

Abstract
The Free Radical Theory of Ageing, was first proposed by Denham Harman in the mid-1950’s, based largely on work conducted by Rebeca Gerschman and Daniel Gilbert. At its core, the Free Radical Theory of Ageing posits that free radical and related oxidants, from the environment and internal metabolism, cause damage to cellular constituents that, over time, result in an accumulation of structural and functional problems. Several variations on the original concept have been advanced over the past six decades, including the suggestion of a central role for mitochondria-derived reactive species, and the proposal of an age-related decline in the effectiveness of protein, lipid, and DNA repair systems. Such innovations have helped the Free Radical Theory of Aging to achieve widespread popularity. Nevertheless, an ever-growing number of apparent ‘exceptions’ to the Theory have seriously undermined its acceptance. In part, we suggest, this has resulted from a rather simplistic experimental approach of knocking-out, knocking-down, knocking-in, or overexpressing antioxidant-related genes to determine effects on lifespan. In some cases such experiments have yielded results that appear to support the Free Radical Theory of Aging, but there are just as many published papers that appear to contradict the Theory. We suggest that free radicals and related oxidants are but one subset of stressors with which all life forms must cope over their lifespans. Adaptive Homeostasis is the mechanism by which organisms dynamically expand or contract the homeostatic range of stress defense and repair systems, employing a veritable armory of signal transduction pathways (such as the Keap1-Nrf2 system) to generate a complex profile of inducible and enzymatic protection that best fits the particular need. Viewed as a component of Adaptive Homeostasis, the Free Radical Theory of Aging appears both viable and robust.
Keywords: Ageing, Stress Adaptation, Signal transduction pathways, Nrf2, Proteasome, Oxidative stress, Protein oxidation and aggregation
Graphical Abstract
INTRODUCTION
Ageing is a universal leveler and a process that has long fascinated humankind. One of the earliest records of our desire to slow or reverse the ageing process may be found in the writings of the 5th century Greek historian Herodotus and, since then, we have continually sought the elusive ‘fountain of youth.’
Indeed, the scientific community has generated over 300 theories in an attempt to explain the driving forces behind ageing [1], but none have, so far, been proven to be universally applicable. Rather, the multitude of extant theories highlights the complexity of the ageing process. One of the most popular postulates is the Free Radical Theory of Ageing, which has become one of the leading explanations to describe the ageing process at the molecular level.
Since its conception in 1956, the Free Radical Theory of Ageing, first hypothesized by Denham Harman [2], has greatly influenced the direction of ageing research for the past half century. Harman’s Free Radical theory is actually based on the original and independent observational work of Rebeca Gershman (from the University of Buenos Aires in Argentina) and Daniel Gilbert (from the University of Rochester) [3]. Gershman and Gilbert first identified the similar characteristics between reactive species produced during cellular respiration and those generated by irradiation: namely, the generation of free radicals, such as the hydroxyl (•OH) and superoxide (O2•−) radicals and the less reactive non-radical oxygen species, hydrogen peroxide (H2O2) [3].
Yet, at the time of Gershman and Gilbert’s observation, little was understood about the specific sites of free radical generation within the cell. We now know that the mitochondria account for one of the primary sources of intracellular reactive oxygen species (~1–5%), due to electron leakage primarily resulting from the electron transport chain [4, 5]. One of the main byproducts is the formation of the highly reactive hydroxyl radical, which can either immediately react and damage neighboring mitochondrial proteins, DNA, and lipids or be converted to the less reactive, but longer-lived superoxide radical [6]. In turn, the superoxide radical can either react with molecules or undergo dismutation to hydrogen peroxide, which is highly dispersive within the cell and is the least reactive mitochondria-generated molecule [7].
Reactive species also arise from peroxisomes, which are the site of fatty acid oxidation. Hydrogen peroxide is the major free radical produced by acyl CoA oxidase in the metabolic breakdown of fatty acids [8], with the majority removed by peroxisome catalase and glutathione peroxidase. However, a small percentage can either leak into the cytosol or form the hydroxyl radical, due to Fenton chemistry. As well, superoxide, a byproduct of xanthine breakdown by xanthine oxidase [9], can either undergo dismutation by Copper-Zinc
Superoxide Dismutase (CuZn SOD), further adding to the peroxisome pool of hydrogen peroxide or react with the nitric oxide radical (a byproduct of peroxisome-mediated amino acid metabolism) to form peroxynitrite [10, 11]. As a result, cellular metabolism was considered the primary culprit for intercellular generation of so-called ‘harmful’ free radicals.
Indeed, Harman’s theory postulated that if these highly unstable and reactive molecules are not removed or neutralized, they could damage proteins, lipids, and DNA either directly or indirectly (via Fenton chemistry) [2, 12]. Thus, Harman first proposed, that ageing occurred because of:
“…..deleterious side attacks of free radicals (which are normally produced in the course of cellular metabolism) on cell constituents and on the connective tissues” [2].
Harman’s theory sparked the idea that complete removal of so-called ‘harmful’ molecules would mitigate cellular damage and, consequently, slow the overall ageing process. Indeed, studies have found that chronic removal of some antioxidant enzymes, such as cytosolic or mitochondrial superoxide dismutases negatively impacts the health-span and/or the lifespan in multiple model organisms [13–15]. However, it should be noted that the reverse is not always true, as multiple studies have shown that chronic over-expression of several antioxidant and detoxification enzymes can actually have little or no impact (and in some circumstances can even be detrimental) [16–20]. Over the years, people have tried to ‘adapt’ the Free radical
Theory of Ageing to accommodate various new, sometimes apparently contradictory findings, including a central role for mitochondria-derived reactive species [6, 21] and age-dependent dysfunction of protein, lipid, and DNA repair systems [22]. Though novel for its time, the Free Radical Theory of Ageing has since come under the scientific ‘firing squad,’ as multiple studies have concluded that it is not a universal phenomenon. For example, naked mole-rats live for some 32 years, yet levels of the reactive oxygen species they experience appear similar to those encountered by their short-lived cousin, the mouse [23]. Similarly, quails and parrots both produce similar levels of reactive oxygen species, yet parrots live five times longer than do quails [24]. Moreover, long-lived species [25, 26] and centenarians [27] show higher amounts of oxidative damage, measured by increased protein carbonyls, when compared to the average living animal or human, which is contradictory to Harman’s theory.
It must be said that our understanding of the chemical and biological implications of free radicals has evolved significantly since 1956. The outdated binary (good or bad) belief, originally proposed by Harman, that free radicals and reactive oxygen species can only be deleterious molecules, while antioxidants and free radical scavengers must always be beneficial molecules, may no longer be sufficient. Indeed, this binary relationship was first tested by Linus Pauling, himself a strong proponent of very high daily-doses of ascorbic acid (Vitamin C) as an anti-ageing and disease intervention [28]. Nevertheless, Pauling’s findings (and those of his followers), are highly controversial and do not demonstrate a definitive benefit [29–31]. Thus, simplistic binary beliefs about free radicals may no longer be an appropriate dogma.
Instead, growing evidence suggests it is the dynamic nature of production and decay, and thus the transient levels of various reactive oxygen and nitrogen species, which can tilt them as either beneficial or harmful. Short-term low concentrations of free radicals and reactive oxygen species make them advantageous (signaling), whereas too much and they become deleterious. Studies in yeast [32] and cell culture [33–40] attest to the beneficial non-damaging, signaling response elicited by transient exposure to H2O2, as low (μM) amounts rather than inhibiting growth (a sign of damage), actually stimulates proliferation, suggesting low amounts of free radicals can be beneficial. Moreover, pretreatment with short-term signaling amounts of a variety of free-radical generators, rather than increasing cellular susceptibility to damage, stimulates transient induction of cellular protective enzymes, indicating their role in elucidating a nuclear response [41–43]. As well evidence extends beyond cell culture models into model organisms, which show similar short-term exposure to low-amounts of free radicals and free-radical generators as protective rather than detrimental [18, 42, 44–47], with evidence suggesting early-life exposure even capable of promoting longevity [48].
Antioxidant and cellular defense enzymes also act as conduits for free radical signaling. Under homeostatic conditions, peroxiredoxins are predicted to breakdown the majority of mitochondrial and cytosolic-derived H2O2: at homeostatic signaling concentrations, peroxiredoxins are capable of continual H2O2 elimination [49, 50]. However, upon elevated H2O2 concentration, leads to peroxiredoxin inactivation and H2O2 accumulation. This triggers a cascade of cellular signaling events, beginning with the oxidation of target proteins and subsequent opening of the cellular defensive response [51]. One of considerable importance being the Nrf2 pathway, wherein modification of the thiol groups on Keap1, initiates Nrf2 nuclear translocation and subsequent activation of the stress protective pathway [52]. Thus, oxidant levels are highly responsible for initiating cellular signaling responses.
Conversely, more is not always better, as too much antioxidants can eliminate free-radical mediated cellular-signaling [51, 53]. Growing evidence shows free radicals are not only important for cellular homeostasis, but loss in their homeostatic balance may be an underlying cause for age-associated diseases, and ageing itself [54–56].
THE FREE RADICAL THEORY OF AGEING
Is More Always Better – and Less Always Worse?
The main premise of the Free Radical Theory argues that ageing is a result of cellular damage accumulation due to free radical attack. Based on this statement, it would appear that at least two outcome predictions should hold true. The first being that if antioxidant and stress-related cellular defenses are removed, then an ‘ageing’ phenotype should appear. Conversely, overexpression of these same enzymes should result in suppression of the ‘ageing’ phenotype and, perhaps even promote lifespan.
Much effort has been invested in addressing the validity of the first predicted outcome, primarily by the chronic knockdown of antioxidant or stress-responsive enzymes. Initial work in baker’s yeast, Saccharomyces cerevisiae (S. cerevisiae), found that elimination of either cytosolic Cu-Zn-Superoxide Dismutase (SOD1) or the mitochondrial Mn-SOD variant (SOD2), the primary enzymes responsible for the breakdown of the superoxide radical, resulted in decreased survival during stationary phase [57]. Yeast survival was even further diminished (presumably a synergistic decline) observed in the SOD1/SOD2 double mutant [57]. Similar outcomes were noted for some other stress responsive enzymes, including the yeast homologues for the 20S Proteasome, Proteinase yscE [58], the ATPase subunit of the 26S Proteasome [59] and the mitochondrial Lon protease, Pim1 [60, 61], all of which are necessary for the removal of oxidized and/or ubiquitin-tagged proteins. Chronic knockdown of the hydrogen peroxide-detoxifying enzyme, catalase, however, actually improved chronological aging in yeast, suggesting that loss of antioxidant enzymes is not universally detrimental, and providing evidence against the Free Radical Theory of Ageing [62].
Parallel work in Drosophila melanogaster (D. melanogaster), or more commonly known as fruit-flies, found that the removal of major antioxidant enzymes, such as the mitochondrial SOD2 [63–65], resulted in shortened lifespan. Similar findings were noted for chronic knockdown of cytosolic SOD1 [66, 67]. Removal of stress-responsive enzymes, such as the 20S proteasome and the mitochondrial Lon protease, also showed decreased survival [18, 45, 68]. Similarly, inactivation of the 20S Proteasome led to shortened lifespan [69]. Conversely, mutants lacking full or partial catalase activity showed no change in survivorship [70].
Studies in the nematode worm, Caenorhabditis elegans (C. elegans), represent an even greater experimental contradiction of the first component of the Free Radical Theory of Ageing. Unlike studies in yeast and fruit-flies, removal of certain stress responsive and antioxidant enzymes had either no impact or, in some instances, were actually beneficial. For example, individual removal of the five superoxide dismutase (SOD) genes caused no change in C. elegans lifespan, and mutants lacking two or three superoxide dismutase genes lived as long as the wild type [71]. Moreover, worms deficient in individual superoxide dismutase genes showed increased mRNA levels in the remaining sod genes [72]. Removal of glutathione transferase did not appear to impact survival at all [73]. Confusingly, removal of the mitochondrial Lon protease resulted in increased lifespan [74], whereas, removal of 20S Proteasome or 26S Proteasome subunits, or their transcriptional activator Nrf2, consistently resulted in shortened lifespan [75–77], even though both Lon and the Proteasome play important roles in clearance of (oxidatively) damaged proteins. In contrast, removal of nuclear-encoded proteins involved in mitochondrial electron transport and oxidative phosphorylation (a primary source of mitochondria-derived O2.− and H2O2) has been linked with increased longevity, which is actually in line with the first predicted outcome of the Free Radical Theory of Aging [78]. These highly conflicting and contradictory outcomes in the worm either highlight a species-specific anomaly, which would certainly challenge the universality of the Free Radical Theory of Ageing, or may hint at even deeper potential flaws in the theory.
