% formamide, 10 mM EDTA, 1 mg/ml xylene cyanol FF and 1 mg/ml bromophenol blue), heated to 95°C for 5 minutes and separated by electrophoresis in a 20% denaturing polyacrylamide and 7 M urea gel. Results were visualized via phosphoimaging (Typhoon, GE Healthcare Life Sciences) and were quantitated via ImageQuant TL software (Molecular Dynamics). The data are presented as the ratio of product to the total radioactivity in the lane.
Figure 1. Paraquat-induced mitochondrial DNA damage is inversely related to OGG1 expression.
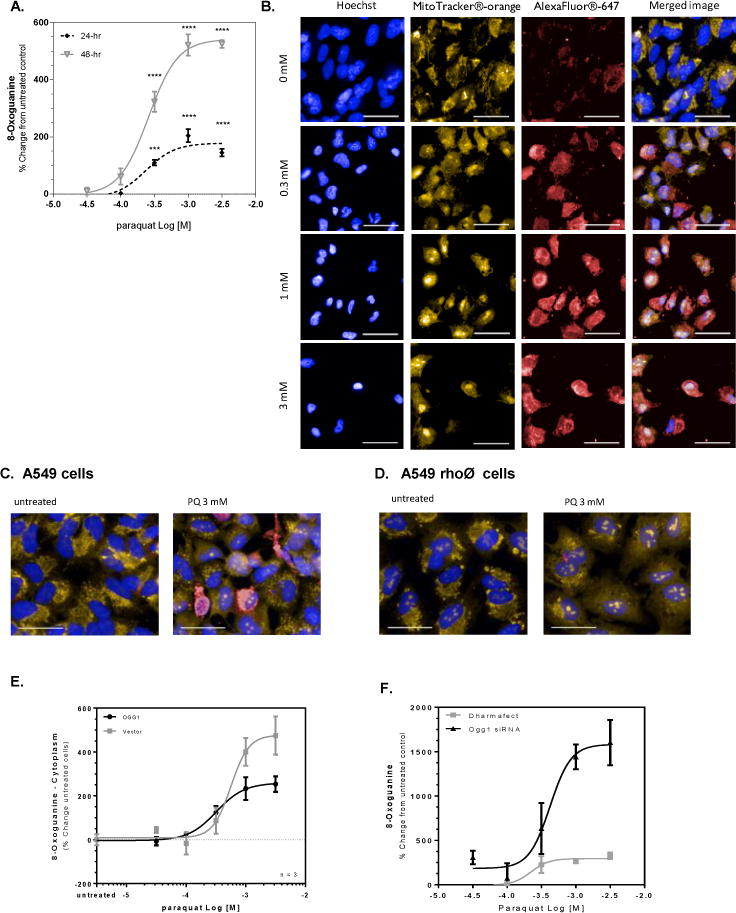
A549 cells were treated with paraquat for 24 or 48 hr and subjected to formaldehyde fixation prior to immunofluorescence detection of 8-oxoguanine. (A) Graphical representation of the percent change in 8-oxoguanine intensity within the cytoplasm region following 24- and 48-hr treated cells with increasing concentrations of paraquat. Significance was determined using a 1-way ANOVA with a Dunnett’s posttest comparing treatment groups to an untreated control (* p<0.05, ** p<0.01, *** p<0.001, **** P<0.0001). B. Changes in 8-oxoguanine are depicted in sample images (Hoechst33342, overlaid blue; MitoTracker® orange, overlaid orange; 8-oxoG, overlaid red) from cells exposed to increasing concentrations of paraquat in panel B (line = 40 μm). (C/D) A549 cells were treated with 50 ng/mL ethidium bromide for 14 population doublings to generate A549rhoφ cells devoid of functional mitochondrial DNA. A549 and A549rhoφ cells were treated with paraquat for 24 hr, followed by staining for 8-oxoG. (C) Sample images of A549 cells from wells containing untreated cells and cells exposed to 3 mM paraquat for 24 hr (line = 40 μm). (D) Sample images like Panel C, but with A549rhoφ cells (line = 40 μm). (E) A549 cells were incubated with 5% v.v. Ogg1-BacMam for 48 hours prior to treatment with varying concentrations of paraquat for 24 hr. The cells were subjected to formaldehyde fixation for immunofluorescence detection of 8-oxoguanine and the percent change in 8-oxoguanine intensity within the mitochondria of the cells from varying concentrations of paraquat compared to control cells. (F) A549 cells were incubated with 100 nM Ogg1 siRNA containing Dharmafect® in serum-free conditions. Following 48-hr incubation, the medium was replaced to complete culture medium and the A549 cells were treated with varying concentrations of paraquat for 24 hr. The cells were stained and imaged for 8-oxoguanine. The figure represents the percent change in 8-oxoguanine intensity within the mitochondria of paraquat treated cells compared to media control.
Preparation of whole cell extracts
Proteins were extracted from cell pellets by homogenizing with an equal cell volume (1XCV) of Buffer I (10 mM Tris-HCl, pH 8.0, 200 mM KCl), followed by addition of another 1XCV of Buffer II (10 mM Tris-HCl, pH 8.0, 200 mM KCl, 2 mM EDTA, 40% glycerol, 0.5% NP-40, 2 mM DTT) and protease inhibitors (1X total volume, ThermoFisher, Rockville, IL). Extracts were incubated at 4°C for 2 hours with rotation, followed by centrifugation at 14,000 rpm at 4°C for 15 minutes. Supernatants were removed to a new tube and protein concentrations were determined (ThermoFisher) followed by storage at −80°C [23].
Mitochondrial isolation
Mitochondria were isolated from cells using the Abcam mitochondrial isolation kit for cultured cells (ab110170) using manufacturers recommended protocol.
In vitro incision assays, cell extracts
Ten micrograms of whole cell or mitochondrial extract were incubated with 48 fmol of 5′ γ-32P-ATP labeled substrate (Figure 2A). The reaction buffer consisted of: 70 mM HEPES-KOH, pH 7.6, 5 mM EDTA, 1mM DTT, 75 mM NaCl, and 10% glycerol and protease inhibitors in 20 μl total volume [22]. Reactions were incubated at 37°C for 30 minutes. Reactions were terminated with 10 μL formamide gel loading dye (96% formamide, 10 mM EDTA, 1 mg/ml xylene cyanol FF and 1 mg/ml bromophenol blue), heated to 95°C for 5 minutes and separated by electrophoresis in a 20% denaturing polyacrylamide and 7M urea gel. Results were visualized via phosphoimaging (Typhoon, GE Healthcare Life Sciences). Results were quantitated via ImageQuant TL software (Molecular Dynamics). Results are presented as the ratio of product to the total radioactivity in the lane.
Figure 2. Purified hOGG1S326C protein incises 8-oxoguanine less efficiently than purified hOGG1 protein.
