

Abstract
BACKGROUND & AIMS
Macrophages contribute to liver disease, but their role in cholestatic liver injury, including primary sclerosing cholangitis (PSC), is unclear. We tested the hypothesis that macrophages contribute to the pathogenesis of, and are therapeutic targets for, PSC.
METHODS
Immune cell profile, hepatic macrophage number, localization and polarization, fibrosis, and serum markers of liver injury and cholestasis were measured in an acute (intrabiliary injection of the IAP antagonist BV6) and chronic (Mdr2−/− mice) mouse model of sclerosing cholangitis (SC). Selected observations were confirmed in liver specimens from PSC patients. Because of the known role of the CCR2/CCL2 axis in monocyte/macrophage chemotaxis, therapeutic effects of the CCR2/5 antagonist cenicriviroc (CVC), or genetic deletion of CCR2 (Ccr2−/− mice) were determined in BV6-injected mice.
RESULTS
We found increased peribiliary proinflammatory (M1-like) and alternatively-activated (M2-like) monocyte-derived macrophages in PSC compared to normal livers. In both SC models, genetic profiling of liver immune cells by nanostring technology identified a predominance of monocytes/macrophages; immunohistochemistry confirmed peribiliary monocyte-derived macrophage recruitment (M1>M2-polarized), which paralleled injury onset and was reversed upon resolution in acute SC mice. PSC, senescent and BV6-treated human cholangiocytes released monocyte chemoattractants (CCL2, IL-8) and macrophage-activating factors in vitro. Pharmacological inhibition of monocyte recruitment by CVC or CCR2 genetic deletion attenuated macrophage accumulation, liver injury and fibrosis in acute SC.
CONCLUSIONS
Peribiliary recruited macrophages are a feature of both PSC and acute and chronic murine SC models. Pharmacologic and genetic inhibition of peribiliary macrophage recruitment decreases liver injury and fibrosis in mouse SC. These observations suggest monocyte-derived macrophages may contribute to the development of SC in mice and in PSC pathogenesis, and support their potential as a therapeutic target.
Keywords: C-C chemokine ligand 2 (CCL2), C-C chemokine receptor 2 (CCR2), cenicriviroc (CVC), cholestatic liver injury, liver fibrosis, macrophages, sclerosing cholangitis
Graphical abstract
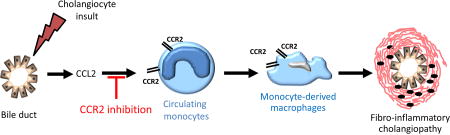
Primary sclerosing cholangitis (PSC) is a progressive cholestatic liver disease, characterized by chronic inflammation of the biliary epithelium and fibrous obliterative cholangitis of the intra- and extra-hepatic bile ducts. Its etiology and pathogenic mechanisms are still largely unknown, which contributes to the lack of effective therapies aside from liver transplantation. PSC patients are at high risk for the development of cirrhosis and its sequela of portal hypertension and end stage liver disease, and cholangiocarcinoma.1 Animal models contribute to our knowledge of disease pathogenesis and malignant transformation of the biliary epithelia, and provide insight regarding new therapeutic strategies. However, given the multifactorial nature of PSC, a single animal model recapitulating all the aspects of the disease may be difficult to achieve. Therefore, the study of multiple models may represent the best approach to investigate the molecular mechanisms of disease pathogenesis.
Mice with targeted disruption of the multidrug resistance gene Mdr2 (Abcb4) (Mdr2−/−) develop chronic and progressive hepatic lesions closely resembling primary sclerosing cholangitis.2, 3 Although no significant association has been found between MDR3 variants (the human orthologue of rodent Mdr2) and PSC susceptibility,4 the Mdr2−/− mouse has been extensively used as an appropriate animal model to study the pathogenesis and progression of sclerosing cholangitis and for the development of anti-fibrotic therapies. We have recently established an acute mouse model of sclerosing cholangitis that develops following the intrabiliary instillation of a single dose of the inhibitor of apoptosis (IAP) antagonist BV6.5 These mice display acute cholestatic injury resembling human sclerosing cholangitis, with histological features consistent with a fibrous cholangiopathy of the interlobular bile ducts, portal and periportal inflammation and fibrosis, elevated serum markers of cholestasis and cholangiographic evidence of intrahepatic biliary strictures and dilatations. These models complement each other for understanding biliary injury with periductular fibrosis.
Macrophages are key regulators of the inflammatory responses. Macrophages are ontologically and functionally diverse. Tissue resident macrophages, such as hepatic Kupffer cells (KC), derive from the yolk sac, whereas recruited macrophages originate from bone marrow-derived monocytes. Their functional diversity relates to their ability to polarize into different functional phenotypes which regulate both the initial inflammatory response and the late healing response. Although several subsets of macrophage phenotypes have been proposed (i.e., M1-like or proinflammatory macrophages; and M2-like or restorative, reparative macrophages), it has not been established whether they are phenotypically distinct populations or, more likely, they derive from the same population with a continuum of phenotypes between the two subtypes regulated by evolving cues in the tissue microenvironment.6 Macrophages are among the first immune cells responding to liver injury and can regulate the fibrogenic response via multiple mechanisms, including phagocytosis of dead cells and secretion of cytokines, chemokines and growth factors, production of matrix metalloproteases (MMPs) and tissue inhibitors of MMP (TIMPs).7 Following liver injury, pro-inflammatory monocyte-derived macrophages are recruited to the injury site in response to the release of chemokines, in particular the CC-chemokine ligand 2/ monocyte chemoattractant protein 1 (CCL2/MCP-1), produced by fibroblasts, resident macrophages, activated cholangiocytes and endothelial cells as part of the inflammatory response. The initial pro-inflammatory response is followed by a resolution phase carried out by macrophages that facilitate tissue healing and remodeling by breaking down extracellular matrix and promoting cell proliferation and hepatic stellate cell (HSC) apoptosis.7
The role of macrophages in the development and resolution of sclerosing cholangitis has not yet been fully elucidated. Therefore, the aim of the present study was to expand our knowledge on the role of macrophages in PSC by utilizing both an acute and a chronic animal model of sclerosing cholangitis, and to determine whether therapeutic inhibition of monocyte/macrophage recruitment may be effective in reducing the biliary injury and fibrosis in these models.