Interestingly, as studies progressed into mice, survival outcomes tended to align more closely to those in the nematode worm. Indeed, mice lacking crucial antioxidant and stress-responsive enzymes showed inconsistencies in lifespan following their removal. The levels of several antioxidant gene products have been diminished or eliminated by knockdown or knockout techniques to generate a variety experimental mice, including sod2+/− [79], gpx4+/− [80], gpx1+/− [13], msrA−/ − [81], sod1−/ − [82], and trx2+/− [82] animals. Of these six genetically modified mice strains, only the sod1−/ − animal, deficient in cytosolic Cu-Zn SOD, exhibited decreased survival. Importantly, even when multiple antioxidant ‘redundancy’ pathways were eliminated, no changes in survival were observed [13, 82]. It should, however, be noted that full knockdown of gpx4 and sod2 were identified as embryonically lethal [83, 84]. It should also be remembered that some of these studies were executed with mixed populations of mice, thus any sex-specific differences may not be evident.
Given the results reviewed above, it should be of little surprise that chronic overexpression is highly organism-dependent. Nevertheless, one can certainly point to quite a large number of studies in which overexpression of antioxidant enzymes resulted in positive effects on survival and/or lifespan. In S. cerevisiae, for example, chronic upregulation of antioxidant and stress-responsive enzymes are largely beneficial. In the case of the proteasome, increased expression leads to enhanced activity and increased replicative lifespan [85], with a similar trend evident upon overexpression of SOD1 and SOD2 [86, 87]. Additionally, chronic upregulation of other detoxification enzymes, including catalase, has provided evidence for improved survival [88, 89]. Similar findings are also evident in mammalian cell culture studies [90–92]. Furthermore, overexpression of cytosolic and mitochondrial forms of SOD [93–96] increased lifespan in D. melanogaster. In one finding, highly active SOD variants in D. melanogaster neurons were associated with exceptional longevity [97]. Enhanced expression of the antioxidant enzymes, glutathione-S-transferase [98], glutathione reductase [99], SOD1 [67] and SOD2 [100], all improved survival in the nematode worm. Finally, overexpression of the beta 5 catalytic subunit of the 20S proteasome resulted in increased survival [101], and indirect proteasome upregulation (achieved through the suppression of germline development) also led to enhanced longevity [102].
Despite the positive results detailed above, little benefit of increased antioxidant gene expression has been found in higher organisms. For example, of the vast majority of lifespan studies completed using transgenic mouse strains, overexpressing antioxidant or oxidant-repair genes, only a small percentage have actually proven beneficial [103, 104]. Indeed, overexpression of the antioxidant genes SOD1, SOD2, GPx4, catalase [19, 82, 105] either individually, or as synergistic pairs (SOD1 + SOD2, or SOD1 + catalase) [19, 82] failed to increase lifespan and, in some instances, actually shortened life [106, 107]. In a similar manner, the forced over-expression of stress-responsive enzymes, rather than being beneficial has often been found to be detrimental. For example, mice transgenic for mitochondrial Lon protease (Lonp1+/+) showed increased tumorigenesis [108].
These lack-luster findings and inconsistencies both across and within species, have led to increased interest in the role of upstream targets. One of the most notable of these is the nuclear factor erythroid 2-related factor 2 (Nrf2), which is a major transcriptional regulator of numerous Phase II detoxification and stress-responsive enzymes [52]. Due to its important role as a master transcriptional regulator, much interest has centered upon understanding the impact of Nrf2 levels upon survival. Yet, even these findings are inconsistent across models. For example, decreased survival has been reported upon Nrf2 suppression in C. elegans [109] and D. melanogaster [110] and overexpression is beneficial to lifespan [45, 111]. Yet, in a mammalian mouse model, Nrf2 knockout resulted in no change in lifespan, but increased sensitivity to oxidative stress [112] and tumorigenesis [113]. Whereas knockout of the Nrf2 cytosolic inhibitor, Keap1, results in death after 20 days [114]. Overall, inconsistences in Nrf2-mediated survival suggest a more nuanced mechanism is at play.
On aggregate, these outcomes suggest that either the binary historical approach in the interpretation of the Free Radical Theory of Ageing is incorrect, with current means of addressing its validity are oversimplified, or that the theory itself may be flawed. We would suggest instead, that these outcomes serve to highlight the adaptability and pathway redundancies that exist, enabling organisms to cope (and even live a normal lifespan), despite significant negative or positive perturbations of specific detoxification or stress-responsive enzymes. Therefore it brings into question, whether weaknesses become evident when the system is perturbed or no longer within its homeostatic range.
System Perturbation: Toppling the Biological House of Cards
A major flaw in addressing the validity of the Free Radical Theory of Ageing, is the widespread approach of forcing a biological system to adapt a new ‘homeostatic’ set-point (through the chronic increase or decrease of a target enzyme) and then assessing the outcome. In so doing, this model overlooks a fundamental component: no organism lives in isolation. Rather, cellular defenses (and their respective enzymes) are continually in a state of transient malleability. In turn, rapid and short-term modulation of enzyme levels, ensures an organism can cope with its environment. In contrast, chronic enzymatic expression prevents small and nuanced changes, which may be necessary for survival.
From this perspective, it should be of little surprise that limited lifespan benefit was found upon chronic expression of target enzymes. Moreover, enzyme manipulation was done in a state of relative non-perturbation (unless ageing itself, is viewed as a stress). The deeper nuances becomes evident when stress resistance was assessed in these studies. For clarity, we define stress resistance as the survivability of an organism when exposed to a high, semi-lethal amount of an oxidant or environmental treatment meant to promote cellular damage and/or death.
Indeed, evidence shows that the removal of enzymes critical for detoxification or protection against oxidative stress are more vulnerable when exposed to these semi-lethal concentrations. For example, fibroblasts from mice deficient in SOD1, though showed few abnormalities and normal lifespan, were highly sensitive to the superoxide generator, paraquat [115–117] and showed higher rates of tissue-specific DNA damage [118], accelerated protein oxidation [119], decreased Proteasome activity [120] and more recently proposed as a model for age-associated frailty [121]. A similar trend occurs in fibroblasts from glutathione peroxidase deficient-mice [122] and cells from mice deficient in mitochondrial SOD2−/+ [123], including an associated loss in mitochondrial aconitase activity and decreased Lon expression [124]. A synergistic effect in heightened oxidative stress sensitivity was noted in a double mutant mouse model of SOD+/− and glutathione peroxidase−/− [125]. Moreover, cells lacking stress-responsive enzymes show increased oxidative-stress sensitivity, including direct knockdown of the Lon protease [126] and the Proteasome [127].
Conversely, lack of an enzyme can result in activation of stress-responsive pathways. This is most prominent in studies manipulating the mitochondrial electron transport chain, one of the primary sources, argued by Harman, to contribute to cellular oxidative stress. Studies in mice lacking surf−/− (an auxiliary protein in the aid of Complex IV), showed a compensatory activation of the unfolded protein response (UPR), leading to increased expression of UPR-associated proteins (including Nrf2, mitochondrial Lon protease, and heme-oxygenase-1) [128]. In a similar trend, mutations resulting in electron transport chain dysfunction, which should promote the generation of free radicals, rather than shortening the lifespan, triggered longevity in worms [78, 129], flies [130], and mice [131].
These differences in stress resistance, evident upon removal of antioxidant and/or stress-responsive enzymes, suggests that Harman’s argument perhaps may not be flawed, rather our scientific approach may be too simplistic. For decades, we have asked the straight-forward and simple question, if we increase or decrease enzyme “X” what is the outcome upon survival? Instead, we should be asking how does an organism adapt to stress and can it be enhanced in the absence or presence of an enzyme in a life-stage dependent manner.
ADAPTIVE HOMEOSTASIS AND AGEING
Although our understanding of the roles of free radicals and other reactive oxygen/nitrogen species has evolved to include important physiological functions in cellular signaling, our theories to adequately describe how such reactive species may affect the ageing process have not kept pace. To address this gap, we propose a new theory that builds on the Free Radical Theory of Ageing, but also includes the dual roles of free radicals and other reactive oxygen/nitrogen species in cellular signaling. Importantly, our new theory is also much broader than the original Free Radical Theory of Ageing, in that it is also applicable to any and all agents or conditions that, at high concentrations, can cause cellular damage. Our new premise is based upon the concept of transient and reversible adaptation, or more aptly coined ‘Adaptive Homeostasis,’ which allows for short-term, transient changes in the homeostatic range of many protective and repair mechanisms. Adaptive Homeostasis has been defined as follows:
“The transient expansion or contraction of the homeostatic range in response to exposure to sub-toxic, non-damaging, signaling molecules or events, or the removal or cessation of such molecules or events.” [132].
Recently, much evidence has come to light to demonstrate both sex-specific [40, 133] and age-associated [54, 134–139] changes in the adaptive homeostatic response, which may more appropriately describe age-dependent changes and susceptibility to diseases. These findings all very clearly point to an age-determined decline in adaptive homeostasis that makes older cells, organisms, animals (and humans?) more sensitive to toxicants, other damaging agents, and harmful environments or conditions.
We, therefore, now formally propose an Adaptive Homeostasis Theory of Ageing, which we define as follows:
“Ageing involves a decline in the ability to activate or signal Adaptive Homeostasis to effect transient, short-term expansion or contraction of the homeostatic ranges of stress-protective systems. Since exposure to damaging, toxic, or stressful molecules and conditions still occurs, the net effect of diminished Adaptive Homeostasis is manifested as deterioration of biological structures and functions, with increasing accumulation of cellular and intracellular damage products in later life and eventual dysfunction, disease, senescence, and death.”
Our purpose in this paper is to demonstrate that the Adaptive Homeostasis Theory of Ageing helps to fill in inconsistences in the Free Radical Theory of Ageing that have become evident over the past few decades, and to provide an alternative and broader theory that can encompass free radicals under its umbrella.
Assessment of the Adaptive Homeostatic Response
Prior work describing adaptation typically refers to the phenomenon as hormesis, wherein concentrations, typically toxic at high doses, become beneficial at lower concentrations [140–142]. Yet this beneficial viewpoint, does not match the original description posited by Southerland (1945), which argued, from a toxicology standpoint, the process in which a toxin or poison could cause sub-lethal damage, leading to an exaggerated repair response [143]. Or more aptly phrased by the 19th century German philosopher, Friedrich Nietzsche “that which does not kill us, makes us stronger.’ This inaccurate, but prominent perception of hormesis, requires it to be distinguished from the adaptive homeostatic response, which involves non-damaging signaling amounts of an oxidant or environment to activate these same pathways [132].
Early studies in bacteria [144, 145] clearly demonstrated that Escherichia coli can undergo regulon-dependent protective adaptive changes in gene expression, with consequent transient increases in stress resistance, following exposure to very low levels of either O2.− (OxyR Regulon) or H2O2 (soxRS Regulon), and we (and others) have successfully repeated these results. We moved on to eukaryotic studies in Saccharomyces cerevisiae yeast and in mammalian cells (that don’t posess regulons) which provided more detailed insights and eventually lead us to propose Adaptive Homeostasis.
Yeast, pretreated with low, non-damaging, amounts of H2O2 and subsequently challenged with a toxic amount of H2O2, showed improved survival compared to yeast only subjected to the challenge dose of H2O2. More importantly, pretreated yeast showed improved growth, even exceeding the density present in control colonies [32]. Later studies, using multiple types of mammalian cell lines (including human) found a consistent and similar phenomenon: pretreated cells showed increased survivorship compared to populations exposed only to an oxidant challenge [146–148]. In addition, cells were found to be capable of undergoing multiple cycles of adaptation, de-adaptation, and re-adaptation [32, 43, 146], suggesting the importance of the adaptive response in day-to-day cellular changes. Demonstration that cells can adapt, de-adapt, and re-adapt is important because it rules out the possibility that we are only selecting for a (possible) pre-existing sub-population of cells with innately high levels of oxidant protection.
The universality of the adaptive homeostatic response has also been tested. Indeed, evidence for its applicability is seen across multiple forms of cellular stress, including heat shock, osmotic stress, caloric restriction, hypoxia, cold shock, exercise training, and more [134, 149]. These findings summarize a unifying theme, wherein organisms rely upon a cadre of synergistic pathways to cope with environments that offer diverse stresses, often in combination. Assessing how an organism adapts to various stresses provides something of a ‘fingerprint’ of where it fits in its environmental niche. From such investigations it appears clear that the ability to rapidly respond to signal-inducing changes is crucial for long-term homeostasis and survival.
