

Abstract
Lipid peroxidation (LPO) products are relatively stable and abundant metabolites, which accumulate in tissues of mammals with aging, being able to modify all cellular nucleophiles, creating protein and DNA adducts including crosslinks. Here, we used cells and mice deficient in the ERCC1-XPF endonuclease required for nucleotide excision repair and the repair of DNA interstrand crosslinks to ask if specifically LPO-induced DNA damage contributes to loss of cell and tissue homeostasis. Ercc1−/− mouse embryonic fibroblasts were more sensitive than wild-type (WT) cells to the LPO products: 4-hydroxy-2-nonenal (HNE), crotonaldehyde and malondialdehyde. ERCC1-XPF hypomorphic mice were hypersensitive to CCl4 and a diet rich in polyunsaturated fatty acids, two potent inducers of endogenous LPO. To gain insight into the mechanism of how LPO influences DNA repair-deficient cells, we measured the impact of the major endogenous LPO product, HNE, on WT and Ercc1−/− cells. HNE inhibited proliferation, stimulated ROS and LPO formation, induced DNA base damage, strand breaks, error-prone translesion DNA synthesis and cellular senescence much more potently in Ercc1−/− cells than in DNA repair-competent control cells. HNE also deregulated base excision repair and energy production pathways. Our observations that ERCC1-deficient cells and mice are hypersensitive to LPO implicates LPO-induced DNA damage in contributing to cellular demise and tissue degeneration, notably even when the source of LPO is dietary polyunsaturated fats.
Keywords: ERCC1-XPF, progeria, lipid peroxidation, 4-hydroxynonenal, senescence, DNA damage, aging
Graphical abstract
1. INTRODUCTION
Oxidative damage to lipids (lipid peroxidation or LPO) is a subject of extensive studies since LPO propagates damage by yielding products that further react with DNA and proteins [1]. The level of LPO products increases with aging in animals and humans [2, 3]. LPO is also elevated in brain matter of patients with neurodegeneration and other age-related diseases [4, 5]. LPO products also accumulate in senescent cells [6]. Thus, there is a strong correlation between LPO, senescence and aging, but full mechanism of LPO contribution to aging is not known.
Polyunsaturated fatty acids (PUFA) are particularly susceptible to oxidative stress-triggered peroxidation, giving rise to divergent products, among which the major are 4-hydroxy-2-nonenal (HNE), malondialdehyde (MDA), crotonaldehyde (CRO), and acrolein (ACR) [7]. These compounds are relatively stable compared to free radicals and can diffuse throughout the cell, attacking other biomolecules, like nucleic acids, proteins and cellular thiols. Thus, they are considered mediators of the toxic effects elicited by oxidative stress.
Concentration of LPO products in the cell is regulated by coordinated function of enzymes, like glutathione-S-transferase (GST), alcohol dehydrogenase (ADH) or aldehyde dehydrogenase (ALDH) [8]. However, the remaining pool of non-detoxified LPO products can react with cellular macromolecules forming: (i) structurally divergent adducts to DNA bases, (ii) adducts to proteins at several amino acids side chains, and due to having more than one reactive group, (iii) DNA-DNA interstrand crosslinks (ICLs), (iv) DNA-protein crosslinks and (v) crosslinks within or between proteins [1, 9–11].
Within the cell the biological effects of LPO products are a mix of effects on DNA and proteins. For example, HNE adducts to DNA bases inhibit replication by prokaryotic and eukaryotic DNA polymerases, as well as transcription in vitro, both by T7 RNA polymerase and by a HeLa cell-free extract, implying that in vivo these lesions could be preferentially removed from the transcribed DNA strand by the TCR (transcription-coupled repair) system [12, 13]. HNE-dG adducts also induce point mutations [12, 14, 15] and are weak stimulators of sister chromatid exchanges (SCEs) in wild-type cells and much stronger in TCR mutants [13]. The major repair/tolerance systems engaged in the cellular response to HNE adducts is NER [14, 15], followed by homologous recombination (HR), and translesion synthesis (TLS) by alternative DNA polymerases [16]. In mammalian cells error-free replication through the HNE-dG adducts is possible by the sequential action of TLS polymerases, Polι and Polκ, in which Polι incorporates correct nucleotide opposite the lesion site and Polκ performs the extension reaction [17]. Unrepaired HNE-DNA adducts induce apoptosis [18]. Interestingly, LPO products at high concentrations were shown to inhibit NER [19] and base excision repair enzymes (BER) [20]. Therefore LPO products should be considered not only as a source of DNA lesions, but also as deregulators of DNA repair pathways.
ERCC1-XPF endonuclease complex is essential for the incision of damaged DNA strand 5′ to the lesion in nucleotide excision repair (NER) as well as the unhooking of DNA strands during repair of interstrand crosslinks (ICLs) [21, 22]. Humans and mice deficient in ERCC1-XPF exhibit symptoms of premature aging and/or developmental abnormalities [23, 24]. Cells derived from affected individuals and mice can be a useful tool in studies of aging and neurodegeneration. There are two models of ERCC1-XPF deficiency, a complete knock-out (Ercc1−/−) and a hypomorphic mutant (Ercc1−/∆), which expresses ~5% of the normal complement of ERCC1-XPF. Ercc1−/− mice live only about four weeks, whereas Ercc1−/∆ have a lifespan of 7 months [21]. Both strains exhibit numerous progeroid symptoms, like premature aging of the hepatobiliary, renal, ocular, neurological, hematopoietic, musculoskeletal, epidermal and endocrine systems [25–29]. ERCC1-deficient mice reflect a human progeroid syndrome caused by mutations in XPF, affecting expression of ERCC1-XPF [24], demonstrating the relevance of these models to human health.
We reasoned that if LPO-induced DNA damage elicits a biological effect, then ERCC1-XPF-deficient cells/organisms should be hypersensitive to LPO products. Genetic depletion of ERCC1-XPF causes a defect in not only NER but also ICL repair, as XPF is also called FANCQ [30, 31], i.e., an enzyme required for the Fanconi anemia pathway of ICL repair.
It was previously shown that mice deficient in one of Fanconi anemia proteins, FANCD2 and aldehyde dehydrogenase 2 (ALDH2), which detoxifies endogenously formed acetaldehyde, display developmental defects, a predisposition to leukemia, and are susceptible to the toxic effects of ethanol, an exogenous source of acetaldehyde [32]. Deficiency of FANCD2 and another isoform of aldehyde dehydrogenase, ADH5, that removes endogenously formed formaldehyde, leads to hematopoietic stem cells depletion, bone marrow failure, karyomegaly and renal damage [33]. Formaldehyde and acetaldehyde are ubiquitous, abundant, highly reactive molecules that are normal byproducts of metabolism now implicated in driving developmental defects and cancer [34, 35].
Here we show that Ercc1−/− cells are more sensitive to long and medium-chain lipid peroxidation products than their wild-type counterparts, and that these aldehydes induce ROS production, cellular senescence and inhibition of Ercc1−/− cellular proliferation. We also show that chronic exposure of Ercc1−/∆ mice with either a sub-lethal dose of CCl4, a known LPO inducer, or a diet enriched in poly-unsaturated fatty acids accelerated the onset of aging-related pathologies and reduced lifespan. These data support the conclusion that LPO can promote aging and does so, at least in part, by inducing DNA and protein damage. Using primary and immortalized MEFs model, we propose the mechanism by which LPO products contribute to the phenotype of ERCC1-deficient animals.
2. MATERIALS AND METHODS
2.2.Chemicals
4-Hydroxy-2-nonenal was synthesized with some minor modifications as described previously [36]. Crotonaldehyde, acrolein, camptothecin, cisplatin, doxorubicin and hydrogen peroxide were purchased from Sigma-Aldrich (Saint Louis, MO). Fresh stocks of malondialdehyde were prepared by acid hydrolysis of 1,1,3,3-tetramethoxypropane (TMP) (Merck Millipore, Darmstad, Germany) as described previously [37].
2.3.Cell culture
The following cell lines were used in this study: (i) wild-type (WT) and Ercc1−/− primary mouse embryonic fibroblasts (MEFs), (ii) Ercc1−/− and control WT SV-40 immortalized MEFs, (iii) Xpa−/− and control WT SV-40 immortalized MEFs. Xpa−/− cell line and its corresponding WT control cell line were a kind gift from Prof. Leon H. F. Mullenders, Leiden University Medical Center, The Netherlands. Primary mouse embryonic fibroblasts (MEFs) were developed independently several times from 13.5-day embryos, as previously described [24] and maintained in a 1:1 mixture of Dulbecco’s modified Eagle’s medium and Ham’s F10 supplemented with 10% fetal bovine serum (HyClone by Thermo Scientific, Waltham, MA), non-essential amino acids (Gibco by Life Technologies, Carlsbad, CA) and penicillin-streptomycin (Gibco). The same medium was used for culturing immortalized MEFs. Cells were grown under standard culture conditions (37°C, 5% CO2) in a humidified incubator with an atmospheric oxygen (immortalized MEFs) or 3% oxygen (primary MEFs).
2.4. Cell viability and proliferation assays
Relative cell viability and proliferation were estimated using the alamarBlue® assay (Invitrogen by Life Technologies, Carlsbad, CA). Immortalized MEFs were seeded in 96-well plate at density of 3,000 cells per well, 24 h before treatment. Treatment with hydrogen peroxide (250-1000 μM), 4-hydroxy-2-nonenal (10-40 μM), malondialdehyde (500-2000 μM), crotonaldehyde (60-240 μM) or acrolein (5-20 μM) was performed for 2 h in serum-free medium for WT and Ercc1−/− cells. Untreated controls were maintained in serum-free medium for the same time. Subsequently, media containing test compounds was removed and fresh growth medium was added together with alamarBlue reagent in an amount equal to 10% of the total volume. Fluorescence intensity was measured at 540 nm excitation and 590 nm emission wavelength on a Synergy HT microplate reader (BioTek, Winooski, VT). Relative cell viability was verified 48 h after treatment and expressed as a ratio of fluorescence values per treatment to fluorescence values in untreated controls. Proliferation of WT and Ercc1−/− fibroblasts exposed to 20 μM HNE was measured 24, 48, 72, 96 and 120 h after treatment and plotted using relative fluorescence units (RFU). At least three independent experiments of viability and proliferation were conducted.
2.5. Measurement of reactive oxygen species
To detect and quantify reactive oxygen species (ROS) induced within cells in response to HNE and MDA CellROX Green Reagent (Invitrogen) was used. This reagent contains a dye, which is weakly fluorescent in a reduced state, but upon oxidation by ROS exhibits green fluorescence and binds to DNA. The assay was conducted following the manufacturer’s protocol. Immortalized MEFs were seeded in a 24-well culture plate, at density of 1.8×104 cells per well and following 24 h were treated with HNE or MDA, as well as a positive control tert-butyl-hydroperoxide (TBHP). MEFs were incubated with the test compounds for 2.5 h after which, the cultures were supplemented with CellROX (5 μM) and DAPI (0.1% (v/v) (Sigma-Aldrich), and imaged on an IX81 Olympus fluorescent microscope with 40× objective. Images were analyzed using ImageJ software. Results were presented as the percent cells that were ROS-positive. At least three independent experiments were performed.
2.6. Measurement of 8-oxo-deoxyguanosine
DNA extraction and DNA hydrolysis to deoxynucleosides was performed using the method described earlier [38, 39]. The Ultra Performance Liquid Chromatography with Tandem Mass Spectrometry (2D-UPLC-MS/MS) analyses were performed as previously describe [38].
2.7. Detection of caspases 3 and 7 activity
To estimate the level of apoptosis, the activity of caspases 3 and 7 was measured by Caspase-Glo 3/7 assay (Promega, Fitchburg, WI). Caspase-Glo 3/7 reagent causes cell lysis, and liberates cellular caspases which cleave luminogenic substrate containing luciferase. Luminescence is proportional to caspase activity. The assay was performed following the manufacturer’s instructions. Cells treatment with HNE or camptothecin (positive control) was carried out for 2 h. Untreated controls were maintained in serum-free medium for the same amount of time. The luminescence was measured on DTX 880 microplate reader (Beckman Coulter, Brea, CA). Luminescence values of untreated controls were used as a reference and set as 100% of caspases 3 and 7 activity. Three independent experiments were performed.
2.8. Detection of dead-cell protease activity
To determine the level of necrosis induced by LPO products, the activity of dead-cell protease was verified using luminescent CytoTox-Glo Cytotoxicity assay (Promega, Fitchburg, WI). This assay uses a luminogenic peptide substrate for dead-cell protease, which is released from cells that have lost membrane integrity. Luminescent signal is generated by luciferase from aminoluciferin – product of substrate cleavage, and is proportional to protease activity liberated from dead-cells. CytoTox-Glo Cytotoxicity assay was conducted according to the manufacturer’s instructions. Attached cells were treated with HNE or hydrogen peroxide (a positive control) for 2 h. Luminescence intensity was measured using a DTX 880 microplate reader (Beckman Coulter). After that cells were lysed, which delivered luminescent signal associated with the total number of cells in each well and was used to normalize the data. At least three independent experiments were performed.
