

Abstract
Background & Aims
Sterile inflammation resulting in alcohol hepatitis (AH) occurs unpredictably after many years of excess alcohol intake. The factors responsible for the development of AH are not known but mitochondrial damage with loss of mitochondrial function are common features. Hcar2 is a G-protein coupled receptor which is activated by β-hydroxybutyrate (BHB), and the relevance of the BHB-Hcar2 pathway in alcoholic liver disease is not known.
Methods
We tested if loss of BHB production can result in increased liver inflammation. We further tested if BHB supplementation can protect in AH through interaction with Hcar2, and the immune and cellular basis for protection.
Results
Humans with AH have reduced hepatic BHB, and inhibition of BHB production in mice aggravated ethanol induced AH, with higher serum ALT levels, increased steatosis and greater neutrophil influx. Conversely supplementation of BHB had the opposite effects with reduced ALT levels, reduced steatosis and neutrophil influx. This therapeutic effect of BHB is dependent on the receptor Hcar2. BHB treatment increased liver IL-10 transcripts, and promoted the M2 phenotype of intrahepatic macrophages. BHB also increased the transcriptional level of M2 related genes in vitro bone marrow derived macrophages. This skewing towards M2 related genes is dependent on lower mitochondrial membrane potential (Δψ) induced by BHB.
Conclusions
Collectively, our data shows that BHB production during excess alcohol consumption has an anti-inflammatory and hepatoprotective role through a Hcar2 dependent pathway, and introduces the concept of metabolite based therapy for AH.
Keywords: β-hydroxybutyrate, therapy, alcohol-induced liver injury, Hcar2
Graphical abstract
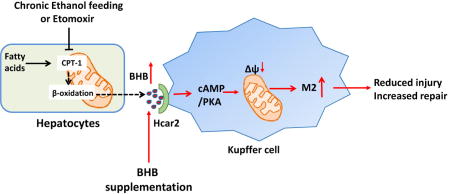
Introduction
Excess alcohol intake has many effects on the liver and can present with several clinical syndromes1. One of the most serious is acute alcoholic hepatitis, which occurs unexpectedly after decades of high levels of alcohol consumption, and is characterized by sterile liver inflammation, jaundice and can progress to a systemic inflammatory response2. There is a mixed inflammatory infiltrate characterized by neutrophils, and up-regulation of a variety of inflammatory cytokines including IL-1β, TNF-α and IL-6. Due to the ubiquitous development of hepatocyte steatosis there has been sustained interest in aspects of mitochondrial function related to lipid metabolism, particularly mitochondrial β-oxidation3. In-vivo and in-vitro models have shown diverse effects of alcohol on mitochondrial biology including abnormal mitochondrial morphology (giant mitochondria), mitochondrial DNA fragmentation, deceased mitochondrial protein synthesis by inhibition of mitochondrial ribosome activity, and oxidation of mitochondrial proteins3. Measurement of the steps in β-oxidation has demonstrated inhibition after acute and chronic alcohol exposure, and this is caused by downregulation of genes involved in fatty acid uptake as well as oxidation4, 5.
To understand the factors that initiate AH a variety of pro-inflammatory changes have been identified, with data supporting a role for increased intestinal permeability allowing bacterial products to enter the hepatic circulation and initiate an inflammatory response6. A compatible hypothesis is that toxic-metabolic injury to hepatocytes from alcohol results in the release of intracellular molecules generically termed damage associated molecular patterns (DAMPs) which initiate, and can then establish a pro-inflammatory loop7. There has been much less attention devoted to identifying the opposite, and equally plausible, scenario of a reduction in anti-inflammatory pathways resulting in AH. As described above, many types of mitochondrial changes induced by alcohol have been identified, but at present there is no link between changes in mitochondrial metabolites and liver inflammation. These considerations raised the possibility that if some of the usual products of mitochondrial metabolism have anti-inflammatory activity, with increasing mitochondrial damage their loss may trigger AH. We focused on the reduction in β-oxidation by alcohol, because one of the products of β-oxidation is β-hydroxybutyric acid (BHB) and is of interest because in addition to being an energy source, BHB is a ligand for the plasma membrane receptor Hcar2 and is known to have immunomodulatory functions via ligating Hcar2, inhibiting inflammasome activation, and via histone deacetylase inhibition8–10. This raises the possibility that ethanol induced reduction in β-oxidation, in addition to resulting in hepatocyte steatosis, may also be contributing to liver inflammation by reduction in BHB induced immunomodulation.
To test the effect of reduced BHB production in the setting of alcohol induced liver injury we applied alcohol gavage with and without chemical inhibition of carnitine palmitoyltransferase 1 (CPT-1), a key step in beta-oxidation. This is a relevant experimental approach because long term consumption of alcohol induces down regulation of CPT-1 and inhibition of beta oxidation11. By using a direct CPT-1 inhibitor we can test the specific effect of inhibition of beta-oxidation and reduced BHB production, independently from the many other effects of chronic alcohol consumption.
Collectively we have shown a novel and protective effect of BHB production in alcohol excess, and importantly linked alcohol induced mitochondrial dysfunction with the development of liver inflammation via metabolite derangement. This opens the possibility that immune modulation via administration of intravenous BHB may be protective in alcoholic hepatitis, and such protective effects have been shown by administration of the HCA1 agonist lactate in acute pancreatitis12.
Materials and Methods
Patients
Alcoholic hepatitis (n=10) liver tissues samples were collected from explants at the time of transplantation at the University Hospital in Lille, France. The diagnosis of AH was based on histological analysis and the presence of hepatocellular necrosis and infiltration of neutrophils1. Control liver tissues were collected from patients undergoing resection for hepatic metastases by the Tissue Procurement Facility at the University of North Carolina at Chapel Hill in Chapel Hill, NC (n=10) or at the University Hospital in Lille, France (n=5). All liver tissue samples were immediatley snap frozen and stored at −80°C until analysis. Experiments were performed with the approval and under accordance with the relevant guidelines establised at the institution of collection. All patients inlcuded in the study gave written informed consent.