Adaptability of the Proteasome and Mitochondrial Lon Protease
The adaptive homeostatic response relies upon multiple stress-responsive pathways and enzymes to ensure appropriate clearance of cellular damage. The 20S Proteasome was one of the first enzymes identified as stress-responsive for protein clearance [34, 41, 150–157]. Although multiple forms of the Proteasome reside simultaneously within the cell, the 20S ‘core’ Proteasome is the predominant form under oxidative duress [158, 159]. The core 20S Proteasome is a barrel-shaped enzyme, comprised of two outer alpha- and two inner beta-rings, with the proteolytic activity contained within the beta rings (β1 has caspase-like activity, β2 has trypsin-like activity, and β5 has chymotrypsin-like activity) [159]. Initial studies identified oxidized proteins as the preferred substrate of the 20S Proteasome [33, 34, 153, 154, 160, 161], with later work showing rapid elevation in proteolytic capacity upon oxidative stress [41, 43, 157].
Activation of the adaptive stress response leads to rapid disassembly of the 26S Proteasome by sequestering of the 19S oxidant-inactivated regulatory caps by HSP70 [162, 163]. HSP70 actually acts in conjunction with a partner enzyme, ECM29, first identified as a proteasome scaffolding protein in yeast [164, 165] and later found to have a conserved role in mammalian cells [166, 167]. This leads to an immediate increase in the cytosolic pool of 20S Proteasomes available to degrade oxidized proteins. HSP70 returns its 19S cargo to 20S Proteasomes (by an unknown mechanism) some 3–5 hours following the initiation of adaptive responses, thus ‘restoring’ 26S Proteasomes which are required over the following 13–15 hours for a fully effective adaptive response in mammalian cells. Importantly, also during the 3–5 hour period of 26S Proteasome disassembly, Nrf2 cannot be degraded by the ubiquitin-26S Proteasome system and Nrf2 levels rapidly rise. At the same time, Nrf2 nuclear translocation and transcriptional binding to Electrophile Responsive Elements (EpRE’s), also called Antioxidant Response Elements (ARE’s) within the upstream regulatory regions of many target genes [52] (including those encoding 20S Proteasome subunits) [41], leads to de novo synthesis and further elevation of the available 20S Proteasome pool [41, 168]. Collectively, these events ensure that the cell has both an immediate (minutes) and long-term (hours) source of available and functionally competent 20S Proteasome, with these responses appearing as an in-phase mediated process [169].
More recently, additional forms of the Proteasome have been found to be involved in the adaptive stress response. The immunoproteasome, first identified for its role in the innate immune response [170, 171], has since been coined the ‘Inducible’ Proteasome [172], as it too, is found to increase in response to oxidative stimuli [156, 157, 170, 173]. In addition, the so called PA28aβ (or 11S) cytoplasmic regulator undergoes increased synthesis and binds to both 20S Proteasomes and Immunoproteasomes which improves their ability to degrade oxidized proteins. Together, these multiple pathways in the Proteasome adaptive response work synergistically to ensure cells and tissues are protected against oxidative insults.
Parallel studies have identified additional proteolytic stress responsive enzymes, one of the most notable being the mammalian mitochondrial LonP1 protease (encoded by the lon gene). First identified in E. coli, as a heat-shock inducible protein (La) [174, 175], Lon was later found to be conserved from yeast (Pim1) [68] to mammals [176], as a characteristic six- (mammal) or seven-membered (yeast) monomeric ring, which degrades oxidized proteins in an ATP-dependent manner [177–179]. Initially discovered as a bacterial stress-inducible enzyme, a conserved role of Lon induction was identified in mammalian cell culture studies in response to multiple types of oxidative stress: including heat shock, hydrogen peroxide, hypoxia, endoplasmic reticulum stress, and serum starvation [37–39, 180–184]. Additional work has identified mammalian LonP1 as an indirect regulator of mitochondrial transcription, as it modulates mitochondrial transcription factor A (TFAM) turnover [108, 185, 186], and more recently its role in the Pink1/Parkin system, crucial in mitophagy [187]. Together, these findings indicate the dynamic role of Lon as highly responsive to the oxidative status and needs of the cell.
Can Model Organisms be Used to Explore and Better Understand Adaptive Homeostasis?
Our understanding of the eukaryotic adaptive stress response is primarily derived from mammalian cell culture studies. Yet, to explore the mechanism(s) and universality of the adaptive homeostatic response in intact organisms, and its relevance to human beings, two caveats need to be met: first, one must be able to experimentally replicate the adaptive homeostatic response, seen in cells, in whole organisms; and second, one must be able to find model organisms in which the pathways for adaptive homeostasis are approachable, dissectible, and strongly related to mammalian/human pathways.
Initial studies used young (3 day old) worms and fruit-flies. Animals were either not pretreated or were pretreated with non-damaging, signal-transducing amounts of H2O2 (10–1000μM) for a short-duration, followed by an activation phase for the adaptive response, and finally application of a toxic (semi-lethal) dose of H2O2 (a 1.6mM solution in worms, and 4.4M in food on Kimwipes in fruit flies) and survival was measured. In both scenarios, pretreated organisms survived longer than those subjected only to the challenge dose [42]. Additionally, similar to the cell-culture studies discussed above, low signaling amounts of H2O2 stimulated increases in 20S Proteasome transcription/translation and activity in the nematode worm [42, 44] and fruit fly [42, 45]. Furthermore, induction of the 20S Proteasome was shown to be Nrf2-dependent [42, 44]. Finally, blocking induction of either the 20S Proteasome or the mitochondrial Lon protease blunted organismal adaptation and survival [18, 44, 45]. These findings indicate that Adaptive Homeostasis is conserved in higher organisms, and also provide us with a model for its assessment.
Assessment of the adaptive homeostatic response further shed light on the divergence in response between the self-fertilizing hermaphroditic worm and the heterogenic fruit-fly. Specifically, the fruit fly provided us with evidence for sex-specific differences in the adaptive response. Indeed, though earlier studies have found lifespan and stress resistant sex differences [188–190], much of the work in this field has been limited to one sex (male) [191]. In contrast, much of our own work on adaptive responses in mammalian cell-cultures are based upon female-derived cell lines [40]. In fruit-flies, 3 day old females were capable of adapting and inducing the 20S Proteasome in response to hydrogen peroxide signaling doses [45], which was found to hold true in multiple fly strains [192]. Additionally, the mitochondrial Lon protease not only showed a female-favored response upon hydrogen peroxide pretreatment, but was the first identification of different isoform patterns in females versus males [18]. Conversely, pretreatment with the superoxide generator, paraquat, found a male-dependent adaptive increase in the amount and proteolytic capacity of the 20S Proteasome [192] and mitochondrial Lon protease [18]. These findings indicate that not only are organisms capable of adaptation, but that the adaptive response is sex- and oxidant-dependent.
More recently, new evidence highlights the translational aspect of adaptive homeostasis in response to environmental factors. One that has gained increasing traction, is assessment of air pollution (specifically from vehicular-derived ‘smog’) and its impact upon human health. Yet, assessing the impact of low-level exposure upon the adaptive response is only in its infancy. To begin to address this gap, we utilized 6 month old C57BL/6 male mice, exposed to ambient air or vehicular-derived nanoparticles for a period of 10 weeks. Lung, liver, and the cerebellum were assessed for changes in Nrf2-responsive enzymes [47]. Interestingly, young males showed induction of Nrf2-phase II responsive genes, compared to controls, suggesting activation of the adaptive homeostatic response [47]. More recently, 6 month old C57BL/6 female mice showed a similar trend, along with activation of the stress responsive enzymes, the mitochondrial Lon protease and the 20S proteasome [193]. Together these findings suggest the conserved role of the adaptive response in a mammalian animal and provide a model in which it may be further tested and explored.
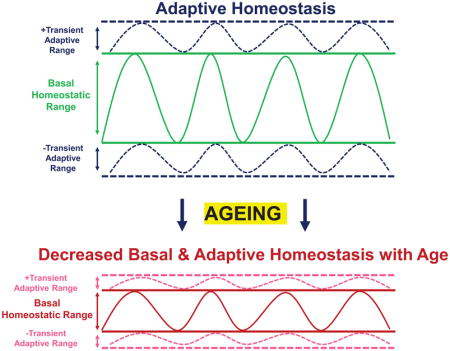
Loss of the Adaptive Homeostatic Response with Age
Young organisms are characterized by the robustness of the adaptive homeostatic response: the reversible expansion or contraction of the homeostatic range, ensuring transient upregulation of protective pathways. However, if adaptation enables young organisms to cope in a dynamic environment, then its antithesis is ageing. Indeed, multiple studies have demonstrated, across multiple signal-transducing mediators of adaptive responses, an age-dependent attenuation [134]. Early work addressing this question showed that high-passage or senescent cells, were no longer capable of transiently activating the adaptive response. The concept of proliferative senescence, first demonstrated by Hayflick and colleagues, in which normal cells (in this case fibroblasts) have a finite number of divisions before they stop dividing [194], offered an excellent model for assessing senescence of the adaptive response. Indeed, high passage fibroblasts not only showed the classical decline in proliferation, and protein synthesis, but also decreased proteolytic capacity [35, 36, 126, 195–201]. Nor was this decline a result of lower amounts of the 20S Proteasome, as levels remained constant [35, 36, 202]. Since then, multiple studies using in vitro cellular senescence models, have revealed a similar decline in proteolytic capacity [202–204]. In addition, the decline in 20S proteolytic capacity holds true in primary human cell types from donors of increasing age [205, 206]. Moreover, fibroblasts from longer-lived species and centenarians show greater retention of 20S proteolytic capacity [207–209] and its regulator Nrf2 [210] into old-age, along with an associated elevation in the amount of the Immunoproteasome [208].
Direct assessment of protein clearance in tissues derived from aged animals reveals a similar decline in proteolytic capabilities. Specifically, human-derived tissues including heart [211], skin [212], and lymphocytes [213], exhibit a clear age-dependent loss of proteolytic capacity. A consistent ageing trend is also evident in tissues from mice [214, 215], rats [216, 217], and primates [208], and this finding is clearly accelerated in the disease states [218–224]. Additionally, studies in mice show a similar decline in Lon activity with age [124, 225], although much less work has focused upon age-associated proteolytic changes of the mitochondrial Lon protease. Together, these findings suggest a globally consistent and significant age-dependent attenuation of proteolytic activity and capacity.
From the evidence presented above, proponents of the Free Radical Theory of Ageing might well argue that the age-dependent loss in protein clearance is a consequence of damage accumulation, most likely arising from a lifetime’s worth of free radical and oxidant exposure. While we do not dispute the importance of free radicals and oxidative stress in ageing processes, we would like to suggest that more nuanced mechanisms are involved, and to offer a somewhat different interpretation. We suggest that the age-dependent decline in proteolytic capacity results from decreasing ability to mount appropriate and sufficient adaptive cellular responses. During ageing, the rate and extent of protein oxidation may not change, or it may actually increase, but cellular clearance of damaged proteins clearly declines [226, 227]. Importantly, however, this intracellular accumulation of aggregates of damaged (often cross-linked) proteins clearly is not a gradual process occurring over the course of a lifespan, but rather a phenomenon that characterizes the final third of life. This was very nicely demonstrated in a paper from Rodney Levine and Earl Stadtman who reviewed several studies, across multiple species, to show that intracellular oxidized protein aggregates do not accumulate during the first two thirds of lifespan but that such aggregates actively accumulate in an exponential fashion in the final third of life [228]. Interestingly, Harman also proposed exponential damage processes in ageing, but he suggested that they begin as early as the second third of life, after age 28, in persons living in Western societies [229].
As noted previously, one important factor in protein aggregation is the 20S Proteasome. Multiple findings suggest that it is not a lack of Proteasome, per se, but rather a diminished pool of functional Proteasome, which contributes to increased aggregation of oxidized proteins because they ‘escape’ degradation. In support of this interpretation, the Proteasome was identified as a major component of the ‘insolubolome,’ or insoluble protein aggregates [230]. Conversely, work in yeast, shows disaggregation of these protein clumps leads to increased proteolytic capacity [231].
Much of the work from our laboratory, and evidence found in the literature, has shown that ageing is associated with late-stage decline in activation of the adaptive response [134]. Unlike early passage cells, which demonstrate a clear response to adaptive signaling (i.e. Nrf2 transcriptional activation, stimulated proliferation, and increased proteolytic capacity), senescent cells show a clear failure or blockage of adaptive homeostasis. Indeed, pretreatment with normally stimulatory (non-damaging) low amounts of an oxidant in high passage cells, produces a sluggish (at best) induction of phase II enzymes and stress-responsive pathways [38, 232]. Moreover, this ageing phenotype could be mimicked if cells were exposed to Nrf2 inducers too frequently, or chronically [43]. Studies using primary human bronchial epithelial cells (HBE cells) from older individuals also exhibit an impaired Nr2-mediated response compared with HBE cells from young people. HBE cells from older individuals, for example, are very poor at inducing Nrf2 target genes such as GCLC, GCLM, and NQO1 following treatment with the known Nrf2 transcriptional activator sulforaphane, in comparison with HBE cells from young people [233].