2.9. Western blot analysis
WT and Ercc1−/− immortalized MEFs at 60% confluence were maintained 2 h in serum-free medium in the presence of 20 μM HNE or 1 μM camptothecin (CPT) and collected by scraping at 2, 6, 12, 24, 48 h following exposure to HNE or media only and 2 and 24 h after CPT (used as a positive control). Cell free extracts were prepared by resuspending the MEFs in cell extraction buffer (10 mM Tris-HCl, pH 7.4, 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM NaF, 20 mM Na4P2O7, 2 mM Na3VO4, 1% Triton X-100, 10% glycerol, 0.1% SDS, 0.5% deoxycholate) supplemented with protease inhibitor cocktail (Complete, Roche, Basel, Switzerland) and 0.25 mM PMSF (Sigma-Aldrich), and incubating on ice for 30 min followed by centrifugation at 16000×g for 15 min. Protein concentration was quantified by the Bradford method (Bio-Rad, Hercules, CA). Cell extracts (25 μg) were electrophoresed and transferred to PVDF membrane (EMD Millipore, Billerica, MA). Blots were incubated with the following primary antibodies: rabbit polyclonal anti-PARP rabbit polyclonal anti-caspase 7, rabbit polyclonal anti-caspase 3, (Cell Signaling Technology, Danvers, MA), mouse monoclonal anti-phospho-histone H2AX (Ser139) (EMD Millipore), mouse monoclonal anti-poly(ADP-ribose) (Trevigen, Gaithersburg, MD), rabbit monoclonal anti-phospho-RPA32 (S4/S8) (Bethyl, Montgomery, TX), mouse monoclonal anti-β-actin (Santa Cruz Biotechnology, Dallas, TX), or mouse monoclonal anti-GAPDH (Abcam, Cambridge, United Kingdom). Primary antibodies were detected with the appropriate secondary HRP-conjugated antibodies (Santa Cruz Biotechnology) and visualized by chemiluminescence using Immobilon Western Chemiluminescent HRP Substrate (Millipore) according to the manufacturer’s instructions. Two independent cell treatments were done and at least two Western blot analyses were carried out for each endpoint.
2.10. Sister chromatid exchange assay
For determination of sister chromatid exchange (SCE) level, cells were seeded in culture vessels at a density of 3×105 WT and 4×105 Ercc1−/− immortalized MEFs per dish. Treatment with 10 μM HNE or 1 μM cisplatin (CDDP) was preformed 24 h following seeding, in serum-free medium, and lasted for 2 h. Untreated controls were incubated in serum-free medium for the same time. Test compounds were washed out and the fibroblasts were cultured for two cell cycle periods (34 h for WT and 38 h for Ercc1−/− MEFs) in fresh growth medium in the presence of 20 μM 5′-bromodeoxyuridine (BrdU, Life Technologies). During the last 90 min of incubation, colcemid (Sigma-Aldrich) was added at final concentration of 0.125 μg/ml. Fibroblasts were then harvested by trypsinization, suspended in KCl hypotonic solution (72.5 mM) for 10 min at 37°C and fixed in a fresh ice cold solution of methanol/acetic acid (3:1). The fixation was repeated at least 3 times. Subsequently, cells were suspended in fixative solution, spread on humidified microscope slides and air-dried. In order to visualize SCEs, slides were stained according to the Fluorescence Plus Giemsa method [40] with some modifications. In general, slides were incubated with 20 μg/ml bis-benzamide water solution (Sigma-Aldrich) for 15 min in darkness, subsequently rinsed with distilled water, and irradiated with UVA light for 45 min (TL Mini Blacklight Blue, Philips, Amsterdam, The Netherlands). Afterwards, slides were kept in 1× SSC solution (0.15 M NaCl, 0.015 M sodium citrate) for 40 min at 50°C, washed and stained with 1% Giemsa solution (Sigma-Aldrich). Pictures of metaphase spreads were captured at 100× objective using an IX81 Olympus microscope and associated software. For each group 30 randomly chosen metaphases were analyzed. Three independent experiments were performed.
2.11. Senescence associated-β-galactosidase assay
To determine the level of senescent cells, activity of β-galactosidase (SA-β-gal) was assessed using the method of Dimri et al. [41]. Briefly, WT and Ercc1−/− primary or immortalized MEFs were seeded in 12-well plates at different densities. The density for untreated controls was 5,000 or 500 cells per well (primary and immortalized MEFs, respectively) or for cells treated with HNE and doxorubicin 10,000 or 6,000 cells per well (primary and immortalized MEFs, respectively). Cells were incubated with 10 or 15 μM HNE or with 25 or 50 nM doxorubicin (a positive control) in growth medium for 24 h at which point the medium was discarded and replaced with fresh media. Then cells were cultured for 5 days, and on the 5th day they were fixed with PBS solution of 0.25% glutaraldehyde and 2% formaldehyde for 5 min at room temperature. Subsequently, fibroblasts were rinsed twice in PBS, stained overnight at 37°C with a solution containing 1 mg/ml 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal), 2.5 mM potassium ferrocyanide, 2.5 mM potassium ferricyanide, 150 mM NaCl, 2 mM MgCl2 and 0.1 M phosphate buffer, pH 6.0, and finally washed twice with PBS. Photos of stained fibroblasts were captured at 40× objective using an IX81 Olympus microscope and the associated software. For each group, 200 randomly chosen cells were analyzed. Three independent experiments were conducted.
2.12. Detection of PCNA ubiquitination
WT and Ercc1−/− immortalized MEFs at 60% confluence were treated with 20 μM HNE in serum-free medium in the presence of 15 μM ML323, an inhibitor of PCNA deubiquination (Axon Medchem, Groningen, The Netherlands). After 2 h, the treatment medium was replaced with a fresh growth medium with 15 μM ML323. Cells were harvested by scraping at 2, 6, 10 h following HNE exposure and 10 h for mock-treated (the longest time of incubation with inhibitor). Simultaneously, UVC-irradiated fibroblasts (60 J/m2) were prepared as a positive control. Cells were cultured for 6 h after exposure to UV light in a growth medium containing 15 μM ML323 and subsequently collected. To prepare chromatin fractions, cells were resuspended in RIPA buffer (Sigma-Aldrich) supplemented with protease inhibitor cocktail (Complete, Roche) and 0.25 mM PMSF (Sigma-Aldrich). The cell suspensions were incubated on ice for 30 min and centrifuged at 15000×g for 15 min. The pellet was collected and washed with RIPA buffer three times followed by adding 2× Laemmli buffer, sonication and boiling. The final solution was used as the chromatin fraction. Western blot analysis was performed using standard protocols. Proteins were separated on 10% SDS-PAGE. The primary antibodies used were mouse monoclonal anti-PCNA (Santa Cruz Biotechnology) and rabbit polyclonal anti-lamin A/C (Santa Cruz Biotechnology).
2.13. RNA extraction and cDNA synthesis
Total RNA was isolated from snap frozen mouse tissues or freshly collected cells using the TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. The quality of total RNA was assessed by agarose gel electrophoresis, Only RNA samples with clearly distinguishable 18S and 28S ribosomal RNA bands and no visible RNA degradation were used. The concentration of RNA samples was ascertained by measuring the optical density at 260 nm. Total RNA (2 μg) from each sample was used to generate cDNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems by Life Technologies; Carlsbad, CA, USA) with oligo(dT)16 primers.
2.14. Real-time PCR using SYBR-Green chemistry
Real-time quantitative PCR assays were carried out on the PikoReal 96 Real-Time PCR System (Thermo Scientific). The primers were designed in the Primer Quest program (http://eu.idtdna.com/PrimerQuest/Home/Index), and are shown in Table 1 (Supplementary material). Each reaction was carried out in a reaction mixture containing: 1× concentrated Real-Time 2× HS-PCR Master Mix SYBR A (A&A Biotechnology, Gdynia, Poland), forward and reverse primers (100 nM each), cDNA template – in three decreasing amounts per well (e.g. 135, 45 and 15 ng) each in duplicate, and water to 10 μl of final volume. Reactions were performed with an initial denaturation step of 95°C 3 min. followed by 45-50 cycles of denaturation (95 °C for 15 sec.) and primer annealing-extension (60°C for 30 sec.). Fluorescence was read at the end of the annealing-extension step of each cycle. After cycling, melting point temperature analysis was performed in the range of 60°C to 95°C with temperature increments of 0.2°C. Evaluation of results quality was based on expected Ct differences among three cDNA amounts as well as product melting curves. Rare outlying results were omitted in calculations. Three concentrations of cDNA allowed to calculate individual efficiencies for each primer pair and normalize all results to one cDNA concentration, common for all genes. The amount of each target gene was calculated by ΔCt method with geometric mean of two common genes Cts as reference [42].
2.15. Tissue and cell-free extracts for base excision repair activities
Tissue and cell-free extracts for measuring BER activities were prepared according to the method of Tanaka et al. [43] modified by Vodenicharov et al. [44]. Biological material used in this study was: snap-frozen tissue fragments of brains, livers and kidneys from WT and Ercc1−/− mice at age 19-21 days and WT and Ercc1−/− immortalized MEFs. Tissue samples were homogenized (grinders, Kimble Chase, Rockwood, TN) in buffer I (10 mM Tris-HCl, pH 7.8, 200 mM KCl) supplemented with 1x protease inhibitor cocktail (Complete, Roche) and 0.25 mM PMSF (Sigma-Aldrich) (10 μl of buffer per 5 mg of tissue). Cells were suspended in this buffer at 106 cells/10 μl. Then, equal volume of buffer II (10 mM Tris-HCl, pH 7.8, 600 mM KCl, 2 mM EDTA, 40% glycerol, 0.2% Nonident P-40, 2 mM DTT) with 1x protease inhibitor cocktail (Complete, Roche) and 0.25 mM PMSF (Sigma-Aldrich) was added to the mixture. Next, the tissue homogenate or cell suspension was shaken at 4°C for 1.5 h and centrifuged at 16000×g for 15 min. The supernatant was recovered, aliquoted and stored at −80°C for further analysis. Protein concentration was measured by the Bradford method using the protein assay reagent (Bio-Rad).
2.16. Substrate oligodeoxynucleotides radiolabeling and annealing
Enzymatic assays of different stages of BER were carried out using oligodeoxynucleotides (34-mers) with a single lesion (Midland Certified Reagent Company, Midland, TX). Sequences of oligonucleotides are given in Table 2 (Supplementary material). Single-stranded oligodeoxynucleotides containing modified nucleotides were 32P-labeled at their 5′ ends in 1× reaction buffer (USB Corporation, Cleveland, OH) containing 50 pmol of the oligodeoxynucleotide, 20 μCi [γ32P]ATP (Hartmann Analytic, Braunschweig, Germany) and 10 units of OptiKinase (USB Corporation). The reaction was carried for 1 h at 37°C. The products were purified on a Micro Bio-Spin Columns with Bio-Gel P-30 (Bio-Rad) according to the manufacturer’s instructions. The labeled oligodeoxynucleotides were annealed to complementary non-labeled oligodeoxynucleotides in a double molar excess in annealing buffer (50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 1 mM DTT). The annealed oligodeoxynucleotides were stored at −20°C at final concentration of 0.5 μM.
2.17. Measurements of base excision repair activities
Activities of BER enzymes were measured in cell-free extracts from WT and Ercc1−/− immortalized MEFs or protein extracts from WT or Ercc1−/− mice tissues using substrates (0.25 pmol per reaction) marked in Supplementary material, Table 2. Reactions were carried for 2 h or 30 min for uracil excision, AP-sites incision and gap filling at 37°C in the following buffers: for 8-oxo-deoxyguanosine (8-oxodG) excision 20 mM HEPES, pH 7.8, 0.5 mM EDTA, 50 mM KCl, 5% glycerol, 2 mM DTT; for 1,N6-ethenoadenine (εA) excision 20 mM HEPES, pH 7.8, 5 mM EDTA, 70 mM KCl, 2 mM MgCl2, 2 mM DTT; for uracil (U) excision 20 mM HEPES, pH 7.8, 2 mM EDTA, 70 mM KCl, 2 mM MgCl2, 2 mM DTT; for AP-site incision 20 mM HEPES, pH 7.8, 5 mM EDTA, 10 mM KCl, 2 mM MgCl2, 2 mM DTT; for gap filling 50 mM HEPES, pH 7.5, 0.5 mM EDTA, 20 mM KCl, 10 mM MgCl2, 2% glycerol, 2 mM DTT) supplemented with 2 mM ATP and 200 μM dCTP; for strand ligation 50 mM HEPES, pH 7.5, 1 mM EDTA, 20 mM KCl, 10 mM MgCl2, 2% glycerol, 2 mM DTT) supplemented with 2 mM ATP.
All reactions were terminated by adding SDS to a final concentration of 0.5% and digestion of proteins for 30 min at 55°C with 250 μg/ml of proteinase K (Sigma). Then the formamide stop solution (92% formamide, 20 mM EDTA, 0.05% bromophenol blue, and 0.05% xylene cyanol) was added in a volume equal to half of the volume of the reaction mixture and samples were heated at 85°C for 5 min. The products were resolved by electrophoresis on 20% polyacrylamide denaturing gels with 7 M urea. Gels were exposed to PhosphorImager screen which was subsequently scanned using Fuji PhosphorImager with FujiFilm FLA7000 software (Fujifilm, Tokyo, Japan). Intensity of bands was quantified with Multi Gauge software (Fujifilm). From each data set, a curve reflecting product rate as a function of the amount of extract protein was plotted and the enzyme activities were calculated from the linear part of the curve. Results were presented as fmol product per h per μg protein (fmol/h/μg protein). Experiments included control samples: a negative control with untreated oligodeoxynucleotides (without cell-free or tissue extracts) and a positive control in which (i) oligodeoxynucleotides were digested with an excess of the damage-specific pure DNA glycosylase/AP-lyase or (ii) radiolabeled oligodeoxynucleotides of an appropriate length were subjected to electrophoresis. All measurements were performed at least in triplicate.
2.18. Measurement of lipid peroxidation
To determine the basic and HNE-induced level of LPO in primary MEFs, Click-iT Lipid Peroxidation Imaging Kit – Alexa Fluor 488 (Invitrogen) was used, which contained linoleamide alkyne (LAA) reagent (alkyne-modified linoleic acid). When incubated with cells, LAA incorporates into cellular membranes. Upon lipid peroxidation, LAA is oxidized and produces hydroperoxides, which subsequently decompose to various α,β-unsaturated aldehydes, which bind to proteins. Proteins with LAA-peroxidation-derived adducts are detected using Click-iT chemistry. Cells were treated with 15 μM HNE for 2 h. The assay was conducted according to instructions supplied with the kit. For nuclear staining and to inhibit photobleaching Vectashield Antifade Mounting Medium with DAPI (Vector Laboratories, Burlingame, CA) was used. Pictures were captured at 40× objective using Olympus IX81 microscope and associated software. The intensity of fluorescence signal was quantified using Fiji software (http://fiji.sc/). Six independent experiments were performed.