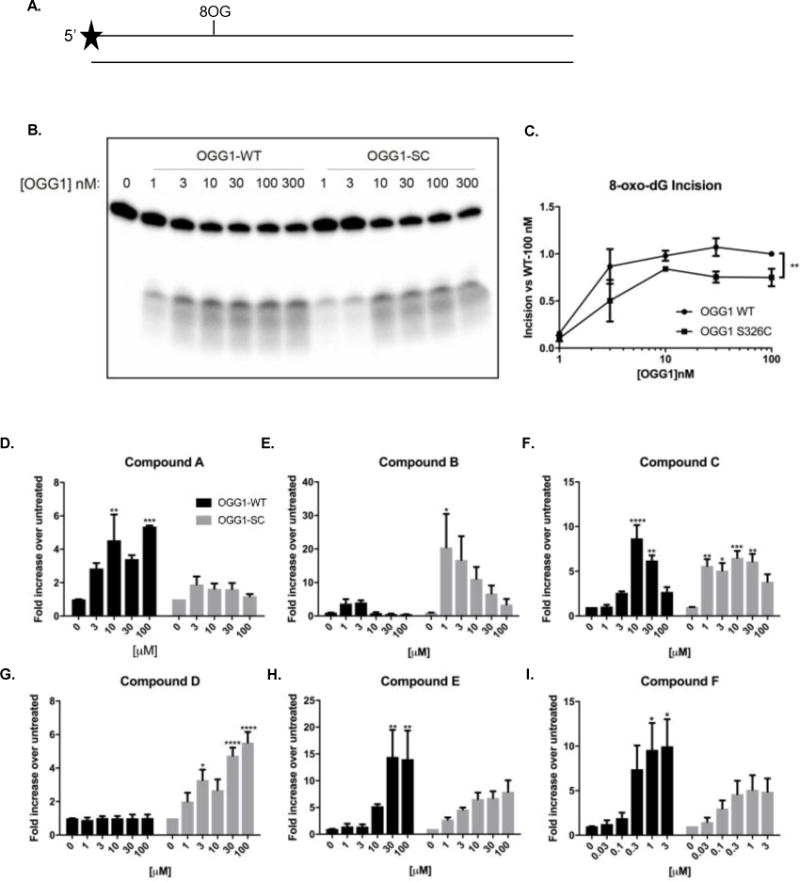
A. Incision of 8-oxodG was assayed using a duplex oligonucleotide containing a single 8-oxodG residue at position 11. Proteins were purified and diluted to indicated concentrations in reaction buffer. B. After 1 hour at 37°C, fragments were separated by denaturing electrophoresis and visualized using phosphoimaging. C. Results were quantitated by comparing incised bands to total radioactivity in lane. D–I. hOGG1wild type (5 nM) or hOGG1S326C (10 nM) were stimulated with compounds: A (D), B (E), C (F), D (G), E (H), or F (I) at the indicated concentrations. Black bars indicate hOGG1wild type and gray bars indicate hOGG1S326C. Asterisks represent statistical differences between untreated and indicated concentration of compound. All experiments were completed three times. Statistical analysis by 2-way ANOVA, with Tukey post-hoc multiple comparisons. (* p<0.05, ** p<0.01, *** p<0.001, **** P<0.0001)
OGG1 MEF stable cell lines
Mouse embryonic fibroblasts (MEFs) lacking Ogg1 were prepared from breeding Ogg1+/− mice, embryos were extracted at day 14 per standard methods [24]. All experiments were approved by the Animal Care and Use Committee and performed in accordance with the Guidelines for the Care and Use of Laboratory Animals (NIH Publication 85-23). Resulting MEF cells were immortalized spontaneously during continuous cell culture. The hOGG1 gene constructs and empty vector control were introduced to the knockout MEFs via retroviral gene transfer (Clontech). Clones were selected by resistance to 400 μg/ml geneticin G418 (Teknova) and isolated via serial dilution. Expression levels of hOGG1 protein in individual clones were monitored periodically by western analysis. Clones were chosen for study based on equal hOGG1 content.
Antibodies
Rabbit polyclonal anti-OGG1 antibody was obtained from GeneTex (#GTX20204, Irvine, CA). Antibody against PCNA (mouse monoclonal # 13–3900) was obtained from ThermoFisher (Waltham, MA). Total OXPHOS rodent western blot antibody cocktail was obtained from Abcam (ab110413, Cambridge, MA).
Flow cytometry analysis of mitochondrial parameters (MEFs)
Cells were plated in at 6-well dish and treated as indicated in text. Cells were harvested for flow cytometry analysis after the incubation time indicated, when cells were approximately 90% confluent. Cells were washed in 1X PBS, trypsinized, and resuspended in DMEM media with 10% FBS and 1% Pen Strep. Cell suspensions were aliquoted into Eppendorf tubes and kept at 37 °C. Cells were treated with 20 nM TMRM (ThermoFisher Scientific), 100 nM Mitotracker Green (ThermoFisher Scientific), DHE (ThermoFisher Scientific), or Mitosox (ThermoFisher Scientific). The same cells were never treated with multiple dyes. Cells treated with TMRM or Mitotracker were incubated for 15 min at 37°C and cells treated with DHE and Mitosox were treated for 30 min at 37°C. Samples were analyzed using an Accuri BD C6 flow cytometer. Live cells were gated and at least 5000 events were recorded per sample. TMRM samples were analyzed at FL2A, Mitotracker samples were analyzed at FL1A, and DHE and Mitosox samples were analyzed at FL3A. Further analysis was conducted using FlowJo software. All experiments included at least three biological replicates.
Cellular oxygen consumption
Oxygen consumption and extracellular acidification rate measurements were performed using the Seahorse XF-24 instrument (Seahorse Biosciences). Cells were seeded into a Seahorse tissue culture plate at a density of 25,000 cells per well and 16 hours later media was changed to unbuffered XF assay media at pH 7.4 (Seahorse Biosciences), supplemented with 11 mM glucose (Sigma-Aldrich), 1 mM sodium-pyruvate, and 1 mM glutamate (Invitrogen). Cells were incubated for 1 h at 37°C at ambient O2 and CO2 concentration before measurements were taken. Respiration was measured in four blocks of 3 × 3 min. The first block measured the basal respiration rate. Next, oligomycin (EMD) was added to a final concentration of 0.1 μM to inhibit complex V and the second block was measured. Then, FCCP (Sigma-Aldrich) was added to a final concentration of 0.5 μM to uncouple respiration and the third block was measured. Finally, antimycin A (Sigma-Aldrich) was added to a final concentration of 0.2 μM to inhibit complex 3 and the last measurements were performed. All compound concentrations used had been optimized for that cell line. Immediately after finishing the measurements, cells were trypsinized and counted using a Coulter counter (Beckman Coulter).
RESULTS
PQ-induced mitochondrial DNA damage is inversely related to OGG1 expression
PQ is an herbicide and environmental toxin banned from many countries but still widely used in Asia. PQ cycles redox equivalents within the mitochondria, producing ROS primarily at Complex I of the respiratory chain [25]. To determine the effects of PQ on the development of 8-oxodG lesions, A549 cells were incubated with ascending concentrations of paraquat (PQ) for a period of 24 or 48 hours. Quantitation of the 8-oxodG was measured using a previously verified antibody against 8-oxodG [26]. Graphical representation (Figure 1A) of optical images obtained in the presence or absence of PQ show that the cellular level of 8-oxodG rose in a concentration-dependent, sigmoidal fashion at both time points with maximal content resulting from 1 mM PQ, and an EC50 value of 0.3 mM. The amount of 8-oxodG was increased by > 2-fold at 48 hours compared to the 24-hour timepoint. The corresponding images obtained at 48 hours post PQ (Figure 1B) demonstrate that the 8-oxodG lesion mostly localizes outside of the nucleus. Further, the merged image of Mito-tracker and 8-oxodG staining suggest that much of the lesion has formed in association with the mitochondrial space, likely mtDNA. Mitochondrial localization of the lesion was confirmed by high-powered laser confocal microscopy (Supplemental Figure 1). In separate experiments, A549 cells were treated with ethidium bromide for 14 population doublings to produce cells devoid of fully competent mtDNA (A549 rho(0) cells), and then exposed to PQ. Application of 3 mM PQ to cells containing fully functional mitochondria resulted in the expected increase of 8-oxodG whereas the A549 rho(0) cells were essentially free of any 8-oxodG despite the exposure to high concentrations of the herbicide (Figure 1C and 1D).
Previous investigations showed that transient over-expression of OGG1 targeted to the mitochondria prevented mtDNA damage resulting from exposure of rat pulmonary artery endothelial cells to the oxidant-generating system, xanthine oxidase plus hypoxanthine [27, 28]. In these studies, overexpression of OGG1 was associated with improved viability despite the presence of excessive superoxide. In the present work, the over-expression of OGG1 protein in A549 cells appeared to show benefit. Despite equivalent seeding procedures, cells overexpressing OGG1 were approximately 70% more abundant under basal conditions than their vector counterparts. PQ induced 8-oxodG was comparable for both cell constructs (EC50 = 0.52 and 0.45 mM, respectively). However, maximal content of the 8-oxodG lesions elicited by PQ (3 mM) was markedly suppressed, by about 50% (p<0.001), in cells overexpressing OGG1 as compared to vector control cells (Figure 1E).
