

Abstract
High-spin iron species with bridging hydrides have been detected in species trapped during nitrogenase catalysis, but there are few general methods of evaluating Fe-H bonds in high-spin multinuclear iron systems. Here, we report an 57Fe nuclear resonance vibrational spectroscopy (NRVS) study on an Fe(μ-H)2Fe model complex, which shows Fe-H stretching vibrations for bridging hydrides at frequencies greater than 1200 cm−1. These isotope-sensitive vibrational bands are not evident in infrared (IR) spectra, showing the power of NRVS for identifying hydrides in this high-spin iron system. Complementary density functional theory (DFT) calculations elucidate the normal modes of the rhomboidal iron-hydride core.
Keywords: nuclear resonance vibrational spectroscopy, density functional theory, iron hydrides, nitrogenases
Graphical abstract
Nuclear Resonance Vibrational Spectroscopy shows high-frequency Fe(II)-H/D isotope-sensitive stretching vibrations that are invisible in infrared spectra, indicating that this technique is useful for exposure of catalytic iron hydrides.
Hydrides of the transition metals hold a prominent position in both inorganic and organic chemistry,[1] and are involved in the mechanisms of hydrogenases[2] and nitrogenases.[3] High-spin metal hydrides[4] are rarely isolated, but are likely to be involved in catalysis with weak-field first-row metal environments[5] and as intermediates in nitrogenase catalysis at iron-sulfur clusters.[3, 6] In a species trapped during proton reduction by nitrogenase, Electron-Nuclear Double Resonance (ENDOR) spectroscopy demonstrated the presence of two iron-bound hydrides in the E4 (“Janus”) intermediate because the species releases two molecules of H2 to return to the resting E0 state.[7] The location of these hydrides is not yet known, and may be important for preparing the paramagnetic FeMo cofactor for N2 binding.[8] Unfortunately, many nitrogenase intermediates are not accessible to ENDOR,[3] and additional methods are needed.
Many common methods have limited effectiveness for characterizing paramagnetic hydride complexes.[9] For example, Nuclear Magnetic Resonance (NMR) spectra of paramagnetic compounds do not show hydride resonances because of fast relaxation.[10] IR spectra of bridging hydride complexes often show weak or undetectably small signals for M-H stretching vibrations.[9] Resonance Raman (RR) spectroscopy has been used for some bridging hydrides,[11] but many reactive polyhydride complexes are susceptible to photolysis.[12] In our extensive experience with diketiminate-supported high-spin iron hydride complexes like the iron(II) species 1 (Figure 1) we have never been able to detect an isotope-sensitive vibration attributable to a bridging hydride using IR or RR.[13] Murray has prepared Fe3H3 species with a related ligand environment and high-spin iron(II) ions, but no direct spectroscopic detection of hydrides was reported.[14] Limberg and Yang have studied related nickel hydride complexes,[15],[16] but neither report assigned hydride bands in the IR spectra. In a recent paper, Ohki has described IR stretching bands for iron-hydride clusters.[17]
Figure 1.
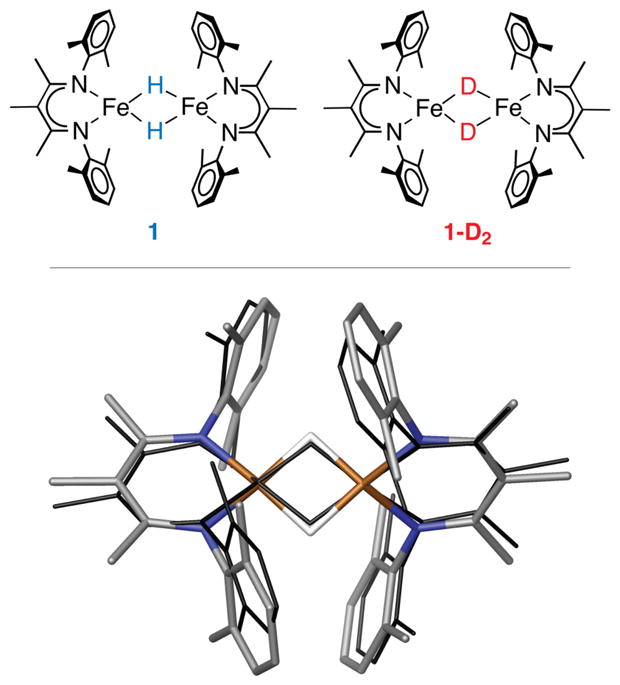
Top: schematic structures of compound 1 and its hydride isotopologue 1-D2 used in this study. Bottom: optimized DFT model of 1 (in element colors) superimposed onto its crystallographic structure (in black wire). Hydrogen atoms except the two bridging hydrides are omitted for clarity.