Metabolomics
Metabolomics was performed at Metabolon, Inc (Durham, NC) where samples were prepared using the automated MicroLab STAR® system from Hamilton Company. After protein removal, sample extracts were divided into fractions for analysis by either RP/UPLC-MS/MS with positive ion mode electrospray ionization (ESI), RP/UPLC-MS/MS with negative ion mode ESI, or HILIC/UPLC-MS/MS with negative ion mode ESI. Raw data was peak-identified using Metabolon’s library of authenticated standards or recurrent unknown entities and QC processed using Metabolon’s hardware and software. Peaks were quantified using area-under-the-curve, normalized in terms of raw area counts, and rescaled to set the median equal to 1.
Study Approval and animal experiments
All animal studies were approved by the Yale University Institutional Animal Care and Use Committee and were performed in accordance with all regulatory standards. All the experimental mice were maintained at 22–23 degrees Celsius under specific pathogen-free conditions on 12-h light/12-h dark cycle with free access to water and standard rodent chow (Harlan Teklad #TD.2018). Male wild-type C57BL/6 (B6) between 8 and 10 weeks old were purchased from Charles River. IL-10 deficient mice were kindly provided by Dr. Susan Kaech. Hcar2−/− mice were kindly provided by Dr. Stefan Offermanns and have been previously described13, 14. To block production of BHB, C57BL/6 mice were pretreated with etomoxir (Sigma-Aldrich, i.p., 20mg/kg) 30 minutes before an acute ethanol gavage (6g/kg). BHB (Santa Cruz Biotechnology, i.p., 3mmol/kg) was given 6 hours post ethanol gavage to test its function during the alcoholic steatohepatitis. Mice were sacrificed 16 hours after ethanol gavage. Plasma ALT, liver histology, neutrophil staining and liver mononuclear cells were analyzed. For the chronic ethanol feeding model, mice were fed with control liquid diet or 5% ethanol liquid diet for 10 days15. On the 11th day, mice on ethanol liquid diet were injected with etomoxir (i.p., 20mg/kg), followed 30 min later by ethanol (5g/kg) gavage. BHB (i.p.,3mmol/kg) was given 6 hours after ethanol gavage, and mice were sacrificed 16 hours after ethanol gavage.
Isolation of liver mononuclear cells
Livers were collected from euthanatized mice, perfused to remove blood and pressed through a 40µm nylon mesh. The filtrate containing nonparenchymal cells was washed once. The cells were resuspended in 40% Percoll (GE Healthcare) and centrifuged at 1260g for 20 minutes at room temperature. The pellet was lysed with ACK lysis buffer and washed once with PBS.
Antibodies and flowcytometry
Antibodies information were provided in Supplementary CTAT Table. Mononuclear cells derived from the livers were incubated with purified rat anti-mouse CD16/CD32 (BD Biosciences) for 30 minutes to block Fc receptors and incubated with a cocktail of antibodies at 4 °C for 30 minutes in the dark and washed with PBS buffer. For staining of IL-10 and iNOS, intracellular staining assays were performed using the fixation/permeabilization kit (BD Biosciences). Flow cytometry was performed on a LSRII (BD Biosciences), and data were analyzed with Flowjo software.
BHB quantification in the liver
Liver tissues were homogenized in phenol red free 1×Hank’s salt solution with Magnesium and Calcium at the concentration at 100mg/500µL. Homogenates were centrifuged at 10,000g for 10 minutes at 4 °C. Supernants were transferred into Amicon 3000K filter tubes and centrifuged at 14,000g for 10 minutes at 4 °C. Eluent were used for BHB quantification by H7587 kit (Pointe Scientific) per manufacturer’s protocol.
Triglyceride quantification in the liver
Triglyceride quantification in the liver were measured using the Triglyceride Colorimetric Assay Kit from Cayman Chemical according to the manufacturer’s protocol.
Immunofluorescence
Hcar2-mRFP reporter mice frozen liver sections were stained with anti-F4/80 and anti-CD32 to identify macrophages and endothelial cells respectively.
BMDM culture and treatment
Bone marrow cells were obtained from Wildtype C57/BL6 and IL-10 deficient mice by flushing femurs and then lysing erythrocytes. Bone marrow cells were cultured with recombinant macrophage colony-stimulating factor (Peprotech, 10ng/mL) in complete RPMI 1640 medium for 7 days to differentiate into macrophage. For M2 polarization, BMDM were cultured with IL-4 (Peprotech, 10ng/mL) and IL-13 (Peprotech, 10ng/mL) for 24 hours or 48hours. BHB (15mM) or control saline were added at the beginning of polarization. BMDM cells were preincubated with Rp-Diastereomer of adenosine-3′,5′-cyclic monophosphothioate (Rp-cAMPS, Sigma-Aldrich) at 100µM) for 30 minutes before BHB treatment. Oligomycin A (5µM) were added at the last 6 hours of the 24 hours culture. For mRNA analysis, BMDM were harvested with TRIZOL. For mitochondrial membrane potential measurement, JC-1 (2µM) were added at the last 30 minutes of culture then washed away. The mitochondrial membrane potential was detected continuously for 6 hours in plate reader under the stimulation of IL-4 and IL-13 with and without BHB.
RNA extraction and qPCR
BMDMs and liver tissues were homogenized with TRIZOL (Invitrogen). cDNA was generated with high capacity cDNA reverse transcription kit (appliedbiosysem). Real-time PCR was performed using 2x SYBR Green supermix (Bio-rad). Expression of β -actin was used to standardize the samples. The expression results were presented as a ratio relative to control. PCR primer sequence were provided in Supplementary CTAT Table.
Luciferase reporter assay
HEK293 cells were seeded in 24-well plates at a density of 3 × 105 cells per well and cultured overnight. The next day, HEK293 cells were transfected with 200ng CRE-luciferase plasmid and 20ng Renilla luciferase plasmid per well using Lipofectamine 2000 according to the manufacturer’s protocol. Six hours after the transfection, IL-4 and IL-13 were added to the culture medium. Control saline, BHB (15mM) or db-cAMP (200µM) was added simultaneously with IL-4 and IL-13. Cells were lysed with passive lysis buffer 24 hours after IL-4 and IL-13 treatment. Supernatants were used to detect luciferase activity.
Statistical analysis
All data are expressed as Mean ± SEM. Animal experimental data (n≧6) were analyzed with nonparametric Wilcoxon rank-sum tests to calculate the P values. BMDM in vitro assay data were from 3 or 4 experiments. Considering the small sample size, the BMDM assay data were analyzed with Student’s t-tests to calculate the P values. Values of P < 0.05 were considered statistically significant. * P<0.05; ** P<0.01; ***P<0.001.