Simpler, model organisms also show an age-related decline in the adaptive homeostatic response. Studies using aged C. elegans worms and D. melanogaster fruit flies, for example, have revealed loss of inducibility of the 20S Proteasome [44, 45] and the mitochondrial Lon protease with age [18]. In addition, aged worms and fruit flies, pretreated with a normally adaptive amount of an oxidant, show little or no increase in stress-resistance in comparison with their young counterparts who adapt very well [18, 44, 45], indicating an age-related loss in adaptation. The ability to modulate protein clearance in response to stress is also diminished in ageing. Whereas young animals, first pretreated with an adaptive amount of an oxidant and then subsequently challenged, are better able to mitigate accumulation of damaged proteins, aged pretreated animals exhibited no such increase in protein clearance [44, 45]. Additionally, studies using 21 month C57BL/6 male and female mice, exposed to low amounts of smog-derived nanoparticulates, showed loss of induction of Phase-II responsive enzymes, in comparison with their young counterparts who adapted very well to the exposures [47, 193].
Another major age-dependent factor that should be noted is that, in all systems so far studied, ageing is associated with an increase in the basal levels of stress-responsive enzymes. This trend is consistent from cell culture to mammalian mouse models [134]. Nor is simply forcing the expression of stress-responsive systems able to restore the adaptive response in old animals, as chronic over-expression of the C. elegans Nrf2 homolog, Skn-1, failed to restore adaptive homeostasis [44]. Similarly, chronic knockdown in D. melanogaster of the Nrf2 cytosolic inhibitor, Keap1, showed improved stress resistance, but did not remediate the adaptive response [45]. When tested in mice, it was again found that baseline levels of Nrf2 increased with age, as did the levels of many Nrf2-inducible gene products (e.g. GCLC, GCLM, NQO1, HO1, Proteasome , and Lon) representing an upward contraction of the basal homeostatic range. In the same old animals, however, adaptive homeostasis declined to negligible levels in comparison with their young counterparts [47, 193].
In search of possible transcriptional players involved in the decline of Nrf2 responsiveness with ageing, we have found Bach1 and c-Myc as potentially viable candidates [47, 193]. Bach1 has been understood for some time to act as a competitive inhibitor of Nrf2-mediated signal transduction by forming heterodimers with MafK and MafG proteins that bind to the EpRE/ARE response elements. Docking of Bach1 sterically blocks the binding of Nrf2, leading to the repression of EpRe/ARE-mediated gene expression and induction [52, 234, 235]. In addition, c-Myc has been revealed as something of a non-traditional Nrf2 inhibitor, by means that have yet to be fully revealed, but which may well involve enhanced Nrf2 nuclear turnover [236]. Importantly, basal levels of both Bach1 and c-Myc have been found to increase with age in mice, and in human HBE cells, with concomitant declines in inducibility of Nrf2-regulated genes [47, 52, 193]. Thus, Bach1 and c-Myc may well represent viable targets for ‘ageing intervention,’ although one must always consider the possibility that these Nrf2 ‘inhibitors’ increase with age for a positive reason, yet to be discerned.
Thus ageing may actually be considered as a quintessential example of the decline in adaptive homeostasis. Ageing results in a compression of the homeostatic range, with chronically high levels of expression of stress-resistant enzymes but, paradoxically, an actual decrease in stress protection. Furthermore, ageing results in a generalized loss of adaptive homeostasis, such that stress-protective systems and enzymes can no longer be appropriately induced by signaling pathways, such as Nrf2, that are highly effective protectants in young animals.
Conclusions
It would seem to us that, although frequently attacked and often badly wounded, the Free Radical Theory of Ageing continues to survive and even evolve. We would suggest, however, that simplistic interpretation or application of the Theory has led to confusion, and to unwarranted negative conclusions. Specifically, it now seems clear that one cannot actually ‘test’ the Theory by simply intervening in one or two biochemical or genetic pathways and expecting that no other adaptive or counterbalancing factors, enzymes, or systems will compensate over the course of a lifetime.
We now realize that multiple protective systems against free radicals and oxidative stress are highly regulated and modulated over periods of minutes to hours, days to weeks, and months to years. Thus, signal transduction pathways such the Keap1-Nrf2 system can respond within minutes to transiently up-regulate antioxidant defenses and damage repair systems. Conversely, repeated or chronic exposure to stresses may blunt long-term defense and repair capabilities [43]. Then again, other interventions, such as exercise training or dietary restriction [132, 134, 237] have been shown to exert longer-lasting (mostly positive) effects that are, nevertheless, still transitory because they depend on maintenance of training or dietary protocols.
Our new understanding of Adaptive Homeostasis [132, 134] now provides a conceptual and mechanistic framework within which to more fully appreciate the implications of the Free Radical Theory of Ageing. In young and middle-aged organisms, Adaptive Homeostasis is proposed as a mechanism by which the effects of variations in numerous environmental and metabolic stresses, including oxidant exposure, are minimized by expanding the range of protective capacities at need. In contrast, aged animals and humans, during the final third of lifespan, experience diminished or dysfunctional adaptive homeostatic capabilities and accumulate stress-induced structural and functional damage at exponential rates. Increased levels of Bach1 and c-Myc may be at least partly responsible for loss of adaptive efficacy in advanced age, due to inhibition of the Nrf2 signaling pathway. Whether or not declines in the effectiveness of other protective signaling pathways also contribute to the ageing phenotype remains to be seen. Viewed then as a facet of Adaptive Homeostasis, along with many other age-related abatements in stress-adaptability, the Free Radical Theory of Ageing once again appears both viable and robust.
Figure 1. Assessment of the Stress Response.

In studies assessing the ability of an organism to withstand an oxidative stress it is important to perturb the homeostatic set-point. (A) Stress resistance is commonly measured to assess tolerance. Biological models (yeast, mammalian cell culture, nematode worms, and fruit flies) are treated with a semi-lethal, high concentration of an oxidant to assess survivability or other markers of stress tolerance. One of the limitations of this approach is the overwhelming biological assault to the system, leading to extensive oxidative damage. Assessment of the adaptive stress response, enables an organism to first activate multiple protective pathways prior to being challenged with a semi-toxic concentration. (B) The adaptive response is induced in multiple biological systems (yeast, mammalian cell culture, nematode worms, and fruit flies) and is carried out by treating the organism with a non-damaging, signaling concentration of an oxidant to stimulate stress-inducible pathways (including those transcriptionally regulated by Nrf2). Once pathways are activated and upregulated, the organism is then challenged with a semi-lethal dose of an oxidant, and subsequently survival and other markers of stress activation are measured.
Figure 2. Age-Dependent Decline of the Adaptive Stress Response.

Early passage cells and young organisms are capable of rapidly and transiently upregulating the adaptive homeostatic response. In turn, keeping damage accumulation, such as protein aggregates, limited and cellular maintenance. However, with age, the inducibility of the adaptive response declines, which is accompanied with a parallel rise in protein aggregation, sluggish cellular signaling and basal rise in inflammation (‘inflammaging’).
Figure 3. Chronic Up- or Downregulation of Target Protein Does Not Extend Lifespan.

Chronic up- or downregulation of multiple stress-responsive proteins have shown little consistent impact upon lifespan across multiple biological models. Forced overexpression (OE) or suppression (RNAi), chronically throughout the lifespan, may limit the ability of transient modulation of target protein to appropriately respond to a stimulus. Instead, by continually upregulating or downregulating a target protein, creates a new physiological set-point, and may limit the dynamic stress-responsive capability within the cellular environment. Thus, many efforts for chronic changes in stress-responsive proteins have shown no impact upon lifespan.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vina J, Borras C, Miquel J. Theories of ageing. IUBMB life. 2007;59(4–5):249–254. doi: 10.1080/15216540601178067. [DOI] [PubMed] [Google Scholar]
- 2.Harman D. Aging: a theory based on free radical and radiation chemistry. 1955 doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 3.Gerschman R, et al. Oxygen poisoning and x-irradiation: a mechanism in common. Science. 1954;119(3097):623–626. doi: 10.1126/science.119.3097.623. [DOI] [PubMed] [Google Scholar]
- 4.Adam-Vizi V. Production of reactive oxygen species in brain mitochondria: contribution by electron transport chain and non–electron transport chain sources. Antioxidants & redox signaling. 2005;7(9–10):1140–1149. doi: 10.1089/ars.2005.7.1140. [DOI] [PubMed] [Google Scholar]
- 5.Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochemical Journal. 1973;134(3):707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress, and aging1. Free Radical Biology and Medicine. 2000;29(3–4):222–230. doi: 10.1016/s0891-5849(00)00317-8. [DOI] [PubMed] [Google Scholar]
- 7.Ott M, et al. Mitochondria, oxidative stress and cell death. Apoptosis. 2007;12(5):913–922. doi: 10.1007/s10495-007-0756-2. [DOI] [PubMed] [Google Scholar]
- 8.Foerster EC, et al. Peroxisomal fatty acid oxidation as detected by H2O2 production in intact perfused rat liver. Biochemical Journal. 1981;196(3):705–712. doi: 10.1042/bj1960705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poole B. Diffusion effects in the metabolism of hydrogen peroxide by rat liver peroxisomes. Journal of theoretical biology. 1975;51(1):149–167. doi: 10.1016/0022-5193(75)90145-9. [DOI] [PubMed] [Google Scholar]
- 10.Pomatto Laura CD, Raynes R, Davies Kelvin JA. The peroxisomal Lon protease LonP2 in aging and disease: functions and comparisons with mitochondrial Lon protease LonP1. Biological Reviews. 2016;92(2):739–753. doi: 10.1111/brv.12253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walker CL, et al. Redox Regulation of Homeostasis and Proteostasis in Peroxisomes. Physiological reviews. 2017;98(1):89–115. doi: 10.1152/physrev.00033.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fridovich I. Oxygen: how do we stand it? Medical Principles and Practice. 2013;22(2):131–137. doi: 10.1159/000339212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y, et al. Mice deficient in both Mn superoxide dismutase and glutathione peroxidase-1 have increased oxidative damage and a greater incidence of pathology but no reduction in longevity. Journals of Gerontology Series A: Biomedical Sciences and Medical Sciences. 2009;64(12):1212–1220. doi: 10.1093/gerona/glp132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Remmen H, et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiological Genomics. 2003;16(1):29–37. doi: 10.1152/physiolgenomics.00122.2003. [DOI] [PubMed] [Google Scholar]
- 15.Sohal RS, Orr WC. The redox stress hypothesis of aging. Free Radical Biology and Medicine. 2012;52(3):539–555. doi: 10.1016/j.freeradbiomed.2011.10.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang W, Hekimi S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS biology. 2010;8(12):e1000556. doi: 10.1371/journal.pbio.1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang TT, et al. Ubiquitous overexpression of CuZn superoxide dismutase does not extend life span in mice. Journals of Gerontology Series A: Biological and Medical Sciences. 2000;55(1):5. doi: 10.1093/gerona/55.1.b5. [DOI] [PubMed] [Google Scholar]
- 18.Pomatto LC, et al. The mitochondrial lon protease is required for age-specific and sex-specific adaptation to oxidative stress. Current Biology. 2017;27(1):1–15. doi: 10.1016/j.cub.2016.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pérez VI, et al. The overexpression of major antioxidant enzymes does not extend the lifespan of mice. Aging cell. 2009;8(1):73–75. doi: 10.1111/j.1474-9726.2008.00449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mele J, et al. Characterization of transgenic mice that overexpress both copper zinc superoxide dismutase and catalase. Antioxidants & redox signaling. 2006;8(3–4):628–638. doi: 10.1089/ars.2006.8.628. [DOI] [PubMed] [Google Scholar]