The increased interest in paramagnetic hydride complexes necessitates the evaluation of other spectroscopic strategies to identify and characterize hydrides. ENDOR spectroscopy of M-H and M-H2 complexes has been used by Hoffman,[18] but is mostly limited to Kramers (half-integer-spin) systems. Inelastic Neutron Scattering (INS) has also been important for detection of hydride vibrations in both molecular species and solids, but is sensitive to vibrational modes of all hydrogen atoms, and is not specific to hydrides.[19]
This paper highlights the utility of 57Fe Nuclear Resonance Vibrational Spectroscopy (NRVS), which takes advantage of resonant nuclear inelastic scattering by 57Fe nuclei.[20][21] The combination of nuclear excitation plus phonon creation/annihilation scattering yields a vibrational spectrum with intensities that are only sensitive to 57Fe nuclear motion and presented as partial vibrational density of states (PVDOS). NRVS lacks the constraints of the IR and Raman selection rules that often make key vibrations undetectable in the latter spectroscopies. The selectivity for motion of a particular isotope (in this case 57Fe) makes it practical to observe NRVS signals in spectral regions that would be otherwise obscured by other modes in IR or Raman spectra.
Fe-H stretching modes present the greatest experimental challenge to NRVS, primarily because the intensity of NRVS signals is proportional to the square of the 57Fe motion in a normal mode, and inversely proportional to its vibrational frequency.[22] In the current study, we utilized long (>24 hour) collection times with focused region of interest scans to overcome these substantial challenges, and to compare diiron bridging hydride stretches in NRVS to a structurally characterized model complex for the first time. Selective deuteration and DFT calculations strengthen the support for the band assignment to Fe-H/D vibrations, and it is particularly notable that these vibrations are not visible in the IR spectrum of a synthetic high-spin iron-hydride complex. This observation highlights the advantages of NRVS for identifying bridging hydrides in high-spin systems and implies that this technique will be useful for other high-spin iron-hydride species including those in the FeMo cofactor of nitrogenase.
This study used LFe(μ-H)2FeL (1), where L = 3-methyl-2,4-bis(2,6-dimethylphenylimido)-3-pentyl (Figure 1, top), which was previously reported in the context of N2 activation.[23] The isotopologue of 1 with deuterium at the bridging positions (1-D2) was prepared by exchange of the hydrides with D2.[13] The relative intensities of the 1H NMR resonances for the ligands were unchanged after deuteration (Figure S1), indicating that D does not exchange into the supporting ligands. The Mössbauer spectrum at 170 K (Figure S3) showed nearly identical doublets with isomer shifts of 0.47 ± 0.02 mm s−1 and quadrupole splittings of 1.25 ± 0.02 mm s−1 for both isotopologues, suggesting a high-spin electronic configuration at the iron sites in 1 and 1-D2. These values are very similar to related high-spin iron(II) hydride and alkyl complexes,[13, 24] and the isomer shift is higher than in known intermediate-spin or low-spin iron(II) compounds.[25] The solid-state magnetization of 1 is consistent with high-spin iron sites (Figure S5).