MATERIALS AND METHODS
Mice
C57BL/6J and FVB/N mice were from Jackson Labs (Bar Harbor, ME). Multidrug resistance 2 knockout mice (on a C57BL/6 background, C57BL/6.Mdr2−/−) were generated by backcrossing FVB/N background Mdr2−/− mice (FVB/N.Mdr2−/−) with C57BL/6 mice for nine generations. Both C57BL/6.Mdr2−/− mice and FVB/N.Mdr2−/− were a gift from Dr. Ronald Oude Elferink, Tytgat Institute, Amsterdam, The Netherlands. Ccr2−/− mice (on a C57BL/6 background) were a gift from Dr. Charles Howe (Mayo Clinic, Rochester, MN). Wild-type C57BL/6J and FVB/N mice were never housed together with Mdr2−/− or Ccr2−/− mice. All mice were maintained in ventilated cages under 12 h light/dark cycles at the Mayo Clinic – Rochester animal facility, with access to water and standard chow diet ad libitum.
Animal studies
All animal experiments were performed in accordance with protocols approved by the Mayo Clinic Institutional Animal Care and Use Committee (IACUC). Six to eight week old wild type and Ccr2−/− mice underwent surgery to receive one intrabiliary instillation of BV6 or saline (control) to induce acute sclerosing cholangitis as previously described.5 In selected experiments, wild type mice were divided into two groups. Each group received either a placebo control (vehicle) or cenicriviroc (CVC, 15 mg/kg body weight) s.c. daily, starting from the day before the intrabiliary instillation (day −1) and continuing for 5 days thereafter (until day 4). The mice were sacrificed at day 5 and blood and liver harvested for histological and biochemical analysis. Mdr2−/− mice were sacrificed and analyzed at 30 to 60 day of age. In selected studies, two groups of C57BL/6.Mdr2−/− mice received either a placebo, or CVC s.c. daily for 2 weeks starting at 45 days of age. At the end of the 2 week treatment, the mice were sacrificed and liver tissue was harvested for histological and biochemical analysis. Dose and mode of administration (intervals, route) were selected based on the results of a pharmacokinetic study of CVC performed at Tobira Therapeutics (subsequently acquired by Allergan) showing high bioavailability by daily s.c. administration. CVC plasma concentrations at day 5 following s.c. daily administration are shown in Suppl. Fig. S1.
Gene expression analysis by NanoString Technology
The prevalence of immune cells in 60-day old C57BL/6.Mdr2−/− (n=4) vs. wild-type (n=4), and BV6-treated (n=5) vs. saline-treated C57BL/6 (n=6) male mouse livers was evaluated by determining expression of a panel of immune cell-related genes using the nCounter Mouse Immunology Gene Expression Code Set (NanoString Technologies, Seattle, USA) according to manufacturer’s instructions. Briefly, 200 ng of total RNA was hybridized with reporter and capture probe sets and quantified in the nCounter (NanoString Technologies). Genes that were significantly over-expressed (p<0.05) in Mdr2−/− mice or BV6-treated wild-type mice by a minimum of two-fold above that of wild-type or saline-treated wild-type mice, respectively, were attributed to immune cell type based on information culled from the database maintained by the Pandey Lab, Johns Hopkins University and The Bioinformatics Lab (http://www.hprd.org). Immune cell types were subsequently hierarchically ranked by number of genes that were over-expressed.
Statistical analysis
Data are expressed as median (or median of fold-changes over control) and IQR, unless otherwise indicated. Statistical analyses were performed with two-tailed Mann-Whitney rank test for unpaired data using GraphPad Prism 7.03 (GraphPad Software Inc., San Diego, CA) and p < 0.05 were considered statistically significant.
RESULTS
Macrophages accumulate in the periportal areas of the livers of PSC patients
We initially examined the number of monocytes/macrophages in PSC liver specimens by immunohistochemistry. Increased infiltration of CD68+ monocytes/macrophages was readily identified in the liver parenchyma of patients with stage 4 PSC compared to normal liver tissue (Fig. 1A). Significant accumulation of monocytes/macrophages was observed within the fibrotic tissue and adjacent to periportal bile ducts (Fig. 1A). Interestingly, peribiliary macrophage accumulation was more pronounced in PSC liver tissue than in liver tissue of primary biliary cholangitis (PBC) or hepatitis C (HCV) patients, despite similar increases in the number of macrophages in the liver parenchyma (Suppl. Fig. S2A, B). In order to determine whether accumulation of monocytes/macrophages varies according to the stage of the disease, we next performed immunofluorescence analysis for CD68 in the liver of patients with stage 1–4 PSC. The number of CD68+ positive cells enhanced both in the parenchyma and in the peribiliary areas as the disease progressed (Fig. 1B). Importantly, we observed a significant increase in the number of CD68+/CCR2+ cells at late stages of the diseases, suggesting this population of accumulating macrophages was largely derived from recruited monocytes (CCR2+) rather than from proliferating resident KC (CCR2−) (Fig. 1C). Both pro-inflammatory (iNOS+) and anti-inflammatory (CD206+) macrophages were detected in progressively larger numbers in the peribiliary area of PSC livers compared to normal livers; however, compared to normal livers, the number of pro-inflammatory macrophages at stage 4 increased by 6.1 fold in PSC livers, as compared to a 3.4-fold increase in anti-inflammatory macrophages, suggesting a predominantly inflammatory reaction (Fig. 1D). Taken together, these findings suggest a role for infiltrating monocytes/macrophages in human PSC and provide a rationale for using animal models to further examine this phenomenon.