By contrast, knockdown of OGG1 by siRNA rendered the cells (siOGG1) more vulnerable to PQ, as demonstrated by increased 8-oxodG levels compared with scrambled controls (siCtrl) (Fig 1F). The magnitude of change in 8-oxoG elicited by 3mM PQ in siOGG1 cells was more than 5-fold greater than that obtained in the scrambled control cells. In addition, the basal levels of 8-oxoG were approximately 200% higher in siOGG1 cells compared with siCtrl cells. Although the number of seeded cells was similar for both conditions, OGG1-knockdown cells grew more slowly, resulting in approximately 25% fewer cells before treatment with PQ as compared to those expressing the fully competent protein or those treated only with the transfection reagent (Dharmafect). Moreover, the concentration of PQ required to decrease the number of cells by 50%, (EC50 value), was significantly lower (p<0.05) for the OGG1 siRNA treated cells than for the control cells (EC50 values were 462 vs 519 mM, respectively). These data demonstrate a greater sensitivity of the cells to PQ when OGG1 content has been diminished. OGG1 reduction verified by immunofluorescence, where cells treated with paraquat demonstrated a significant reduction in OGG1 content within the cells, including within the mitochondria (data not shown).
Purified hOGG1S326C is less efficient in 8-oxodG incision than WT hOGG1
In vitro oligonucleotide incision assays were used to examine basal OGG1 activity, wild type versus the common hOGG1S326C variant. A duplexed, 5′-labeled oligonucleotide substrate (Figure 2A) containing a unique 8-oxodG adduct was used under reducing conditions to eliminate sulfide bonds between variant molecules. However, even without oxidizing conditions, the S326C variant was less active than wild type (Figure 2B), suggesting that dimer formation is not the only limitation for hOGG1S326C function. Quantitation of activity (Figure 2C) translates to a 40.8% decrease across all concentrations of the enzyme (area under curve, 70.32 vs. 41.61). These results are consistent with those found by others comparing purified OGG1 incision activity [17, 29]. Abundant evidence of this deficiency and associated pathology stimulated our interest to try and rescue the glycosylase activity of the S326C variant by small molecule intervention.
Characterization of small molecule activators of Ogg1
Small molecule activators were used to determine whether enhancement of Ogg1 activity would benefit cells upon challenge with PQ or make up for the catalytic deficiencies in hOGG1S326C. Identification of small molecules using a fluorescence-based 8-oxoGua-DNA cleavage assay within the GlaxoSmithKline chemical collection yielded “tool” molecules, low potency and lacking appropriate pharmacokinetic properties, to further explore whether OGG1 activation would be beneficial.
Several OGG1 activators were evaluated for their ability to stimulate the in vitro activity of purified OGG1 wild type and S326C variant. Of six compounds tested (Table 1), four could enhance the activity of both wild-type and S326C OGG1 (Figure 2D–H) using the current paradigm. Compounds varied in both specificity for the OGG1 variant and effect. Compound A, at 100 μM, maximally stimulated wild-type OGG1 5.4-fold, but only marginally affected the S326C variant. In contrast, compound D, at 100 μM, increased the activity of S326C, 5.5-fold over untreated, but had no effect on wild-type OGG1. Compounds B, C, E, and F activated both OGG1 proteins. Compound F was very potent, increasing activity of both proteins at concentrations as low as 0.1 μM. These tool compounds demonstrate the potential for direct stimulation of OGG1-mediated 8-oxodG incision.
To determine the effects of the OGG1 activators on functional cellular processes, compounds were evaluated in two cellular models: PQ-induced 8-oxoG in A549 cells and in a MEF model. In the latter case, stable cell lines lacking endogenous mouse Ogg1 were created to express either hOGG1wild type or hOGG1S326C. Figures 3A and 3B show the 8-oxoG concentration response curve elicited by PQ treatment in A549 cells and the modulation of these values in the presence of the small molecule activators. When the compounds (3–100 uM) were incubated with the cells up to 4 hours prior to PQ (0.6 mM) exposure, attenuation of the development of 8-oxoG resulted. The lone exception was compound C. The compound did not induce a concentration-dependent change in 8-oxoG. Representative images clearly depict a rise in red fluorescence associated with the PQ-induced increase of 8-oxoG in the absence of the compounds (Fig. 3B). By comparison, there was a reduced level of red emission in images obtained from cells that were incubated with the activators prior to PQ exposure.
Figure 3. OGG1 activators attenuate paraquat-induced oxidative damage in A549 cells.
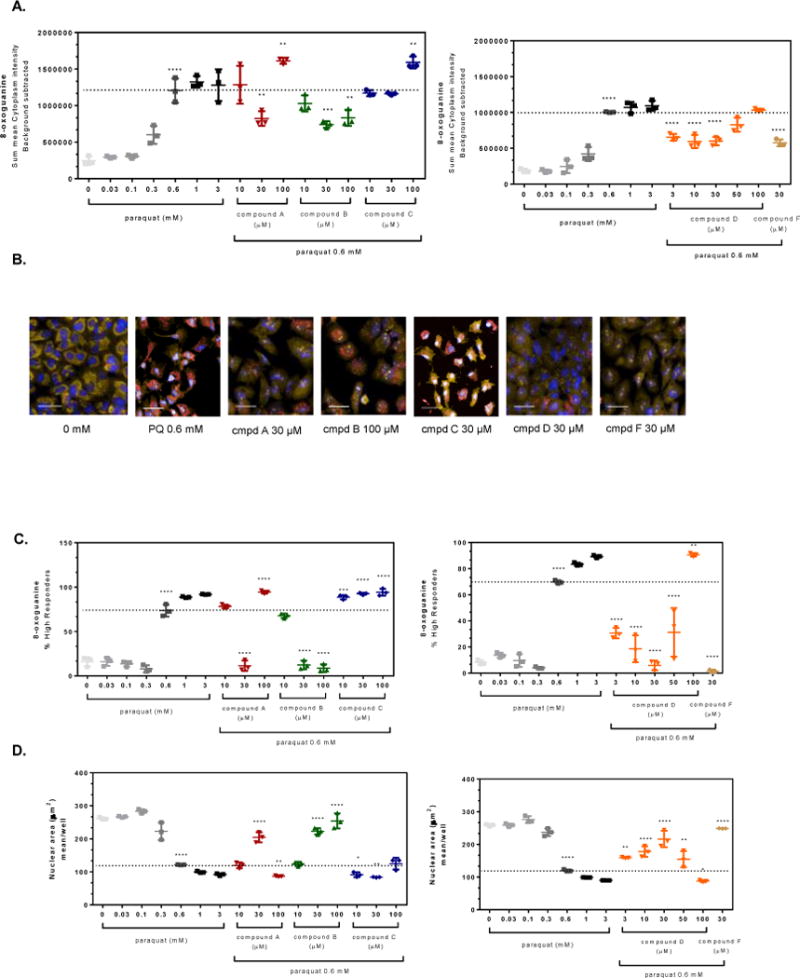
A549 cells were pre-treated for 4 hr with small molecule OGG1 activators or 0.1% DMSO prior to addition of 0.6 mM paraquat for 48 hr. The cells were stained and imaged for 8-oxoG content and the average fluorescence intensity within the cell was normalized based on the number of cells imaged. (A) Graphical representations of the change in 8-oxoG intensity within the cytoplasm of cells compared to 0.6 mM paraquat treated cells (dashed line). (B) Representative images of cells from panel A depict changes in mitochondrial 8-oxoG staining (Hoechst 33342, overlaid blue; MitoTracker® orange, overlaid orange; 8-oxoG-AleaFluor®-647, overlaid red; line = 40 μm). (C) The percent of the cell populations labeled as high responders is shown for A549 cells pre-incubated with OGG1 activators (4 hr) prior to exposure to a single concentration of paraquat (0.6 mM, dashed line) for 48 hr. (D) The images from panel A were analyzed further by examining morphology changes. The mean nuclear area (dashed line indicates no treatment, 0.6 mM PQ) for A549 cells was graphed depicting a change in overall cell health. Significance was determined using a 1-way ANOVA with a Dunnett’s posttest comparing paraquat alone treated groups to untreated control (0 mM) and OGG1 activator treated groups to 0.6 mM paraquat control (** p<0.01, *** p<0.001, **** p<0.0001). Data shown are from a representative experiment, performed three times.