Results
Liver BHB concentration in alcoholic hepatitis
We initially wanted to confirm if the basic assumption that BHB concentration is lower in livers with alcoholic hepatitis was true. Supplementary Table 1 shows the clinical characteristics of the control and alcoholic hepatitis groups. Liver BHB concentration was assayed and there was a significant reduction in liver BHB concentration in the alcoholic hepatitis livers (Fig.1A).
Fig. 1. Low liver BHB in humans with alcoholic hepatitis and a combination of etomoxir and ethanol induces liver injury in mice.

(A) BHB levels from normal and alcoholic hepatitis human livers. Data are shown as means +/− SEM. Control, n = 15; AH, n=10. (B–H) Mice were injected i.p. with etomoxir (20mg/kg) or saline. 30 minutes later they were gavaged with PBS or ethanol (6g/kg). Plasma and livers were harvested 16 hours after ethanol gavage. (B) Representative liver histology. (C) Statistical analysis of steatosis scores. U.D. Undetectable. (D) Statistical analysis of liver TG levels. (E) Plasma ALT. (F) Statistical results of neutrophil (CD45+CD11b+Ly6G+) numbers in the liver by flow cytometry. (G) Representative images of Ly6B+ neutrophils in the liver. (H) Statistical analysis of Ly6B+ neutrophils in the liver by immunohistochemistry. C–H, n=6–8 per group. (I) Mice were injected i.p. with etomoxir (20mg/kg) or saline. 30 minutes later they were gavaged with PBS or ethanol (6g/kg). Livers were harvested 6 hours after ethanol gavage and the liver BHB levels were detected (n=7~8 per group). Data were analyzed with nonparametric Wilcoxon rank-sum tests.* P<0.05; ** P<0.01; ***P<0.001.
Combination of the beta-Oxidation Inhibitor etomoxir and ethanol induced liver injury
Having confirmed that BHB levels are lower in alcoholic hepatitis we next wanted to directly test if a reduction in BHB production can exacerbate alcohol induced liver injury. To specifically test the role of BHB in alcohol induce live injury, and to isolate this from all of the other changes associated with alcohol consumption, we chose to use etomoxir an inhibitor of β-oxidation. In the absence of ethanol, etomoxir did not cause liver steatosis, injury or inflammation as shown by normal liver histology (Fig.1B and C) and normal plasma ALT levels (Fig.1E). Consistent with previous report16, a binge of ethanol resulted in mild accumulation of small lipid droplets in the liver (Fig.1B and C) and moderate elevation of plasma ALT (Fig.1E). An acute dose of ethanol in the presence of etomoxir resulted in a significantly greater lipid accumulation in hepatocytes (Fig.1B and C), and significantly plasma higher ALT (Fig.1E). Etomoxir and ethanol separately upregulated the triglyceride levels in the liver slightly (Fig.1D), while the combination of etomoxir and ethanol elevated hepatic TG levels (Fig.1D). Neutrophil infiltration is a key marker of alcoholic steatohepatitis, and we quantified the number of neutrophils (CD11b+Ly6G+) in the liver 16 hours after ethanol gavage by flow cytometry. Etomoxir or ethanol treatments alone had no effect on the number of neutrophils (Fig.1F). However, the combination of etomoxir and ethanol resulted in a significantly increased number of neutrophils in the liver (Fig.1F). Immunochemistry confirmed the increase in neutrophil infiltration in the livers of mice treated with etomoxir and ethanol (Fig.1G and H). In addition, we also found the combination of etomoxir and ethanol significantly elevated the mRNA levels of inflammatory cytokines such as il6 and tnf alpha but not il1 beta in the liver (Supplementary Fig.1). Etomoxir has been shown to decrease BHB levels in humans, and our goal in using etomoxir was to reduce liver BHB concentrations17, 18. We found a single etomoxir injection decreased BHB levels in murine livers (Fig.1I). Because ethanol is metabolized to BHB, liver BHB concentrations were higher after an ethanol binge, which is consistent with previous studies showing BHB levels are higher is alcoholics in the early stages of alcohol consumption, and in alcohol fed mice19, 20. As predicted by the mechanism of action of etomoxir, BHB levels were lower in mice treated with etomoxir and ethanol as compared to ethanol alone (Fig.1I). Collectively, combination of etomoxir and ethanol induced the key features of alcoholic steatohepatitis, as shown in higher plasma ALT, greater hepatic fatty acid accumulation and more neutrophil influx, accompanied by decreased BHB levels in the liver, which is consistent with the BHB alteration in the liver of patients with alcoholic hepatitis.
BHB Rescued the liver injury Induced by Etomoxir and Ethanol
The liver inflammation induced by alcohol after inhibition of β-oxidation by etomoxir could be due to metabolic changes other than a reduction in BHB. To confirm the role of BHB in the increased liver inflammation caused by inhibition of β-oxidation the experiment was repeated but with supplementation of BHB. Mice received intraperitoneal BHB 6 hours after ethanol gavage, and liver injury was assessed 10hrs after BHB injection. As shown in Fig.2A BHB supplementation did not affect the liver injury caused by ethanol gavage alone, presumably because as shown in Fig.1I BHB levels are already elevated. However, addition of BHB significantly decreased the plasma ALT levels of mice treated with etomoxir and ethanol (Fig.2A). Histological examination and TG measurement of liver tissues revealed BHB attenuated steatosis and TG levels in the liver caused by etomoxir and ethanol (Fig.2B, C and D). BHB supplementation also decreased neutrophil infiltration (Fig. 2E, F and G). Collectively these data demonstrate that inhibition of β-oxidation results in lower hepatic BHB levels (Fig 1I), and BHB supplementation protects from the increase in ethanol induced steatohepatitis due to inhibition of β-oxidation (Fig. 2). This strongly supports the hypothesis that inhibition of β-oxidation worsens liver inflammation and injury by reducing hepatic BHB levels.
Fig. 2. BHB protected mice from etomoxir and ethanol induced liver injury.