Fig. 1. Macrophages accumulate in the peribiliary areas of the livers of PSC patients.

(A) CD68 immunohistochemistry on healthy and cirrhotic-stage PSC livers (20×); quantification of CD68+ area. Arrows: bile ducts. (B) CD68 (total macrophages), CK19 (cholangiocytes) co-immunofluorescence (40×); quantification of CD68+ cells (n=4 normal, n=3 PSC). (C) CD68, CK19 and CCR2 (recruited macrophages) co-immunofluorescence (40×); quantification of CCR2+ cells (n=3/group) (D) CK19, iNOS (pro-inflammatory macrophages) and CD206 (anti-inflammatory macrophages) co-immunofluorescence (63×); quantification of iNOS+ and CD206+ cells (4 normal; 3 PSC). Higher magnifications of the dotted areas are shown in the lower row. *p<0.05; **p<0.005 (Mann-Whitney).
Increased expression of monocyte/macrophage-associated markers in the liver of mouse models of sclerosing cholangitis
We next sought to determine whether the above observations in human PSC could be confirmed in animal models of sclerosing cholangitis. For these studies, we utilized both an acute (BV6 biliary instillation)5 and a chronic (Mdr2−/−) mouse model of biliary injury. To characterize the immune cell populations in the livers of these two murine models of sclerosing cholangitis, we first performed differential analysis of gene expression of 561 immunology-related mouse genes utilizing a NanoString platform. Our results identified 55 genes with statistically significant upregulation of ≥ 2 folds in the livers of BV6-injected mice compared to saline-injected controls, and 26 genes in the Mdr2−/− mouse livers compared to wild-type livers (Suppl. Table S1 and Table S2). Overexpressed genes were subsequently attributed to different immune cell populations. Remarkably similar results were obtained in the two animal models, with the largest number of overexpressed genes associated with monocytes and/or macrophages (Fig. 2A, B). In particular, in the liver of BV6-injected mice, out of the 20 most highly expressed genes, 16 (80%) were specifically expressed in monocytes/macrophages (Suppl. Table S1). Ccl2 was the second most highly induced gene (7.6-fold) in the livers of BV6-injected mice, whereas the macrophage attractant chemokine C-C motive ligand 6 (Ccl6) and 3 (Ccl3) were also expressed > 3-fold compared to controls (3.9 and 3.1, respectively). Similarly, in the Mdr2−/− mouse livers, 10 out of the 20 (50%) most highly expressed genes were specifically expressed in monocytes/macrophages (Suppl. Table S2). C-C chemokine receptor type 2 (Ccr2), the main receptor for CCL2, was the third most highly expressed gene (4.3-fold). These data are consistent with a pathogenic role for monocytes and macrophages in sclerosing cholangitis in mice.
Fig. 2. Expression of monocyte/macrophage-associated genes is enhanced in the liver of two mouse models of sclerosing cholangitis.

Prevalence of immune cells in the liver of (A) BV6-treated vs. saline-injected C57BL/6 and (B) C57BL/6.Mdr2−/− vs. wild-type mice evaluated by gene expression using NanoString technology. The graphs depict genes significantly over-expressed (p<0.05) in BV6-treated wild-type mice or Mdr2−/− mice by a minimum of two-fold above that of saline-injected wild-type or wild-type mice, respectively. The genes were attributed to immune cell types and hierarchically ranked by number of over-expressed genes.
Macrophages accumulate in the peribiliary areas of the livers of two murine models of sclerosing cholangitis
Given the results of our gene expression analysis, we next verified whether macrophage accumulation was evident in the peribiliary areas of the mouse models. Immunohystochemistry for CD68 displayed significant enrichment of CD68+ cells both in the parenchyma and around the portal bile ducts of BV6-injected mice compared to saline-injected control mice at the peak of the injury (day 5)5 (Fig. 3A, B). Notably, macrophage accumulation paralleled the onset of the injury (day 3) and was completely reversed with the resolution of the injury (day 21) (Fig. 3B). In a similar way, parenchymal and peribiliary accumulation of CD68+ cells was observed in sixty-days old C57BL6.Mdr2−/− mice compared to wild-type (Fig. 3C) and even more strikingly in the parental strain FVB.Mdr2−/−, which displayed a more severe phenotype (Suppl. Fig. S3A, B). In line with the observations in human PSC samples, macrophage accumulation directly correlated with the age-dependent severity of the phenotype (Suppl. Fig. S3C, D). At day 5, iNOS+ macrophages were increased by 8.5-fold in the periportal areas of BV6-injected mice compared to controls, whereas CD206+ macrophages were increased by 2.5-fold, consistent with a predominantly pro-inflammatory response (Fig. 3D). Similarly, the increase in the number of pro-inflammatory iNOS+ macrophages was larger than that of anti-inflammatory CD206+ macrophages in the livers of both C57BL6.Mdr2−/− and FVB.Mdr2−/− mice compared to their wild-type controls (35.7-fold iNOS+ vs. 6.8-fold CD206+, and 184.5-fold iNOS+ vs. 12.1-fold CD206+, respectively)(Fig. 3E and Supplemental S3E,F). Interestingly, in larger bile ducts of both mouse and human injured livers, macrophages (mostly CD206+) appear to infiltrate the biliary epithelium and/or directly associate with its luminal side (Fig. 3F). Finally, as observed in human PSC livers, recruited monocyte-derived macrophages rather than replicating KC were the main source of accumulating macrophages, as assessed by immunofluorescence co-staining for CCR2 (recruited macrophages) and Clec4F (KC) (Fig. 4A). These results were further confirmed by flow cytometry analysis showing that infiltrating monocyte-derived macrophages, defined as CD45+F4/80+CD11bhi, were significantly increased in the liver of BV6-treated mice and C57BL6.Mdr2−/− mice compared to their respective saline-injected and wild type controls, whereas KC, defined as CD45+F4/80+CD11binter were unchanged (Fig. 4B).8 Similarly, recruited inflammatory CD45+F4/80+Ly6Chi monocytes/macrophages were also elevated in the liver of diseased mice (Fig. 4B).9 Taken together, these observations associate inflammatory monocyte-derived macrophages with both chronic and acute sclerosing cholangitis in mice.