- 21.Beckman KB, Ames BN. The free radical theory of aging matures. Physiological reviews. 1998;78(2):547–581. doi: 10.1152/physrev.1998.78.2.547. [DOI] [PubMed] [Google Scholar]
- 22.Pacifici RE, Davies KJ. Protein, lipid and DNA repair systems in oxidative stress: the free-radical theory of aging revisited. Gerontology. 1991;37(1–3):166–180. doi: 10.1159/000213257. [DOI] [PubMed] [Google Scholar]
- 23.Lewis KN, et al. The naked mole-rat response to oxidative stress: just deal with it. Antioxidants & redox signaling. 2013;19(12):1388–1399. doi: 10.1089/ars.2012.4911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacRae SL, et al. DNA repair in species with extreme lifespan differences. Aging (Albany NY) 2015;7(12):1171. doi: 10.18632/aging.100866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blazej A, et al. High oxidative damage levels in the longest-living rodent, the naked mole rat. Aging Cell. 2006;5(6):463–471. doi: 10.1111/j.1474-9726.2006.00237.x. [DOI] [PubMed] [Google Scholar]
- 26.Brys K, Vanfleteren JR, Braeckman BP. Testing the rate-of-living/oxidative damage theory of aging in the nematode model Caenorhabditis elegans. Experimental Gerontology. 2007;42(9):845–851. doi: 10.1016/j.exger.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 27.Nagase M, et al. Increased oxidative stress and coenzyme Q10 deficiency in centenarians. Journal of Clinical Biochemistry and Nutrition. 2018:17–124. doi: 10.3164/jcbn.17-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cameron E, Pauling L. Supplemental ascorbate in the supportive treatment of cancer: Prolongation of survival times in terminal human cancer. Proceedings of the National Academy of Sciences. 1976;73(10):3685–3689. doi: 10.1073/pnas.73.10.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vina J, et al. The free radical theory of aging revisited: the cell signaling disruption theory of aging. Antioxidants & redox signaling. 2013;19(8):779–787. doi: 10.1089/ars.2012.5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller ER, 3rd, et al. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Ann Intern Med. 2005;142(1):37–46. doi: 10.7326/0003-4819-142-1-200501040-00110. [DOI] [PubMed] [Google Scholar]
- 31.Lee DH, et al. Does supplemental vitamin C increase cardiovascular disease risk in women with diabetes? Am J Clin Nutr. 2004;80(5):1194–200. doi: 10.1093/ajcn/80.5.1194. [DOI] [PubMed] [Google Scholar]
- 32.Davies JM, Lowry CV, Davies KJ. Transient adaptation to oxidative stress in yeast. Archives of Biochemistry and Biophysics. 1995;317(1):1–6. doi: 10.1006/abbi.1995.1128. [DOI] [PubMed] [Google Scholar]
- 33.Grune T, Reinheckel T, Davies K. Degradation of oxidized proteins in mammalian cells. The FASEB Journal. 1997;11(7):526–534. [PubMed] [Google Scholar]
- 34.Grune T, Reinheckel T, Davies KJ. Degradation of oxidized proteins in K562 human hematopoietic cells by proteasome. Journal of Biological Chemistry. 1996;271(26):15504–15509. doi: 10.1074/jbc.271.26.15504. [DOI] [PubMed] [Google Scholar]
- 35.Sitte N, et al. Protein oxidation and degradation during cellular senescence of human BJ fibroblasts: part II—aging of nondividing cells. The FASEB Journal. 2000;14(15):2503–2510. doi: 10.1096/fj.00-0210com. [DOI] [PubMed] [Google Scholar]
- 36.Sitte N, et al. Protein oxidation and degradation during cellular senescence of human BJ fibroblasts: part I—effects of proliferative senescence. The FASEB Journal. 2000;14(15):2495–2502. doi: 10.1096/fj.00-0209com. [DOI] [PubMed] [Google Scholar]
- 37.Ngo JK, Davies KJ. Mitochondrial Lon protease is a human stress protein. Free Radical Biology and Medicine. 2009;46(8):1042–1048. doi: 10.1016/j.freeradbiomed.2008.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ngo JK, et al. Impairment of lon-induced protection against the accumulation of oxidized proteins in senescent wi-38 fibroblasts. Journals of Gerontology Series A: Biomedical Sciences and Medical Sciences. 2011;66(11):1178–1185. doi: 10.1093/gerona/glr145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ngo JK, Pomatto LC, Davies KJ. Upregulation of the mitochondrial Lon Protease allows adaptation to acute oxidative stress but dysregulation is associated with chronic stress, disease, and aging. Redox biology. 2013;1(1):258–264. doi: 10.1016/j.redox.2013.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pomatto LC, Tower J, Davies KJ. Sexual Dimorphism and Aging Differentially Regulate Adaptive Homeostasis. The Journals of Gerontology: Series A. 2017 doi: 10.1093/gerona/glx083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pickering AM, et al. Nrf2-dependent induction of proteasome and Pa28aβ regulator are required for adaptation to oxidative stress. Journal of Biological Chemistry. 2012;287(13):10021–10031. doi: 10.1074/jbc.M111.277145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pickering AM, et al. A conserved role for the 20S proteasome and Nrf2 transcription factor in oxidative stress adaptation in mammals, Caenorhabditis elegans and Drosophila melanogaster. The Journal of Experimental Biology. 2013;216(4):543. doi: 10.1242/jeb.074757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pickering AM, et al. Oxidative stress adaptation with acute, chronic, and repeated stress. Free Radical Biology and Medicine. 2013;55:109–118. doi: 10.1016/j.freeradbiomed.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raynes R, et al. Aging and SKN-1-dependent Loss of 20S Proteasome Adaptation to Oxidative Stress in C. elegans. Journals of Gerontology Series A: Biomedical Sciences and Medical Sciences. 2016;72(2):143–151. doi: 10.1093/gerona/glw093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pomatto LC, et al. The age-and sex-specific decline of the 20s proteasome and the Nrf2/CncC signal transduction pathway in adaption and resistance to oxidative stress in Drosophila melanogaster. Aging (Albany NY) 2017;9(4):1153. doi: 10.18632/aging.101218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pomatto LC, et al. AGING ATTENUATES REDOX ADAPTIVE HOMEOSTASIS AND PROTEOSTASIS IN FEMALE MICE EXPOSED TO TRAFFIC-DERIVED NANOPARTICLES (‘VEHICULAR SMOG’) Free Radical Biology and Medicine. 2018 doi: 10.1016/j.freeradbiomed.2018.04.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang H, et al. Nrf2-regulated phase II enzymes are induced by chronic ambient nanoparticle exposure in young mice with age-related impairments. Free Radical Biology and Medicine. 2012;52(9):2038–2046. doi: 10.1016/j.freeradbiomed.2012.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Obata F, Fons CO, Gould AP. Early-life exposure to low-dose oxidants can increase longevity via microbiome remodelling in Drosophila. Nature communications. 2018;9(1):975. doi: 10.1038/s41467-018-03070-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cox AG, Winterbourn CC, Hampton MB. Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling. Biochemical Journal. 2010;425(2):313–325. doi: 10.1042/BJ20091541. [DOI] [PubMed] [Google Scholar]
- 50.Wood ZA, Poole LB, Karplus PA. Peroxiredoxin Evolution and the Regulation of Hydrogen Peroxide Signaling. Science. 2003;300(5619):650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 51.Sies H. Role of metabolic H2O2 generation Redox signaling and oxidative stress. Journal of Biological Chemistry. 2014;289(13):8735–8741. doi: 10.1074/jbc.R113.544635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang H, Davies KJ, Forman HJ. Oxidative stress response and Nrf2 signaling in aging. Free Radical Biology and Medicine. 2015;88:314–336. doi: 10.1016/j.freeradbiomed.2015.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ristow M, Schmeisser S. Extending life span by increasing oxidative stress. Free Radical Biology and Medicine. 2011;51(2):327–336. doi: 10.1016/j.freeradbiomed.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 54.Epel ES, Lithgow GJ. Stress biology and aging mechanisms: toward understanding the deep connection between adaptation to stress and longevity. Journals of Gerontology Series A: Biomedical Sciences and Medical Sciences. 2014;69(Suppl_1):S10–S16. doi: 10.1093/gerona/glu055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rollins JA, et al. Assessing health span in Caenorhabditis elegans: lessons from short-lived mutants. Journals of Gerontology Series A: Biomedical Sciences and Medical Sciences. 2017;72(4):473–480. doi: 10.1093/gerona/glw248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Labbadia J, et al. Mitochondrial stress restores the heat shock response and prevents proteostasis collapse during aging. Cell reports. 2017;21(6):1481–1494. doi: 10.1016/j.celrep.2017.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Longo VD, Gralla EB, Valentine JS. Superoxide dismutase activity is essential for stationary phase survival in Saccharomyces cerevisiae Mitochondrial production of toxic oxygen species in vivo. Journal of Biological Chemistry. 1996;271(21):12275–12280. doi: 10.1074/jbc.271.21.12275. [DOI] [PubMed] [Google Scholar]
- 58.Heinemeyer W, et al. Proteinase yscE, the yeast proteasome/multicatalytic-multifunctional proteinase: mutants unravel its function in stress induced proteolysis and uncover its necessity for cell survival. The EMBO Journal. 1991;10(3):555–562. doi: 10.1002/j.1460-2075.1991.tb07982.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kruegel U, et al. Elevated proteasome capacity extends replicative lifespan in Saccharomyces cerevisiae. PLoS genetics. 2011;7(9):e1002253. doi: 10.1371/journal.pgen.1002253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Erjavec N, et al. Deletion of the mitochondrial Pim1/Lon protease in yeast results in accelerated aging and impairment of the proteasome. Free Radical Biology and Medicine. 2013;56(Supplement C):9–16. doi: 10.1016/j.freeradbiomed.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 61.Suzuki CK, et al. Requirement for the yeast gene LON in intramitochondrial proteolysis and maintenance of respiration. Science. 1994;264(5156):273–277. doi: 10.1126/science.8146662. [DOI] [PubMed] [Google Scholar]
- 62.Mesquita A, et al. Caloric restriction or catalase inactivation extends yeast chronological lifespan by inducing H2O2 and superoxide dismutase activity. Proceedings of the National Academy of Sciences. 2010;107(34):15123–15128. doi: 10.1073/pnas.1004432107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kirby K, et al. RNA interference-mediated silencing of Sod2 in Drosophila leads to early adult-onset mortality and elevated endogenous oxidative stress. Proceedings of the National Academy of Sciences. 2002;99(25):16162–16167. doi: 10.1073/pnas.252342899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martin I, et al. Sod2 knockdown in the musculature has whole-organism consequences in Drosophila. Free Radical Biology and Medicine. 2009;47(6):803–813. doi: 10.1016/j.freeradbiomed.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Duttaroy A, et al. A Sod2 null mutation confers severely reduced adult life span in Drosophila. Genetics. 2003;165(4):2295–2299. doi: 10.1093/genetics/165.4.2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wicks S, et al. Hypoxia rescues early mortality conferred by superoxide dismutase deficiency. Free Radical Biology and Medicine. 2009;46(2):176–181. doi: 10.1016/j.freeradbiomed.2008.09.036. [DOI] [PubMed] [Google Scholar]
- 67.Melov S, et al. Extension of life-span with superoxide dismutase/catalase mimetics. Science. 2000;289(5484):1567–1569. doi: 10.1126/science.289.5484.1567. [DOI] [PubMed] [Google Scholar]
- 68.Van Dyck L, Pearce DA, Sherman F. PIM1 encodes a mitochondrial ATP-dependent protease that is required for mitochondrial function in the yeast Saccharomyces cerevisiae. Journal of Biological Chemistry. 1994;269(1):238–242. [PubMed] [Google Scholar]
- 69.Tsakiri EN, et al. Proteasome dysfunction in Drosophila signals to an Nrf2-dependent regulatory circuit aiming to restore proteostasis and prevent premature aging. Aging cell. 2013;12(5):802–813. doi: 10.1111/acel.12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Orr WC, Arnold LA, Sohal RS. Relationship between catalase activity, life span and some parameters associated with antioxidant defenses in Drosophila melanogaster. Mechanisms of Ageing and Development. 1992;63(3):287–296. doi: 10.1016/0047-6374(92)90006-y. [DOI] [PubMed] [Google Scholar]
- 71.Doonan R, et al. Against the oxidative damage theory of aging: superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans. Genes & development. 2008;22(23):3236–3241. doi: 10.1101/gad.504808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Van Raamsdonk JM, Hekimi S. Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans. PLoS genetics. 2009;5(2):e1000361. doi: 10.1371/journal.pgen.1000361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Leiers B, et al. A stress-responsive glutathione S-transferase confers resistance to oxidative stress in Caenorhabditis elegans. Free Radical Biology and Medicine. 2003;34(11):1405–1415. doi: 10.1016/s0891-5849(03)00102-3. [DOI] [PubMed] [Google Scholar]