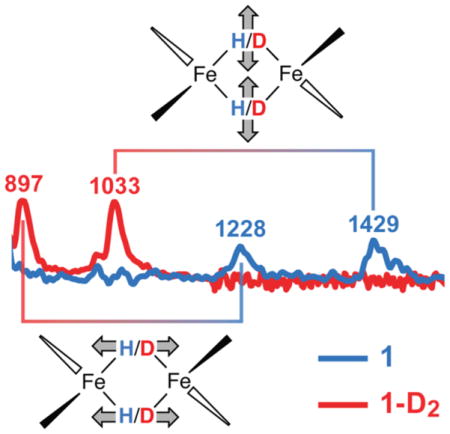
IR spectra collected on solids showed no detectable differences between 1 and 1-D2 (Figure S2), despite the excellent signal/noise ratio of the spectra. In contrast, NRVS on 1 (Figure 2a) showed distinct bands at 1429 and 1228 cm−1 that shift in 1-D2 to 1033 and 897 cm−1 that are not seen in the IR spectra. As expected for a harmonic oscillator where H/D binds a much heavier atom, the ratio of energies between the isotopologues for the NRVS features is close to (Chart 1). The energies of the higher-frequency bands are reminiscent of carbonyl-supported metal-hydride clusters, which have isotope-sensitive IR signals in the frequency range 1150 ± 300 cm−1.[9, 26] In agreement with the literature precedents, there is sharpening in the iron-deuterium modes of 1-D2 relative to 1, which may be attributed to the reduced anharmonicity of the lower-energy M-D stretching modes.[11] The intensity of the Fe-D NRVS bands is further enhanced by the greater 57Fe motion in these modes in the deuteride variant (1-D2) versus the modes in the hydride (1). In the <880 cm−1 region dominated by Fe-N motions (see the corresponding DFT normal mode animations in Supporting Information), the two isotopologues share nearly identical features.
Figure 2.

57Fe-PVDOS vibrational energies in 1 and 1-D2 characterized by NRVS experiments (a) and DFT calculations (b). DFT-computed modes of predominantly hydride motion character are further revealed via 2×H−/D−-specific PVDOS and vibrational mode assignments with respect to the Fe(μ-H)2Fe core of 1 (c). For the DFT 57Fe-PVDOS, stick-style spectra are additionally shown (b). The ×5 intensity insets display 57Fe-PVDOS spectra >430 cm−1, where equivalences between the observed and computed bands are indicated by broken vertical lines. Data for 1 are in blue, and for 1-D2 are in red, including the band positions; the bands labeled in gray are common in both isotopologues. Tentatively assigned weak NRVS features are in semi-transparent colors. The combined low- and high-energy region-of-interest (ROI) NRVS scans for 1 are additionally provided in Figure S6.
Chart 1.

Arrow-style graphic representation of the principal iron-hydride normal modes along with their computed frequencies (DFT) and corresponding observed (NRVS) band positions (cm−1) for 1 (in blue) and 1-D2 (in red). Frequencies for 1-D2 projected from 1 based on H-to-D isotopic mass shift and tentatively assigned Fe-H-Fe wagging NRVS features are in semi-transparent colors. Only the core Fe sites and their first-shell ligand atoms are shown. Animations of the DFT normal modes are available in Supporting Information.
In order to assign the normal modes, we used density functional theory (DFT) calculations employing the BP86 functional. DFT has challenges in this system because of several issues: (i) the location of the hydrides is not determined accurately from X-ray crystallography, and DFT optimizations showed a relatively flat potential energy surface, (ii) samples of 1 undergo a phase change at ~150 K that influences the Mössbauer spectrum (Figure S4) and the relative orientation of the diketiminates and the Fe(μ-H)2Fe core, and (iii) SQUID magnetometry data fit best to a model where the zero-field splitting D is larger than the exchange coupling J, and therefore there is a dense manifold of states (see Supporting Information). However, DFT is the only method that can accommodate frequency calculations on molecules of this size. With these caveats, we used geometry optimization of several possible electronic states and modelling schemes (see Tables S1–S2 and Figures S7–S10). The open-shell singlet state of 1/1-D2 bearing a broken-symmetry[27] character yielded the best agreement between peak positions in the calculated versus observed 57Fe-PVDOS spectra. The quality of the spectroscopic agreement is evident in Figure 2, where the bottom section (c) gives the calculated frequencies of the hydride-specific normal modes, and above these (b) lie the simulated NRVS spectra. The calculated vibrational energies of the bands are similar to the ones observed, despite some overlap in the broad experimental signals. The best-fit DFT-optimized structure (Figure 1, bottom) also reproduces the crystallographic Fe-Fe and Fe-N distances (Tables S1–S2). There are differences in the relative orientation of the diketiminate ligands and the Fe(μ-H)2Fe core between the low-temperature structure of 1 and the DFT-optimized structure, and our analysis assumes that these do not influence the high-energy vibrations. Future studies will examine this issue in more detail using other spectroscopic methods and neutron crystallography.