Fig. 3. Macrophages accumulate in the peribiliary areas of BV6-injected mouse livers.

(A) CD68 liver immunohistochemistry on BV6- or saline-injected mice at day 5 (20×); quantification of CD68+ (saline n=5; BV6 n=4; 3–6 areas/sample). (B) CD68 liver immunohistochemistry on untreated mice (normal) or BV6-injected mice at day 3, 5, 7 or 21 (20×); quantification of CD68+ area (normal, day 7: n=3; day 3, day 5: n=4; day 21: n=5; 3–6 areas/sample). (C) CD68 liver immunohistochemistry on sixty-day old wild-type and C57BL/6.Mdr2−/− female mice (10×). (D)(E) iNOS, CD206 and EpCAM or CK19 (cholangiocytes) liver co-immunofluorescence of (D) saline- or BV6-injected mice (day 5) and (E) sixty-day old wild-type and C57BL/6.Mdr2−/− female mice; quantification of iNOS+ and CD206+ cells (BV6 n=3/group; wild type n=4 females; Mdr2−/− n=3 females). (F) Macrophage and cholangiocyte co-immunofluorescence of BV6-treated (left), Mdr2−/− (center) and PSC livers (right). Arrows: macrophages. ***p<0.001; ****p<0.0001 (Mann-Whitney).
Fig. 4. Macrophages accumulate in the peribiliary areas of Mdr2−/− mouse livers.

(A) Clec4F (KC), CCR2 and CK19 liver co-immunofluorescence on normal and BV6-injected mice (day 3–21); quantification of Clec4F+ and CCR2+ cells (n=3/group). (B) Representative flow cytometric plots showing CD45+F4/80+ liver macrophage population, CD45+F4/80+CD11bhi monocyte-derived macrophages (MoMF), CD45+F4/80+CD11bint Kupffer cells (KC) and CD45+F4/80+Ly6Chi inflammatory macrophages in the liver of saline- or BV6-injected mice (day 5), and sixty-day old C57BL/6.Mdr2−/− mice. Quantification shown on the right (3 saline, 5 BV6, 4 C57BL/6, 4 Mdr2−/−). *p<0.05 (Mann-Whitney).
Cholangiocyte-derived chemokines and cytokines contribute to the recruitment of monocytes/macrophages to the portal areas
Chemokines such as CCL2/MCP-1 and CCL20/MIP3α recruit circulating monocytes into the injured tissue by stimulating their respective receptors, CCR2 and C-C chemokine receptor type 6 (CCR6).10 Our previous studies demonstrated that IL6, IL8 and CCL2 are transcriptionally upregulated in BV6-treated human cholangiocytes in vitro and in the livers of BV6-injected mice.5 Therefore, we first measured secretion of IL-6, IL-8 and CCL2/MCP-1 into the conditioned media of H69 cells treated with BV6, and found that all three proteins were significantly elevated (Fig. 5A). Similarly, IL-8, IL-6 and CCL2/MCP-1 are also increased in lipopolysaccharide (LPS)-treated normal human cholangiocytes after 10 days (a model of experimentally-induced cellular senescence), as well as in isolated PSC cholangiocytes11, 12 (Fig. 5B, C). To establish whether release of the chemokines IL-8 and CCL2/MCP-1 by injured cholangiocytes can promote migration of monocytes/macrophages, we exposed the human monocytic cell line THP-1 to conditioned media obtained from either BV6-treated H69 cells, or isolated PSC cholangiocytes, or LPS-treated NHC, in the presence or absence of specific neutralizing antibodies against IL-8 or CCL2/MCP-1, and quantitatively determined their migration in a Boyden chamber assay. THP-1 cells demonstrated increased migration when incubated with conditioned media from BV6-treated H69 compared to media from untreated H69 (Fig. 5D). Likewise, exposure to conditioned media from isolated PSC cholangiocytes or LPS-treated cholangiocytes promoted THP-1 migration compared to vehicle-treated cholangiocytes (Fig. 5D). To further validate these results, we performed a migration assay utilizing a static, two chamber-microfluidic device where NHC or PSC cholangiocytes and THP-1 cells were plated in adjacent chambers separated by microgrooves. The number of THP-1 cells that had migrated to the cholangiocyte chamber after 24 hr. was significantly larger in the presence of PSC cholangiocytes than NHC (Fig. 5E and Suppl. Fig. S4). Notably, THP-1 migration was significantly inhibited by neutralization of either CCL2/MCP-1 or IL-8 (Fig. 5D). Moreover, incubation with the conditioned media triggered activation of THP-1 cells, as demonstrated by transcriptional upregulation of markers such as IL-1β and IL-6 (Fig. 5F, G); as expected, IL-8 or CCL2/MCP-1 neutralization had no effect on THP-1 activation, implicating these chemokines specifically in promoting monocyte migration, but not their activation. Neutralization of IL-6 also did not affect THP-1 activation (Fig. 5F, G). It is important to note that gene expression analysis by RNAseq also identified other macrophage chemotactic proteins, such as CCL20/MIP3α, chemokine (C-X-C motif) ligand 2 (CXCL2)/MIP2α and CXCL3/MIP2β, as significantly upregulated in cholangiocytes isolated from PSC patients compared to normal cholangiocytes (Fig. 5B), and their secretion into the conditioned media was confirmed by ELISA (Fig. 5C). Therefore, it is likely that multiple inflammatory chemokines and cytokines may be involved in recruitment and activation of monocytes/macrophages in the pathogenesis of PSC. These results confirm that both activated and senescent cholangiocytes release pro-inflammatory cytokines and chemokines that can directly promote recruitment and activation of monocytes/macrophages.