- 74.Reis-Rodrigues P, et al. Proteomic analysis of age-dependent changes in protein solubility identifies genes that modulate lifespan. Aging cell. 2012;11(1):120–127. doi: 10.1111/j.1474-9726.2011.00765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ghazi A, Henis-Korenblit S, Kenyon C. Regulation of Caenorhabditis elegans lifespan by a proteasomal E3 ligase complex. Proceedings of the National Academy of Sciences. 2007;104(14):5947–5952. doi: 10.1073/pnas.0700638104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stout GJ, et al. Insulin/IGF-1-mediated longevity is marked by reduced protein metabolism. Molecular systems biology. 2013;9(1):679. doi: 10.1038/msb.2013.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.An JH, Blackwell TK. SKN-1 links C. elegans mesendodermal specification to a conserved oxidative stress response. Genes & development. 2003;17(15):1882–1893. doi: 10.1101/gad.1107803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lee SS, et al. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nature genetics. 2003;33(1):40–48. doi: 10.1038/ng1056. [DOI] [PubMed] [Google Scholar]
- 79.Van Remmen H, et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiological genomics. 2003;16(1):29–37. doi: 10.1152/physiolgenomics.00122.2003. [DOI] [PubMed] [Google Scholar]
- 80.Ran Q, et al. Reduction in glutathione peroxidase 4 increases life span through increased sensitivity to apoptosis. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences. 2007;62(9):932–942. doi: 10.1093/gerona/62.9.932. [DOI] [PubMed] [Google Scholar]
- 81.Salmon AB, et al. Lack of methionine sulfoxide reductase A in mice increases sensitivity to oxidative stress but does not diminish life span. The FASEB Journal. 2009;23(10):3601–3608. doi: 10.1096/fj.08-127415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pérez VI, et al. Is the oxidative stress theory of aging dead? Biochimica et Biophysica Acta (BBA) - General Subjects. 2009;1790(10):1005–1014. doi: 10.1016/j.bbagen.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yant LJ, et al. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radical Biology and Medicine. 2003;34(4):496–502. doi: 10.1016/s0891-5849(02)01360-6. [DOI] [PubMed] [Google Scholar]
- 84.Zelko IN, Mariani TJ, Folz RJ. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radical Biology and Medicine. 2002;33(3):337–349. doi: 10.1016/s0891-5849(02)00905-x. [DOI] [PubMed] [Google Scholar]
- 85.Kruegel U, et al. Elevated proteasome capacity extends replicative lifespan in Saccharomyces cerevisiae. PLoS Genet. 2011;7(9):e1002253. doi: 10.1371/journal.pgen.1002253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Harris N, et al. MnSOD overexpression extends the yeast chronological (G 0) life span but acts independently of Sir2p histone deacetylase to shorten the replicative life span of dividing cells. Free Radical Biology and Medicine. 2003;34(12):1599–1606. doi: 10.1016/s0891-5849(03)00210-7. [DOI] [PubMed] [Google Scholar]
- 87.Fabrizio P, et al. SOD2 functions downstream of Sch9 to extend longevity in yeast. Genetics. 2003;163(1):35–46. doi: 10.1093/genetics/163.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fabrizio P, et al. Regulation of longevity and stress resistance by Sch9 in yeast. Science. 2001;292(5515):288–290. doi: 10.1126/science.1059497. [DOI] [PubMed] [Google Scholar]
- 89.Fabrizio P, et al. Sir2 blocks extreme life-span extension. Cell. 2005;123(4):655–667. doi: 10.1016/j.cell.2005.08.042. [DOI] [PubMed] [Google Scholar]
- 90.Kapetanou M, et al. Proteasome activation enhances stemness and lifespan of human mesenchymal stem cells. Free Radical Biology and Medicine. 2017;103:226–235. doi: 10.1016/j.freeradbiomed.2016.12.035. [DOI] [PubMed] [Google Scholar]
- 91.Lu L, et al. Ameliorating replicative senescence of human bone marrow stromal cells by PSMB5 overexpression. Biochemical and biophysical research communications. 2014;443(4):1182–1188. doi: 10.1016/j.bbrc.2013.12.113. [DOI] [PubMed] [Google Scholar]
- 92.Hwang JS, et al. Age-associated decrease in proteasome content and activities in human dermal fibroblasts: restoration of normal level of proteasome subunits reduces aging markers in fibroblasts from elderly persons. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences. 2007;62(5):490–499. doi: 10.1093/gerona/62.5.490. [DOI] [PubMed] [Google Scholar]
- 93.Curtis C, et al. Transcriptional profiling of MnSOD-mediated lifespan extension in Drosophila reveals a species-general network of aging and metabolic genes. Genome biology. 2007;8(12):R262. doi: 10.1186/gb-2007-8-12-r262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sun J, et al. Induced overexpression of mitochondrial Mn-superoxide dismutase extends the life span of adult Drosophila melanogaster. Genetics. 2002;161(2):661–672. doi: 10.1093/genetics/161.2.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Parkes TL, et al. Extension of Drosophila lifespan by overexpression of human SOD1 in motorneurons. Nature genetics. 1998;19(2):171–174. doi: 10.1038/534. [DOI] [PubMed] [Google Scholar]
- 96.Sun J, Tower J. FLP recombinase-mediated induction of Cu/Zn-superoxide dismutase transgene expression can extend the life span of adult Drosophila melanogaster flies. Molecular and cellular biology. 1999;19(1):216–228. doi: 10.1128/mcb.19.1.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Aigaki T, Seong K-h, Matsuo T. Longevity determination genes in Drosophila melanogaster. Mechanisms of ageing and development. 2002;123(12):1531–1541. doi: 10.1016/s0047-6374(02)00089-1. [DOI] [PubMed] [Google Scholar]
- 98.Ayyadevara S, et al. Lifespan extension in hypomorphic daf-2 mutants of Caenorhabditis elegans is partially mediated by glutathione transferase CeGSTP2-2. Aging cell. 2005;4(6):299–307. doi: 10.1111/j.1474-9726.2005.00172.x. [DOI] [PubMed] [Google Scholar]
- 99.Lüersen K, et al. The glutathione reductase GSR-1 determines stress tolerance and longevity in Caenorhabditis elegans. PloS one. 2013;8(4):e60731. doi: 10.1371/journal.pone.0060731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cabreiro F, et al. Increased life span from overexpression of superoxide dismutase in Caenorhabditis elegans is not caused by decreased oxidative damage. Free Radical Biology and Medicine. 2011;51(8):1575–1582. doi: 10.1016/j.freeradbiomed.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chondrogianni N, et al. 20S proteasome activation promotes life span extension and resistance to proteotoxicity in Caenorhabditis elegans. The FASEB Journal. 2015;29(2):611–622. doi: 10.1096/fj.14-252189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Vilchez D, et al. RPN-6 determines C. elegans longevity under proteotoxic stress conditions. Nature. 2012;489(7415):263–268. doi: 10.1038/nature11315. [DOI] [PubMed] [Google Scholar]
- 103.Ladiges W, et al. Lifespan extension in genetically modified mice. Aging cell. 2009;8(4):346–352. doi: 10.1111/j.1474-9726.2009.00491.x. [DOI] [PubMed] [Google Scholar]
- 104.Schriner SE, et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308(5730):1909–11. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 105.Jang YC, et al. Overexpression of Mn superoxide dismutase does not increase life span in mice. Journals of Gerontology Series A: Biomedical Sciences and Medical Sciences. 2009;64(11):1114–1125. doi: 10.1093/gerona/glp100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jaarsma D, et al. Human Cu/Zn superoxide dismutase (SOD1) overexpression in mice causes mitochondrial vacuolization, axonal degeneration, and premature motoneuron death and accelerates motoneuron disease in mice expressing a familial amyotrophic lateral sclerosis mutant SOD1. Neurobiology of disease. 2000;7(6):623–643. doi: 10.1006/nbdi.2000.0299. [DOI] [PubMed] [Google Scholar]
- 107.Wang L, et al. Wild-type SOD1 overexpression accelerates disease onset of a G85R SOD1 mouse. Human molecular genetics. 2009;18(9):1642–1651. doi: 10.1093/hmg/ddp085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Quirós PM, et al. ATP-dependent Lon protease controls tumor bioenergetics by reprogramming mitochondrial activity. Cell reports. 2014;8(2):542–556. doi: 10.1016/j.celrep.2014.06.018. [DOI] [PubMed] [Google Scholar]
- 109.Tullet J, et al. The SKN 1/Nrf2 transcription factor can protect against oxidative stress and increase lifespan in C. elegans by distinct mechanisms. Aging Cell. 2017 doi: 10.1111/acel.12627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bishop NA, Guarente L. Two neurons mediate diet-restriction-induced longevity in C. elegans. Nature. 2007;447(7144):545–549. doi: 10.1038/nature05904. [DOI] [PubMed] [Google Scholar]
- 111.An JH, et al. Regulation of the Caenorhabditis elegans oxidative stress defense protein SKN-1 by glycogen synthase kinase-3. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(45):16275–16280. doi: 10.1073/pnas.0508105102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhu H, et al. Role of Nrf2 signaling in regulation of antioxidants and phase 2 enzymes in cardiac fibroblasts: protection against reactive oxygen and nitrogen species-induced cell injury. FEBS letters. 2005;579(14):3029–3036. doi: 10.1016/j.febslet.2005.04.058. [DOI] [PubMed] [Google Scholar]
- 113.Pearson KJ, et al. Nrf2 mediates cancer protection but not prolongevity induced by caloric restriction. Proceedings of the National Academy of Sciences. 2008;105(7):2325–2330. doi: 10.1073/pnas.0712162105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wakabayashi N, et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nature genetics. 2003;35(3):238–245. doi: 10.1038/ng1248. [DOI] [PubMed] [Google Scholar]
- 115.Watanabe K, et al. Superoxide dismutase 1 loss disturbs intracellular redox signaling, resulting in global age-related pathological changes. BioMed research international. 2014;2014 doi: 10.1155/2014/140165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Watanabe K, et al. Sod1 loss induces intrinsic superoxide accumulation leading to p53-mediated growth arrest and apoptosis. International journal of molecular sciences. 2013;14(6):10998–11010. doi: 10.3390/ijms140610998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Huang TT, et al. Superoxide-mediated cytotoxicity in superoxide dismutase-deficient fetal fibroblasts. Archives of biochemistry and biophysics. 1997;344(2):424–432. doi: 10.1006/abbi.1997.0237. [DOI] [PubMed] [Google Scholar]
- 118.Wanagat J, et al. Skeletal muscle mitochondrial DNA deletions are not increased in CuZn-superoxide dismutase deficient mice. Experimental Gerontology. 2015;61(Supplement C):15–19. doi: 10.1016/j.exger.2014.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhang Y, et al. A new role for oxidative stress in aging: The accelerated aging phenotype in Sod1−/− mice is correlated to increased cellular senescence. Redox biology. 2017;11:30–37. doi: 10.1016/j.redox.2016.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Homma T, et al. SOD1 deficiency decreases proteasomal function, leading to the accumulation of ubiquitinated proteins in erythrocytes. Archives of biochemistry and biophysics. 2015;583:65–72. doi: 10.1016/j.abb.2015.07.023. [DOI] [PubMed] [Google Scholar]
- 121.Deepa SS, et al. A new mouse model of frailty: the Cu/Zn superoxide dismutase knockout mouse. GeroScience. 2017;39(2):187–198. doi: 10.1007/s11357-017-9975-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Merry TL, et al. High-fat-fed obese glutathione peroxidase 1-deficient mice exhibit defective insulin secretion but protection from hepatic steatosis and liver damage. Antioxidants & redox signaling. 2014;20(14):2114–2129. doi: 10.1089/ars.2013.5428. [DOI] [PubMed] [Google Scholar]
- 123.Scheurmann J, et al. Mice with heterozygous deficiency of manganese superoxide dismutase (SOD2) have a skin immune system with features of “inflamm-aging”. Archives of dermatological research. 2014;306(2):143–155. doi: 10.1007/s00403-013-1389-7. [DOI] [PubMed] [Google Scholar]
- 124.Bota DA, Van Remmen H, Davies KJ. Modulation of Lon protease activity and aconitase turnover during aging and oxidative stress. FEBS letters. 2002;532(1–2):103–106. doi: 10.1016/s0014-5793(02)03638-4. [DOI] [PubMed] [Google Scholar]