(A–G) Mice were given an i.p. injection of BHB (3mmol/kg body weight) 6 hours after ethanol gavage and livers were collected 10 hours later. (A) Plasma ALT (n=8 per group for the EtOH gavage group with and without BHB; n=11~12 for the EtOH plus etomoxir group). (B) Representative liver histology. (C) Statistical analysis of steatosis scores (n=11~12 each group). (D) Statistical analysis of liver TG levels. (n=11~12 each group) (E) Statistical results of neutrophil (CD45+CD11b+Ly6G+) numbers in the liver (n=8~9 per group). (F) Representative images of Ly6B+ neutrophils in the liver. (G) Statistical analysis of Ly6B+ neutrophils in the liver by immunohistochemistry (n=10~11 per group). Data were analyzed with nonparametric Wilcoxon rank-sum tests.* P<0.05; ** P<0.01; ***P<0.001.
The Therapeutic Effect of BHB is Dependent on Hcar2
BHB has immunomodulatory actions that are dependent, and independent of the plasma membrane receptor Hcar28–10. If Hcar2 is required for BHB mediated immunomodulation in the liver, then unlike wild-type mice, supplementation of BHB in Hcar2 deficient mice will not protect from etomoxir and alcohol mediated liver injury. This was tested and BHB did not decrease hepatocyte steatosis (Fig.3A and B) or hepatic TG levels (Fig.3C) or plasma ALT levels (Fig. 3D) in Hcar2−/− mice. Intrahepatic neutrophil numbers were also analyzed, and no significant difference was observed in the number neutrophil in the livers in Hcar2 deficient mice treated with and without BHB (Fig.3E). These results demonstrate that the protective effect of BHB on alcohol-induced liver injury is dependent on Hcar2.
Fig. 3. Expression of Hcar2 is required for the protective effect of BHB.

Male Hcar2 deficient mice were treated with etomoxir and ethanol. Control saline (CT) or BHB were injected 6 hours after ethanol gavage. Plasma and livers were collected 10 hours later. (A) Representative liver histology (n=10~11 per group). (B) Statistical analysis of steatosis scores (n=10~11 per group). (C) Statistical analysis of liver TG levels (n=8 per group). (D) Plasma ALT (n=10~11 per group). (E) Statistical results of neutrophil (CD45+CD11b+Ly6G+) numbers in the liver (n=8 per group). Data were analyzed with nonparametric Wilcoxon rank-sum tests.
BHB Increased IL-10 and M2 Macrophages in the Liver
Having established that BHB can protect the liver from alcohol induced inflammation we wanted to investigate the cellular mechanism by which this occurs. We compared the hepatic mRNA levels of several cytokines known to be important in alcohol mediated liver injury1 in the ethanol alone group and the combination group of ethanol and etomoxir with and without BHB (Fig. 4). We found that the combination of ethanol and etomoxir resulted in significant elevations of liver mRNA levels of il6 and il10 (Fig. 4). IL-6 has been recently shown to play an inflammatory role in alcoholic steatohepatitis through the IL-6–p47phox–oxidative stress pathway in neutrophils21. BHB didn’t change hepatic mRNA levels of these with ethanol gavage alone. We also examined the effect of BHB on liver mRNA levels of the above cytokines with the combination of etomoxir and ethanol gavage. There was no difference in mRNA levels of IL-1β, IL-4, IL-6, IL-17, and TNF-α in BHB and control treated livers, however mRNA levels of IL-10 were significantly higher in the livers of BHB treated mice (Fig. 4). This was of interest because IL-10 has been shown to be anti-inflammatory and hepatoprotective in alcoholic liver disease, and polymorphisms in promoter region resulting in low expression of IL-10 have been shown to be associated with advanced alcoholic liver disease22.
Fig. 4. BHB increased il10 mRNA levels in the liver.

Mice were injected i.p. with Etomoxir (20mg/kg) or saline. 30 minutes later they were gavaged with ethanol (6g/kg). Mice were given an i.p. injection of BHB (3mmol/kg body weight) 6 hours after ethanol gavage and livers were collected 10 hours later. mRNA levels of cytokines in the livers were detected. n=8~11 per group. Data were analyzed with nonparametric Wilcoxon rank-sum tests. * P<0.05. U.D. Undetectable.
The in vivo experiments above are important in establishing integrated consequences of the various manipulations of high levels of alcohol, inhibition of β-oxidation and supplementation with BHB, but they are not cell specific. To obtain mechanistic insights it is necessary to move to in vitro single cell population experiments. As we had already identified a requirement for Hcar2 for the hepatoprotective effects of BHB a first step was to obtain the cellular localization of Hcar2 in the liver. This was done by imaging the liver in a reporter mouse expressing mRFP under the control of the Hcar2 promotor, after staining for F4/80 (macrophages and Kupffer cells), and separately staining for CD32 (endothelium)14. Merging of the F4/80 and CD32 images with Hcar2 clearly shows perfect overlap with F4/80+ cells, and no overlap with CD32+ cells (Fig.5A).
Fig. 5. Hcar2 is expressed on liver macrophages and BHB promoted an M2 macrophage phenotype in the liver.

(A) Immunofluorescence of Hcar2 expression in the liver. Blue, DAPI; red(mRFP), Hcar2; Green, F4/80 or CD32. (B&C) Wild-type mice were injected i.p. with Etomoxir (20mg/kg) or saline. 30 minutes later they were gavaged with ethanol (6g/kg). Mice were given an i.p. injection of BHB (3mmol/kg body weight) 6 hours after ethanol gavage and livers were collected 10 hours later. (B) Expression of M1 markers on liver macrophages (CD45+CD11b+F4/80+) by flowcytometry. (C) Expression of M2 markers on liver macrophages (CD45+CD11b+F4/80+) by flow cytometry. (Quantified expression values of M1 and M2 markers on macrophage in the liver (n=7 per group). ΔMFI=(Mean Fluoresce Intensity)positive staining – (Mean Fluoresce Intensity)isotype staining. Data were analyzed with nonparametric Wilcoxon ranksum tests.* P<0.05; ** P<0.01.