2.19. Measurement of ATP
The level of cellular ATP was measured using the luminescent CellTiter-Glo assay (Promega). The assay relies on the properties of thermostable luciferase, which generates a stable “glow-type” luminescent signal by mono-oxygenation of luciferin in the presence of Mg2+, ATP and molecular oxygen. Luminescent signal is proportional to the amount of ATP present. Twenty-four h before treatment WT and Ercc1−/− immortalized MEFs were seeded in triplicate in 96-well plates at density of 4500 cells per well and incubated with increasing concentrations of HNE (5 – 40 μM) in serum-free medium for 2 h. Then the procedure was performed according to manufacturer instructions. Luminescence was recorded using PARADIGM microplate reader (Beckman Coulter). The relative ATP level was counted as a percent of luminescence values from wells with treated cells divided by luminescence values from wells with untreated controls. At least three independent experiments were conducted.
2.20. Measurement of NAD+ and NADH
The cellular levels of oxidized (NAD+) and reduced (NADH) forms of nicotinamide adenine dinucleotide were detected using NAD/NADH-Glo assay kit (Promega). The assay provides NAD cycling enzyme, which converts NAD+ to NADH. In the presence of NADH, reductase enzymatically reduces a proluciferin reductase substrate to luciferin. Luciferin is detected using luciferase, and the amount of light produced is proportional to the amount of NAD+ and NADH in a sample. Separate detection of NAD+ and NADH is possible due to their differential stability at acidic and basic conditions. Oxidized forms are selectively destroyed by heating in basic solution, while reduced forms are not stable in acidic solution. WT and Ercc1−/− immortalized MEFs were seeded in 96-well plate, at density of 7,500 cells per well, in triplicate for each experimental point and allowed to adhere and grow for 24 h. The MEFs were incubated for 2 h in serum-free medium with varying concentrations of HNE (5 - 40 μM). After medium removal and growth in the fresh medium for next 2 h cells were lysed using a mixture of PBS, 0.1 M NaOH and 0.5% DTAB (dodecyltrimethylammonium bromide). Each lysate was divided in two parts, one of them was assigned to basic treatment (in 0.1 M NaOH with and 0.5% DTAB) and the other one to acid treatment (in 0.13 M HCl). Both variants were incubated for 15 min at 60°C and subsequently cooled to room temperature. To neutralize the acid and base-treated samples they were supplemented with Tris-HCl pH 7.4. Next, NAD/NADH-Glo reagent was added to all wells and after 45 min at room temperature, luminescence was measured using PARADIGM microplate reader (Beckman Coulter). The results were presented as the relative NAD+ and NADH levels (percent of luminescence values from wells with treated cells divided by luminescence values from wells with untreated controls). Three independent experiments were performed.
2.21. Measurement of GSH-Px and GST activities
Enzymatic activities of glutathione peroxidase (GSH-Px) and glutathione S-transferase (GST) were determined in cell-free extracts of WT and Ercc1−/− immortalized MEFs. At 80% confluence were washed with 1× PBS, collected by scraping and pelleted. Cells were sonicated on ice in ice-cold buffer (25 mM Tris–HCl, pH 7.5, 25 mM NaCl, 2 mM EDTA, 10% glycerol) with protease inhibitor cocktail (Complete, Roche). Cells lysates were centrifuged for 15 min, 14000×g, at 4°C. The supernatants were aliquoted and stored at −80°C. Protein concentration was measured by the Bradford method using Bio-Rad reagent.
Glutathione S-transferase activity was measured using 1 mM 1-chloro-2,4-dinitrobenzene (CDNB) as an electrophilic substrate for transferase activity according to the method established by Habig et al. [45]. Selenium-dependent glutathione peroxidase activity was determined with 0.25 mM hydrogen peroxide and selenium-independent activity with 1.5 mM cumene hydroperoxide using the method of Paglia and Valentine [46] with modifications of Lawrence and Burk [47]. All activities were expressed as μmol of reaction product per min per mg of protein from cell extracts.
2.22. Mice
NER-deficient mice (Ercc1−/−, Ercc1−/∆, Xpa−/−) were generated and genotyped as previously described [24, 48–51]. Ercc1−/∆ mice were generated by breeding Ercc1+/− and Ercc1+/Δ mice in inbred C57Bl/6J and FVB/n backgrounds, respectively, to create a cohort of mice that were in an f1 hybrid background yet genetically identical. The mice were genotyped by PCR as previously described [49]. Animal husbandry and experimental procedures were approved by the University of Pittsburgh IACUC.
2.23. CCl4 treatment of animals
WT, Xpa−/− and Ercc1−/∆ mice were injected subcutaneously with 0.5 ml/kg CCl4, prepared as a 1:1 suspension in olive oil, twice a week for five weeks. Treatment began when the mice were 10 weeks of age. Animals were monitored daily and weighed twice a week. Mice were sacrificed when they were considered terminal or experienced reduced spontaneous activity. Tissues were isolated for histological analysis.
2.24. Measurement of lipid hydroperoxides in tissue
Lipid peroxidation level in the liver of CCl4-exposed mice was estimated by measuring lipid hydroperoxides using a commercially available kit (Cayman Chemical, Ann Arbor Michigan, #705002) according to the manufacturer’s instructions (n=3 mice per group).
2.25. Diet
Special diets were ordered from Harlan-Teklad. The chow pellets contained a standard formula of macronutrients (20.6% protein, 1.7% crude fiber, 3.5 kcal/g energy density) and necessary micronutrients. The only difference was that the fat component of the diet was substituted with 12.4% polyunsaturated fats derived from safflower oil (PUFA diet). The control diet contained the same amount of saturated fat derived from coconut oil with a small component of polyunsaturated fat (<0.6% linoleic acid, which is essential).
2.26. Histological staining and histopathology
Tissues were fixed in 10% formalin overnight, embedded in paraffin and sectioned using a microtome. Subsequently, sections were stained by standard procedures with hematoxylin and eosin (H&E), Masson’s trichrome and periodic acid Schiff (PAS). Slides of liver, kidney and pancreas sections were scored by a veterinary pathologist who performed analyses blindly in respect to the genotype and treatment of the animals. The scoring was based on a three point system with 0 being unaffected or no pathology and 3 being the most severe pathology.
2.27. Statistical analysis
All presented data are the mean ± SD, unless otherwise indicated. Statistical analysis was performed using the Statistica computer software STATISTICA (version 10.0, StatSoft, Inc.). Parameters were tested using non-parametric Mann-Whitney’s U-test or Student’s t-test one- or two-tailed.
3. RESULTS
3.2.ERCC1-deficient cells and mice are hypersensitive to LPO
ERCC1-deficient mice are characterized by accumulation of oxidative DNA lesions [28, 52, 53]. Among them are adducts of LPO products, which are repaired by NER [14, 15]. In order to gain insight into the mechanism of cell response to LPO in Ercc1−/− background, we investigated the sensitivity of WT and Ercc1−/− immortalized mouse embryonic fibroblasts (MEFs) to hydrogen peroxide and selected LPO products. Ercc1−/− cells were more sensitive to hydrogen peroxide (Fig. 1A), HNE (Fig. 1B), MDA (Fig. 1C) and CRO (Fig. 1D), with HNE being the most toxic. Interestingly, ACR was equally toxic to WT and ERCC1-deficient cells (Fig. 1E). ACR, in contrast to HNE, MDA and CRO, was not demonstrated to form DNA ICLs [54–56], or their level is below the detection limits. Similarly, XPA-deficient cells, defective in NER but not ICL repair were not hypersensitive to HNE (Fig. 1F), and XPA protein is not directly involved in ICLs unhooking, while ERCC1-XPF complex is engaged.
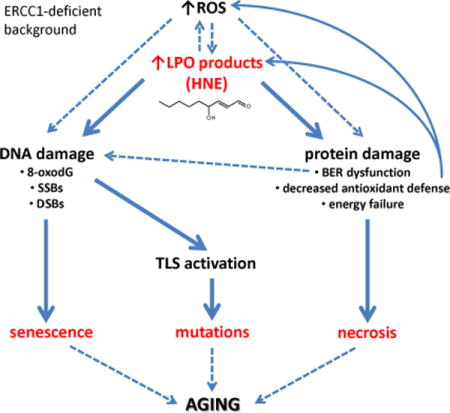
Fig. 1. Ercc1−/− MEFs are hypersensitive to hydrogen peroxide and long chain LPO products, the latter significantly inhibiting proliferation of Ercc1−/− MEFs.
(A – F) Viability curves. To assess the relative sensitivity, wild-type and Ercc1−/− immortalized MEFs were incubated with increasing concentrations of hydrogen peroxide (H2O2) (A), 4-hydroxy-2-nonenal (HNE) (B), malondialdehyde (MDA) (C), crotonaldehyde (CRO) (D) and acrolein (ACR) (E) for 2 h in serum-free medium. Additionally, Xpa−/− immortalized MEFs together with their WT counterparts were subjected to HNE treatment as described above (F). Relative cell viability was measured by alamarBlue assay on the second day after treatment. (G, H) Proliferation curves. Similarly, proliferation of wild-type (G) and Ercc1−/− (H) immortalized MEFs was assessed after 2 h treatment with 20 μM HNE in serum-free medium using alamarBlue assay. Measurements were taken each day for 5 days. One hundred percent confluency was observed when readouts achieved about 300 RFU. Each point of curves represents the mean ± SD of at least three independent experiments done in triplicate. Structures of LPO products are shown above graphs. RFU, relative fluorescence units.
The in vitro and in vivo ERCC1-XPF is critical for the repair of LPO-induced DNA lesions, among which crosslinks are probably the critical ones. To investigate this further, we focused on the most toxic compound, HNE. HNE transiently inhibited proliferation of WT immortalized MEFs, however on the third day following HNE treatment cells already attained as high confluence as untreated cells (Fig. 1G). In contrast, recovery of ERCC1-deficient cells was significantly delayed (Fig. 1H). Even 5 days after HNE treatment, the growth of cells was significantly inhibited and their density did not reach plateau, which in untreated Ercc1−/− cells, like in WT, occurred within 48 h.
To determine if LPO elicits the same effect in vivo, we challenged mice with a common environmental exposure that induces LPO, carbon tetrachloride (CCl4). CCl4 is reduced in the liver, to the trichloromethyl free radical, which in the presence of oxygen is converted into a peroxy-radical [57]. Both of these radicals are capable of oxidizing lipids. Ercc1−/∆ mice, which are defective in NER and ICLs repair, their wild-type (WT) littermates, and Xpa−/− mice defective only in NER were chronically exposed to CCl4 (0.5 ml/kg) biweekly via subcutaneous injection for five weeks (cumulative dose was 5 ml/kg) (Fig. 2A). The mice were monitored and sacrificed when the Ercc1−/∆ mice were considered end-stage. CCl4 treatment of the ERCC1-deficient mice resulted in a significantly decreased median lifespan compared to untreated Ercc1−/∆ mice (Figs 2B and supplementary S1, p=0.0032, Log-rank (Mantel-Cox) test); namely 50% of CCl4 treated mice survived 19 weeks, while untreated 22 weeks, 20% of treated mice survived 22 weeks, while untreated 28 weeks, however the end stage of life was 29 weeks for both CCl4 treated and untreated animals (Fig. 2B). The same dose had no detrimental effect on WT or Xpa−/− mice. These data support the hypothesis that unrepaired LPO-induced ICLs have a negative health impact, which may result in lifespan shortening.
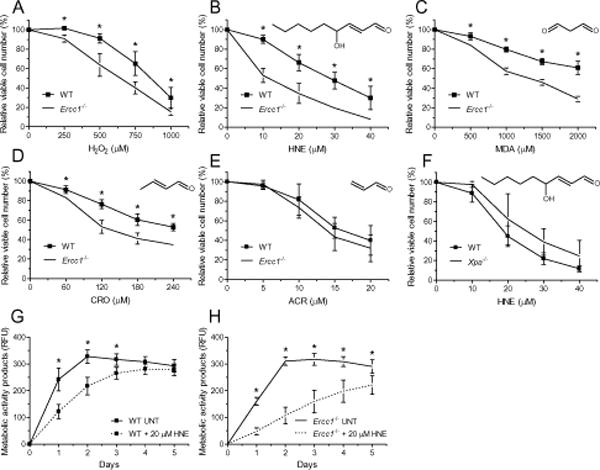
Fig. 2. Chronic exposure to low doses of CCl4 promotes lipid peroxidation and decreases lifespan of Ercc1−/∆ mice.
(A) A scheme of experiment: CCl4 was administered to wild-type, Ercc1−/∆ and Xpa−/− mice by subcutaneous injection of 0.5 ml/kg, twice a week for five weeks, beginning at 10 weeks of age. (B) Survival curve of mice after treatment with CCl4. Graphed are WT mice + CCl4 (n=12; green), Xpa−/− mice + CCl4 (n=8; red), untreated Ercc1−/∆ mice (n=10; light purple), and Ercc1−/∆ mice + CCl4 (n=13; dark purple). See also Fig. S1. (C) Lipid hydroperoxides (LOOH) measured by ELISA assay in liver tissue isolated from untreated WT (light green), untreated Ercc1−/∆ (light purple), CCl4-treated WT (dark green) and CCl4-treated Ercc1−/∆ mice (dark purple). Values indicted the mean for 3 mice per group ± S.E.M. and were evaluated using one-tailed Student’s t-test (*, p<0.05; **, p<0.01). (D) H&E stained liver sections from WT and Ercc1−/∆ mice treated with CCl4 or untreated (10× objective, top; 40× objective, bottom). Black arrows indicate areas of nodular regeneration; white arrow illustrates areas of necrosis. (E) Masson’s trichrome stained liver sections from WT and Ercc1−/∆ mice treated or untreated with CCl4 (20× objective). Blue indicates areas of fibrosis. (F) Periodic acid Schiff (PAS) stained liver sections from WT and Ercc1−/∆ mice CCl4 treated or untreated (20× objective). The dark purple indicates areas of polysaccharide deposition.