In the idealized D2 point group (which was computationally explored using the symmetrized minimal model 1M as shown in Figures S11–12 and Chart S1), there are four (μ-H/D)2 hydride normal modes with Fe-H stretching, with irreducible representations A + B1 + B2 + B3, which all involve 57Fe motion. These are expressed as calculated normal modes at 1474 (A), 1405 (B2), 1252 (B1), and 1243 (B3) in 1 (Chart 1). The high-frequency calculated ‘breathing’ mode at 1474 cm−1 has out-of-phase hydride stretching motion perpendicular to the Fe-Fe axis and is similar in energy to a calculated mode at 1405 cm−1 that is an in-phase hydride stretching motion. The pair of isotope-sensitive calculated modes at 1252 and 1243 cm−1 are out-of-phase and in-phase hydride motions parallel to the Fe-Fe axis. In the observed spectra, the two members of each pair are not clearly resolved from one another, although the experimental peaks have asymmetric shapes and shoulders that suggest overlap of several bands (Figure 2a). Lower in energy lie numerous ligand-based vibrations, including the Fe-H-Fe wagging modes (out-of-plane deformations of the Fe(μ-H)2Fe core). The calculations suggest that these wagging modes are screened by NRVS bands produced by other modes with larger 57Fe displacements. We tentatively assign the H-variant wagging modes calculated at 570/445 cm−1 with NRVS features observed at 587/455 cm−1 in 1 (Figures 2a–b, Chart 1), and the lower frequency wagging modes calculated at 407/326 cm−1 in 1-D2 are masked completely. In contrast to 1, the Fe(II)-H-Fe(II)/Ni(II) wagging and Fe(II)-H bending modes of low-spin iron(II) hydrides are not screened by other bands in the NRVS spectra of e.g. [NiFe]- and [FeFe]-hydrogenases and their bimetallic model complexes, and were identified at higher energies of 670–800 cm−1.[22a, 28]
The current results are relevant to proposed hydride intermediates in the catalytic mechanism of nitrogenase, because the iron atoms have high-spin electronic configurations as is typical in iron-sulfur clusters like the FeMo cofactor. Hoffman has used a combination of ENDOR and cryoannealing to establish the presence of two hydrides in the E4 state.[7] The reversibility of H2 loss from this dihydride species was established from deuterium scrambling studies.[29] Since bridging hydrides – perhaps part of an Fe(μ-H)2Fe unit like in 1 – are present in an intermediate of nitrogenase, our results indicate the frequency range that may be expected for modes of an Fe-H/D-Fe unit when the iron ions have a high-spin electronic configuration. It is surprising to find that the range of stretching frequencies observed in 1 is similar to the frequencies for carbonyl-supported hydride clusters of second- and third-row transition metals with low-spin electronic configurations, even though high-spin iron(II) has occupied metal-ligand σ* orbitals. Overall, the results described here provide a guide for establishing the presence of high-spin iron(II) hydride intermediates. An advantage is that NRVS can be used for EPR-silent species, and even when EPR spectra are available, NRVS provides complementary information that can constrain DFT models for hydride intermediates.
Supplementary Material
Acknowledgments
We thank Eckhard Bill for magnetic measurements. This work was supported by NIH grants GM-116463 (to S.F.M.), GM-65313 (to P.L.H.) and GM-65440 (to S.P.C.), and by the Cluster of Excellence EXC 314 “Unifying Concepts in Catalysis” initiative of DFG (to V.P.). P.L.H. was supported by a Bessel Award from the Alexander von Humboldt Foundation. The NRVS work was performed at SPring-8 (proposal 2014B0103, 2016A1154, 2016B1347) and APS (proposals 39192). We also thank Drs. Y. Yoda (SPring-8) and M. Hu/J. Zhao/E. Alp (APS) for the beamtime assistance.