The localization of Hcar2 on liver tissue macrophages, and the increase in IL-10 expression in total liver suggested that BHB was modifying the liver macrophage phenotype. Tissue macrophages are well known for their plasticity, and at extreme ends of polarization are classified as proinflammatory M1 or anti-inflammatory M2. This is recognized to be over simplistic as there are over 3500 differentially regulated genes in tissue macrophages and there is no single M1 or a single M2 phenotype23. Within these recognized limitations, the M1 and M2 phenotypes still provide a convenient shorthand for classification of tissue macrophages and we used flowcytometric analysis of markers associated with a pro-inflammatory M1 like phenotype (CD80, CCR7, TLR2/4, TLR9 and iNOS), which did not show any difference after BHB treatment (Fig.5B). In contrast, markers associated with an anti-inflammatory M2 like phenotype such as ER-MP23, CD206, ST2 and IL-10 were all upregulated by BHB (Fig.5C). Collectively, these data show that Hcar2 is expressed on liver macrophages, and during ethanol induced liver injury BHB elevated IL-10 expression in whole liver, and enhanced the M2 phenotype in the liver macrophage population.
BHB Promotes M2 Macrophage Polarization through Activation of cAMP Pathway and Reduction of Mitochondrial Membrane Potential
To confirm the effect of BHB on the M2 macrophage phenotype and to gain mechanistic understanding of the pathways involved, we induced M2 polarization in conventional bone marrow derived macrophages (BMDMs) and compared gene expressions with and without BHB. Under M2 polarization condition (IL-4 and IL-13), expression of il10 and arginase 1 was significantly upregulated by BHB in BMDMs at 48 hours (Fig.6A), which is consistent with the alteration of macrophage phenotype in vivo. We also examined an earlier time point and found the mRNA of il10, arginase 1 and ym1 had already been upregulated at 24 hours (Fig.6B). Expression of other M2 markers such as cd163, fizz and mr was comparable with and without BHB at 24 hours (Fig.6B). Because IL-10 itself could enhance M2 polarization, to investigate whether BHB promoted the M2 polarization directly or due to an IL-10 mediated pathway, we examined the effect of BHB on M2 polarization of IL-10 deficient BMDMs. As shown in Fig.6C, BHB upregulated M2 marker such as arginase 1 and ym1 in the absence of IL-10, demonstrating an IL-10 independent effect of BHB on macrophage polarization.
Fig. 6. BHB promoted M2 gene expression through decreasing mitochondrial membrane potential.

Bone marrow derived macrophages (BMDMs) from wild-type mice were cultured under M2 condition (IL-4 and IL-13) for 48 hours (A) and 24 hours (B) with and without BHB (15mM). BMDMs from IL-10 deficient mice were cultured under M2 condition (IL-4 and IL-13) for 24 hours (C) with and without BHB (15mM). (A and B) mRNA levels of M2 associated gene were detected in WT BMDMs. (C) mRNA levels of M2 associated gene were detected in IL-10 deficient BMDMs. (D) cAMP response element-luciferase activity in HEK cells treated under M2 condition for 24 hours with addition of control saline (CT), BHB or dbCAMP separately. (E) mRNA levels of M2 associated genes were detected WT BMDM cultured under M2 condition (IL-4 and IL-13) and BHB for 24 hours with and without Rp-cAMPS (100µM). (F) Mitochondrial membrane potentials were measured in WT BMDMs under M2 condition (IL-4 and IL-13) for 24 hours with and without BHB (15mM). (G) mRNA levels of M2 associated gene were detected in WT BMDMs treated with BHB (15mM) and oligomycin A (5µM) to maintain the mitochondrial membrane potential. Data are mean and SEM from three or four independent experiments. Data were analyzed with Student’s t-tests. * P<0.05; ** P<0.01; ***P<0.001.
Since a requirement for the receptor Hcar2 had already been established we considered the downstream signaling pathways. In macrophages Hcar2 activation is known to increase cAMP levels via a conventional adenylate cyclase/PKA pathway24. This was of interest because increase in cAMP has been shown to regulate macrophage function by promoting M2 like functionality25. To confirm the role of BHB driven cAMP, a cAMP stimulated CREB-reporter system was used. HEK cells were transfected with CRE/promoter-luciferase plasmid and treated with IL-4 and IL-13 alone or in the presence of BHB, or stable cAMP analogue (db-cAMP). Treatment with IL-4 and IL-13 alone induced significant upregulation of CREB activity (Fig.6D). Addition of BHB further increased the activity of luciferase under treatment of IL-4 and IL-13, confirming a BHB/cAMP pathway (Fig.6D). Rp-cAMPS, a specific membrane-permeable inhibitor of cAMP pathway by competitively binding the cAMP-induced activation of cAMP-dependent protein kinase (PKA)26, decreased the upregulation of il10, arginase1 and ym1 induced by BHB (Fig 6E).
BHB mediated cAMP can result in many intracellular changes. We were interested in reports that the cAMP-dependent phosphorylation of cytochrome c oxidase with PKA switched on the allosteric ATP inhibition of cytochrome c oxidase and kept the mitochondrial membrane potential (Δψ) low27, 28. Mitochondrial membrane potential is generated by a proton gradient existing across the mitochondrial inner membrane29. Previous studies showed maintenance of the mitochondrial membrane potential is associated with the development of the M1 phenotype, and loss with an M2 phenotype30. Consistently, we found BHB decreased mitochondrial membrane potential in IL-4 and IL-13 treated BMDMs at 24 hours (Fig.6F). To test if the decrease in mitochondrial membrane potential was required for the ability of BHB to induce an M2 phenotype, the mitochondrial membrane potential was artificially maintained by oligomycin A, which maintains the mitochondrial membrane potential by inhibiting the activity of ATP synthase. As previously BHB resulted in an increase in IL-10 and other M2 genes, but this did not occur in the presence of oligomycin A (Fig.6G).
BHB Protected from the alcohol-induced liver injury Induced by Chronic Ethanol feeding
To investigate if BHB is protective in chronic ethanol feeding model, mice were fed with an ethanol liquid diet for 10 days. On the 11th day of ethanol diet feeding, to replicate the decreased BHB levels present in the alcoholic patients (Fig. 1A), we injected mice with etomoxir, followed 30 minutes later by an ethanol gavage (5g/kg). BHB were i.p. injected 6 hours after ethanol gavage. We found BHB reduced hepatic steatosis (Fig.7A and B), total liver triglyceride (Fig.7C), and plasma ALT (Fig.7D). In summary, we showed BHB could protect from chronic alcohol-induced liver injury.
Figure 7. BHB protected liver injury induced by chronic alcohol feeding and binge.