To ensure that CCl4 was inducing LPO in our experimental conditions, lipid hydroperoxides (LOOH) were measured in liver of Ercc1−/∆ mice and their WT littermates using an ELISA assay. Surprisingly, untreated mutant animals had a 7-fold higher LOOH level compared to their WT littermates (Fig. 2C). After treatment with CCl4, the level of lipid hydroperoxides was unchanged in WT mice. LOOH level was ~1.5-fold higher after CCl4 treatment of Ercc1−/∆ mice, but this was not statistically significant (Fig. 2C, p=0.25 Student’s one-tailed t-test).
To determine to what extent CCl4-induced LPO affected the liver of WT and DNA repair-deficient mice, tissue sections were analyzed for histologic changes in a blinded fashion. Hepatocytes from WT mice treated with CCl4 exhibited signs of increased nuclear size and focal necrosis, both changes associated with aging [28, 58]. In addition, Ercc1−/∆ liver samples contained hepatocytes that were extremely enlarged with dense eosinophilic cytoplasm (Fig. 2D). There was significantly more hepatocellular necrosis and degeneration, but no increase in immune cell infiltrates were detected in the ERCC1-deficient liver samples compared to CCl4-treated WT mice. There were also foci of nodular regeneration evident in the Ercc1−/∆ liver sections with frequent oval cell hyperplasia, often stemming from portal areas.
The liver sections from WT and Ercc1−/∆ littermate pairs treated with CCl4, were stained with Masson’s trichrome and periodic acid Schiff (PAS), to show fibrosis and glycogen deposition, respectively. Increased portal fibrosis was evident in the treated Ercc1−/∆ mice compared to treated WT littermates by the dark blue staining with Masson’s trichrome (Fig. 2E). In addition, the CCl4-treated Ercc1−/∆ liver samples showed high levels of glycogen deposition (Fig. 2F). Necrosis, nodular regeneration, fibrosis and steatosis were greater in the treated Ercc1−/∆ mice compared to treated WT mice, consistent with their hypersensitivity to CCl4.
The untreated WT animals show normal liver histology with no evidence of hepatic fibrosis or glycogen inclusions. Pathologic changes detected in progeroid Ercc1−/Δ mice included focal degeneration and necrosis with neutrophilic inflammation, a diffuse increase in sinusoidal lining cells, karyomegaly, with intranuclear inclusions, mild periportal fibrosis and glycogen deposition [28] (Fig. 2D-F).
3.3. Oxidative stress and lipid peroxidation are elevated in ERCC1-deficient cells
LPO products, such as HNE and MDA may affect electron flow and production of ATP in mitochondria [59], which is anticipated to increase oxidative stress in the cell [60]. In untreated immortalized cells, the ROS level was similar in WT and Ercc1−/− cells. HNE, MDA or TBHP increased ROS formation in both cell lines in a dose-dependent fashion (Fig. 3A). However, only at lower HNE and MDA concentrations the increase was greater in ERCC1-deficient than in WT cells. Such changes might reflect random modifications of proteins with LPO products, and with higher reactive aldehydes concentrations their toxicity is very big, which may mask mitochondrial ROS formation. In primary cells ROS level measured with the same method was higher even in untreated Ercc1−/− than in WT cells. These results were confirmed using DCFDA and MitoSOX to measure ROS (Fig. S2). Similarly to immortalized cells, HNE and TBHP increased the level of ROS in primary cells of both genotypes, but it was significantly higher in Ercc1−/− than in WT background (Fig. 3B). Increased ROS was suppressed by the addition of the radical scavenger XJB-5-131 [61] to cell cultures (Fig. S3A).
Fig. 3. LPO products induce oxidative stress and accelerate LPO in Ercc1−/− cells most probably due to reduced activities of GSH-dependent enzymes.
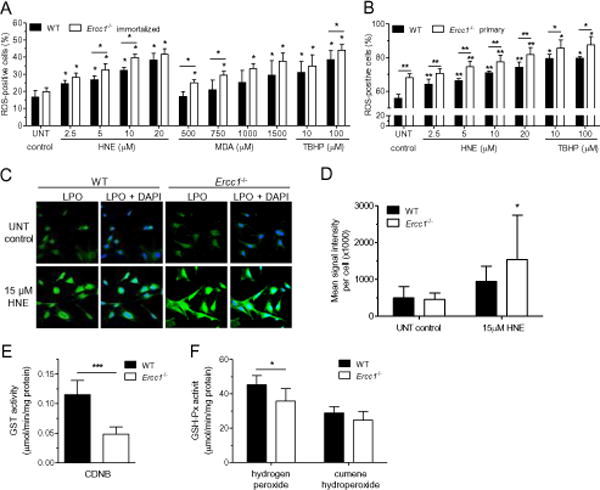
(A, B) ROS level. Percentage of ROS-positive WT and Ercc1−/− immortalized (A) or primary (B) cells was measured after 2 h incubation with increasing concentrations of HNE, MDA (only immortalized) or tert-butyl hydroperoxide (TBHP), here used as a positive control. Data bars are the mean ± SD of at least three independent experiments evaluated using the Mann-Whitney U-test (*, p<0.05 vs. untreated control, unless otherwise indicated). (C, D) The level of lipid peroxidation. LPO was measured with a linoleamide alkyne (LAA) detection kit as a LAA-peroxidation-derived protein modifications in wild-type and Ercc1−/− primary MEFs. Analysis was performed on untreated cells or cells exposed for 2 h to HNE. (C) Representative fluorescent microscopy images. Green color represents LPO, blue color – DAPI staining in nuclei. Images were captured at 40× magnification. (D) Quantification of LPO shown in C, graphed as a mean signal intensity per cell ± SD of six independent experiments. The statistical analysis was done by Mann-Whitney U-test (*, p<0.05). (E, F) Activities of antioxidant enzymes. Activity of glutathione S-transferase was measured using 1-chloro-2,4-dinitrobenzene (CDNB) as a substrate (E) and glutathione peroxidase (GSH-Px) using hydrogen peroxide or cumene hydroperoxide (F) as substrates. All of them were measured in cell-free extracts of WT and Ercc1−/− immortalized MEFs. Data bars are the mean ± SD of at least eight biological repeats. Statistical analysis was done using Mann-Whitney U-test (*, p<0.05; ***, p<0.001).
The increased ROS induced further LPO. LPO was measured with linoleamide alkyne (LAA) detection kit as LAA-peroxidation-derived protein modifications in untreated and HNE-treated primary MEFs (Fig. 3C and B). In response to HNE, primary ERCC1-deficient cells produced more LPO products than WT cells, which confirmed increased oxidative stress in Ercc1−/− cells. Thus, increased level of LPO products in ERCC1-deficient background induce ROS formation, which in turn leads to LPO. Increased LPO was observed also in vivo in liver of Ercc1−/∆ mice (see Fig. 2C and [28]).
The main pathway for elimination of some LPO products, e.g. HNE, is its conjugation with glutathione and removal from the cell via ATP-dependent RLIP76 transporter [62, 63]. It was shown that deficiency of glutathione transferases is related to increase of HNE level in cells [64]. This prompted us to measure glutathione-related antioxidant enzymes activity in immortalized MEFs. Glutathione transferase activity was severely decreased in Ercc1−/− MEFs (Fig. 3C). Activity of selenium-dependent glutathione peroxidase was also lower in ERCC1-deficient than in WT MEFs (this isoform uses inorganic substrates, like hydrogen peroxide, and can decrease ROS level in the cell). However, the activity of selenium-independent isoform, using organic substrates, e.g. cumene hydroperoxide, was similar in both cell types (Fig. 3D). This suggests that in ERCC1-deficient cells, ROS and LPO levels may be increased due to decreased antioxidant buffering capacity.
3.4. DNA damage and repair under increased oxidative stress and LPO in ERCC1-deficient background
3.4.1. DNA damage
To assess the extent of oxidative stress and DNA damage we measured a direct oxidative DNA lesion, 8-oxo-deoxyguanosine (8-oxodG) in the DNA of primary ERCC1-proficient and -deficient MEFs grown in different oxygen conditions, 3% oxygen, which is normoxic for primary MEFs, and 21% oxygen (hyperoxia). In both oxygen concentrations the level of 8-oxodG was higher in ERCC1-deficient than in -proficient cells, however the differences were more profound in 21% (Fig. 4A). Thus, oxidative stress is present in Ercc1−/− fibroblasts under basic conditions and escalates further in cells challenged with high oxygen.
Fig. 4. Oxidative stress and LPO generate high level of DNA damage in Ercc1−/− cells.
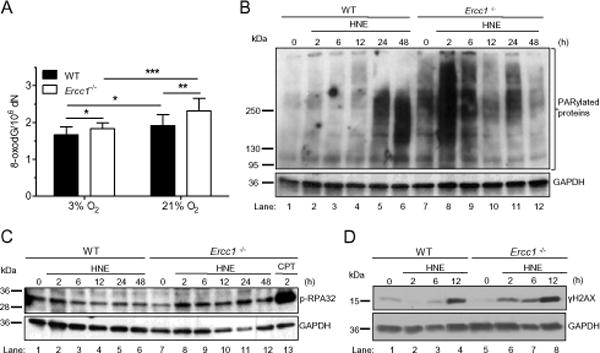
(A) 8-OxodG level. The level of a marker of oxidative DNA damage, 8-oxo-deoxyguanosine (8-oxodG), in the DNA of wild type and Ercc1−/− primary MEFs cultured under normoxia (3%) or hyperoxia (21%) conditions. To measure 8-oxodG the Ultra Performance Liquid Chromatography with Tandem Mass (2D-UPLC-MS/MS) was applied. Statistical analysis was done using Mann-Whitney U-test (*, p <0.05). (B – D) Assessment of DNA breaks. Markers of DNA single strand breaks (SSBs) identified as protein PARylation (B) and RPA32 protein phosphorylation (C), as well as DNA double strand breaks (DSBs) identified as phosphorylation of H2AX (D) were estimated by Western blotting in wild-type and Ercc1−/− immortalized MEFs after exposure to HNE. Cells were incubated with 20 μM HNE for 2 h in serum-free medium and collected for analysis at the indicated time points. Detection of GAPDH was used to confirm equal protein loading on all presented blots.
As an indirect measure of DNA damage in response to HNE, poly(ADP-ribose) polymerase 1 (PARP1) activity and phosphorylation of H2AX were measured. PARP1 is activated by single-(SSB) and double-strand DNA breaks (DSB), and the enzyme adds poly(ADP-ribose) (PAR) chains to proteins. We detected protein PARylation by Western blot. In WT immortalized cells, a minor increase of protein PARylation was observed up to 12 h following HNE treatment (Fig. 4B, lanes 2-4), but it increased dramatically at 24 and 48 h post-HNE treatment (Fig. 4B, lanes 5 and 6). The timing of the increased PARylation in WT cells corresponded with onset of apoptosis following HNE exposure (see paragraph 2.4.2 an Fig. 6I).
Fig. 6. Energy failure and types of death induced by HNE in Ercc1−/− and WT MEFs.
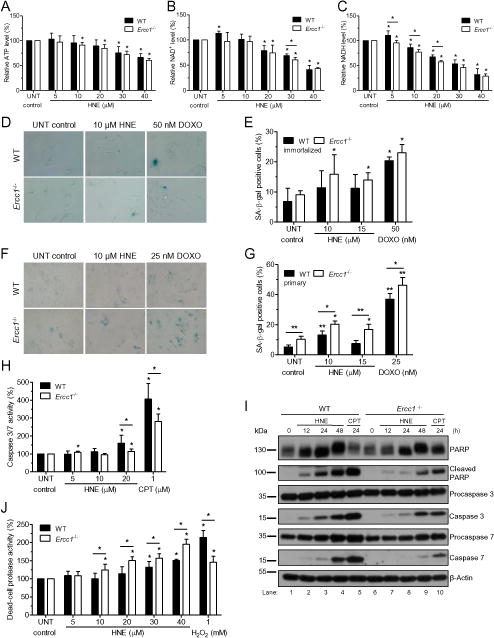
(A – C) ATP, NAD+, and NADH levels. The levels of ATP (A), NAD+ (B) and NADH (C) measured in wild-type and Ercc1−/− immortalized MEFs after 2 h of treatment with different doses of HNE followed by 2 h culture in fresh growth medium. Data bars are the mean ± SD of at least three independent experiments. Statistical analysis was done using Mann-Whitney U-test (*, p<0.05). (D – G) Senescence. Senescence was assayed by staining WT and Ercc1−/− cells with X-gal to measure senescence-associated beta-galactosidase activity (SA-β-gal) 5 days after 2 h treatment with different doses of HNE or doxorubicin (DOXO), which was used as a positive control. Representative images of SA-β-gal staining of immortalized and primary MEFs are shown on panels D and F, respectively. Blue color represents senescent cells. Images were captured at 40× microscope objective. Panels E and G show percent of wild-type and Ercc1−/− SA-β-gal positive cells, immortalized (E) and primary (G). (H, I) Apoptosis. Apoptosis of wild-type and Ercc1−/− immortalized MEFs was estimated 24 h after genotoxic treatments by measuring the activity of caspases 3 and 7 in response to 2 h exposure to drug (H) or by verifying the levels of cleaved PARP1, caspases 3 and 7 and their precursors in response to 20 μM HNE at different time points after 2 h exposure (I). Detection of β-actin was applied to confirm equal protein loading. In both assays, pro-apoptotic agent, camptothecin (CPT) was used as a positive control. (J) Necrosis. Necrosis was tested by measuring the activity of dead-cell protease directly after 2 h treatment with indicated concentrations of HNE. Hydrogen peroxide was used here as a positive control. All data bars are the mean ± SD of at least two or three independent experiments done in duplicate or triplicate. Statistical analysis was done using Mann-Whitney U-test (*, p<0.05; **, p<0.01 vs. untreated control, unless otherwise indicated).