- 125.Van Remmen H, et al. Multiple deficiencies in antioxidant enzymes in mice result in a compound increase in sensitivity to oxidative stress. Free Radical Biology and Medicine. 2004;36(12):1625–1634. doi: 10.1016/j.freeradbiomed.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 126.Bota DA, Ngo JK, Davies KJ. Downregulation of the human Lon protease impairs mitochondrial structure and function and causes cell death. Free Radical Biology and Medicine. 2005;38(5):665–677. doi: 10.1016/j.freeradbiomed.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 127.Maharjan S, et al. Mitochondrial impairment triggers cytosolic oxidative stress and cell death following proteasome inhibition. Scientific reports. 2014:4. doi: 10.1038/srep05896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Pulliam DA, et al. Complex IV-deficient Surf1−/− mice initiate mitochondrial stress responses. Biochemical Journal. 2014;462(2):359–371. doi: 10.1042/BJ20140291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Walter L, et al. The homeobox protein CEH-23 mediates prolonged longevity in response to impaired mitochondrial electron transport chain in C. elegans. PLoS biology. 2011;9(6):e1001084. doi: 10.1371/journal.pbio.1001084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Copeland JM, et al. Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Current Biology. 2009;19(19):1591–1598. doi: 10.1016/j.cub.2009.08.016. [DOI] [PubMed] [Google Scholar]
- 131.Liu X, et al. Evolutionary conservation of the clk-1-dependent mechanism of longevity: loss of mclk1 increases cellular fitness and lifespan in mice. Genes & development. 2005;19(20):2424–2434. doi: 10.1101/gad.1352905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Davies KJA. Adaptive homeostasis. Molecular Aspects of Medicine. 2016;49(Supplement C):1–7. doi: 10.1016/j.mam.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Honjoh S, et al. The Sexual Dimorphism of Dietary Restriction Responsiveness in Caenorhabditis elegans. Cell Reports. 2017;21(13):3646–3652. doi: 10.1016/j.celrep.2017.11.108. [DOI] [PubMed] [Google Scholar]
- 134.Pomatto LC, Davies KJ. The role of declining adaptive homeostasis in ageing. The Journal of Physiology. 2017 doi: 10.1113/JP275072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sallam N, Laher I. Exercise modulates oxidative stress and inflammation in aging and cardiovascular diseases. Oxidative medicine and cellular longevity. 2016;2016 doi: 10.1155/2016/7239639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Current biology. 2014;24(10):R453–R462. doi: 10.1016/j.cub.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Loboda A, et al. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cellular and Molecular Life Sciences. 2016;73(17):3221–3247. doi: 10.1007/s00018-016-2223-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zhou L, et al. Aging-related decline in the induction of Nrf2-regulated antioxidant genes in human bronchial epithelial cells. Redox biology. 2018;14:35–40. doi: 10.1016/j.redox.2017.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhou L, Zhang H, Forman HJ. Age related alteration of the antioxidant/inflammatory axis in human lung epithelial cells in response to nanoparticle challenge. Free Radical Biology and Medicine. 2017;112:59. [Google Scholar]
- 140.Gems D, Partridge L. Stress-response hormesis and aging:“that which does not kill us makes us stronger”. Cell metabolism. 2008;7(3):200–203. doi: 10.1016/j.cmet.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 141.Rattan SI. The future of aging interventions: aging intervention, prevention, and therapy through hormesis. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences. 2004;59(7):B705–B709. doi: 10.1093/gerona/59.7.b705. [DOI] [PubMed] [Google Scholar]
- 142.Calabrese EJ, et al. What is hormesis and its relevance to healthy aging and longevity? Biogerontology. 2015;16(6):693–707. doi: 10.1007/s10522-015-9601-0. [DOI] [PubMed] [Google Scholar]
- 143.Southam CM. Effects of extract of western red-cedar heartwood on certain wood-decaying fungi in culture. 1943 [Google Scholar]
- 144.Christman MF, Storz G, Ames BN. OxyR, a positive regulator of hydrogen peroxide-inducible genes in Escherichia coli and Salmonella typhimurium, is homologous to a family of bacterial regulatory proteins. Proceedings of the National Academy of Sciences. 1989;86(10):3484–3488. doi: 10.1073/pnas.86.10.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Demple B. Redox signaling and gene control in the Escherichia coli soxRS oxidative stress regulon—a review. Gene. 1996;179(1):53–57. doi: 10.1016/s0378-1119(96)00329-0. [DOI] [PubMed] [Google Scholar]
- 146.Wiese AG, Pacifici RE, Davies KJA. Transient Adaptation to Oxidative Stress in Mammalian Cells. Archives of Biochemistry and Biophysics. 1995;318(1):231–240. doi: 10.1006/abbi.1995.1225. [DOI] [PubMed] [Google Scholar]
- 147.Davies KJ. The broad spectrum of responses to oxidants in proliferating cells: a new paradigm for oxidative stress. IUBMB life. 1999;48(1):41–47. doi: 10.1080/713803463. [DOI] [PubMed] [Google Scholar]
- 148.Chen Z-H, et al. Adaptation to hydrogen peroxide enhances PC12 cell tolerance against oxidative damage. Neuroscience letters. 2005;383(3):256–259. doi: 10.1016/j.neulet.2005.04.022. [DOI] [PubMed] [Google Scholar]
- 149.Olsen A, Vantipalli MC, Lithgow GJ. Lifespan extension of Caenorhabditis elegans following repeated mild hormetic heat treatments. Biogerontology. 2006;7(4):221. doi: 10.1007/s10522-006-9018-x. [DOI] [PubMed] [Google Scholar]
- 150.Grune T, et al. Proteolysis in cultured liver epithelial cells during oxidative stress Role of the multicatalytic proteinase complex, proteasome. Journal of Biological Chemistry. 1995;270(5):2344–2351. doi: 10.1074/jbc.270.5.2344. [DOI] [PubMed] [Google Scholar]
- 151.Shringarpure R, et al. Ubiquitin conjugation is not required for the degradation of oxidized proteins by proteasome. Journal of Biological Chemistry. 2003;278(1):311–318. doi: 10.1074/jbc.M206279200. [DOI] [PubMed] [Google Scholar]
- 152.Grune T, Davies KJ. The proteasomal system and HNE-modified proteins. Molecular aspects of medicine. 2003;24(4):195–204. doi: 10.1016/s0098-2997(03)00014-1. [DOI] [PubMed] [Google Scholar]
- 153.Shringarpure R, Grune T, Davies K. Protein oxidation and 20S proteasome-dependent proteolysis in mammalian cells. Cellular and Molecular Life Sciences CMLS. 2001;58(10):1442–1450. doi: 10.1007/PL00000787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Davies KJ. Degradation of oxidized proteins by the 20S proteasome. Biochimie. 2001;83(3):301–310. doi: 10.1016/s0300-9084(01)01250-0. [DOI] [PubMed] [Google Scholar]
- 155.Reinheckel T, et al. Differential impairment of 20S and 26S proteasome activities in human hematopoietic K562 cells during oxidative stress. Archives of Biochemistry and Biophysics. 2000;377(1):65–68. doi: 10.1006/abbi.2000.1717. [DOI] [PubMed] [Google Scholar]
- 156.Pickering AM, et al. The immunoproteasome, the 20S proteasome and the PA28aβ proteasome regulator are oxidative-stress-adaptive proteolytic complexes. Biochemical Journal. 2010;432(3):585–595. doi: 10.1042/BJ20100878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Pickering AM, Davies KJ. Differential roles of proteasome and immunoproteasome regulators Pa28aβ, Pa28 and Pa200 in the degradation of oxidized proteins. Archives of biochemistry and biophysics. 2012;523(2):181–190. doi: 10.1016/j.abb.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Pickering AM, Davies KJ. Degradation of damaged proteins-the main function of the 20S proteasome. Progress in molecular biology and translational science. 2012;109:227. doi: 10.1016/B978-0-12-397863-9.00006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Raynes R, Pomatto LC, Davies KJ. Degradation of oxidized proteins by the proteasome: distinguishing between the 20S, 26S, and immunoproteasome proteolytic pathways. Molecular aspects of medicine. 2016;50:41–55. doi: 10.1016/j.mam.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Grune T, et al. Selective degradation of oxidatively modified protein substrates by the proteasome. Biochemical and biophysical research communications. 2003;305(3):709–718. doi: 10.1016/s0006-291x(03)00809-x. [DOI] [PubMed] [Google Scholar]
- 161.GRUNE T, et al. Ezrin turnover and cell shape changes catalyzed by proteasome in oxidatively stressed cells. The FASEB journal. 2002;16(12):1602–1610. doi: 10.1096/fj.02-0015com. [DOI] [PubMed] [Google Scholar]
- 162.Grune T, et al. HSP70 mediates dissociation and reassociation of the 26S proteasome during adaptation to oxidative stress. Free Radical Biology and Medicine. 2011;51(7):1355–1364. doi: 10.1016/j.freeradbiomed.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Reeg S, et al. The molecular chaperone Hsp70 promotes the proteolytic removal of oxidatively damaged proteins by the proteasome. Free Radical Biology and Medicine. 2016;99:153–166. doi: 10.1016/j.freeradbiomed.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wani PS, et al. Phosphorylation of the C-terminal tail of proteasome subunit a7 is required for binding of the proteasome quality control factor Ecm29. Scientific reports. 2016;6:27873. doi: 10.1038/srep27873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wang X, et al. Regulation of the 26S proteasome complex during oxidative stress. Sci Signal. 2010;3(151):ra88. doi: 10.1126/scisignal.2001232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wang X, et al. The proteasome-interacting Ecm29 protein disassembles the 26S proteasome in response to oxidative stress. Journal of Biological Chemistry. 2017;292(39):16310–16320. doi: 10.1074/jbc.M117.803619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Haratake K, et al. KIAA0368-deficiency affects disassembly of 26S proteasome under oxidative stress condition. The Journal of Biochemistry. 2016;159(6):609–618. doi: 10.1093/jb/mvw006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Jang J, et al. Nrf2, a Regulator of the Proteasome, Controls Self-Renewal and Pluripotency in Human Embryonic Stem Cells. STEM CELLS. 2014;32(10):2616–2625. doi: 10.1002/stem.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Desvergne A, et al. Circadian modulation of proteasome activity and accumulation of oxidized protein in human embryonic kidney HEK 293 cells and primary dermal fibroblasts. Free Radical Biology and Medicine. 2016;94(Supplement C):195–207. doi: 10.1016/j.freeradbiomed.2016.02.037. [DOI] [PubMed] [Google Scholar]
- 170.Johnston-Carey HK, Pomatto LC, Davies KJ. The immunoproteasome in oxidative stress, aging, and disease. Critical reviews in biochemistry and molecular biology. 2016;51(4):268–281. doi: 10.3109/10409238.2016.1172554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Basler M, Kirk CJ, Groettrup M. The immunoproteasome in antigen processing and other immunological functions. Current opinion in immunology. 2013;25(1):74–80. doi: 10.1016/j.coi.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 172.Grimm S, et al. Advanced-glycation-end-product-induced formation of immunoproteasomes: involvement of RAGE and Jak2/STAT1. Biochemical Journal. 2012;448(1):127–139. doi: 10.1042/BJ20120298. [DOI] [PubMed] [Google Scholar]
- 173.Seifert U, et al. Immunoproteasomes preserve protein homeostasis upon interferon-induced oxidative stress. Cell. 2010;142(4):613–624. doi: 10.1016/j.cell.2010.07.036. [DOI] [PubMed] [Google Scholar]
- 174.Chung CH, Goldberg AL. The product of the lon (capR) gene in Escherichia coli is the ATP-dependent protease, protease La. Proceedings of the National Academy of Sciences. 1981;78(8):4931–4935. doi: 10.1073/pnas.78.8.4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Chin DT, et al. Sequence of the lon gene in Escherichia coli. A heat-shock gene which encodes the ATP-dependent protease La. Journal of Biological Chemistry. 1988;263(24):11718–11728. [PubMed] [Google Scholar]
- 176.Wang N, et al. A human mitochondrial ATP-dependent protease that is highly homologous to bacterial Lon protease. Proceedings of the National Academy of Sciences. 1993;90(23):11247–11251. doi: 10.1073/pnas.90.23.11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Bota DA, Davies KJ. Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP-stimulated mechanism. Nature cell biology. 2002;4(9):674–680. doi: 10.1038/ncb836. [DOI] [PubMed] [Google Scholar]
- 178.Waxman L, Goldberg AL. Protease La, the lon gene product, cleaves specific fluorogenic peptides in an ATP-dependent reaction. Journal of Biological Chemistry. 1985;260(22):12022–12028. [PubMed] [Google Scholar]
- 179.Cha SS, et al. Crystal structure of Lon protease: molecular architecture of gated entry to a sequestered degradation chamber. The EMBO journal. 2010;29(20):3520–3530. doi: 10.1038/emboj.2010.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Gibellini L, et al. Silencing of mitochondrial Lon protease deeply impairs mitochondrial proteome and function in colon cancer cells. The FASEB Journal. 2014;28(12):5122–5135. doi: 10.1096/fj.14-255869. [DOI] [PubMed] [Google Scholar]