References
- 1.Norton JR, Sowa J. Chem Rev. 2016;116:8315–8317. doi: 10.1021/acs.chemrev.6b00441. [DOI] [PubMed] [Google Scholar]
- 2.Schilter D, Camara JM, Huynh MT, Hammes-Schiffer S, Rauchfuss TB. Chem Rev. 2016;116:8693–8749. doi: 10.1021/acs.chemrev.6b00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoffman BM, Lukoyanov D, Yang Z-Y, Dean DR, Seefeldt LC. Chem Rev. 2014;114:4041–4062. doi: 10.1021/cr400641x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peruzzini M, Poli R. Recent Advances in Hydride Chemistry. Elsevier; New York: 2001. [Google Scholar]
- 5.Crossley SWM, Obradors C, Martinez RM, Shenvi RA. Chem Rev. 2016;116:8912–9000. doi: 10.1021/acs.chemrev.6b00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Čorić I, Holland PL. J Am Chem Soc. 2016;138:7200–7211. doi: 10.1021/jacs.6b00747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Igarashi RY, Laryukhin M, Dos Santos PC, Lee H-I, Dean DR, Seefeldt LC, Hoffman BM. J Am Chem Soc. 2005;127:6231–6241. doi: 10.1021/ja043596p. [DOI] [PubMed] [Google Scholar]; b) Lukoyanov D, Barney BM, Dean DR, Seefeldt LC, Hoffman BM. Proc Natl Acad Sci USA. 2007;104:1451–1455. doi: 10.1073/pnas.0610975104. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lukoyanov D, Yang ZY, Duval S, Danyal K, Dean DR, Seefeldt LC, Hoffman BM. Inorg Chem. 2014;53:3688–3693. doi: 10.1021/ic500013c. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Lukoyanov D, Khadka N, Yang ZY, Dean DR, Seefeldt LC, Hoffman BM. J Am Chem Soc. 2016;138:10674–10683. doi: 10.1021/jacs.6b06362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dance I. Inorg Chem. 2013;52:13068–13077. doi: 10.1021/ic401818k. [DOI] [PubMed] [Google Scholar]
- 9.Kaesz HD, Saillant RB. Chem Rev. 1972;72:231–281. [Google Scholar]
- 10.Ming L-J. Nuclear magnetic resonance of paramagnetic metal centers in proteins and synthetic complexes. In: Que L, editor. Physical Methods in Bioinorganic Chemistry. University Science Books; Sausalito, CA: 2000. [Google Scholar]
- 11.Shafaat HS, Weber K, Petrenko T, Neese F, Lubitz W. Inorg Chem. 2012;51:11787–11797. doi: 10.1021/ic3017276. [DOI] [PubMed] [Google Scholar]
- 12.Perutz RN, Procacci B. Chem Rev. 2016;116:8506–8544. doi: 10.1021/acs.chemrev.6b00204. [DOI] [PubMed] [Google Scholar]
- 13.Dugan TR, Bill E, MacLeod KC, Brennessel WW, Holland PL. Inorg Chem. 2014;53:2370–2380. doi: 10.1021/ic4013137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee Y, Anderton KJ, Sloane FT, Ermert DM, Abboud KA, García-Serres R, Murray LJ. J Am Chem Soc. 2015;137:10610–10617. doi: 10.1021/jacs.5b05204. [DOI] [PubMed] [Google Scholar]
- 15.Pfirrmann S, Limberg C, Herwig C, Knispel C, Braun B, Bill E, Stösser R. J Am Chem Soc. 2010;132:13684–13691. doi: 10.1021/ja106266v. [DOI] [PubMed] [Google Scholar]
- 16.Dong Q, Zhao Y, Su Y, Su J-H, Wu B, Yang X-J. Inorg Chem. 2012;51:13162–13170. doi: 10.1021/ic301392p. [DOI] [PubMed] [Google Scholar]
- 17.Araake R, Sakadani K, Tada M, Sakai Y, Ohki Y. J Am Chem Soc. 2017;139:5596–5606. doi: 10.1021/jacs.7b01965. [DOI] [PubMed] [Google Scholar]
- 18.a) Lee Y, Kinney RA, Hoffman BM, Peters JC. J Am Chem Soc. 2011;133:16366–16369. doi: 10.1021/ja207003m. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chiang KP, Scarborough CC, Horitani M, Lees NS, Ding K, Dugan TR, Brennessel WW, Bill E, Hoffman BM, Holland PL. Angew Chem, Int Ed. 2012;51:3658–3662. doi: 10.1002/anie.201109204. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kinney RA, Saouma CT, Peters JC, Hoffman BM. J Am Chem Soc. 2012;134:12637–12647. doi: 10.1021/ja303739g. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Gunderson WA, Suess DLM, Fong H, Wang X, Hoffmann CM, Cutsail GE, III, Peters JC, Hoffman BM. J Am Chem Soc. 2014;136:14998–15009. doi: 10.1021/ja508117h. [DOI] [PubMed] [Google Scholar]