There was a much greater degree of protein PARylation in untreated Ercc1−/− immortalized cells compared to WT controls. The amount of PARylated proteins increased dramatically at 2 h after HNE treatment, and then gradually decreased to the level found in untreated cells by 12 h after exposure to HNE (Fig. 4B, lanes 8-10). This earlier activation of PARylation in Ercc1−/− cells compared to WT is consistent with HNE inducing more ROS in Ercc1−/− cells (Fig. 3A and B), and thereby more oxidative DNA damage repaired by BER (8-oxodG, Fig. 4A), which generates SSBs in the DNA (Fig. 4B and C).
To get an insight into the types of DNA breaks induced by HNE, we investigated phosphorylation of RPA protein, as a measure of SSBs, and γH2AX histone, as a marker of DSBs. No dramatic change in RPA phosphorylation was observed in WT cells treated with HNE (Fig. 4C). However, already 6 h after treatment of cells with HNE, γH2AX appeared, and lasted up to 12 h of the study (Fig. 4D). The level of RPA phosphorylation was not increased in untreated Ercc1−/− cells compared to WT controls. However, in Ercc1−/− cells the level of p-RPA32 increased appreciably already 2 h following HNE treatment and remained elevated for at least 48 hours post-treatment (Fig. 4D, lanes 8-12). Similarly, γH2AX was not increased in untreated Ercc1−/− cells compared to WT controls, but increased more rapidly than in WT cells in response to HNE treatment. Thus markers of DNA damage increase more rapidly and robustly in Ercc1−/− cells than in WT cells, suggesting that HNE might be inducing more damage in the mutant cells and these DNA lesions are not well repaired.
3.4.2. Base excision repair
LPO products can directly modify and change activities of DNA repair proteins, for example, BER is disrupted by LPO product, HNE [20]. Since LPO was increased in ERCC1-deficient mice and HNE-treated cells, we investigated BER activities in MEFs and tissues of Ercc1−/− and WT mice. 8-OxodG, εA and U excision, incision of AP sites, single nucleotide gap filling, and DNA ligation were measured using cell extracts and a modified duplex DNA substrate. Activity at each step of BER was similar in ERCC1-deficient and WT immortalized MEFs (Fig. S4).
In stark contrast, in all tissues studied (brain, liver and kidney), the rate of DNA ligation was significantly decreased in Ercc1−/− mice compared to WT (Fig. 5A-F). Therefore, not only a defect in NER and ICL repair, but also impaired BER may contribute to LPO sensitivity in ERCC1-deficient mice.
Fig. 5. HNE induces promutagenic imbalance in BER and TLS in Ercc1−/− cells and mice.
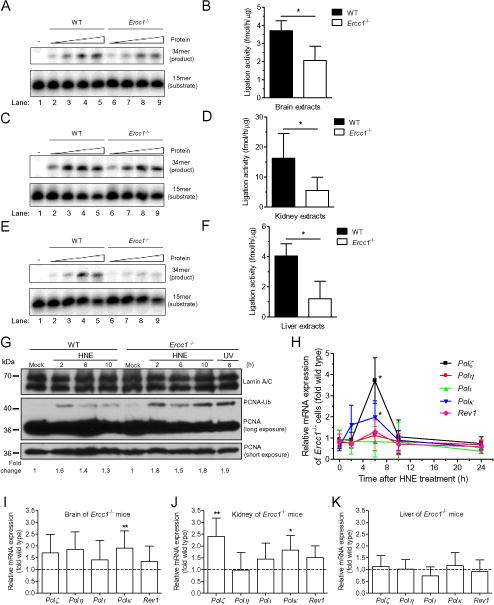
(A –F) Ligation step of BER. (A, C, E) SSB ligation shown by sealing of oligodeoxynucleotides by extracts from mice tissues. PhosphorImages of typical gels showing DNA ligation by tissue extracts from brains (A), kidneys (C) and livers (E) of wild-type and Ercc1−/− mice. Radiolabeled double-stranded oligodeoxynucleotide with a nick was used as a substrate. The reaction conditions and products analyses are described in Materials and Methods. (B, D, F) Quantitative analysis of (A, C, E respectively) showed as a fmol of product per h per μg of protein. Data are presented as mean ± SD (n=3 for WT, n=4 for Ercc1−/−) and were evaluated using the Mann-Whitney U-test (*, p<0.05). (G) Activation of TLS polymerases. Monoubiquitination of PCNA, that mediates the switch from replicative to translesion synthesis DNA polymerases, was estimated by Western blotting in wild-type and Ercc1−/− immortalized MEFs after exposure to HNE. Cells were incubated with 20 μM HNE for 2 h in serum-free medium and collected for assay in indicated time points. UV light was used as a positive control. Detection of lamin A/C was applied to confirm equal protein loading. (H) Transcriptions of TLS polymerases in cells. The ratio of mRNA level of TLS polymerases: Pol ζ, Pol η, Pol ι, Pol κ and Rev1 in Ercc1−/− immortalized MEFs to that in WT control cells. mRNA level was verified in different time intervals after 2 h treatment with 20 μM HNE in serum-free medium. Each point of curves represents the mean ± SD of results obtained from at least three independent experiments. Increase of the transcription is statistically significant for Pol κ and Pol ζ 6 h after the beginning of HNE-treatment vs. untreated control, as calculated by Mann-Whitney U-test with p<0.05. (I, J, K) Transcriptions of TLS polymerases in tissue. mRNA expression levels of TLS polymerases: Pol ζ, Pol η, Pol ι, Pol κ and Rev1 in brains (I), kidneys (J) and livers (K) of 3 week-old Ercc1−/− mice relative to WT littermates (n≥5 per group). Data are presented as a mean ± SD. Statistical analysis was done using Mann-Whitney U-test (*, p<0.05; **, p<0.01 for Ercc1−/− vs. WT).
3.4.3. Translesion synthesis
Another important mechanism that leads to increased genome instability is activation of translesion (TLS) DNA polymerases, which can bypass DNA lesions accumulated in ERCC1-deficient mice, but often in an error-prone manner. TLS is activated by monoubiquitylation of PCNA on K164, which facilitates exchange of error-free replicative polymerase for error-prone TLS DNA polymerase [65]. We measured activation of TLS polymerases in WT and Ercc1−/− MEFs in response to HNE by Western blot. PCNA monoubiquitylation, as indicated by PCNA size during electrophoretic migration, was increased in cells treated with HNE for 2 – 10 h post-treatment (Fig. 5G). Interestingly, activation was greater in Ercc1−/− MEFs compared to WT, again consistent with HNE inducing more DNA damage and increasing genome instability in the mutant cells.
TLS polymerases are also regulated by HNE at the transcription level [66]. Thus, we measured the level of mRNAs of TLS polymerases following 2 and 6 h treatment of cells with HNE. TLS polymerase transcription was greater in ERCC1-deficient than in WT MEFs (Fig. 5H). The ratio of polymerase mRNA in Ercc1−/− MEFs to that in WT was 3.7 for Polζ, 2 for Polκ, 1.3 for Rev1 and 1.1 for Polη. The level of Polι transcript was similar in both genotypes. Interestingly, HNE also induced transcription of some TLS polymerases in Xpa−/− MEFs, which are missing NER but not ICL repair (Fig. S5). However, their profile was different than in ERCC1-XPF deficient cells. In Xpa−/− MEFs transcription of two TLS polymerases was increased over that in WT: Polζ – 1.9-fold and Polκ – 3.1-fold. These results are consistent with Rev1 and Polη having roles in ICL repair [67, 68], Our results may also suggest that Pol ζ plays an important role in the protection of human cells by carrying out TLS across bulky adducts (LPO adducts) and cross-links, as suggested by Suzuki and coworkers [69].
Interestingly, mRNA level of TLS polymerases appeared increased in tissues of Ercc1−/− compared to WT mice. However, statistically significant differences were observed only for Polκ in brain and Polκ and Polζ in kidney (Fig. 5I and J) Surprisingly, no change in the level of TLS polymerase mRNA was observed in liver of Ercc1−/− mice (Fig. 5K). Thus, there is evidence of persistent DNA damage in tissues of ERCC1-deficient mice, which are bypassed by TLS polymerases.
It is also worth to note that homologous recombination is either similar or slightly decreased in ERCC1-deficient mice in comparison to WT ones, as judged by sister chromatid exchanges frequency (for description see Supplementary and Fig. S6). Thus deficiency of error-proof BER, NER and HR repair systems, and in the same time, increase of error-prone TLS tremendously decreases genome stability in ERCC1-deficient mice under chronic oxidative stress and lipid peroxidation.
3.5. Physiological consequences of LPO in an ERCC1-deficient background
3.5.1. Possible energy failure
Poly(ADP-ribose) polymerase consumes cellular NAD+ for protein PARylation, and thus might upset the energy balance of cells. We measured the level of ATP, NAD+ and NADH in HNE-treated immortalized cells. Indeed, treatment of cells with HNE decreased the level of all studied energetic carriers both in WT and ERCC1-deficient cells (Fig. 6A-C). Particularly dramatic was decrease of NADH level. However, statistically significant differences between genotypes were found only for NADH after treatment with HNE (5-20 μM) (Fig. 6C). Taking into consideration that LPO level is much higher in liver tissues of ERCC1-deficient than proficient mice (Fig. 2C), LPO might have a more detrimental effect on bioenergetics in ERCC1-deficient mice compared to WT mice.
3.5.2. Senescence and cell death
To investigate further the cellular response to LPO, we measured in WT and Ercc1−/− cells senescence, apoptosis and necrosis in response to a wide range of HNE concentrations (5-40 μM), occurring in mammals under oxidative stress [1]. HNE induced senescence in immortalized (Fig. 6D, E) and in primary (Fig. 6F and G) Ercc1−/− MEFs. It is worth noting that significantly more senescent cells were observed in primary cell populations compared to immortalized ones. Also only in Ercc1−/− primary MEFs the number of senescent cells both untreated and treated was higher compared to WT (Fig. 6F and G, Fig. S3). Notably, addition of a radical scavenger XJB-5-131 to the cultures suppressed ROS and signs of cellular senescence (Fig. S3).
Apoptosis was assessed by measuring the activity of caspases 3 and 7 in immortalized MEFs. Beginning at 20 μM HNE caspase activity was significantly increased in both cell lines, but to a lesser extent in Ercc1−/− cells than in WT (Fig. 6H). A control genotoxic agent, camptothecin, elicited a similar result, lower apoptosis in Ercc1−/− than in WT cells. To verify this, we did Western blot detection of procaspases and caspases 3 and 7 at different time intervals following treatment of cells with 20 μM HNE. In WT cells, cleaved caspase 3 and 7 were detected by 12 h after a 2 h treatment of cells with HNE, and with time the quantity of cleaved forms increased (Fig. 6I, lanes 2-4). Caspase cleavage was lower in Ercc1−/− cells in response to the same HNE concentration and time of treatment (Fig. 6I, lanes 7-9). Similar pattern was observed for cleavage of poly(ADP-ribose) polymerase 1 (PARP1), a protein which is a substrate for caspase 3 proteolysis (Fig. 6I). Cleaved form of PARP1 appeared 12 h following HNE treatment and increased with time, again more in WT controls than in Ercc1−/− mutants.
Since Ercc1−/− cells were more sensitive to HNE than WT, we measured the activity of proteases released from cells as a measure of necrosis. In both cell types HNE induced necrosis, however significantly more effectively in Ercc1−/− than in WT immortalized cells (Fig. 6J). These results suggest that in response to LPO product HNE, Ercc1−/− cells undergo necrosis and apoptosis, however necrosis is a major pathway of cell death. Summarizing, LPO severely affects proliferation of ERCC1-deficient cells by inducing senescence and necrosis, and to a lesser extent, apoptosis.
3.6. Dietary PUFAs decrease lifespan of Ercc1−/∆ mice
An important source of LPO in mammals is dietary fats. Dietary polyunsaturated fatty acids (PUFAs), harboring conjugated double bonds, are particularly prone to LPO [70], and diet rich in PUFAs, such as soybean, corn, sunflower or safflower oil can promote endogenous lipid peroxidation [71]. Therefore, by feeding mice a diet enriched with PUFAs derived from safflower oil, it is possible to increase endogenous LPO.
Mice were fed either a diet high in PUFAs (safflower oil) or an isocaloric, control diet, in which the PUFAs were replaced with saturated fats (coconut oil), starting at 21 days of age (at weaning) and continued throughout their entire lifespan. Mice were sacrificed when they were terminal, showing decreased spontaneous movement. Maximum lifespan was significantly reduced in PUFA-fed Ercc1−/∆ mice compared to mice fed the control diet (Fig. 7A and B). In addition, Ercc1−/∆ mice that were fed the diet high in PUFAs exhibited increased aging-associated symptoms, including hind-limb muscle wasting, decreased grooming and increased kyphosis (Fig. 7C). Importantly, there was not a difference in the weights of mice on the special diets compared to those fed normal mouse chow, indicating that difference in lifespan could not be attributed to caloric differences (Fig. S7).
Fig. 7. Dietary PUFAs decrease the lifespan of Ercc1−/∆ mice.