The changes in 8-oxodG that were stimulated by PQ were not evenly distributed across all cells within a given cell culture well. It was not apparent what the cause of this heterogeneity was, but some cells were affected more than others. PQ must be transported into cells due to its structure (di-quaternary amine), which does not readily diffuse across cell membranes and thus may have introduced a given level of heterogeneity. To correct for this phenomenon, a single-cell analysis was constructed in which the shift in the mean frequency distribution of 8-oxodG fluorescence intensity for each cell was compared to the mean value across the untreated cell population in the absence of PQ (histograms of the frequency distributions are not shown). This analysis yielded a cell population termed “high responders” which refers to those cells that express high levels of 8-oxodG (Figure 3C). In the absence of PQ, 10–15% of the cells were classified in this category whereas the relative numbers in the PQ-treated population of cells increased significantly (at 0.6, 1 and 3 mM, p<0.001). With this analysis, compounds A, B, D and F prevented the PQ-induced shift in the rise of 8-oxodG high responders at 0.6 mM PQ. For compounds B, D, and F at 30 μM, there was essentially no PQ-induced shift to the 8-oxodG high responder category.
Corresponding with 8-oxodG development, the nuclear area was also adversely affected by PQ in a concentration-dependent manner (Figure 3D). The images show that as the nucleus diminishes in size, it appears as a brighter blue and even white as the Hoechst dye intercalates into the DNA. Quantification of these changes show that the nuclear area was decreased, by more than 50% at 0.6 mM PQ. In the presence of the compounds (excluding Compound C, which was also ineffective against 8-oxodG), this nuclear condensation was prevented in a concentration-dependent fashion for compound B or in a bell-shaped response for compounds A and D (compound F was tested at only one concentration, 30 μM, but prevented nuclear condensation).
Given the impact of PQ on the development of oxidatively generated lesions, especially within the mtDNA, it was possible that mitochondrial function was similarly affected. When PQ was incubated with the A549 cells, it lowered mitochondrial membrane potential (MMP) as measured by changes in fluorescence emission associated with the aggregate:monomer ratio of JC-1 dye (Figure 4A). Also, shown in Figure 4A is the response of cells to the uncoupler, carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP, 3 uM), which was used as a positive control to define complete collapse of MMP. When the cells were pre-treated with compound D, it partially prevented the fall in MMP, at least when determined at 24 hours, post-incubation with 0.3 mM PQ (Figure 4B). Compound F did not protect against the PQ effect on MMP (the other compounds were not evaluated).
Figure 4. OGG1 activators protect against paraquat-induced loss of mitochondrial membrane potential in A549 cells.
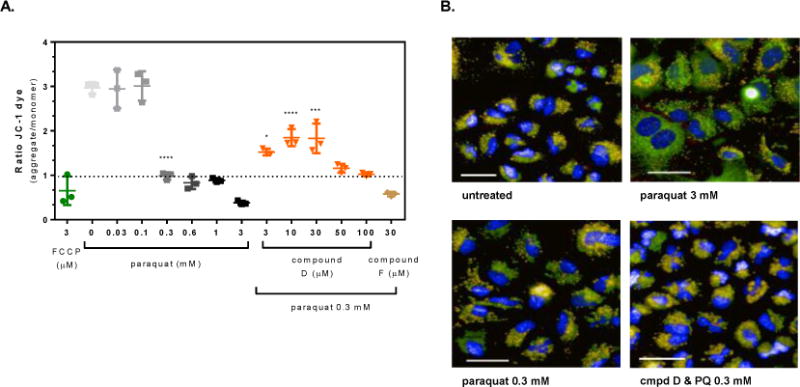
A549 cells were pre-treated for 4 hours with the OGG1 small molecule activators or 0.1% DMSO prior to exposure to 0.3 mM paraquat for 24 hr. Mitochondrial membrane potential was measured from images captured as described in the methods. (A) Graphical representation of the change in the JC-1 ratio with increasing concentration of paraquat and cells pre-treated with the OGG1 activators prior to exposure to a single concentration of paraquat (0.3 mM) for 24 hr. (B) Representative images from untreated, 3 mM paraquat, 0.3 mM paraquat, and Compound D (30 μM) with 0.3 mM paraquat exposed cells (Hoechst 33342, overlaid blue; JC-1 monomer, overlaid green; JC-1 aggregate, overlaid orange; CellMask™ Deep Red, overlaid red; line = 40 μm). Significance was determined using a 1-way ANOVA with a Dunnett’s posttest comparing paraquat alone treated groups to untreated control (0 mM) and OGG1 activator treated groups to 0.3 mM paraquat control (* p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001).
Mitochondrial OGG1S326C is as abundant as, but less active than, wild-type OGG1
Ogg1−/− MEF cells were generated by the mating of heterozygous mice as described in methods. MEFs were passaged continuously in culture until they spontaneously immortalized. MEFs were considered immortalized after a period of slow growth, followed by continuous exponential growth [30]. Spontaneously immortalized Ogg1−/− MEF cells were transduced with cDNA of wild-type hOGG1, hOGG1S326C, or a vector control and subsequently single-cell cloned. Clones that expressed equal levels of OGG1 protein were selected. Clones were randomly selected for the vector control cells that express no OGG1 protein. Because the transduced construct is a cDNA, the mitochondrial splice variant of OGG1 is not produced. However, mitochondrial isolation revealed that the ectopic protein is also expressed in the mitochondria. BER occurs in both the nuclear and mitochondrial compartments. Because we previously found that mtDNA was particularly vulnerable to 8-oxodG accumulation in mice lacking Ogg1 [4], we compared mitochondrial preparations of each cell line for OGG1 protein content by western blot and found the wild type and S326C variant have equal levels of OGG1, and no OGG1 expression in vector control cells (Figure 5A). Next, we compared mitochondrial extracts for 8-oxodG incision activity and found that the S326C variant was 30% as active as wild type (Figure 5B). In contrast, 8-oxodG incision activity in whole cell extracts was comparable between the two cell lines (Figure 5B), suggesting that the variant is only deficient in 8-oxodG incision activity in the mitochondria, perhaps due to pro-oxidant environment in the mitochondria versus the nucleus [31]. To promote the generation of ROS in the mitochondria, we exposed cells to rotenone, an inhibitor of complex I of the electron transport chain [32]. We expected the variant mtOGG1 to be less active in the presence of rotenone as it has been reported that OGG1S326C is less active when oxidized [16], but we found that both wild-type and variant mtOGG1 were both inhibited by rotenone treatment (Figure 5B). Because mtOGG1wild type has more basal activity than the serine variant, after rotenone treatment, it retained more activity upon rotenone treatment, suggesting vulnerability following oxidative stress for individuals expressing only the serine allele.
Figure 5. Incision of 8-oxodG is less efficient in mitochondria isolated from hOGG1S326C cells than hOGG1 cells.
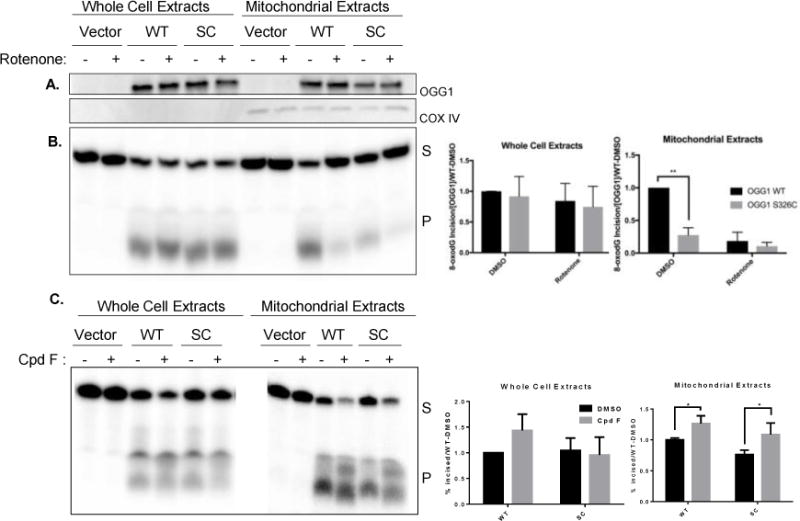
A. Cells were grown in the presence of DMSO (0.1%) or rotenone (5 μM) for 24 hours and then harvested. Western blot demonstrates equal expression of hOGG1 protein in hOGG1S326C and hOGG1 cells and no hOGG1 expression in vector cells. COX IV expression is shown as a mitochondrial marker. B. Cell extracts were diluted to protein equal concentrations in reaction buffer and incubated with substrate shown in Figure 1A. Fragments were separated by denaturing electrophoresis and visualized using phosphoimaging. Graphs show quantitation of incised band relative to total radioactivity in lane. The percent incised was divided by the OGG1 content in each sample and normalized to activity of hOGG1wild type extracts in each experiment. Significance was determined by 2-way ANOVA. C. Cells were grown in the presence of DMSO (0.1%) or compound F (15 μM) and harvested after 24 hours. Incision of 8-oxodG was assessed as above. All experiments included three biological repeats. Significance was determined by 2-way ANOVA.