Mice were fed with control (Control) liquid diet or 5% ethanol (EtOH) liquid diet for 10 days. On the 11th day, mice on ethanol liquid diet were injected with etomoxir (i.p., 20mg/kg). Ethanol (5g/kg) was gavaged to mice on alcoholic liquid diet 30 minutes after etomoxir injection. BHB (i.p., 3mmol/kg) or Control saline (CT) was given 6 hours after ethanol gavage. Plasma and livers were harvested 16 hours after ethanol gavage. (A) Representative liver histology. (B) Statistical analysis of steatosis scores. U.D. Undetectable. (C) Statistical analysis of liver TG levels. (D) Plasma ALT. (A–D, n=8~10 per group). Data were analyzed with nonparametric Wilcoxon rank-sum tests. * P<0.05.
Discussion
The association of chronic excess alcohol intake with liver disease is known very widely. It is less well appreciated that long term excess alcohol intake is associated with a range of clinical presentations. Among these alcoholic hepatitis (AH) is of great importance due to its high mortality, and the lack of understanding of its etiology1. Clinically AH develops unpredictably over weeks, after many years of high levels of alcohol consumption. AH progresses with jaundice, abdominal pain, fever and frequently progressing to a systemic inflammatory response syndrome with multi organ failure. 28-day mortality is high and ranges between 15–25%, and despite multiple clinical trials an effective therapy has not been identified1. A key feature in this lack of progress in therapeutics is a lack of understanding of why individuals with chronic high alcohol consumption unpredictably develop a life-threatening sterile inflammatory response in the liver.
Our study has directly tested the concept that loss of the anti-inflammatory metabolite BHB is a contributing factor to liver inflammation in AH. The livers of patients with severe AH have significantly lower BHB levels than controls (Fig 1A), making it possible that low BHB have a mechanistic role. Direct evidence for a role for BHB was subsequently demonstrated in a series of in vivo experiments, using a chemical inhibitor of β-oxidation, which reduced liver BHB and increased inflammation in alcohol induced injury (Fig. 1B–H). Supplemented BHB reduced the increased inflammatory response caused by inhibiting β-oxidation, and this effect of BHB supplementation was lost in Hcar2 knockout mice (Fig 2, 3). This provides strong evidence for a novel anti-inflammatory role for BHB via Hcar2, and suggests a pro-inflammatory switch linked to the loss of BHB produced by mitochondria. Excess alcohol intake along with poor food intake is well known to result in a state of alcoholic ketoacidosis with elevated BHB levels31. As an interesting clinical correlate, alcoholic ketoacidosis and AH do not appear to occur at the same time.
The requirement for Hcar2 for the anti-inflammatory effects of BHB was unexpected as BHB has been shown to have anti-inflammatory effects by direct inhibition of inflammasome activation, independent of its known activation of Hcar2. In the Hcar2 independent pathways BHB inhibits the NLRP3 inflammasome by preventing K(+) efflux and reducing ASC oligomerization and speck formation10. This has been shown to have important in vivo anti-inflammatory effects including inhibiting neutrophil activation in gout32. The Hcar2 reporter mice placed Hcar2 on liver macrophage populations, and on these cells BHB did not broadly inhibit macrophage activation but stimulated a switch of the well-recognized M1/M2 spectrum towards M2. This is a more nuanced affect because although moving away from M1 would have resulted in loss of pro-inflammatory functions, the switch to M2 results in gain of functions such as tissue repair and remodeling. In addition to promoting expression of M2 genes such as arg1 and ym1, il10 expression was also elevated by BHB. IL-10 itself can promote M2 polarization, and by using IL-10 deficient mice we demonstrated an IL-10 independent effect of BHB in stimulating the M2 phenotype.
Activation of Hcar2 is known to activate a cAMP pathway and we were interested in the ability of cAMP to down-regulate the mitochondrial membrane potential33. This is relevant due to the requirement for a high mitochondrial membrane potential for the maintenance of an M1 phenotype and a low mitochondrial membrane potential for an M2 phenotype30. BHB, along with switching towards an M2 phenotype, resulted in a decrease in the mitochondrial membrane potential (Fig.6F). Artificially maintaining a high mitochondrial membrane potential with the ATPase inhibitor oligomycin blocked the switch to the M2 phenotype (Fig.6G).
Collectively this study identifies a novel anti-inflammatory role of BHB in alcohol-induced liver injury. This is significant as it provides a link between the metabolic changes known to occur from chronic alcohol consumption, and the subsequent development of liver inflammation. It also opens the possibility that derangements in other metabolites such as succinate, citrate and lactate, with known effects on sterile inflammation, may be contributing to liver inflammation30, 34, 35. Despite showing an important anti-inflammatory effect of BHB in AH, this study does not identify if a decrease in BHB concentrations is the switch responsible for a change in the liver from a low level of chronic inflammation to the increased inflammatory state in AH. If this is the case, one possibility is that there is a gradual decrease in mitochondrial function and loss of BHB production which eventually reaches a tipping point. Another possibility is that the BHB- Hcar2 pathway is downstream of one of the known pathways such as EtOH induced endotoxemia36. This is attractive because LPS concentrations in the range known to be present in alcoholic liver disease result in inhibition of β-oxidation and reduction in BHB production in muscle cells37, 38. If the same is true for hepatocytes then LPS, in addition to being directly pro-inflammatory for macrophages, may also be stimulating hepatic inflammation by reducing production of BHB hepatocytes.
There is extensive experience on therapeutic elevation of serum BHB levels, ranging from refractory epilepsy to genetic disorders of metabolism39. Starvation and high fat/low protein diets are not an option in patients with alcoholic hepatitis as they may not have the capacity to make BHB. Supplemental BHB however may be viable option. Oral administration of the sodium salt of BHB in doses of 80–900mg/Kg/day was sufficient to achieve blood levels of BHB of 0.19–0.36mM and produced a therapeutic effect in children with acyl CoA dehydrogenase deficiency40. Intravenous administration of BHB in healthy subjects and septic patients increased serum levels to 2mM and did not have any adverse effects41. More recently lactate, the ligand for GPR81 (closely related to Hcar2), was found to reduce C-reactive protein (CRP) levels and the systemic inflammatory response syndrome in a clinical trial of acute pancreatitis12. There are significant similarities between AH and pancreatitis in the sterile inflammatory pathways that are activated, and these data point towards a new metabolite based antiinflammatory approach in AH7, 42.
Supplementary Material
Lay summary.