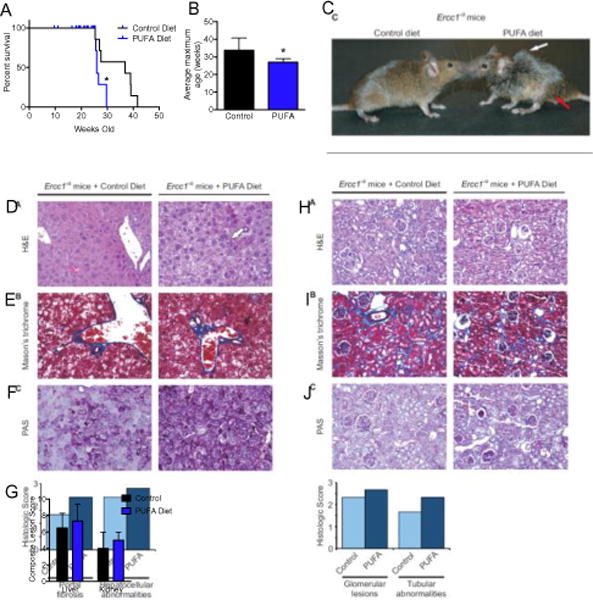
(A) Kaplan-Meier survival curve of Ercc1−/∆ mice fed a diet rich in PUFAs compared to a control diet that contained saturated fats. p=0.018, Log-rank Mantel-Cox test. (B) The average maximum lifespan was calculated using the mean age of the oldest 20% of mice on each diet [95] (n=6 per group; p=0.016, two-tailed Student’s t-test). (C) Photograph of control- or PUFA-fed Ercc1−/∆ mice at 20 weeks of age. The white arrow illustrates kyphosis and the red arrow indicates hind-limb muscle wasting. (D) H&E stained liver sections from control- or PUFA-fed Ercc1−/∆ mice (20× objective). White arrow illustrates necrosis. (E) Masson’s trichrome stained liver sections from control- or PUFA-fed Ercc1−/∆ mice (20× objective). Blue indicates areas of fibrosis. (F) Periodic acid Schiff (PAS) stained liver sections from control- or PUFA-fed Ercc1−/∆ mice (20× objective). Dark purple indicates areas of glycogen deposition. (G) The histogram indicates the average pathologic score assigned indicating the extent of portal fibrosis and hepatocellular changes in each group (n=3 mice per group). The scores were assigned by a veterinary pathologist who was blinded as to the diet of the mice. (H) H&E stained kidney sections from control- or high PUFA-fed Ercc1−/∆ mice (20× objective). (I) Masson’s trichrome stained kidney sections from control- or high PUFA-fed Ercc1−/∆ mice (20× objective). (J) Periodic acid Schiff (PAS) stained kidney sections from control- or high PUFA-fed Ercc1−/∆ mice (20× objective). (K) The histogram indicates the average pathologic score assigned indicating the extent of glomerulosclerosis and changes in the renal tubular epithelium for each group (n=3 mice per group). The scores were assigned by a veterinary pathologist who was blinded as to the diet of the mice.
A veterinary pathologist, blinded to the treatment group, examined the tissue sections for histologic changes and scored the severity. Hematoxylin and eosin stained liver sections revealed portal fibrosis, necrosis and pyknotic nuclei in liver sections from Ercc1−/∆ mice fed the PUFA diet compared to Ercc1−/∆ mice fed the control diet (Fig. 7D). Portal fibrosis was also evident by staining Ercc1−/∆ livers with Masson’s trichrome (Fig. 7E). The severity of portal fibrosis was scored on average 30% higher (more severe) in ERCC1-deficient mice fed the diet high in PUFAs compared to mutant mice fed the control diet (Fig. 7G). Additionally, Ercc1−/∆ mice fed the diet high in PUFAs had an increase in the amount of PAS staining (glycogen storage) compared to Ercc1−/∆ mice on the control diet (Fig. 7F). The liver sections were also scored for portal fibrosis and hepatocellular abnormalities. Hepatocellular abnormalities were more severe in mutant mice fed the high PUFA diet compared to the control diet (Fig. 7G). Taken together, these data indicate that dietary PUFA can promote aging-associated degenerative changes in the liver [72].
Kidneys from PUFA-fed Ercc1−/∆ mice showed a loss of renal epithelial tubular cells and increased glomerulosclerosis compared to control-fed Ercc1−/∆ mice (Fig. 7H). However, Masson’s trichrome stain of kidney cortex did not show differences in fibrosis between animals on the two diets (Fig. 7I). In contrast, PAS stain revealed an increase in extra-cellular matrix accumulation around the glomeruli in mutant mice fed the high PUFA diet (Fig. 7J). When the kidney sections were scored for histopathology, there was an increase in both glomerular lesions and tubular abnormalities in the Ercc1−/∆ mice fed a high PUFA diet compared to mice fed the control diet (Fig. 7K). These data demonstrate that dietary PUFAs can promote aging-related degenerative changes in the kidney.
4. DISCUSSION
The products of LPO constitute a class of reactive aldehydes which are implicated in the etiology of diverse diseases, as well as potentially in aging [7]. To specifically investigate the biological impact of LPO-induced DNA damage, we employed ERCC1-XPF deficient cells and mice, lacking in NER and ICL repair. NER system is required to repair numerous types of bulky adducts including adducts caused by reactive aldehydes [7]. Thus, as expected, Ercc1−/∆ mice have elevated levels of oxidized DNA bases, like 8,5′-cyclopurines [53] and d(G[8-5]C) intrastrand crosslinks [52]. The animals also have an increased level of LPO products, namely lipid hydroperoxides, acrolein and lipofuscin [28]. This may reflect the fact that the Ercc1−/∆ mice age prematurely, and LPO is increased in old age [73]. Therefore, Ercc1−/∆ mice have evidence of increased LPO and LPO-induced damage, but how this contributes to their premature aging phenotype is not clear.
In vitro, we investigated the effects of LPO on ERCC1-deficient cells. We found that Ercc1−/− MEFs are more sensitive to HNE, CRO and MDA than WT cells, but not to ACR (Fig. 1A-E). In contrast, NER-deficient Xpa−/− MEFs were not hypersensitive to HNE (Fig. 1F). Since ACR is not known to form ICLs, unlike the other LPO products tested [54–56], and XPA is not required for ICL repair, these results suggest that LPO-induced ICLs may be particularly toxic lesions when not repaired.
To test this in vivo, we compared the sensitivity of WT, Ercc1−/∆ and Xpa−/− mice to CCl4, a potent inducer of LPO and common environmental contaminant. At a dose that was sublethal to WT and Xpa−/− mice, CCl4 significantly decreased the median lifespan of Ercc1−/∆ mice (Fig. 2A and B, Fig. S1, p=0.0032). Reduced lifespan of CCl4-treated ERCC1-deficient mice was accompanied with very high level of lipid hydroperoxides (LOOH) in their livers, about 10-fold higher than in control, CCl4-treated littermates (Fig. 2C). Interestingly, the level of LOOH was already strongly elevated in mutant mice without any treatment (7-fold, vs. untreated WT animals). Additionally, CCl4 induced karyomegaly, nodular regeneration, necrosis, fibrosis, steatosis and glycogen deposition in liver of Ercc1−/∆ mice, signs of tissue damage that are often associated with aging (Fig. 2D-F). These in vivo data further support the conclusion that LPO can have toxic consequences in particular if repair of DNA damage is not possible.
Loss of regenerative capacity, cellular senescence and cell death are implicated in aging and age-related degenerative diseases [74]. HNE inhibits cell proliferation to a significantly greater degree in Ercc1−/− MEFs than in WT (Fig. 1G and H). Even 5 days following treatment of cells with 20 μM HNE, the cells did not fully recover from growth arrest. In blood plasma, HNE concentration ranges from 0.1 to 3 μM, and under oxidative stress increases to 10 μM. HNE is more abundant in cells, and its highest concentration measured directly in human cells is 46 μM [1]. The following factors may contribute to the permanent growth inhibition induced by HNE in Ercc1−/− MEFs: (i) cell senescence (Fig. 6D-G), (ii) necrosis (Fig. 6J), (iii) increased ROS and LPO (Fig. 3A-D), (iv) increased DNA damage including SSBs, DSBs, and oxidative lesions repaired by NER, BER or ICL repair machinery (Fig. 4), (v) decreased repair capacity and increased mutagenesis driven by TLS polymerases bypassing DNA lesions (Fig. 5G and H), (vi) altered energetics, particularly NADH depletion (Fig. 6A-C).
The question arises: how does nuclear DNA damage (for example in the absence of ERCC1-dependent DNA repair) trigger increased ROS? A recent study demonstrated that in progeroid Ercc1−/∆ mice and aged WT mice, both of which have increased oxidative DNA damage [53], there are mitochondrial abnormalities, increased xanthine oxidase and NADPH oxidase activity, and decreased antioxidant buffering capacity, all of which likely contribute to the increased ROS [75]. Furthermore, reduced DNA repair leads to accelerated accumulation of DNA damage in vivo [53], which is a major driver of cellular senescence [74]. Indeed, Ercc1−/∆ mice have accelerated accumulation of senescent cells [75]. Senescent cells secrete pro-inflammatory factors as part of their secretory phenotype [76, 77]. Indeed, these pro-inflammatory factors are detected in serum and tissues of Ercc1−/∆ mice [75, 78]. This secretory phenotype activates both the innate and adaptive immune system [79]. Activated macrophages secrete reactive oxygen and nitrogen species, the latter being a main source of lipid peroxidation [80]. It was demonstrated that reactive species secreted by activated immune cells damage DNA of neighbor cells [81]. In response to one of major LPO products, HNE, a higher percent of senescent cells was observed in the population of ERCC1-deficient than in WT cells (Fig. 6D–G). Senescing cells are unable to divide and are characterized by a greater volume, increased lysosomal β-galactosidase activity [82] and lipofuscin accumulation [83]. Ercc1−/− and Ercc1−/∆ mice have evidence of increased senescence compared to age-matched WT mice [84]. The increased senescence in DNA repair-deficient Ercc1−/− MEFs is most logically a direct consequence of unrepaired DNA damage induced by LPO. However, other factors may contribute, for example, increased ROS production (Fig. 3A and B) leading to increased LPO (Fig. 3C and D) and a vicious cycle of increasing oxidative stress. Increased oxidative stress in Ercc1−/∆ mice was recently confirmed by measurement of superoxide anion (via oxidation of hydroethidine to its O2-specific product, 2-hydroxyethidium (2-OH-E+), which was assessed by HPLC coupled to electrochemical detection and validated by electron paramagnetic resonance (EPR) spin trapping [85], as well as measurement of free radicals detected by immuno-spin trapping with the nitrone EPR spin trap 5,5-20 dimethyl-1-pyrroline-N-oxide (DMPO), and HNE adducts to proteins [75]. In that study, Ercc1−/∆ mice were chronically treated with a mitochondrial-targeted radical scavenger, which has been demonstrated to significantly reduce oxidation of the mitochondrial phospholipid cardiolipin [86]. This was sufficient to suppress oxidative damage, senescence and age-related pathology [75], implicating oxidative stress as the ultimate driver of cellular and physiological decline. The data herein implicate LPO products as an important component of that oxidative stress.
Importantly, in ERCC1-deficient MEFs LPO products and their precursors are neutralized with much lower efficiency than in WT cells, since the activity of glutathione transferase and selenium-dependent glutathione peroxidase, the enzymes responsible for removing major LPO products from the cells [87], are decreased (Fig. 3E and F). Although previous study by Niedernhofer and coworkers [24] showed that detoxification-related genes are rather up than downregulated in ERCC1-deficient background, LPO products may react with enzymes and affect their activity, so upregulated transcription of antioxidant-related genes is not sufficient to provide the proper level of defense against ROS and LPO products. Indeed, we have found a higher level of HNE-protein adducts in livers of ERCC1-deficient than proficient mice [75]. This might result in decrease of antioxidant enzymes, and some BER steps (ligation, Fig. 5A-F).
HNE, MDA and other long chain LPO products bind to the DNA and form adducts, which block replication or/and trigger mutations, and are removed from the DNA mainly by the NER system [10]. In ERCC1-deficient cells HNE-DNA adducts may be recognized by NER proteins, but the lesion is not excised due to the lack of ERCC1-XPF endonuclease activity. Although incision at 3′-side of the lesion by XPG endonuclease may occur generating SSBs, but without catalytic activity of ERCC1-XPF this incision is very inefficient, yet still possible [88]. We observed a massive protein PARylation, widely recognized as a marker of SSBs, as soon as 2 h following treatment of Ercc1−/− cells with HNE (Fig. 4B). Therefore, we suggest that these SSBs were formed due to one-side incision of XPG and/or increased oxidative stress in Ercc1−/− cells producing lesions repaired by BER. This is supported by higher 8-oxo-deoxyguanosine level in Ercc1−/− MEFs than in WT cells grown both in 3% and in 21% of oxygen (Fig. 4A). SSBs were repaired up to 12 h after HNE treatment, since the level of protein PARylation returned to baseline at that time after HNE treatment. In Ercc1−/− cells there was also evidence of increased DSBs in response to HNE, as histone γH2AX levels were greater in ERCC1-deficient than in proficient cells (Fig. 4D). Protein PARylation did not increase significantly until 24-48 h after HNE treatment in WT cells (Fig. 4B). This coincides with appearance of apoptosis markers (cleaved PARP1 and caspase 3 and 7, Fig. 6I) and is likely explained by an increase in DSBs due to induction of apoptosis [89].
SSBs and protein PARylation lead to depletion of NAD+/NADH and ATP resulting in energetic failure in cells [90]. We observe such phenomenon in both genotypes of MEFs (Fig. 6A-C), but in Ercc1−/− cells at low HNE concentrations the level of NADH was significantly lower than in WT (Fig. 6C). Since LPO is elevated in ERCC1-deficient cells and tissues (Fig. 2C, Fig 3C and D), more dramatic energy failure may occur in deficient animals compared to WT. For example, under conditions in which HNE level in cells of WT mice would be 5 μM, in Ercc1−/− mice HNE concentration would reach 35 μM. Taking into account these differences the level of ATP in Ercc1−/− cells would reach only 65 % of that in WT cells, the level of NAD+ 50 % of that in WT cells, and the level of NADH only 35 % of that in WT cells (Fig. 6A-C). Thus, ERCC1-deficient mice grown in the same conditions as WT animals, could suffer from the energy failure.
Energy failure could contribute to differences between genotypes in the mode of cell death induced by HNE. HNE efficiently induces apoptosis in WT but not Ercc1−/− cells (Fig. 6H and I). This is consistent with the transcriptome of Ercc1−/− mice, indicating suppression of pro-apoptotic pathways [24], which is also characteristic of senescent cells [91].