- 181.Cheng C, et al. Overexpression of Lon contributes to survival and aggressive phenotype of cancer cells through mitochondrial complex I-mediated generation of reactive oxygen species. Cell death & disease. 2013;4(6):e681. doi: 10.1038/cddis.2013.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Bernstein SH, et al. The mitochondrial ATP-dependent Lon protease: a novel target in lymphoma death mediated by the synthetic triterpenoid CDDO and its derivatives. Blood. 2012;119(14):3321–3329. doi: 10.1182/blood-2011-02-340075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Hori O, et al. Transmission of cell stress from endoplasmic reticulum to mitochondria. The Journal of cell biology. 2002;157(7):1151–1160. doi: 10.1083/jcb.200108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Fukuda R, et al. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129(1):111–122. doi: 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- 185.Matsushima Y, Goto Y-i, Kaguni LS. Mitochondrial Lon protease regulates mitochondrial DNA copy number and transcription by selective degradation of mitochondrial transcription factor A (TFAM) Proceedings of the National Academy of Sciences. 2010;107(43):18410–18415. doi: 10.1073/pnas.1008924107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Lu B, et al. Phosphorylation of human TFAM in mitochondria impairs DNA binding and promotes degradation by the AAA+ Lon protease. Molecular cell. 2013;49(1):121–132. doi: 10.1016/j.molcel.2012.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Thomas RE, et al. PINK1-Parkin pathway activity is regulated by degradation of PINK1 in the mitochondrial matrix. PLoS genetics. 2014;10(5):e1004279. doi: 10.1371/journal.pgen.1004279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Holzenberger M, et al. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421(6919):182–187. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- 189.Fontana L, Partridge L, Longo VD. Extending healthy life span—from yeast to humans. science. 2010;328(5976):321–326. doi: 10.1126/science.1172539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Partridge L, Piper MD, Mair W. Dietary restriction in Drosophila. Mechanisms of ageing and development. 2005;126(9):938–950. doi: 10.1016/j.mad.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 191.Shah K, McCormack CE, Bradbury NA. Do you know the sex of your cells? American Journal of Physiology-Cell Physiology. 2014;306(1):C3–C18. doi: 10.1152/ajpcell.00281.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Pomatto LC, et al. Sexual Dimorphism in Oxidant-Induced Adaptive Homeostasis in Multiple Wild-Type D. melanogaster Strains. Archives in Biochemistry and Biophysics. 2017;636:57–70. doi: 10.1016/j.abb.2017.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Pomatto LC, et al. Aging Attenuates Adaptive Homeostasis and Proteostasis in Female Mice Exposed to Traffic- Free Radical Biology and Medicine. doi: 10.1016/j.freeradbiomed.2018.04.574. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Hayflick L. THE LIMITED IN VITRO LIFETIME OF HUMAN DIPLOID CELL STRAINS. Exp Cell Res. 1965;37:614–36. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- 195.Grune T, et al. Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and ‘aggresomes’ during oxidative stress, aging, and disease. The international journal of biochemistry & cell biology. 2004;36(12):2519–2530. doi: 10.1016/j.biocel.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 196.Sitte N, et al. Proteasome inhibition by lipofuscin/ceroid during postmitotic aging of fibroblasts. The FASEB Journal. 2000;14(11):1490–1498. doi: 10.1096/fj.14.11.1490. [DOI] [PubMed] [Google Scholar]
- 197.Petropoulos I, et al. Increase of oxidatively modified protein is associated with a decrease of proteasome activity and content in aging epidermal cells. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences. 2000;55(05):B220–B227. doi: 10.1093/gerona/55.5.b220. [DOI] [PubMed] [Google Scholar]
- 198.Ott C, et al. Reduced autophagy leads to an impaired ferritin turnover in senescent fibroblasts. Free Radical Biology and Medicine. 2016;101(Supplement C):325–333. doi: 10.1016/j.freeradbiomed.2016.10.492. [DOI] [PubMed] [Google Scholar]
- 199.Capasso S, et al. Changes in autophagy, proteasome activity and metabolism to determine a specific signature for acute and chronic senescent mesenchymal stromal cells. Oncotarget. 2015;6(37):39457–39468. doi: 10.18632/oncotarget.6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kapetanou M, et al. Proteasome activation enhances stemness and lifespan of human mesenchymal stem cells. Free Radical Biology and Medicine. 2017;103(Supplement C):226–235. doi: 10.1016/j.freeradbiomed.2016.12.035. [DOI] [PubMed] [Google Scholar]
- 201.Sander CS, et al. Photoaging is associated with protein oxidation in human skin in vivo. Journal of Investigative Dermatology. 2002;118(4):618–625. doi: 10.1046/j.1523-1747.2002.01708.x. [DOI] [PubMed] [Google Scholar]
- 202.Li Y, et al. Alterations of activity and intracellular distribution of the 20S proteasome in ageing retinal pigment epithelial cells. Experimental gerontology. 2008;43(12):1114–1122. doi: 10.1016/j.exger.2008.08.052. [DOI] [PubMed] [Google Scholar]
- 203.Merker K, Grune T. Proteolysis of oxidised proteins and cellular senescence. Experimental Gerontology. 2000;35(6):779–786. doi: 10.1016/s0531-5565(00)00140-6. [DOI] [PubMed] [Google Scholar]
- 204.Cavinato M, et al. UVB-induced senescence of human dermal fibroblasts involves impairment of proteasome and enhanced autophagic activity. The Journals of Gerontology: Series A. 2017;72(5):632–639. doi: 10.1093/gerona/glw150. [DOI] [PubMed] [Google Scholar]
- 205.Caballero M, et al. Effects of donor age on proteasome activity and senescence in trabecular meshwork cells. Biochemical and biophysical research communications. 2004;323(3):1048–1054. doi: 10.1016/j.bbrc.2004.08.195. [DOI] [PubMed] [Google Scholar]
- 206.Serrano R, et al. Linked p-FOXO4, PSMD11, and proteasomal function deficiency in human oa chondrocytes drives differentiation loss. Osteoarthritis and Cartilage. 2017;25:S170. [Google Scholar]
- 207.Rodriguez KA, et al. Altered composition of liver proteasome assemblies contributes to enhanced proteasome activity in the exceptionally long-lived naked mole-rat. PloS one. 2012;7(5):e35890. doi: 10.1371/journal.pone.0035890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Pickering AM, Lehr M, Miller RA. Lifespan of mice and primates correlates with immunoproteasome expression. The Journal of Clinical Investigation. 2015;125(5):2059–2068. doi: 10.1172/JCI80514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Chondrogianni N, et al. Fibroblast cultures from healthy centenarians have an active proteasome. Experimental gerontology. 2000;35(6):721–728. doi: 10.1016/s0531-5565(00)00137-6. [DOI] [PubMed] [Google Scholar]
- 210.Lewis KN, et al. Regulation of Nrf2 signaling and longevity in naturally long-lived rodents. Proceedings of the National Academy of Sciences. 2015;112(12):3722–3727. doi: 10.1073/pnas.1417566112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Bulteau A-L, Szweda LI, Friguet B. Age-dependent declines in proteasome activity in the heart. Archives of biochemistry and biophysics. 2002;397(2):298–304. doi: 10.1006/abbi.2001.2663. [DOI] [PubMed] [Google Scholar]
- 212.Bulteau AL, Petropoulos I, Friguet B. Age-related alterations of proteasome structure and function in aging epidermis. Experimental Gerontology. 2000;35(6):767–777. doi: 10.1016/s0531-5565(00)00136-4. [DOI] [PubMed] [Google Scholar]
- 213.Carrard G, et al. Impact of ageing on proteasome structure and function in human lymphocytes. The International Journal of Biochemistry & Cell Biology. 2003;35(5):728–739. doi: 10.1016/s1357-2725(02)00356-4. [DOI] [PubMed] [Google Scholar]
- 214.Breusing N, et al. Inverse correlation of protein oxidation and proteasome activity in liver and lung. Mechanisms of Ageing and Development. 2009;130(11):748–753. doi: 10.1016/j.mad.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 215.Baehr LM, et al. Age-related deficits in skeletal muscle recovery following disuse are associated with neuromuscular junction instability and ER stress, not impaired protein synthesis. Aging (Albany NY) 2016;8(1):127–146. doi: 10.18632/aging.100879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Gohlke S, et al. Molecular alterations in proteasomes of rat liver during aging result in altered proteolytic activities. AGE. 2014;36(1):57–72. doi: 10.1007/s11357-013-9543-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Dasuri K, et al. Aging and dietary restriction alter proteasome biogenesis and composition in the brain and liver. Mechanisms of ageing and development. 2009;130(11):777–783. doi: 10.1016/j.mad.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Scott MR, et al. Protein expression of proteasome subunits in elderly patients with schizophrenia. Neuropsychopharmacology. 2016;41(3):896–905. doi: 10.1038/npp.2015.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Seo H, Sonntag KC, Isacson O. Generalized brain and skin proteasome inhibition in Huntington's disease. Annals of neurology. 2004;56(3):319–328. doi: 10.1002/ana.20207. [DOI] [PubMed] [Google Scholar]
- 220.van Rijt SH, et al. Acute cigarette smoke exposure impairs proteasome function in the lung. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2012;303(9):L814–L823. doi: 10.1152/ajplung.00128.2012. [DOI] [PubMed] [Google Scholar]
- 221.Collier TJ, Kanaan NM, Kordower JH. Ageing as a primary risk factor for Parkinson's disease: evidence from studies of non-human primates. Nat Rev Neurosci. 2011;12(6):359–366. doi: 10.1038/nrn3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.St McNaught PK, et al. Altered Proteasomal Function in Sporadic Parkinson's Disease. Experimental Neurology. 2003;179(1):38–46. doi: 10.1006/exnr.2002.8050. [DOI] [PubMed] [Google Scholar]
- 223.Agholme L, et al. Proteasome inhibition induces stress kinase dependent transport deficits— Implications for Alzheimer's disease. Molecular and Cellular Neuroscience. 2014;58:29–39. doi: 10.1016/j.mcn.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 224.Bonet-Costa V, Pomatto LC-D, Davies KJ. The Proteasome and Oxidative Stress in Alzheimer's Disease. Antioxidants & redox signaling. 2016;25(16):886–901. doi: 10.1089/ars.2016.6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Bakala H, et al. Changes in rat liver mitochondria with aging. The FEBS Journal. 2003;270(10):2295–2302. doi: 10.1046/j.1432-1033.2003.03598.x. [DOI] [PubMed] [Google Scholar]
- 226.Grune T, et al. Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and ‘aggresomes’ during oxidative stress, aging, and disease. The International Journal of Biochemistry & Cell Biology. 2004;36(12):2519–2530. doi: 10.1016/j.biocel.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 227.SITTE N, et al. Proteasome inhibition by lipofuscin/ceroid during postmitotic aging of fibroblasts. The FASEB Journal. 2000;14(11):1490–1498. doi: 10.1096/fj.14.11.1490. [DOI] [PubMed] [Google Scholar]
- 228.Levine RL, Stadtman ER. Oxidative modification of proteins during aging. Experimental gerontology. 2001;36(9):1495–1502. doi: 10.1016/s0531-5565(01)00135-8. [DOI] [PubMed] [Google Scholar]
- 229.Harman D. Free radical theory of aging: an update. Annals of the New York Academy of Sciences. 2006;1067(1):10–21. doi: 10.1196/annals.1354.003. [DOI] [PubMed] [Google Scholar]
- 230.Reis-Rodrigues P, et al. Proteomic analysis of age-dependent changes in protein solubility identifies genes that modulate lifespan. Aging Cell. 2012;11(1):120–127. doi: 10.1111/j.1474-9726.2011.00765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Andersson V, et al. Enhancing protein disaggregation restores proteasome activity in aged cells. Aging (Albany NY) 2013;5(11):802–812. doi: 10.18632/aging.100613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Jung T, et al. Age-related differences in oxidative protein-damage in young and senescent fibroblasts. Archives of Biochemistry and Biophysics. 2009;483(1):127–135. doi: 10.1016/j.abb.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 233.Zhou L, et al. Aging-related decline in the induction of Nrf2-regulated antioxidant genes in human bronchial epithelial cells. Redox Biology. 2018;14(Supplement C):35–40. doi: 10.1016/j.redox.2017.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Sun J, et al. Heme regulates the dynamic exchange of Bach1 and NF-E2-related factors in the Maf transcription factor network. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(6):1461–1466. doi: 10.1073/pnas.0308083100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Dhakshinamoorthy S, et al. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD (P) H: quinone oxidoreductase 1 gene expression and induction in response to antioxidants. Journal of Biological Chemistry. 2005;280(17):16891–16900. doi: 10.1074/jbc.M500166200. [DOI] [PubMed] [Google Scholar]
- 236.Levy S, Forman HJ. C-Myc is a Nrf2-interacting protein that negatively regulates phase II genes through their electrophile responsive elements. IUBMB life. 2010;62(3):237–246. doi: 10.1002/iub.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Sachdev S, Davies KJ. Production, detection, and adaptive responses to free radicals in exercise. Free Radical Biology and Medicine. 2008;44(2):215–223. doi: 10.1016/j.freeradbiomed.2007.07.019. [DOI] [PubMed] [Google Scholar]