Alcoholic hepatitis (AH) is a life-threatening condition with no approved therapy that occurs unexpectedly in people who take excess alcohol. The liver makes many metabolites, and we demonstrate that loss of one such metabolite β-hydroxybutyrate (BHB) occurs in patients with AH, that this loss can increase alcohol-induced liver injury, and BHB can protect from alcohol-induced liver injury via a receptor on liver macrophages. This opens the possibility of metabolite based therapy for AH.
Highlights.
-
*
Liver BHB concentration is low in human AH, and reducing BHB in mice worsens liver injury after alcohol.
-
*
BHB supplementation reduced alcohol induced liver injury in wild-type but not Hcar2 deficient mice.
-
*
BHB supplementation increased IL-10 levels and increased the M2 macrophage phenotype.
-
*
Development of the M2 phenotype is dependent on reduction of mitochondrial membrane potential by BHB.
Acknowledgments
This study was funded by VA Merit award and NIH grants 5U01AA021912-02 (to R. B. and W. Z. M); P30 pilot grant (to X. O.); K08DK092281 (to R. H.). The Yale Liver Center core facilities were funded by NIH by grant DK P30-034989.
Financial support:
This study was funded by VA Merit award and NIH grants 2R56DK0 5U01AA021912-02 (R. Bataller and W. Mehal); P30 pilot grant (X. Ouyang); K08DK092281 (R. Hoque). The Yale Liver Center core facilities were funded by NIH by grant DK P30-034989.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: none
AUTHOR CONTRIBUTION
Yonglin Chen, Xinshou Ouyang, Stefan Offermanns, Laurent Dubuquoy, Alexander Louvet, Philippe Mathurin, Veronica Massey, Bernd Schnabl, Ramon A Bataller, Wajahat Zafar Mehal: Hypothesis generation, conceptual design, data analysis and manuscript preparation.
Yonglin Chen, Xinshou Ouyang, Rafaz Hoque, Sarah Tonack, Irma Garcia-Martinez and Muhammad Yousaf conducting experiments and data analysis.
References
- 1.Stickel F, Datz C, Hampe J, Bataller R. Pathophysiology and Management of Alcoholic Liver Disease: Update 2016. Gut Liver. 2017;11:173–188. doi: 10.5009/gnl16477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mandrekar P, Bataller R, Tsukamoto H, Gao B. Alcoholic hepatitis: Translational approaches to develop targeted therapies. Hepatology. 2016;64:1343–1355. doi: 10.1002/hep.28530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nassir F, Ibdah JA. Role of mitochondria in alcoholic liver disease. World J Gastroenterol. 2014;20:2136–2142. doi: 10.3748/wjg.v20.i9.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clugston RD, Jiang H, Lee MX, Piantedosi R, Yuen JJ, Ramakrishnan R, et al. Altered hepatic lipid metabolism in C57BL/6 mice fed alcohol: a targeted lipidomic and gene expression study. J Lipid Res. 2011;52:2021–2031. doi: 10.1194/jlr.M017368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crabb DW, Liangpunsakul S. Alcohol and lipid metabolism. J Gastroenterol Hepatol. 2006;21(Suppl 3):S56–60. doi: 10.1111/j.1440-1746.2006.04582.x. [DOI] [PubMed] [Google Scholar]
- 6.Orman ES, Odena G, Bataller R. Alcoholic liver disease: pathogenesis, management, and novel targets for therapy. J Gastroenterol Hepatol. 2013;28(Suppl 1):77–84. doi: 10.1111/jgh.12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kubes P, Mehal WZ. Sterile inflammation in the liver. Gastroenterology. 2012;143:1158–1172. doi: 10.1053/j.gastro.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 8.Chang PV, Hao L, Offermanns S, Medzhitov R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci U S A. 2014;111:2247–2252. doi: 10.1073/pnas.1322269111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity. 2014;40:128–139. doi: 10.1016/j.immuni.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med. 2015;21:263–269. doi: 10.1038/nm.3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peng Z, Borea PA, Varani K, Wilder T, Yee H, Chiriboga L, et al. Adenosine signaling contributes to ethanol-induced fatty liver in mice. J Clin Invest. 2009;119:582–594. doi: 10.1172/JCI37409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu BU, Hwang JQ, Gardner TH, Repas K, Delee R, Yu S, et al. Lactated Ringer's solution reduces systemic inflammation compared with saline in patients with acute pancreatitis. Clin Gastroenterol Hepatol. 2011;9:710–717. e711. doi: 10.1016/j.cgh.2011.04.026. [DOI] [PubMed] [Google Scholar]
- 13.Taggart AK, Kero J, Gan X, Cai TQ, Cheng K, Ippolito M, et al. (D)-beta-Hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA-G. J Biol Chem. 2005;280:26649–26652. doi: 10.1074/jbc.C500213200. [DOI] [PubMed] [Google Scholar]
- 14.Hanson J, Gille A, Zwykiel S, Lukasova M, Clausen BE, Ahmed K, et al. Nicotinic acid- and monomethyl fumarate-induced flushing involves GPR109A expressed by keratinocytes and COX-2-dependent prostanoid formation in mice. J Clin Invest. 2010;120:2910–2919. doi: 10.1172/JCI42273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model) Nat Protoc. 2013;8:627–637. doi: 10.1038/nprot.2013.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ding WX, Li M, Chen X, Ni HM, Lin CW, Gao W, et al. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology. 2010;139:1740–1752. doi: 10.1053/j.gastro.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gatta B, Zuberbuehler C, Arnold M, Aubert R, Langhans W, Chapelot D. Acute effects of pharmacological modifications of fatty acid metabolism on human satiety. Br J Nutr. 2009;101:1867–1877. doi: 10.1017/S0007114508143604. [DOI] [PubMed] [Google Scholar]
- 18.Kahler A, Zimmermann M, Langhans W. Suppression of hepatic fatty acid oxidation and food intake in men. Nutrition. 1999;15:819–828. doi: 10.1016/s0899-9007(99)00212-9. [DOI] [PubMed] [Google Scholar]
- 19.Elliott S, Smith C, Cassidy D. The post-mortem relationship between beta-hydroxybutyrate (BHB), acetone and ethanol in ketoacidosis. Forensic Sci Int. 2010;198:53–57. doi: 10.1016/j.forsciint.2009.10.019. [DOI] [PubMed] [Google Scholar]