- 19.Parker SF, Lennon D, Albers PW. Appl Spectr. 2011;65:1325–1341. [Google Scholar]
- 20.Scheidt WR, Durbin SM, Sage JT. J Inorg Biochem. 2005;99:60–71. doi: 10.1016/j.jinorgbio.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 21.Wang H, Alp EE, Yoda Y, Cramer SP. A Practical Guide for Nuclear Resonance Vibrational Spectroscopy (NRVS) of Biochemical Samples and Model Compounds. In: Fontecilla-Camps JC, Nicolet Y, editors. Metalloproteins: Methods and Protocols. Springer - Humana Press; 2014. pp. 125–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.a) Ogata H, Kramer T, Wang H, Schilter D, Pelmenschikov V, van Gastel M, Neese F, Rauchfuss TB, Gee LB, Scott AD, Yoda Y, Tanaka Y, Lubitz W, Cramer SP. Nature Commun. 2015;6:7890. doi: 10.1038/ncomms8890. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pelmenschikov V, Guo Y, Wang H, Cramer SP, Case DA. Faraday Disc. 2011;148:409–420. doi: 10.1039/c004367m. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Bergmann U, Sturhahn W, Linn DE, Jenney FE, Adams MWW, Rupnik K, Hales BJ, Alp EE, Mayse A, Cramer SP. J Am Chem Soc. 2003;125:4016–4017. doi: 10.1021/ja028767+. [DOI] [PubMed] [Google Scholar]
- 23.Rodriguez MM, Bill E, Brennessel WW, Holland PL. Science. 2011;334:780–783. doi: 10.1126/science.1211906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andres H, Bominaar E, Smith JM, Eckert NA, Holland PL, Münck E. J Am Chem Soc. 2002;124:3012–3025. doi: 10.1021/ja012327l. [DOI] [PubMed] [Google Scholar]
- 25.Gütlich P, Bill E, Trautwein AX. Mössbauer Spectroscopy and Transition Metal Chemistry. Springer Verlag; Berlin Heidelberg: 2011. [Google Scholar]
- 26.a) Mays MJ, Simpson RNF. J Chem Soc A. 1968:1444–1447. [Google Scholar]; b) Bercaw JE, Brintzinger HH. J Am Chem Soc. 1969;91:7301–7306. [Google Scholar]; c) Johnson BFG, Lewis J, Williams IG. J Chem Soc A. 1970:901–903. [Google Scholar]; d) Knox SAR, Koepke JW, Andrews MA, Kaesz HD. J Am Chem Soc. 1975;97:3942–3947. [Google Scholar]; e) Koepke JW, Johnson JR, Knox SAR, Kaesz HD. J Am Chem Soc. 1975;97:3947–3952. [Google Scholar]
- 27.Noodleman L, Case DA. Adv Inorg Chem. 1992;38:423–470. [Google Scholar]
- 28.a) Reijerse EJ, Pham CC, Pelmenschikov V, Gilbert-Wilson R, Adamska-Venkatesh A, Siebel JF, Gee LB, Yoda Y, Tamasaku K, Lubitz W, Rauchfuss TB, Cramer SP. J Am Chem Soc. 2017;139:4306–4309. doi: 10.1021/jacs.7b00686. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Carlson MR, Gray DL, Richers CP, Wang W, Zhao PH, Rauchfuss TB, Pelmenschikov V, Pham CC, Gee LB, Wang H, Cramer SP. Inorg Chem. 2018;57:1988–2001. doi: 10.1021/acs.inorgchem.7b02903. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Pelmenschikov V, Birrell JA, Pham CC, Mishra N, Wang H, Sommer C, Reijerse E, Richers CP, Tamasaku K, Yoda Y, Rauchfuss TB, Lubitz W, Cramer SP. J Am Chem Soc. 2017;139:16894–16902. doi: 10.1021/jacs.7b09751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang Z-Y, Khadka N, Lukoyanov D, Hoffman BM, Dean DR, Seefeldt LC. Proc Natl Acad Sci USA. 2013;110:16327–16332. doi: 10.1073/pnas.1315852110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.