Fig. 5. Active and senescent cholangiocytes release monocyte chemoattractants and macrophage-activating factors.

(A) ELISA on supernatants of H69 cells treated with DMEM + BV6 (n=3 in quintuplicate). (B) Transcriptomic profiles of H69, NHC and isolated PSC cholangiocytes by high-throughput NGS. (C) ELISA on supernatants from isolated PSC cholangiocytes (n=3) or NHC + LPS (n=3). RPKM: reads/kilobase of transcript/million mapped reads. (D) THP-1 cell transmembrane migration following exposure for 6 hours to conditioned media as in (A), or (C), ± neutralizing IL-8 or CCL2 antibodies. THP-1 exposed to DMEM used as controls (DMEM)(BV6 n=3; NHC+LPS n=3; PSC n=4 at different times from PSC-1). RFU: relative fluorescent units. (E) THP-1 cell migration in a dual-chamber microfluidic device after 24 hr. co-culture (PSC1, black symbols, PSC2, grey symbols, n=3 each). (F) Gene expression in THP-1 cells incubated with conditioned media as in (A) or (C) (PSC1, black symbols, n=6; PSC2, grey symbols, n=3). *p<0.05; **p<0.005; ***p<0.0005; ****p<0.0001 (Mann-Whitney).
Sclerosing cholangitis-associated liver injury in mice is ameliorated by either pharmacological or genetic disruption of the CCR2/CCL2 axis
Given the potential role of the CCR2/CCL2 axis in the recruitment of monocyte/macrophages during biliary injury and their significant increased gene expression in the liver of the two mouse models (Suppl. Table S1 and S2), we sought to verify whether pharmacological or genetic inhibition of CCR2 was protective in our animal models. Treatment with the dual CCR2/CCR5 antagonist cenicriviroc (CVC) strongly reduced monocyte/macrophage recruitment and liver fibrosis in BV6-injected mice compared to vehicle-treated, BV6-injected controls (Fig. 6A, B). Periportal fibrosis and infiltration of CCR2+ monocytes/macrophages were also attenuated in sixty-day old C57BL6.Mdr2−/− mice after two weeks of treatment with CVC compared to vehicle-treated C57BL6.Mdr2−/− mice (Suppl. Fig 5A–D). Serum bile acids and total bilirubin levels were also significantly decreased in BV6-injected, CVC-treated mice, but serum ALT levels were unaffected, which may suggest a more specific effect on the cholestatic liver injury rather than on the hepatocyte injury (Fig. 6C). Consistently, reduced mRNA expression of Ccr2, Ccl2/Mcp1, Cd68 (macrophage markers), Tnfα (pro-inflammatory macrophage marker) and Ym1 (anti-inflammatory macrophage marker) confirmed markedly attenuated infiltration and activation of both pro-inflammatory and anti-inflammatory monocytes/macrophages in the livers of CVC-treated compared to vehicle-treated BV6-injected mice (Fig. 6D). Reduced mRNA expression of Tgfβ and α-smooth muscle actin (Acta2) were also consistent with decreased fibrosis (Fig. 6D). In mice, CCR2 is also expressed on a small subpopulation of T lymphocytes (2–15%) and natural killer T cells, with a significant expansion of this population occasionally observed in inflammatory conditions.13 To confirm that CVC-mediated attenuation of the biliary injury was mainly due to impaired recruitment of CCR2+ monocytes/macrophages, and not CCR2+ lymphocytes, we performed flow cytometry analysis of livers from saline- or BV6-injected mice to determine the extent of CCR2+ infiltrating macrophages (F4/80+) and lymphocytes (CD3+) in either condition. We observed that whereas the total number of CCR2+ cells increased in the livers of BV6-injected mice, the percentage of CD3+/CCR2+ lymphocytes and F4/80+/CCR2+ macrophages did not change with the treatment (Fig. 6E). However, the CD3+/CCR2+ lymphocyte population represented a substantially smaller percentage (<3–4%) of the total liver CD45+CCR2+ population compared to F4/80+/CCR2+ macrophages (40–65%), both in controls and BV6-injected mice. Lack of change in percentage of F4/80+ and CD3+ cells following BV6 administration implies that both populations of cells are recruited into the liver equally. In addition, there was no increase in the mRNA expression of CCR5 or its ligand CCL5/RANTES in the livers of BV6-injected mice compared to saline-injected controls, suggesting that infiltration of CCR5+ cells did not contribute significantly to the BV6-induced biliary injury (Fig. 6F). Consistently, mice genetically deficient in CCR2 (Ccr2−/−) were also resistant to the BV6-induced biliary injury and fibrosis, and displayed normal liver histology (Fig. 7A, B). Serum markers of liver injury and cholestasis were also significantly reduced in Ccr2−/− mice compared to wild type and were not significantly different from their saline-injected controls (Fig. 7C). In a similar fashion, upregulation of markers of inflammation, macrophage accumulation and polarization (Ccl2/Mcp-1, Cd68, Ly6c, Tnfα, Il1β, Ym1 and Il10), and fibrosis (Tgfβ, Coll1a1) was only observed in the livers of BV6-treated wild type, but not Ccr2−/− mice (Fig. 7D). Collectively, these findings suggest that recruited CCR2+ monocyte-derived macrophages are an important source of inflammatory mediators and may play a pivotal role in acute and chronic sclerosing cholangitis in mice.
Fig. 6. Pharmacologic inhibition of CCR2-dependent monocyte recruitment attenuates biliary injury.