- 20.Kang X, Zhong W, Liu J, Song Z, McClain CJ, Kang YJ, et al. Zinc supplementation reverses alcohol-induced steatosis in mice through reactivating hepatocyte nuclear factor-4alpha and peroxisome proliferator-activated receptor-alpha. Hepatology. 2009;50:1241–1250. doi: 10.1002/hep.23090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li M, He Y, Zhou Z, Ramirez T, Gao Y, Gao Y, et al. MicroRNA-223 ameliorates alcoholic liver injury by inhibiting the IL-6-p47phox-oxidative stress pathway in neutrophils. Gut. 2017;66:705–715. doi: 10.1136/gutjnl-2016-311861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grove J, Daly AK, Bassendine MF, Gilvarry E, Day CP. Interleukin 10 promoter region polymorphisms and susceptibility to advanced alcoholic liver disease. Gut. 2000;46:540–545. doi: 10.1136/gut.46.4.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin J, Hu Y, Nunez S, Foulkes AS, Cieply B, Xue C, et al. Transcriptome-Wide Analysis Reveals Modulation of Human Macrophage Inflammatory Phenotype Through Alternative Splicing. Arterioscler Thromb Vasc Biol. 2016;36:1434–1447. doi: 10.1161/ATVBAHA.116.307573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gaidarov I, Chen X, Anthony T, Maciejewski-Lenoir D, Liaw C, Unett DJ. Differential tissue and ligand-dependent signaling of GPR109A receptor: implications for anti-atherosclerotic therapeutic potential. Cell Signal. 2013;25:2003–2016. doi: 10.1016/j.cellsig.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 25.Luan B, Yoon YS, Le Lay J, Kaestner KH, Hedrick S, Montminy M. CREB pathway links PGE2 signaling with macrophage polarization. Proc Natl Acad Sci U S A. 2015;112:15642–15647. doi: 10.1073/pnas.1519644112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dostmann WR. (RP)-cAMPS inhibits the cAMP-dependent protein kinase by blocking the cAMP-induced conformational transition. FEBS Lett. 1995;375:231–234. doi: 10.1016/0014-5793(95)01201-o. [DOI] [PubMed] [Google Scholar]
- 27.Bender E, Kadenbach B. The allosteric ATP-inhibition of cytochrome c oxidase activity is reversibly switched on by cAMP-dependent phosphorylation. FEBS Lett. 2000;466:130–134. doi: 10.1016/s0014-5793(99)01773-1. [DOI] [PubMed] [Google Scholar]
- 28.Kadenbach B. Intrinsic and extrinsic uncoupling of oxidative phosphorylation. Biochim Biophys Acta. 2003;1604:77–94. doi: 10.1016/s0005-2728(03)00027-6. [DOI] [PubMed] [Google Scholar]
- 29.Chen LB. Mitochondrial membrane potential in living cells. Annu Rev Cell Biol. 1988;4:155–181. doi: 10.1146/annurev.cb.04.110188.001103. [DOI] [PubMed] [Google Scholar]
- 30.Mills EL, Kelly B, Logan A, Costa AS, Varma M, Bryant CE, et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell. 2016;167:457–470. e413. doi: 10.1016/j.cell.2016.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Noor NM, Basavaraju K, Sharpstone D. Alcoholic ketoacidosis: a case report and review of the literature. Oxf Med Case Reports. 2016;2016:31–33. doi: 10.1093/omcr/omw006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goldberg EL, Asher JL, Molony RD, Shaw AC, Zeiss CJ, Wang C, et al. beta-Hydroxybutyrate Deactivates Neutrophil NLRP3 Inflammasome to Relieve Gout Flares. Cell Rep. 2017;18:2077–2087. doi: 10.1016/j.celrep.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maldonado EN, Patnaik J, Mullins MR, Lemasters JJ. Free tubulin modulates mitochondrial membrane potential in cancer cells. Cancer Res. 2010;70:10192–10201. doi: 10.1158/0008-5472.CAN-10-2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O'Neill LA. A critical role for citrate metabolism in LPS signalling. Biochem J. 2011;438:e5–6. doi: 10.1042/BJ20111386. [DOI] [PubMed] [Google Scholar]
- 35.Hoque R, Farooq A, Ghani A, Gorelick F, Mehal WZ. Lactate reduces liver and pancreatic injury in Toll-like receptor- and inflammasome-mediated inflammation via GPR81-mediated suppression of innate immunity. Gastroenterology. 2014;146:1763–1774. doi: 10.1053/j.gastro.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nolan JP. The role of intestinal endotoxin in liver injury: a long and evolving history. Hepatology. 2010;52:1829–1835. doi: 10.1002/hep.23917. [DOI] [PubMed] [Google Scholar]
- 37.Frisard MI, McMillan RP, Marchand J, Wahlberg KA, Wu Y, Voelker KA, et al. Toll-like receptor 4 modulates skeletal muscle substrate metabolism. Am J Physiol Endocrinol Metab. 2010;298:E988–998. doi: 10.1152/ajpendo.00307.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frisard MI, Wu Y, McMillan RP, Voelker KA, Wahlberg KA, Anderson AS, et al. Low levels of lipopolysaccharide modulate mitochondrial oxygen consumption in skeletal muscle. Metabolism. 2015;64:416–427. doi: 10.1016/j.metabol.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Veech RL. The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot Essent Fatty Acids. 2004;70:309–319. doi: 10.1016/j.plefa.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 40.Van Hove JL, Grunewald S, Jaeken J, Demaerel P, Declercq PE, Bourdoux P, et al. D,L-3-hydroxybutyrate treatment of multiple acyl-CoA dehydrogenase deficiency (MADD) Lancet. 2003;361:1433–1435. doi: 10.1016/S0140-6736(03)13105-4. [DOI] [PubMed] [Google Scholar]
- 41.Beylot M, Chassard D, Chambrier C, Guiraud M, Odeon M, Beaufrere B, et al. Metabolic effects of a D-beta-hydroxybutyrate infusion in septic patients: inhibition of lipolysis and glucose production but not leucine oxidation. Crit Care Med. 1994;22:1091–1098. doi: 10.1097/00003246-199407000-00007. [DOI] [PubMed] [Google Scholar]
- 42.Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, et al. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest. 2012;122:3476–3489. doi: 10.1172/JCI60777. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.