We have previously found that BER pathway is highly impaired by HNE, most probably due to dysregulation of enzymes activities caused by protein modification [20]. DNA ligation, the last step of BER is particularly sensitive to HNE. Indeed, DNA ligation step was significantly impaired in brain, liver and kidney of Ercc1−/− mice (Fig. 5A-F). This may be caused by chronic exposure to LPO in vivo, compared to in vitro conditions, where no differences in DNA ligation between WT and ERCC1-deficient cells were detected (Fig. S4K and L).
ICLs, unrepaired bulky adducts, some oxidative lesions and SSBs can all inhibit replication. Stalled replication forks may trigger exchange of replicative DNA polymerases for error-prone DNA polymerases (TLS polymerases) of Y family, a process stimulated by PCNA monoubiquitylation [92]. Indeed, even in untreated Ercc1−/− cells PCNA monoubiquitylation was greater than in WT MEFs (Fig. 5G). HNE enhanced PCNA monoubiquitylation both in Ercc1−/− and WT cells, but much more efficiently in an ERCC1-deficient background. This suggests increased induction of mutations in ERCC1-deficient mice in response to LPO products.
TLS polymerases may collaborate to bypass HNE-dG adducts in an error-free mechanism. The mostly studied HNE-dG adduct inhibits all DNA polymerases. Polι is, however, able to introduce dC opposite HNE-dG and then dissociate. Polκ, in turn, may extend DNA synthesis yielding a non-mutagenic event [17]. Polζ, Polκ and Rev1 expression are increased in Ercc1−/− MEFs compared to WT (Fig. 5H). Polζ is the first TLS polymerase to exchange with the replicative DNA polymerase and is a major source of mutations [93]. Furthermore, Polζ participates in repair of ICLs and bulky adducts, such as HNE-DNA monoadducts [94]. Thus, increased use of TLS polymerases in Ercc1−/− MEFs might be a protective mechanism against strongly cytotoxic long-chain reactive aldehyde adducts to DNA bases, but as well would result in increase in mutation frequency. We also investigated mRNA level of TLS polymerases in tissues of Ercc1−/− mice (Fig. 5I-K). To our surprise, slightly elevated mRNA levels of all studied TLS polymerases (ζ, η, ι, κ, Rev1) were found in brain and kidney (except Polη), but not in the liver. Failure to detect increased TLS transcripts in liver may reflect the fact that the mice were euthanized close to the end of their lifespan and these mice die of liver failure [84].
5. CONCLUSIONS
In summary, we find that LPO products are highly toxic to DNA repair-deficient Ercc1−/− cells. A major endogenous LPO product, HNE, induces cell senescence and necrosis. The unique sensitivity of ERCC1-deficient cells implicates LPO-induced ICLs and to a lesser extent bulky adducts in driving these cell fates. However, we also demonstrate that LPO promotes ROS (due to uncoupling mitochondrial electron transport and oxidative phosphorylation, as well as decrease of antioxidant enzyme activities), the indirect production of DNA damage such as oxidized DNA bases, strand breaks, and altered responses to genotoxic stress (reduced BER, most probably due to direct effect of LPO on BER proteins regulation, increased TLS bypass, and energy failure), which likely also contribute to determining cell fate. In vivo, DNA repair-deficient mice were hypersensitive to two sources of environmentally-driven LPO, which accelerated the onset of age-related degenerative changes in tissues and shortened lifespan. Importantly, one of these LPO sources was dietary polyunsaturated fatty acids which are consumed regularly in a western diet. Our data implicate LPO-induced DNA damage as playing a causal role in promoting age-related degenerative changes.
Supplementary Material
HIGHLIGHTS.
ERCC1-deficient mice have increased oxidative stress and LPO
LPO induced DNA and protein damage causes senescence and necrosis in Ercc1−/− cells
HNE induces promutagenic imbalance in BER and TLS in Ercc1−/− mice
Differential antioxidative defense and energy production in Ercc1−/− cells
LPO contributes to premature aging and morbidity
Acknowledgments
Experiments were carried out with the use of CePT infrastructure financed by the European Union – the European Regional Development Fund (Innovative economy 2007 – 2013, Agreement POIG.02.02.00-14-024/08-00). The authors would like to thank Prof. Leon H. F. Mullenders for providing Xpa−/− cells and Aleksander Chlebowski for his help in microscopy studies.
FUNDING
This work was supported by the Polish Ministry of Science and Higher Education, grant. N N303 819540 (BT) and by the National Institutes of Health, grants P01-AG043376 (LJN), R01-ES016114 (LJN), P30-AG024837 (LJN), R03-CA121411 (LJN), K99/R00-AG049126 (AUG) and the Ellison Medical Foundation AG-NS-0303-05 (LJN).
Abbreviations
- 8-oxodG
8-oxo-deoxyguanosine
- ACR
acrolein
- BER
base excision repair
- CPT
camptothecin
- CRO
crotonaldehyde
- DSB
double strand break
- HNE
4-hydroxy-2-nonenal
- HR
homologous recombination
- ICL
DNA interstrand crosslink
- LPO
lipid peroxidation
- MDA
malondialdehyde
- MEFs
mouse embryonic fibroblasts
- NAD(H)
nicotinamide adenine dinucleotide
- NER
nucleotide excision repair
- PAR
poly(ADP-ribose)
- PUFA
polyunsaturated fatty acids
- ROS
reactive oxygen species
- SSB
single strand break
- TBHP
tert-butyl-hydroperoxide
- TCR
transcription-coupled repair
- TLS
translesion synthesis
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTION
JC, MN, PW designed the study and showed sensitivity of MEFs to LPO products, senescence, aging, cell death types, LPO, DNA damage and repair; JMC was engaged in qPCR; DDL, DK, BG, AB-K studied ROS formation and antioxidant enzymes activity; KK studied LPO effect in XPA cells; MB and ES helped with BER activities; BT, LJN, JC and ES designed the study and prepared parts of manuscript; ARR and HLS did all mice work; LHR was engaged in histopathology studies; MJY and AUG were engaged in studies on primary cells; EZ, DG and RO studied the level of 8-oxodG.
CONFLICT OF INTEREST
None declared
References
- 1.Poli G, et al. 4-hydroxynonenal: a membrane lipid oxidation product of medicinal interest. Med Res Rev. 2008;28(4):569–631. doi: 10.1002/med.20117. [DOI] [PubMed] [Google Scholar]
- 2.Ando K, Beppu M, Kikugawa K. Evidence for accumulation of lipid hydroperoxides during the aging of human red blood cells in the circulation. Biol Pharm Bull. 1995;18(5):659–63. doi: 10.1248/bpb.18.659. [DOI] [PubMed] [Google Scholar]
- 3.Honma T, et al. Aging decreases antioxidant effects and increases lipid peroxidation in the Apolipoprotein E deficient mouse. J Clin Biochem Nutr. 2013;52(3):234–40. doi: 10.3164/jcbn.12-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bradley MA, Markesbery WR, Lovell MA. Increased levels of 4-hydroxynonenal and acrolein in the brain in preclinical Alzheimer disease. Free Radic Biol Med. 2010;48(12):1570–6. doi: 10.1016/j.freeradbiomed.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Busciglio J, Yankner BA. Apoptosis and increased generation of reactive oxygen species in Down’s syndrome neurons in vitro. Nature. 1995;378(6559):776–9. doi: 10.1038/378776a0. [DOI] [PubMed] [Google Scholar]
- 6.Evangelou K, et al. Robust, universal biomarker assay to detect senescent cells in biological specimens. Aging Cell. 2017;16(1):192–197. doi: 10.1111/acel.12545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Winczura A, Zdzalik D, Tudek B. Damage of DNA and proteins by major lipid peroxidation products in genome stability. Free Radic Res. 2012;46(4):442–59. doi: 10.3109/10715762.2012.658516. [DOI] [PubMed] [Google Scholar]
- 8.Siems W, Grune T. Intracellular metabolism of 4-hydroxynonenal. Mol Aspects Med. 2003;24(4–5):167–75. doi: 10.1016/s0098-2997(03)00011-6. [DOI] [PubMed] [Google Scholar]
- 9.Chung FL, et al. Formation of trans-4-hydroxy-2-nonenal- and other enal-derived cyclic DNA adducts from omega-3 and omega-6 polyunsaturated fatty acids and their roles in DNA repair and human p53 gene mutation. Mutat Res. 2003;531(1–2):25–36. doi: 10.1016/j.mrfmmm.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 10.Nair U, Bartsch H, Nair J. Lipid peroxidation-induced DNA damage in cancer-prone inflammatory diseases: a review of published adduct types and levels in humans. Free Radic Biol Med. 2007;43(8):1109–20. doi: 10.1016/j.freeradbiomed.2007.07.012. [DOI] [PubMed] [Google Scholar]
- 11.Niedernhofer LJ, et al. Malondialdehyde, a product of lipid peroxidation, is mutagenic in human cells. J Biol Chem. 2003;278(33):31426–33. doi: 10.1074/jbc.M212549200. [DOI] [PubMed] [Google Scholar]
- 12.Kowalczyk P, et al. Long-chain adducts of trans-4-hydroxy-2-nonenal to DNA bases cause recombination, base substitutions and frameshift mutations in M13 phage. Mutat Res. 2004;550(1–2):33–48. doi: 10.1016/j.mrfmmm.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 13.Maddukuri L, et al. Cockayne syndrome group B protein is engaged in processing of DNA adducts of lipid peroxidation product trans-4-hydroxy-2-nonenal. Mutat Res. 2009;666(1–2):23–31. doi: 10.1016/j.mrfmmm.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Janowska B, et al. Nucleotide excision repair and recombination are engaged in repair of trans-4-hydroxy-2-nonenal adducts to DNA bases in Escherichia coli. Int J Biol Sci. 2009;5(6):611–20. doi: 10.7150/ijbs.5.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng Z, et al. Mutational spectrum and genotoxicity of the major lipid peroxidation product, trans-4-hydroxy-2-nonenal, induced DNA adducts in nucleotide excision repair-proficient and -deficient human cells. Biochemistry. 2003;42(25):7848–54. doi: 10.1021/bi034431g. [DOI] [PubMed] [Google Scholar]
- 16.Janowska B, et al. Role of damage-specific DNA polymerases in M13 phage mutagenesis induced by a major lipid peroxidation product trans-4-hydroxy-2-nonenal. Mutat Res. 2012;729(1–2):41–51. doi: 10.1016/j.mrfmmm.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 17.Wolfle WT, et al. Replication past a trans-4-hydroxynonenal minor-groove adduct by the sequential action of human DNA polymerases iota and kappa. Mol Cell Biol. 2006;26(1):381–6. doi: 10.1128/MCB.26.1.381-386.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dalleau S, et al. Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance. Cell Death Differ. 2013;20(12):1615–30. doi: 10.1038/cdd.2013.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feng Z, Hu W, Tang MS. Trans-4-hydroxy-2-nonenal inhibits nucleotide excision repair in human cells: a possible mechanism for lipid peroxidation-induced carcinogenesis. Proc Natl Acad Sci U S A. 2004;101(23):8598–602. doi: 10.1073/pnas.0402794101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Winczura A, et al. Lipid peroxidation product 4-hydroxy-2-nonenal modulates base excision repair in human cells. DNA Repair (Amst) 2014;22:1–11. doi: 10.1016/j.dnarep.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 21.Gregg SQ, Robinson AR, Niedernhofer LJ. Physiological consequences of defects in ERCC1-XPF DNA repair endonuclease. DNA Repair (Amst) 2011;10(7):781–91. doi: 10.1016/j.dnarep.2011.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niedernhofer LJ, et al. The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol Cell Biol. 2004;24(13):5776–87. doi: 10.1128/MCB.24.13.5776-5787.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jaspers NG, et al. First reported patient with human ERCC1 deficiency has cerebro-oculo-facio-skeletal syndrome with a mild defect in nucleotide excision repair and severe developmental failure. Am J Hum Genet. 2007;80(3):457–66. doi: 10.1086/512486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Niedernhofer LJ, et al. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature. 2006;444(7122):1038–43. doi: 10.1038/nature05456. [DOI] [PubMed] [Google Scholar]
- 25.Chen Q, et al. DNA damage drives accelerated bone aging via an NF-kappaB-dependent mechanism. J Bone Miner Res. 2013;28(5):1214–28. doi: 10.1002/jbmr.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dolle ME, et al. Broad segmental progeroid changes in short-lived Ercc1(-/Delta7) mice. Pathobiol Aging Age Relat Dis. 2011;1 doi: 10.3402/pba.v1i0.7219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goss JR, et al. Premature aging-related peripheral neuropathy in a mouse model of progeria. Mech Ageing Dev. 2011;132(8–9):437–42. doi: 10.1016/j.mad.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gregg SQ, et al. A mouse model of accelerated liver aging caused by a defect in DNA repair. Hepatology. 2012;55(2):609–21. doi: 10.1002/hep.24713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spoor M, et al. Accelerated loss of hearing and vision in the DNA-repair deficient Ercc1(delta/-) mouse. Mech Ageing Dev. 2012;133(2–3):59–67. doi: 10.1016/j.mad.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 30.Bogliolo M, et al. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am J Hum Genet. 2013;92(5):800–6. doi: 10.1016/j.ajhg.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhagwat N, et al. XPF-ERCC1 participates in the Fanconi anemia pathway of cross-link repair. Mol Cell Biol. 2009;29(24):6427–37. doi: 10.1128/MCB.00086-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garaycoechea JI, et al. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature. 2012;489(7417):571–5. doi: 10.1038/nature11368. [DOI] [PubMed] [Google Scholar]