(A) Liver sections of BV6-injected mice (5 days) ± CVC or vehicle (20×). Arrows: bile ducts. pv: portal vein. Quantification of CD68+ cells on the right. (B) Liver hydroxyproline content. (C) Serum ALT, total BA and total bilirubin. (D) Macrophage infiltration and hepatic fibrosis markers by gene expression in whole livers. BV6 (n=8), BV6+vehicle (n=11), BV6+CVC (n=9). (E) Representative flow cytometric plots and quantification of CD45+CCR2+, CD45+CCR2+F4/80+ (macrophages) and CD45+CCR2+CD3+ (lymphocytes) liver cells. Saline (n=3); BV6 (n=5). (F) Ccr5 and Ccl5/Rantes gene expression. *p<0.05; **p<0.005 (Mann-Whitney).
Fig. 7. Genetic disruption of the CCR2/CCL2 axis attenuates biliary injury.

(A) Liver sections of saline- or BV6-injected wild-type and Ccr2−/− mice at day 5 (20×). Arrows: bile ducts. pv: portal vein. (B) Liver hydroxyproline content. (C) ALT, total BA and total bilirubin. (D) Macrophage infiltration markers and fibrosis markers by gene expression in whole livers. Wild-type: saline (n=8), BV6 (n=8); Ccr2−/−: saline (n=4), BV6 (n=5). *p<0.05, **p<0.005 (Mann-Whitney).
DISCUSSION
The results of this study provide mechanistic insights regarding the contribution of macrophages in the pathogenesis of sclerosing cholangitis. The principal findings indicate that: (i) CD68+ bone marrow-derived macrophages accumulate in the peribiliary region and infiltrate the fibrotic tissue in both an acute and a chronic mouse model of sclerosing cholangitis, and in human PSC livers; (ii) as compared to healthy livers, the number of pro-inflammatory (“M1-like”) macrophages increases in the fibrotic livers of both animal models and in human PSC livers; (iii) activated and senescent cholangiocytes release chemokines promoting monocyte/macrophage recruitment in vitro; and (iv) inhibition of monocyte-derived macrophage recruitment through genetic or pharmacological depletion of CCR2 prevents biliary injury and fibrosis. Our data support the development of therapeutic strategies aimed to reduce early recruitment of circulating monocytes in the treatment of sclerosing cholangitis.
Hepatic macrophages are critical regulators of liver homeostasis, inflammation and fibrosis through their ability to engulf apoptotic and necrotic cells, and secrete pro-inflammatory cytokines and chemokines that recruit and activate other inflammatory cells. Secretion of pro-fibrotic mediators, such as transforming growth factor beta (TGFβ) and platelet-derived growth factor (PDGF), by macrophages promote proliferation and differentiation of hepatic stellate cells into collagen-producing myofibroblasts.14, 15 Likewise, macrophages are also critically involved in the resolution of fibrosis and inflammation, by promoting myofibroblast apoptosis, degrading extracellular matrix components and engulfing cellular debris that could perpetuate inflammation. Due to this dual role in regulating inflammation and fibrosis, therapeutic depletion of macrophages in liver diseases may have beneficial or deleterious effects depending on the stage of the disease, as it has been demonstrated in several murine models of acute liver injury.8, 16–20 The role of recruited macrophages in chronic cholestatic liver diseases remains obscure. Macrophage accumulation has been demonstrated in a mouse model of xenobiotic-induced chronic cholangiopathy resembling human sclerosing cholangitis, and their depletion by liposome-encapsulated clodronate effectively attenuates fibrogenesis and ductular reaction.21 In line with these studies, our data clearly demonstrate that the number of macrophages is increased both in the parenchyma and in the peribiliary regions of the liver in two distinct mouse models of sclerosing cholangitis. The study of these two models has allowed us to compare and contrast our findings in a chronic and an acute condition, and has revealed remarkable similarities with regard to macrophage involvement in both stages of biliary injury. First, gene expression analysis identified a predominance of monocyte/macrophage-associated genes among those over-expressed in the liver of both mouse models. Second, peribiliary macrophage accumulation corresponds to the progression of the injury both in the acute and in the chronic model. And finally, at the peak of the injury, recruited pro-inflammatory CD45+F4/80+Ly6C+ macrophages are increased in the liver, and iNOS+ macrophages are evident around the bile ducts. These results also mirror what we observed in human PSC, underlining the clinical relevance of our observations.
An important question in the pathogenesis of PSC and other cholangiopathies is whether the inflammatory response is initiated directly by the cholangiocytes or by pre-existing stromal cells in the peribiliary plexus. In our studies, normal human cholangiocytes treated with BV6, or isolated PSC cholangiocytes (which exhibit features of cellular senescence),11, 12 or normal cholangiocytes exposed to LPS (an experimental model of cellular senescence)11 release monocyte-chemotactic factors, including CCL2/MCP-1 and IL-8, that promote monocyte migration in vitro. Despite being unable to expand these studies in a larger number of PSC cholangiocytes isolates from multiple patients, due to the limited availability of these cells and their senescent phenotype, monocyte/macrophage activation, and CCL2/MCP-1- and/or IL-8-dependent monocyte chemotaxis toward the diseased cholangiocytes was determined by employing PSC-derived cholangiocytes from two unique patients. However, we acknowledge that both our migration assays in the Boyden chambers and in the microfluidic devices only measure chemotaxis of THP-1 cells toward cholangiocytes, but do not address their adhesion to endothelial cells and extravasation, which would require a flow-based assay. IL6, IL8 and CCL2 are transcriptionally up-regulated in human cholangiocytes following treatment with BV6.5 In addition, IL-8, IL-6 and CCL2/MCP-1 are part of the senescence-associated secretory phenotype (SASP) and are significantly enriched in the conditioned media from senescent cholangiocytes treated with LPS, as well as from isolated PSC cholangiocytes.11, 12 IL6, IL8 and CCL2 gene expression is regulated by the transcription factor NF-κB, whose activation we have previously demonstrated in our experimental paradigms (cholangiocytes treated with BV6 or LPS)5,22 and has also been indirectly implicated in PSC.23–25 Thus, we speculate that activation of NF-κB may represent a common pathway to which cholangiocyte insults converge to generate a similar fibro-inflammatory phenotype.