Because compound F stimulated the activity of purified OGG1 and the variant at relatively low concentrations, we treated cells with 15 μm compound F to determine if the level of activity by the variant could also be improved in cells. We found that both the wild type and S326C variant were stimulated by compound F, and that the level of activity of the variant was fully restored to values achieved from the wild-type extracts (Figure 5C).
Cellular and mitochondrial superoxide are reduced by expression of hOGG1 but not hOGG1S326
To determine the health of the mitochondria of each cell line, we measured four mitochondrial parameters: mitochondrial membrane potential (TMRM), mitochondrial content (Mitotracker Green), cellular superoxide (DHE), and mitochondrial superoxide (MitoSox). We observed no statistical difference in membrane potential among the three cells lines (Figure 6A). Wild-type complementation appeared to increase mitochondrial content, although it only reached statistical significance in comparison to the S326C variant-complemented cells. However, the differences in cellular superoxide were quite striking. The S326C variant-expressing cells appear very similar to cells expressing only the vector. Cells expressing wild-type hOGG1 show an almost 50% decrease in cellular superoxide compared with either of the others (Figure 6A). These cells also showed a smaller (~20%) decrease in mitochondrial superoxide, when compared with the vector. The variant cells were not statistically different from the vector under any parameter.
Figure 6. MEFs expressing hOGG1 demonstrate marked differences in mitochondrial parameters from those expressing no OGG1 or hOGG1S326C.
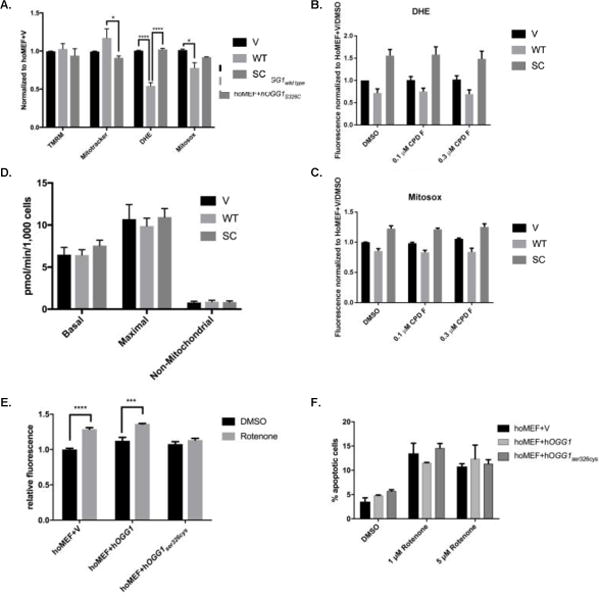
A. Cells were analyzed for mitochondrial parameters via flow cytometry. Stains for mitochondrial membrane potential (TMRM), mitochondrial content (Mitotracker Green), total cellular ROS (DHE) or mitochondrial ROS (Mitosox Red) were added to 90% confluent cells and fluorescence was measured in appropriate channels using a flow cytometer. All results were normalized to vector control. Results analyzed by 2-way ANOVA. B–C. Cells were grown in the presence of 0.1% DMSO, 0.1 μM, or 0.3 μM compound F for 24 hours. Measurements of mitochondrial ROS (B) and cellular ROS (C) were analyzed via flow cytometry and normalized to DMSO treated vector cells. D. Cells were analyzed via Seahorse XF24 for basal oxygen consumption, maximal oxygen consumption, and non-mitochondrial oxygen consumption, and results from three independent experiments were graphed and analyzed by 1-way ANOVA. E. Cells were grown in the presence of 0.1% DMSO or 1 μM rotenone and analyzed for mitophagy using a pH sensitive dye via flow cytometry. Results were normalized to vector DMSO control and compared for significance by 2-way ANOVA. F. Cells were grown in the presence of 0.1% DMSO, 1 μM rotenone, or 5 μM rotenone and analyzed via flow cytometry after 24 hours. Results analyzed by 2-way ANOVA. All experiments were performed repeated three times. Mean results were graphed with standard error of the mean for error bars.
To determine if we could stimulate the variant to more closely approximate wild-type OGG1 with regards to mitochondrial parameters, we treated cells with compound F and measured mitochondrial parameters. We found no significant changes in cellular or mitochondrial superoxide after treatment with compound F in any of the cell lines (Figures 6B and 6C).
To further examine the mitochondrial phenotype of S326C variant-expressing cells, we performed mitochondrial bioenergetics analysis on all three cell lines and found no difference in basal, maximal or non-mitochondrial oxygen consumption rate (Figure 6D). Additionally, it has been reported that OGG1 is important for overall mitochondrial health [33]. Because of this, we explored the possibility of altered mitophagy, a process essential for a healthy population of mitochondria. All three cell lines displayed equal basal mitophagy levels, but vector and wild-type OGG1 cells showed increased mitophagy following rotenone treatment, approximately 30% over basal conditions. Interestingly, the variant-expressing cells did not increase mitophagy over basal levels after rotenone treatment, suggesting a possible defect in mitophagy in response to agents that damage mitochondria (Figure 6E). Finally, we examined apoptosis among the cell lines and again found that although there was an overall effect of rotenone treatment (p=0.0355) there was no statistical difference in either basal or induced apoptosis among the three cell lines Figure 6F).
DISCUSSION
A decrease in the ability to repair DNA results in persistent lesions that can result in mutagenesis and altered transcription. Individuals homozygous for the common OGG1 variant, Ser326Cys, are at increased risk of disease in multiple organ systems. The risks associated with this variant tend to be age-related illnesses such as cancer [11–13], cardiovascular disease [15] and macular degeneration [14]. The risk is compounded by environmental exposure such as smoking and pollution [9, 34, 35]. Asian populations have the highest allele frequency of the Ser326Cys variant and increased environmental risk due to substantial pollution [36–38]. There are over 250 publications demonstrating a correlation between this variant and significant disease risk. To our knowledge, this is the first publication to demonstrate rescue of 8-oxodG incision activity to wild-type levels in cells expressing hOGG1S326C.
Both exogenous and endogenous sources of DNA damage can overwhelm cellular health if not repaired efficiently. Oxidative damage is easily bypassed by replicative and specialized polymerases, but not without substantial risk of mutagenesis. We demonstrate that the mitochondria are a target of the environmental toxicant, PQ, and that the ensuing damage sustained from oxidative stress is at least partially determined by OGG1 expression (Figure 1). Moreover, the adverse changes brought about by the mitochondrial toxin were prevented in the presence of a small molecule OGG1 activator. Importantly, some of these benefits were also obtained when using cells containing the common variant allele, termed S326C. These findings support a series of investigations using cultured cells [27, 28, 39] that demonstrate that mitochondrial-targeted overexpression of OGG1 ameliorates oxidative stress-mediated pathophysiological consequences such as cytotoxicity and apoptosis. Although it cannot be ruled out that these cellular benefits obtained in the presence of increased DNA glycosylase content may have been by mechanisms other than enhanced DNA repair [40], the data suggest that improved mtDNA integrity provides a basis to counter disease(s) related to oxidative conditions. Recent literature (reviewed by [41]) suggests that pharmacological enhancement of DNA repair may be a viable clinical strategy to, for example, prevent lung ischemia reperfusion injury in response to graft transplant dysfunction. Although more work is necessary to improve the qualities of the pharmacophores used in the present study, these pre-clinical data are consistent with the notion that improved DNA repair, particularly, mtDNA repair, could benefit oxidative stress-related pathophysiology.