- 33.Pontel LB, et al. Endogenous Formaldehyde Is a Hematopoietic Stem Cell Genotoxin and Metabolic Carcinogen. Mol Cell. 2015;60(1):177–88. doi: 10.1016/j.molcel.2015.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Falnes PO, Klungland A, Alseth I. Repair of methyl lesions in DNA and RNA by oxidative demethylation. Neuroscience. 2007;145(4):1222–32. doi: 10.1016/j.neuroscience.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 35.Walport LJ, Hopkinson RJ, Schofield CJ. Mechanisms of human histone and nucleic acid demethylases. Curr Opin Chem Biol. 2012;16(5–6):525–34. doi: 10.1016/j.cbpa.2012.09.015. [DOI] [PubMed] [Google Scholar]
- 36.Komisarski M, Kaczmarska Z, Kusmierek JT. Practical highly enantioselective synthesis of (R)- and (S)-(E)-4-hydroxynon-2-enal. Acta Biochim Pol. 2009;56(1):189–93. [PubMed] [Google Scholar]
- 37.Li G, Li L, Yin D. A novel observation: melatonin’s interaction with malondiadehyde. Neuro Endocrinol Lett. 2005;26(1):61–6. [PubMed] [Google Scholar]
- 38.Gackowski D, et al. Accurate, Direct, and High-Throughput Analyses of a Broad Spectrum of Endogenously Generated DNA Base Modifications with Isotope-Dilution Two-Dimensional Ultraperformance Liquid Chromatography with Tandem Mass Spectrometry: Possible Clinical Implication. Anal Chem. 2016;88(24):12128–12136. doi: 10.1021/acs.analchem.6b02900. [DOI] [PubMed] [Google Scholar]
- 39.Modrzejewska M, et al. Vitamin C enhances substantially formation of 5-hydroxymethyluracil in cellular DNA. Free Radic Biol Med. 2016;101:378–383. doi: 10.1016/j.freeradbiomed.2016.10.535. [DOI] [PubMed] [Google Scholar]
- 40.Wolff S, Perry P. Differential Giemsa staining of sister chromatids and the study of chromatid exchanges without autoradiography. Chromosoma. 1974;48(4):341–53. doi: 10.1007/BF00290991. [DOI] [PubMed] [Google Scholar]
- 41.Dimri GP, et al. A Biomarker That Identifies Senescent Human-Cells in Culture and in Aging Skin in-Vivo. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(20):9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vandesompele J, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3(7) doi: 10.1186/gb-2002-3-7-research0034. RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanaka M, Lai JS, Herr W. Promoter-selective activation domains in Oct-1 and Oct-2 direct differential activation of an snRNA and mRNA promoter. Cell. 1992;68(4):755–67. doi: 10.1016/0092-8674(92)90150-b. [DOI] [PubMed] [Google Scholar]
- 44.Vodenicharov MD, et al. Base excision repair is efficient in cells lacking poly(ADP-ribose) polymerase 1. Nucleic Acids Res. 2000;28(20):3887–96. doi: 10.1093/nar/28.20.3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Habig WH, Pabst MJ, Jakoby WB. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J Biol Chem. 1974;249(22):7130–9. [PubMed] [Google Scholar]
- 46.Paglia DE, Valentine WN. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J Lab Clin Med. 1967;70(1):158–69. [PubMed] [Google Scholar]
- 47.Lawrence RA, Burk RF. Glutathione peroxidase activity in selenium-deficient rat liver. Biochem Biophys Res Commun. 1976;71(4):952–8. doi: 10.1016/0006-291x(76)90747-6. [DOI] [PubMed] [Google Scholar]
- 48.Weeda G, et al. Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Curr Biol. 1997;7(6):427–39. doi: 10.1016/s0960-9822(06)00190-4. [DOI] [PubMed] [Google Scholar]
- 49.Ahmad A, et al. ERCC1-XPF endonuclease facilitates DNA double-strand break repair. Mol Cell Biol. 2008;28(16):5082–92. doi: 10.1128/MCB.00293-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Vries A, et al. Increased susceptibility to ultraviolet-B and carcinogens of mice lacking the DNA excision repair gene XPA. Nature. 1995;377(6545):169–73. doi: 10.1038/377169a0. [DOI] [PubMed] [Google Scholar]
- 51.de Waard MC, et al. Age-related motor neuron degeneration in DNA repair-deficient Ercc1 mice. Acta Neuropathol. 2010;120(4):461–75. doi: 10.1007/s00401-010-0715-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang J, et al. Endogenous formation and repair of oxidatively induced G[8-5 m]T intrastrand cross-link lesion. Nucleic Acids Res. 2012;40(15):7368–74. doi: 10.1093/nar/gks357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang J, et al. The oxidative DNA lesions 8,5′-cyclopurines accumulate with aging in a tissue-specific manner. Aging Cell. 2012;11(4):714–6. doi: 10.1111/j.1474-9726.2012.00828.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cho YJ, et al. Stereospecific formation of interstrand carbinolamine DNA cross-links by crotonaldehyde- and acetaldehyde-derived alpha-CH3-gamma-OH-1,N2-propano-2′-deoxyguanosine adducts in the 5′-CpG-3′ sequence. Chem Res Toxicol. 2006;19(2):195–208. doi: 10.1021/tx050239z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang H, et al. DNA cross-link induced by trans-4-hydroxynonenal. Environ Mol Mutagen. 2010;51(6):625–34. doi: 10.1002/em.20599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Niedernhofer LJ, et al. Temperature-dependent formation of a conjugate between tris(hydroxymethyl)aminomethane buffer and the malondialdehyde-DNA adduct pyrimidopurinone. Chem Res Toxicol. 1997;10(5):556–61. doi: 10.1021/tx960191c. [DOI] [PubMed] [Google Scholar]
- 57.Kadiiska MB, et al. Biomarkers of oxidative stress study: are plasma antioxidants markers of CCl(4) poisoning? Free Radic Biol Med. 2000;28(6):838–45. doi: 10.1016/s0891-5849(00)00198-2. [DOI] [PubMed] [Google Scholar]
- 58.Chipchase MD, O’Neill M, Melton DW. Characterization of premature liver polyploidy in DNA repair (Ercc1)-deficient mice. Hepatology. 2003;38(4):958–66. doi: 10.1053/jhep.2003.50421. [DOI] [PubMed] [Google Scholar]
- 59.Serviddio G, et al. Uncoupling protein-2 (UCP2) induces mitochondrial proton leak and increases susceptibility of non-alcoholic steatohepatitis (NASH) liver to ischaemia-reperfusion injury. Gut. 2008;57(7):957–65. doi: 10.1136/gut.2007.147496. [DOI] [PubMed] [Google Scholar]
- 60.Lee JY, et al. 4-Hydroxynonenal induces vascular smooth muscle cell apoptosis through mitochondrial generation of reactive oxygen species. Toxicol Lett. 2006;166(3):212–21. doi: 10.1016/j.toxlet.2006.07.305. [DOI] [PubMed] [Google Scholar]
- 61.Fink MP, et al. Hemigramicidin-TEMPO conjugates: novel mitochondria-targeted anti-oxidants. Biochem Pharmacol. 2007;74(6):801–9. doi: 10.1016/j.bcp.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 62.Awasthi S, et al. Novel function of human RLIP76: ATP-dependent transport of glutathione conjugates and doxorubicin. Biochemistry. 2000;39(31):9327–34. doi: 10.1021/bi992964c. [DOI] [PubMed] [Google Scholar]
- 63.Sharma R, et al. RLIP76 is the major ATP-dependent transporter of glutathione-conjugates and doxorubicin in human erythrocytes. Arch Biochem Biophys. 2001;391(2):171–9. doi: 10.1006/abbi.2001.2395. [DOI] [PubMed] [Google Scholar]
- 64.Ayyadevara S, et al. Lifespan and stress resistance of Caenorhabditis elegans are increased by expression of glutathione transferases capable of metabolizing the lipid peroxidation product 4-hydroxynonenal. Aging Cell. 2005;4(5):257–71. doi: 10.1111/j.1474-9726.2005.00168.x. [DOI] [PubMed] [Google Scholar]
- 65.Chen J, Bozza W, Zhuang Z. Ubiquitination of PCNA and its essential role in eukaryotic translesion synthesis. Cell Biochem Biophys. 2011;60(1–2):47–60. doi: 10.1007/s12013-011-9187-3. [DOI] [PubMed] [Google Scholar]
- 66.Zhou J, et al. Overexpression of DNA polymerase iota (Poliota) in esophageal squamous cell carcinoma. Cancer Sci. 2012;103(8):1574–9. doi: 10.1111/j.1349-7006.2012.02309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Roy U, et al. The structure and duplex context of DNA interstrand crosslinks affects the activity of DNA polymerase eta. Nucleic Acids Res. 2016;44(15):7281–91. doi: 10.1093/nar/gkw485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Budzowska M, et al. Regulation of the Rev1-pol zeta complex during bypass of a DNA interstrand cross-link. EMBO J. 2015;34(14):1971–85. doi: 10.15252/embj.201490878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Suzuki T, et al. The role of DNA polymerase zeta in translesion synthesis across bulky DNA adducts and cross-links in human cells. Mutat Res. 2016;791-792:35–41. doi: 10.1016/j.mrfmmm.2016.08.004. [DOI] [PubMed] [Google Scholar]
- 70.Buege JA, Aust SD. Microsomal lipid peroxidation. Methods Enzymol. 1978;52:302–10. doi: 10.1016/s0076-6879(78)52032-6. [DOI] [PubMed] [Google Scholar]
- 71.Jump DB. The biochemistry of n-3 polyunsaturated fatty acids. J Biol Chem. 2002;277(11):8755–8. doi: 10.1074/jbc.R100062200. [DOI] [PubMed] [Google Scholar]
- 72.Thoolen B, et al. Proliferative and nonproliferative lesions of the rat and mouse hepatobiliary system. Toxicol Pathol. 2010;38(7 Suppl):5S–81S. doi: 10.1177/0192623310386499. [DOI] [PubMed] [Google Scholar]
- 73.Pratico D. Lipid peroxidation and the aging process. Sci Aging Knowledge Environ. 2002;2002:50re5. doi: 10.1126/sageke.2002.50.re5. [DOI] [PubMed] [Google Scholar]
- 74.Childs BG, et al. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. 2015;21(12):1424–35. doi: 10.1038/nm.4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Robinson AR, et al. Spontaneous DNA damage to the nuclear genome promotes senescence and aging. Redox Biology. 2018 doi: 10.1016/j.redox.2018.04.007. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Coppe JP, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6(12):2853–68. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Davalos AR, et al. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. 2010;29(2):273–83. doi: 10.1007/s10555-010-9220-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yousefzadeh MJ, et al. Circulating levels of monocyte chemoattractant protein-1 as a potential measure of biological age in mice and frailty in humans. Aging Cell. 2018;17(2) doi: 10.1111/acel.12706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jeon OH, et al. Senescent cells and osteoarthritis: a painful connection. J Clin Invest. 2018;128(4):1229–1237. doi: 10.1172/JCI95147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Firth CA, et al. Macrophage mediated protein hydroperoxide formation and lipid oxidation in low density lipoprotein are inhibited by the inflammation marker 7,8-dihydroneopterin. Biochim Biophys Acta. 2008;1783(6):1095–101. doi: 10.1016/j.bbamcr.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 81.Dizdaroglu M, et al. Modification of DNA bases in chromatin of intact target human cells by activated human polymorphonuclear leukocytes. Cancer Res. 1993;53(6):1269–72. [PubMed] [Google Scholar]
- 82.Sikora E, Bielak-Zmijewska A, Mosieniak G. Cellular senescence in ageing, age-related disease and longevity. Curr Vasc Pharmacol. 2014;12(5):698–706. doi: 10.2174/1570161111666131219094045. [DOI] [PubMed] [Google Scholar]
- 83.Salmonowicz H, Passos JF. Detecting senescence: a new method for an old pigment. Aging Cell. 2017;16(3):432–434. doi: 10.1111/acel.12580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Selfridge J, et al. Correction of liver dysfunction in DNA repair-deficient mice with an ERCC1 transgene. Nucleic Acids Res. 2001;29(22):4541–50. doi: 10.1093/nar/29.22.4541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zielonka J, Vasquez-Vivar J, Kalyanaraman B. Detection of 2-hydroxyethidium in cellular systems: a unique marker product of superoxide and hydroethidine. Nat Protoc. 2008;3(1):8–21. doi: 10.1038/nprot.2007.473. [DOI] [PubMed] [Google Scholar]
- 86.Ji J, et al. Lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury. Nat Neurosci. 2012;15(10):1407–13. doi: 10.1038/nn.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Singhal SS, et al. Antioxidant role of glutathione S-transferases: 4-Hydroxynonenal, a key molecule in stress-mediated signaling. Toxicol Appl Pharmacol. 2015;289(3):361–70. doi: 10.1016/j.taap.2015.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Staresincic L, et al. Coordination of dual incision and repair synthesis in human nucleotide excision repair. EMBO J. 2009;28(8):1111–20. doi: 10.1038/emboj.2009.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Beck C, et al. Poly(ADP-ribose) polymerases in double-strand break repair: Focus on PARP1, PARP2 and PARP3. Exp Cell Res. 2014 doi: 10.1016/j.yexcr.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 90.Altmeyer M, Hottiger MO. Poly(ADP-ribose) polymerase 1 at the crossroad of metabolic stress and inflammation in aging. Aging-Us. 2009;1(5):458–469. doi: 10.18632/aging.100052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhu Y, et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. 2015;14(4):644–58. doi: 10.1111/acel.12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Andersen PL, Xu F, Xiao W. Eukaryotic DNA damage tolerance and translesion synthesis through covalent modifications of PCNA. Cell Res. 2008;18(1):162–73. doi: 10.1038/cr.2007.114. [DOI] [PubMed] [Google Scholar]
- 93.Sharma S, Helchowski CM, Canman CE. The roles of DNA polymerase zeta and the Y family DNA polymerases in promoting or preventing genome instability. Mutat Res. 2013;743–744:97–110. doi: 10.1016/j.mrfmmm.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sharma S, Canman CE. REV1 and DNA polymerase zeta in DNA interstrand crosslink repair. Environ Mol Mutagen. 2012;53(9):725–40. doi: 10.1002/em.21736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhou B, et al. Midlife gene expressions identify modulators of aging through dietary interventions. Proc Natl Acad Sci U S A. 2012;109(19):E1201–9. doi: 10.1073/pnas.1119304109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.