Our current results strongly suggest that a cholangiocyte insult may be sufficient to trigger an inflammatory cascade initiated and, perhaps, perpetuated by recruited macrophages. Indeed, inhibition of monocyte/macrophage recruitment at the onset of the injury, either by genetic or pharmacological inhibition of CCR2 (CVC), significantly blunted the inflammatory and fibrotic response caused by the intrabiliary injection of BV6, suggesting that macrophages are required in the initial stages of the injury. CVC also ameliorated liver fibrosis when administered to diseased Mdr2−/− mice, indicating a potential use for the treatment of established cholangitis. Despite the fact that other macrophage-chemotactic proteins are released by senescent cholangiocytes or cholangiocytes exposed to BV6 in vitro, inhibition of CCR2 was sufficient to reduce the biliary injury, suggesting the CCL2/CCR2 axis is predominantly responsible for the recruitment of circulating monocyte-derived macrophages. Based on the results of this and other pre-clinical studies showing the efficacy of CVC treatment in several models of kidney and liver fibrosis,26, 27 CVC is currently being tested in a phase 2 clinical trial for the treatment of PSC (Study 652-205, NCT02653625).
In summary, we have demonstrated that macrophages are associated with both acute and chronic sclerosing cholangitis in mice, and inhibition of monocyte-derived macrophage recruitment decreases liver injury and fibrosis. These observations need to be interpreted with caution as CCR2/CCL2 manipulation in these studies was not specific to monocytes/macrophages and other inflammatory cell types could have been affected. Nonetheless, these findings provide a rationale for the examination of CCR2/CCL2 inhibitors in clinical trials for the treatment of PSC.
Supplementary Material
LAY SUMMARY.
Primary sclerosing cholangitis (PSC) is an inflammatory liver disease which often progresses to liver failure. The cause of the disease is unclear and therapeutic options are limited. Therefore, we explored the role of white blood cells termed macrophages in PSC given their frequent contribution to other human inflammatory diseases. Our results implicate macrophages in PSC and PSC-like diseases in mice. More importantly, we found that pharmacologic inhibition of macrophage recruitment to the liver reduces PSC-like liver injury in the mouse. These exciting observations highlight potential new strategies to treat PSC.
Highlights.
Peribiliary macrophages are increased in PSC and animal models of PSC
Both M1-like and M2-like peribiliary macrophages are increased
Genetic and pharmacologic CCR2 inhibition restrains monocyte recruitment
CCR2 inhibition reduces fibrosis and cholestasis in animal models of PSC
These studies support the use of CCR2 inhibitors in human PSC
Acknowledgments
The authors thank Allergan for kindly providing pure cenicriviroc. The secretarial assistance of Ms. Courtney Hoover is much appreciated.
Financial support: This work was supported by NIH grants DK63947 (to GJG), DK57993 (to NFL), DK107255 (to AR), P30DK084567 (to the optical microscopy core of the Mayo Clinic Center for Cell Signaling in Gastroenterology), the PSC Partners Seeking a Cure Foundation (to MEG), and the Chris M. Carlos and Catharine Nicole Jockisch Carlos Foundation for PSC.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: At the time of the study, P.V. was an employee of Tobira (subsequently acquired by Allergan), which provided the CCR2/CCR5 antagonist cenicriviroc. All other authors have declared no conflict of interest related to this study.
Authors contributions:
M.E.G.: study design, data acquisition and analysis, manuscript drafting
C.E.T.: study design, data acquisition and analysis, manuscript revision
A.K.: data acquisition and analysis, manuscript revision
S.F.B.: data acquisition and analysis
M.J.L.P.: data acquisition
S.P.O.: data acquisition and analysis
P.L.S.: data acquisition
Y.G.: data acquisition
P.V.: manuscript revision
A.R.: data acquisition
N.F.L.: study design, data analysis, manuscript revision
G.J.G.: study design, data analysis, manuscript revision
References
- 1.Eaton JE, Talwalkar JA, Lazaridis KN, Gores GJ, Lindor KD. Pathogenesis of primary sclerosing cholangitis and advances in diagnosis and management. Gastroenterology. 2013;145(3):521–536. doi: 10.1053/j.gastro.2013.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fickert P, Fuchsbichler A, Wagner M, Zollner G, Kaser A, Tilg H, et al. Regurgitation of bile acids from leaky bile ducts causes sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology. 2004;127(1):261–274. doi: 10.1053/j.gastro.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 3.Popov Y, Patsenker E, Fickert P, Trauner M, Schuppan D. Mdr2 (Abcb4)−/− mice spontaneously develop severe biliary fibrosis via massive dysregulation of pro- and antifibrogenic genes. J Hepatol. 2005;43(6):1045–1054. doi: 10.1016/j.jhep.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 4.Rosmorduc O, Hermelin B, Boelle PY, Poupon RE, Poupon R, Chazouilleres O. ABCB4 gene mutations and primary sclerosing cholangitis. Gastroenterology. 2004;126(4):1220–1222. doi: 10.1053/j.gastro.2004.02.042. author reply 1222–1223. [DOI] [PubMed] [Google Scholar]