To this end, targeting of mtDNA repair is limited by those pathways in operation within the mitochondrial compartment and those that affect the removal of oxidatively generated lesions. Choosing base excision repair served both purposes. OGG1 was chosen for a target of activation based on early evidence suggesting that the activity of the protein could be modulated in the presence of the guanine analogs, 8-bromoguanine or 8-aminoguanine [18]. The authors showed that the cleaved 8-oxoG residue was not catapulted out of the enzyme-substrate complex, but rather, it remained in the recognition pocket during the β-lyase step. Structural data indicated that the oxoG nucleobase served as a co-factor in catalysis and the reaction was termed “product-assisted” catalysis. This was the first evidence of this type of reaction scheme; however, this type of enzyme-mediated event would not lend itself to drug discovery. Consequently, efforts were initiated to explore whether small molecules were a viable alternative (subject of a separate publication in preparation) and culminated in finding a set of small molecule activators described herein.
OGG1 activity must be optimal to combat endogenous and environmental sources of DNA damage. The common variant allele of OGG1, S326C, has reduced capacity for DNA repair and results in risk to individuals homozygous for the allele. We find that cells have an increase in cellular and mitochondrial superoxide. It was observed that the half-maximal effective concentrations of PQ were similar across the OGG1 siRNA treatment groups. These data suggest that the mechanism of action for PQ is unchanged, but in the absence of OGG1 activity, the 8-oxodG lesion begins to accumulate with time. Moreover, the current work support previously published reports demonstrating the beneficial and deleterious effects of, respectively, increasing or decreasing OGG1 content within the cell [42] [43].
While it has been shown previously that molecules such as 8-bromoguanine can exert a feed-forward effect by activating 8-oxodG DNA repair through OGG1 [18], it has not been previously shown that a small molecule OGG1 activator could have an impact on reducing overall oxidative DNA damage. A drug screening effort identified potential OGG1 activators considered to be early stage “tool” molecules. These compounds were capable of activating and accelerating repair of PQ-induced 8-oxodG by almost 60%. Furthermore, some of the compounds were shown to have a functional impact by blocking PQ-induced mitochondrial membrane depolarization as assessed by JC-1 aggregation. This lent support for the OGG1 activators to have a functional impact, through the reduction of PQ-induced mitochondrial DNA damage whereby mitochondrial membrane potential could remain intact.
In our ex vivo studies with MEFs expressing no OGG1, wild-type OGG1, or the S326C variant, we found that compared with no OGG1, the wild-type OGG1 cells had lower cellular and mitochondrial superoxide, but the S326C expressing cells were not significantly different than cells with no OGG1 at all (Figure 6A). Additionally, we find that compound F did not reduce cellular and mitochondrial superoxide in any of the cell lines (Figures 6B and 6C), but the increase in mitochondrial 8-oxo-dG incision provided by compound F (Figure 5C) provides a mechanism to cope with the genotoxic effects of increased superoxide in S326C-expressing cells.
In response to mitochondrial damaging agents, such as rotenone, cells increase mitophagy to eliminate damaged mitochondria. Curiously, both the wild-type and vector cells responded as expected to rotenone (increased mitophagy) but the variant cells did not (Figure 6E). This raises the possibility that hOGG1S326C may participate in preventing efficient mitophagy. More research is needed to fully explain the mechanism.
Over time, wild type and Cys/Cys genotype differences may lead to larger cellular problems. Our data suggest that due to an increased basal level of superoxide, variant cells are particularly vulnerable to an oxidative event, which may result in residual damage (Figure 6). Of note, the variant allele is often attributed to increased lung cancer and COPD disease progression [10, 44]. Because the lungs are continually exposed to elevated levels of oxidative stress, both endogenously and through exposure to environmental toxins such as PQ, small changes are compounded, especially over a human lifetime.
Increasingly, personalized medicine offers the promise of identifying patients at particular risk for disease, based on complex combinations of genetic variants as well as environmental factors. For instance, it has been shown that there is an interactional effect of the variant genotype with cigarette smoking [34, 35]. For individuals with the Cys/Cys genotype, it is particularly important to avoid cigarette smoking and poor air quality. The compounds developed here demonstrate that small molecule intervention can potentially modulate this risk by increasing the activity of the protein, thus providing protection in patients at genetic and environmental risk. Previously, we demonstrated that OGG1 is important for recovery from stroke [45]. These compounds may also prove to be beneficial to boost the activity of OGG1 following a stroke. Similarly, several labs have identified OGG1 as an important factor following lung injury [46, 47]. Indeed, it has consistently been seen that oxidative stress plays a critical role in both risk of and recovery from heart attack [48], stroke [49], Parkinson’s disease [50], diabetes [51], and many other age-associated diseases [52–54]. The future availability of small molecules that enhance the activity of protective enzymes could provide a benefit to patients genetically or environmentally predisposed to such diseases.
Supplementary Material
HIGHLIGHTS.
This manuscript demonstrates that the OGG1 S326C variant has a mitochondrial defect in both 8-oxo-dG incision activity and ROS accumulation.
We demonstrate in cells expressing only the human OGG1 variant, the 8-oxo-dG incision defect can be rescued by small molecule intervention.
We demonstrate in human cells that stimulation of OGG1 by small molecule intervention decreases 8-oxo-dG accumulation in response to exogenous oxidation.
Acknowledgments
The authors would like to thank Drs. Arina Perez and Nima Fakouri for their critical review of this paper. They would also like to thank Alicia Davis for her contributions of method development and review of the manuscript and Yong Jiang and Michelle Kimberland for generation of reagents.
FUNDING
This research was supported in part by the Intramural Research Program of the NIH, National Institute on Aging and by GlaxoSmithKline.
Funding for open access charge: National Institute on Aging/#AG 000727
Abbreviations
- 8-oxodG
8-oxo-7,8-dihydro-2′-deoxyguanosine
- 8-oxoGua
8-oxo-7,8-dihydroguanine, BER, base excision repair
- AP
apurinic/apyrimidic
- APE1
AP endonuclease-1
- DMSO
dimethyl sulfoxide
- FRET
fluorescence resonance energy transfer
- MEF
mouse embryonic fibroblast
- MMP
mitochondrial membrane potential
- PQ
paraquat
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST
The authors state that they have no conflicts of interest to report.