- 5.Guicciardi ME, Krishnan A, Bronk SF, Hirsova P, Griffith TS, Gores GJ. Biliary tract instillation of a SMAC mimetic induces TRAIL-dependent acute sclerosing cholangitis-like injury in mice. Cell Death Dis. 2017;8(1):e2535. doi: 10.1038/cddis.2016.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Italiani P, Boraschi D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front Immunol. 2014;5:514. doi: 10.3389/fimmu.2014.00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. 2010;30(3):245–257. doi: 10.1055/s-0030-1255354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mossanen JC, Krenkel O, Ergen C, Govaere O, Liepelt A, Puengel T, et al. Chemokine (C-C motif) receptor 2-positive monocytes aggravate the early phase of acetaminophen-induced acute liver injury. Hepatology. 2016;64(5):1667–1682. doi: 10.1002/hep.28682. [DOI] [PubMed] [Google Scholar]
- 9.Yang J, Zhang L, Yu C, Yang XF, Wang H. Monocyte and macrophage differentiation: circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark Res. 2014;2(1):1. doi: 10.1186/2050-7771-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11(11):762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tabibian JH, O'Hara SP, Splinter PL, Trussoni CE, LaRusso NF. Cholangiocyte senescence by way of N-ras activation is a characteristic of primary sclerosing cholangitis. Hepatology. 2014;59(6):2263–2275. doi: 10.1002/hep.26993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tabibian JH, Trussoni CE, O'Hara SP, Splinter PL, Heimbach JK, LaRusso NF. Characterization of cultured cholangiocytes isolated from livers of patients with primary sclerosing cholangitis. Lab Invest. 2014;94(10):1126–1133. doi: 10.1038/labinvest.2014.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mack M, Cihak J, Simonis C, Luckow B, Proudfoot AE, Plachy J, et al. Expression and characterization of the chemokine receptors CCR2 and CCR5 in mice. J Immunol. 2001;166(7):4697–4704. doi: 10.4049/jimmunol.166.7.4697. [DOI] [PubMed] [Google Scholar]
- 14.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115(2):209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friedman SL, Arthur MJ. Activation of cultured rat hepatic lipocytes by Kupffer cell conditioned medium. Direct enhancement of matrix synthesis and stimulation of cell proliferation via induction of platelet-derived growth factor receptors. J Clin Invest. 1989;84(6):1780–1785. doi: 10.1172/JCI114362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morita A, Itoh Y, Toyama T, Fujii H, Nishioji K, Kirishima T, et al. Activated Kupffer cells play an important role in intra-hepatic Th1-associated necro-inflammation in Concanavalin A-induced hepatic injury in mice. Hepatol Res. 2003;27(2):143–150. doi: 10.1016/s1386-6346(03)00206-7. [DOI] [PubMed] [Google Scholar]
- 17.Holt MP, Cheng L, Ju C. Identification and characterization of infiltrating macrophages in acetaminophen-induced liver injury. J Leukoc Biol. 2008;84(6):1410–1421. doi: 10.1189/jlb.0308173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115(1):56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitchell C, Couton D, Couty JP, Anson M, Crain AM, Bizet V, et al. Dual role of CCR2 in the constitution and the resolution of liver fibrosis in mice. Am J Pathol. 2009;174(5):1766–1775. doi: 10.2353/ajpath.2009.080632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ide M, Kuwamura M, Kotani T, Sawamoto O, Yamate J. Effects of gadolinium chloride (GdCl(3)) on the appearance of macrophage populations and fibrogenesis in thioacetamide-induced rat hepatic lesions. J Comp Pathol. 2005;133(2–3):92–102. doi: 10.1016/j.jcpa.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 21.Best J, Verhulst S, Syn WK, Lagaisse K, van Hul N, Heindryckx F, et al. Macrophage Depletion Attenuates Extracellular Matrix Deposition and Ductular Reaction in a Mouse Model of Chronic Cholangiopathies. PLoS One. 2016;11(9):e0162286. doi: 10.1371/journal.pone.0162286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O'Hara SP, Splinter PL, Trussoni CE, Gajdos GB, Lineswala PN, LaRusso NF. Cholangiocyte N-Ras protein mediates lipopolysaccharide-induced interleukin 6 secretion and proliferation. J Biol Chem. 2011;286(35):30352–30360. doi: 10.1074/jbc.M111.269464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Strazzabosco M, Fiorotto R, Cadamuro M, Spirli C, Mariotti V, Kaffe E, et al. Pathophysiologic implications of innate immunity and autoinflammation in the biliary epithelium. Biochim Biophys Acta. 2018;1864(4 Pt B):1374–1379. doi: 10.1016/j.bbadis.2017.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fiorotto R, Scirpo R, Trauner M, Fabris L, Hoque R, Spirli C, et al. Loss of CFTR affects biliary epithelium innate immunity and causes TLR4-NF-kappaB-mediated inflammatory response in mice. Gastroenterology. 2011;141(4):1498–1508. 1508 e1491–1495. doi: 10.1053/j.gastro.2011.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yokoyama T, Komori A, Nakamura M, Takii Y, Kamihira T, Shimoda S, et al. Human intrahepatic biliary epithelial cells function in innate immunity by producing IL-6 and IL-8 via the TLR4-NF-kappaB and -MAPK signaling pathways. Liver Int. 2006;26(4):467–476. doi: 10.1111/j.1478-3231.2006.01254.x. [DOI] [PubMed] [Google Scholar]
- 26.Lefebvre E, Moyle G, Reshef R, Richman LP, Thompson M, Hong F, et al. Antifibrotic Effects of the Dual CCR2/CCR5 Antagonist Cenicriviroc in Animal Models of Liver and Kidney Fibrosis. PLoS One. 2016;11(6):e0158156. doi: 10.1371/journal.pone.0158156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Puengel T, Krenkel O, Kohlhepp M, Lefebvre E, Luedde T, Trautwein C, et al. Differential impact of the dual CCR2/CCR5 inhibitor cenicriviroc on migration of monocyte and lymphocyte subsets in acute liver injury. PLoS One. 2017;12(9):e0184694. doi: 10.1371/journal.pone.0184694. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.