References
- 1.Hoeijmakers JHJ. DNA Damage, Aging, and Cancer. New England Journal of Medicine. 2009;361(15):1475–1485. doi: 10.1056/NEJMra0804615. [DOI] [PubMed] [Google Scholar]
- 2.Croteau DL, Bohr VA. Repair of Oxidative Damage to Nuclear and Mitochondrial DNA in Mammalian Cells. Journal of Biological Chemistry. 1997;272(41):25409–25412. doi: 10.1074/jbc.272.41.25409. [DOI] [PubMed] [Google Scholar]
- 3.Wallace SS. Base excision repair: A critical player in many games. DNA Repair. 2014;19:14–26. doi: 10.1016/j.dnarep.2014.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Souza-Pinto NC, Eide L, Hogue BA, Thybo T, Stevnsner T, Seeberg E, Klungland A, Bohr VA. Repair of 8-oxodeoxyguanosine lesions in mitochondrial dna depends on the oxoguanine dna glycosylase (OGG1) gene and 8-oxoguanine accumulates in the mitochondrial dna of OGG1-defective mice. Cancer Res. 2001;61(14):5378–81. [PubMed] [Google Scholar]
- 5.Shibutani S, Takeshita M, Grollman AP. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 1991;349(6308):431–434. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- 6.Pinz KG, Shibutani S, Bogenhagen DF. Action of mitochondrial DNA polymerase at sites of base loss or oxidative damage. Journal of Biological Chemistry. 1995;270(16):9202–9206. doi: 10.1074/jbc.270.16.9202. [DOI] [PubMed] [Google Scholar]
- 7.McCulloch SD, Kokoska RJ, Garg P, Burgers PM, Kunkel TA. The efficiency and fidelity of 8-oxo-guanine bypass by DNA polymerases δ and η. Nucleic Acids Research. 2009;37(9):2830–2840. doi: 10.1093/nar/gkp103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuraoka I, Endou M, Yamaguchi Y, Wada T, Handa H, Tanaka K. Effects of Endogenous DNA Base Lesions on Transcription Elongation by Mammalian RNA Polymerase II: IMPLICATIONS FOR TRANSCRIPTION-COUPLED DNA REPAIR AND TRANSCRIPTIONAL MUTAGENESIS. Journal of Biological Chemistry. 2003;278(9):7294–7299. doi: 10.1074/jbc.M208102200. [DOI] [PubMed] [Google Scholar]
- 9.Hung RJ, Hall J, Brennan P, Boffetta P. Genetic Polymorphisms in the Base Excision Repair Pathway and Cancer Risk: A HuGE Review. American Journal of Epidemiology. 2005;162(10):925–942. doi: 10.1093/aje/kwi318. [DOI] [PubMed] [Google Scholar]
- 10.Duan WX, Hua RX, Yi W, Shen LJ, Jin ZX, Zhao YH, Yi DH, Chen WS, Yu SQ. The association between OGG1 Ser326Cys polymorphism and lung cancer susceptibility: a meta-analysis of 27 studies. PloS one. 2012;7(4):e35970. doi: 10.1371/journal.pone.0035970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Z, Gan L, Nie W, Geng Y. The OGG1 Ser326Cys polymorphism and the risk of esophageal cancer: a meta-analysis. Genet Test Mol Biomarkers. 2013;17(10):780–5. doi: 10.1089/gtmb.2013.0224. [DOI] [PubMed] [Google Scholar]
- 12.Xu ZG, Yu L, Zhang XY. Association between the hOGG1 Ser326Cys polymorphism and lung cancer susceptibility: a meta-analysis based on 22,475 subjects. Diagnostic pathology. 2013;8(1):144. doi: 10.1186/1746-1596-8-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yan Y, Chen X, Li T, Li M, Liang H. Association of OGG1 Ser326Cys polymorphism and pancreatic cancer susceptibility: evidence from a meta-analysis. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2013:1–6. doi: 10.1007/s13277-013-1317-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Synowiec E, Blasiak J, Zaras M, Szaflik J, Szaflik JP. Association between polymorphisms of the DNA base excision repair genes MUTYH and hOGG1 and age-related macular degeneration. Experimental Eye Research. 2012;98(0):58–66. doi: 10.1016/j.exer.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 15.Corella D, Ramirez-Sabio JB, Coltell O, Ortega-Azorin C, Estruch R, Martinez-Gonzalez MA, Salas-Salvado J, Sorli JV, Castaner O, Aros F, Garcia-Corte FJ, Serra-Majem L, Gomez-Gracia E, Fiol M, Pinto X, Saez GT, Toledo E, Basora J, Fito M, Cofan M, Ros E, Ordovas JM. Effects of the Ser326Cys Polymorphism in the DNA Repair OGG1 Gene on Cancer, Cardiovascular, and All-Cause Mortality in the PREDIMED Study: Modulation by Diet. J Acad Nutr Diet. 2018 doi: 10.1016/j.jand.2017.09.025. [DOI] [PubMed] [Google Scholar]
- 16.Bravard A, Vacher M, Moritz E, Vaslin L, Hall J, Epe B, Radicella JP. Oxidation status of human OGG1-S326C polymorphic variant determines cellular DNA repair capacity. Cancer Res. 2009;69(8):3642–9. doi: 10.1158/0008-5472.CAN-08-3943. [DOI] [PubMed] [Google Scholar]
- 17.Hill JW, Evans MK. Dimerization and opposite base-dependent catalytic impairment of polymorphic S326C OGG1 glycosylase. Nucleic Acids Res. 2006;34(5):1620–32. doi: 10.1093/nar/gkl060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fromme JC, Bruner SD, Yang W, Karplus M, Verdine GL. Product-assisted catalysis in base-excision DNA repair. Nature Structural & Molecular Biology. 2003;10(3):204–211. doi: 10.1038/nsb902. [DOI] [PubMed] [Google Scholar]
- 19.Rumsey WL, Bolognese B, Davis AB, Flamberg PL, Foley JP, Katchur SR, Kotzer CJ, Osborn RR, Podolin PL. Effects of airborne toxicants on pulmonary function and mitochondrial DNA damage in rodent lungs. Mutagenesis. 2017;32(3):343–353. doi: 10.1093/mutage/gew063. [DOI] [PubMed] [Google Scholar]
- 20.Torres-Gonzalez M, Gawlowski T, Kocalis H, Scott BT, Dillmann WH. Mitochondrial 8-oxoguanine glycosylase decreases mitochondrial fragmentation and improves mitochondrial function in H9C2 cells under oxidative stress conditions. American Journal of Physiology-Cell Physiology. 2014;306(3):C221–C229. doi: 10.1152/ajpcell.00140.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McNulty DE, Claffee BA, Huddleston MJ, Kane JF. Mistranslational errors associated with the rare arginine codon CGG in Escherichia coli. Protein expression and purification. 2003;27(2):365–374. doi: 10.1016/s1046-5928(02)00610-1. [DOI] [PubMed] [Google Scholar]
- 22.Canugovi C, Maynard S, Bayne AC, Sykora P, Tian J, de Souza-Pinto NC, Croteau DL, Bohr VA. The mitochondrial transcription factor A functions in mitochondrial base excision repair. DNA Repair (Amst) 2010;9(10):1080–9. doi: 10.1016/j.dnarep.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akbari M, Otterlei M, Pena-Diaz J, Aas PA, Kavli B, Liabakk NB, Hagen L, Imai K, Durandy A, Slupphaug G, Krokan HE. Repair of U/G and U/A in DNA by UNG2-associated repair complexes takes place predominantly by short-patch repair both in proliferating and growth-arrested cells. Nucleic Acids Res. 2004;32(18):5486–98. doi: 10.1093/nar/gkh872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu J. Current Protocols in Molecular Biology. John Wiley & Sons, Inc; 2001. Preparation, Culture, and Immortalization of Mouse Embryonic Fibroblasts. [DOI] [PubMed] [Google Scholar]
- 25.Cochemé HM, Murphy MP. Complex I is the major site of mitochondrial superoxide production by paraquat. Journal of biological chemistry. 2008;283(4):1786–1798. doi: 10.1074/jbc.M708597200. [DOI] [PubMed] [Google Scholar]
- 26.Ramamoorthy M, Sykora P, Scheibye-Knudsen M, Dunn C, Kasmer C, Zhang Y, Becker KG, Croteau DL, Bohr VA. Sporadic Alzheimer disease fibroblasts display an oxidative stress phenotype. Free Radical Biology and Medicine. 2012;53(6):1371–1380. doi: 10.1016/j.freeradbiomed.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dobson AW, Grishko V, LeDoux SP, Kelley MR, Wilson GL, Gillespie MN. Enhanced mtDNA repair capacity protects pulmonary artery endothelial cells from oxidant-mediated death. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2002;283(1):L205–L210. doi: 10.1152/ajplung.00443.2001. [DOI] [PubMed] [Google Scholar]
- 28.Ruchko M, Gorodnya O, LeDoux SP, Alexeyev MF, Al-Mehdi AB, Gillespie MN. Mitochondrial DNA damage triggers mitochondrial dysfunction and apoptosis in oxidant-challenged lung endothelial cells. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2005;288(3):L530–L535. doi: 10.1152/ajplung.00255.2004. [DOI] [PubMed] [Google Scholar]
- 29.Sidorenko VS, Grollman AP, Jaruga P, Dizdaroglu M, Zharkov DO. Substrate specificity and excision kinetics of natural polymorphic variants and phosphomimetic mutants of human 8-oxoguanine-DNA glycosylase. The FEBS journal. 2009;276(18):5149–62. doi: 10.1111/j.1742-4658.2009.07212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Di Micco R, Cicalese A, Fumagalli M, Dobreva M, Verrecchia A, Pelicci PG, di Fagagna FdA. DNA damage response activation in mouse embryonic fibroblasts undergoing replicative senescence and following spontaneous immortalization. Cell Cycle. 2008;7(22):3601–3606. doi: 10.4161/cc.7.22.7152. [DOI] [PubMed] [Google Scholar]
- 31.Go YM, Jones DP. Redox compartmentalization in eukaryotic cells. Biochimica et biophysica acta. 2008;1780(11):1273–1290. doi: 10.1016/j.bbagen.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, Robinson JP. Mitochondrial Complex I Inhibitor Rotenone Induces Apoptosis through Enhancing Mitochondrial Reactive Oxygen Species Production. Journal of Biological Chemistry. 2003;278(10):8516–8525. doi: 10.1074/jbc.M210432200. [DOI] [PubMed] [Google Scholar]
- 33.Panduri V, Liu G, Surapureddi S, Kondapalli J, Soberanes S, de Souza-Pinto NC, Bohr VA, Budinger GRS, Schumacker PT, Weitzman SA, Kamp DW. Role of mitochondrial hOGG1 and aconitase in oxidant-induced lung epithelial cell apoptosis. Free Radical Biology and Medicine. 2009;47(6):750–759. doi: 10.1016/j.freeradbiomed.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ji G, Yan L, Liu W, Qu J, Gu A. OGG1 Ser326Cys polymorphism interacts with cigarette smoking to increase oxidative DNA damage in human sperm and the risk of male infertility. Toxicol Lett. 2013;218(2):144–9. doi: 10.1016/j.toxlet.2013.01.017. [DOI] [PubMed] [Google Scholar]
- 35.Hashimoto T, Uchida K, Okayama N, Imate Y, Suehiro Y, Hamanaka Y, Ueyama Y, Yamashita H, Hinoda Y. Interaction of OGG1 Ser326Cys polymorphism with cigarette smoking in head and neck squamous cell carcinoma. Molecular Carcinogenesis. 2006;45(5):344–348. doi: 10.1002/mc.20140. [DOI] [PubMed] [Google Scholar]
- 36.Yin P, Chen R, Wang L, Meng X, Liu C, Niu Y, Lin Z, Liu Y, Liu J, Qi J. Ambient Ozone Pollution and Daily Mortality: A Nationwide Study in 272 Chinese Cities. Environmental health perspectives. 2017;125(11):117006–117006. doi: 10.1289/EHP1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pinichka C, Makka N, Sukkumnoed D, Chariyalertsak S, Inchai P, Bundhamcharoen K. Burden of disease attributed to ambient air pollution in Thailand: A GIS-based approach. PLOS ONE. 2017;12(12):e0189909. doi: 10.1371/journal.pone.0189909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao L, Liang HR, Chen FY, Chen Z, Guan WJ, Li JH. Association between air pollution and cardiovascular mortality in China: a systematic review and meta-analysis. Oncotarget. 2017;8(39):66438. doi: 10.18632/oncotarget.20090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rachek LI, Grishko VI, Musiyenko SI, Kelley MR, LeDoux SP, Wilson GL. Conditional targeting of the DNA repair enzyme hOGG1 into mitochondria. Journal of Biological Chemistry. 2002;277(47):44932–44937. doi: 10.1074/jbc.M208770200. [DOI] [PubMed] [Google Scholar]
- 40.Kim SJ, Cheresh P, Williams D, Cheng Y, Ridge K, Schumacker PT, Weitzman S, Bohr VA, Kamp DW. Mitochondria-targeted Ogg1 and Aconitase-2 Prevent Oxidant-induced Mitochondrial DNA Damage in Alveolar Epithelial Cells. The Journal of biological chemistry. 2014;289(9):6165–76. doi: 10.1074/jbc.M113.515130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tan YB, Mulekar S, Gorodnya O, Weyant MJ, Zamora MR, Simmons JD, Machuka T, Gillespie MN. Pharmacologic Protection of Mitochondrial DNA Integrity May Afford a New Strategy for Suppressing Lung Ischemia-Reperfusion Injury. Annals of the American Thoracic Society. 2017;14(Supplement 3):S210–S215. doi: 10.1513/AnnalsATS.201706-438MG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruchko MV, Gorodnya OM, Zuleta A, Pastukh VM, Gillespie MN. The DNA glycosylase Ogg1 defends against oxidant-induced mtDNA damage and apoptosis in pulmonary artery endothelial cells. Free Radical Biology and Medicine. 2011;50(9):1107–1113. doi: 10.1016/j.freeradbiomed.2010.10.692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Su YH, Lee YL, Chen SF, Lee YP, Hsieh YH, Tsai JH, Hsu JL, Tian WT, Huang W. Essential role of β-human 8-oxoguanine DNA glycosylase 1 in mitochondrial oxidative DNA repair. Environmental and molecular mutagenesis. 2013;54(1):54–64. doi: 10.1002/em.21742. [DOI] [PubMed] [Google Scholar]
- 44.da Silva AL, da Rosa HT, Karnopp TE, Charlier CF, Ellwanger JH, Moura DJ, Possuelo LG, Valim AR, Guecheva TN, Henriques JA. Evaluation of DNA damage in COPD patients and its correlation with polymorphisms in repair genes. BMC Med Genet. 2013;14(1):93. doi: 10.1186/1471-2350-14-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu D, Croteau DL, Souza-Pinto N, Pitta M, Tian J, Wu C, Jiang H, Mustafa K, Keijzers G, Bohr VA. Evidence that OGG1 glycosylase protects neurons against oxidative DNA damage and cell death under ischemic conditions. Journal of Cerebral Blood Flow & Metabolism. 2010;31(2):680–692. doi: 10.1038/jcbfm.2010.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dobson AW, Grishko V, LeDoux SP, Kelley MR, Wilson GL, Gillespie MN. Enhanced mtDNA repair capacity protects pulmonary artery endothelial cells from oxidant-mediated death. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2002;283(1):L205–L210. doi: 10.1152/ajplung.00443.2001. [DOI] [PubMed] [Google Scholar]
- 47.Hashizume M, Mouner M, Chouteau JM, Gorodnya OM, Ruchko MV, Potter BJ, Wilson GL, Gillespie MN, Parker JC. Mitochondrial-targeted DNA repair enzyme 8-oxoguanine DNA glycosylase 1 protects against ventilator-induced lung injury in intact mice. American journal of physiology Lung cellular and molecular physiology. 2013;304(4):L287–97. doi: 10.1152/ajplung.00071.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li H, Horke S, Förstermann U. Oxidative stress in vascular disease and its pharmacological prevention. Trends in Pharmacological Sciences. 2013;34(6):313–319. doi: 10.1016/j.tips.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 49.Chamorro Á, Dirnagl U, Urra X, Planas AM. Neuroprotection in acute stroke: targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. The Lancet Neurology. 2016;15(8):869–881. doi: 10.1016/S1474-4422(16)00114-9. [DOI] [PubMed] [Google Scholar]
- 50.Hwang O. Role of Oxidative Stress in Parkinson’s Disease. Exp Neurobiol. 2013;22(1):11–17. doi: 10.5607/en.2013.22.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pitocco D, Tesauro M, Alessandro R, Ghirlanda G, Cardillo C. Oxidative stress in diabetes: implications for vascular and other complications. International journal of molecular sciences. 2013;14(11):21525–21550. doi: 10.3390/ijms141121525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barzilai A, Rotman G, Shiloh Y. ATM deficiency and oxidative stress: a new dimension of defective response to DNA damage. DNA Repair. 2002;1(1):3–25. doi: 10.1016/s1568-7864(01)00007-6. [DOI] [PubMed] [Google Scholar]
- 53.Cheresh P, Kim SJ, Tulasiram S, Kamp DW. Oxidative stress and pulmonary fibrosis. Biochimica et biophysica acta. 2013;1832(7):1028–40. doi: 10.1016/j.bbadis.2012.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang X, Wang W, Li L, Perry G, Lee H-g, Zhu X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2014;1842(8):1240–1247. doi: 10.1016/j.bbadis.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.