

Key Clinical Message
This case highlights an important lesson for laboratory genetic testing. Geneticists and Genetic Counselors should be aware that although rare, mosaic variegated aneuploidy should be considered if mosaic aneuploidies are observed on karyotype, particularly in the context of short stature.
Keywords: CEP57, genetic testing, mosaic variegated aneuploidy syndrome, whole exome sequencing
1. INTRODUCTION
Mosaic variegated aneuploidy (MVA) syndrome is a rare disorder presenting with short stature, intellectual disability, and mosaic aneuploidies. Herein, we report a patient with MVA diagnosed via whole exome sequencing after two normal karyotype results. MVA should be considered if mosaic aneuploidies are observed on karyotype.
Mosaic variegated aneuploidy (MVA) syndrome is a rare disorder characterized by mosaic aneuploidies of many chromosomes.1 The inheritance is autosomal recessive and caused by biallelic mutations in TRIP13, BUB1B, or CEP57, which establish the spindle assembly checkpoint, stabilize mitotic spindle attachment, and stabilize mitotic microtubule attachment, respectively.1, 2, 3, 4, 5, 6, 7 The phenotype of MVA is nonspecific and includes growth retardation, intrauterine growth restriction (IUGR), microcephaly, facial dysmorphia, and intellectual disability (ID).1, 2, 3, 8 The nonspecific phenotype can result in patients being underdiagnosed. Additionally, although mosaic aneuploidies should be observed on karyotype, they may be dismissed as an artifact.9 In this report, we describe a patient with MVA caused by biallelic mutations in CEP57 who was undiagnosed until whole exome sequencing (WES).
2. CLINICAL REPORT
The patient was an 11‐year 6‐month‐old Pakistani female who presented for an initial endocrine evaluation of short stature. She was born at 38 weeks of gestation to consanguineous parents. She had past diagnoses of IUGR, failure to thrive, developmental delay, mild ID, acanthosis nigricans, and precocious puberty. A physician in Pakistan treated her with leuprolide depot, growth hormone, and metformin for precocious puberty, idiopathic short stature, and acanthosis nigricans, respectively. Her physical examination revealed a prominent forehead and nose, frontal bossing, triangular face, and mild rhizomelic shortening of upper limbs. Her height was 51 inches (<3rd percentile), weighed 76 lbs (19th percentile), and BMI of 76th percentile. High‐resolution karyotype revealed a 46, XX karyotype.
3. MOLECULAR ANALYSIS
Informed consent was obtained from the parents and approved by the local institutional review board prior to molecular analysis. Based on the family history of consanguinity and unaffected parents, we suspected an autosomal recessive pattern of inheritance for which we performed WES. We hypothesized that the causal variant would be a rare, nonsynonymous, or frameshift homozygous variant. We excluded all variants with a minor allele frequency (MAF) >0.001 in the 1000 Genomes database (http://www.1000genomes.org) or in our internal exome database. There were 34 homozygous variants that fit these criteria. Review of each identified gene revealed only one that has previously been associated with growth retardation. This variant is a frameshift mutation in CEP57 (c.697delA, p.Lys235Argfs*31) and is not listed in dbSNP or ClinVar but is present in the Exome Aggregation Consortium database (exac.broadinstitute.org) with a MAF of 2.576e−5. Truncating mutations in CEP57 have been reported to cause MVA (OMIM 614114).2, 3 Sanger sequencing of CEP57 in the patient and her parents confirmed that the patient had this homozygous frameshift mutation, while her parents each had a heterozygous frameshift mutation (Figure 1).
Figure 1.
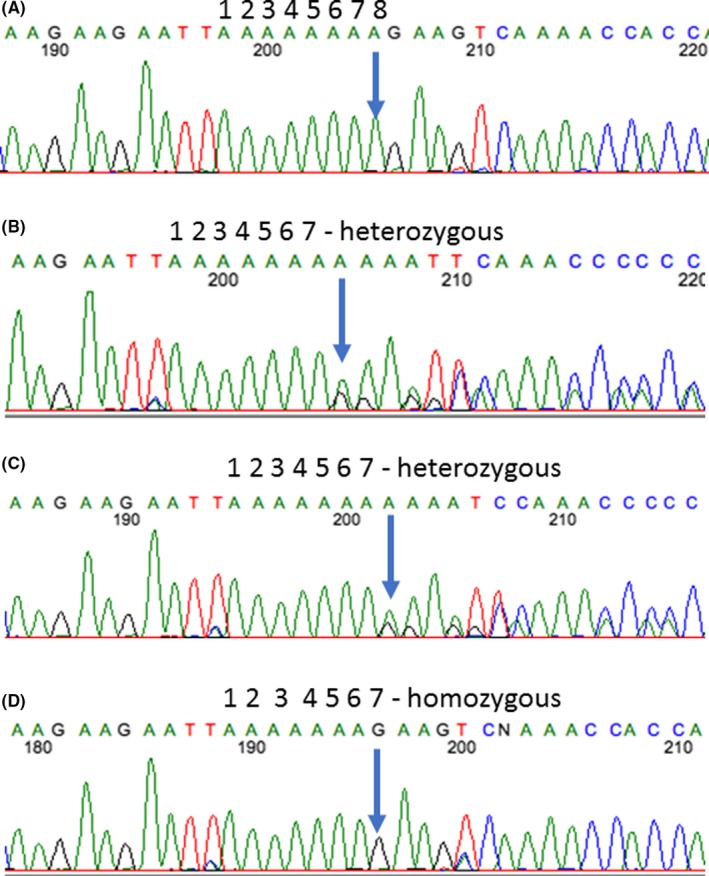
Sanger sequencing results confirm the CEP57 frameshift mutation. Chromatograms of a control wild‐type sample (A) as well as the patient's mother (B), father (C), and the patient (D) confirm that the patient has a homozygous frameshift variant in CEP57 (c.697delA, p.Lys235Argfs*31), while both of her parents are heterozygous for the same variant. The deleted nucleotide is indicated by an arrow
Due to WES findings, we ordered a repeat karyotype from peripheral blood mononuclear cells. Surprisingly, the results from the clinical laboratory were normal 46,XX. After requesting a re‐review, 17 of 22 cells were 46,XX, while 5 cells had unique aneuploidies that the laboratory initially interpreted as artifacts (Figure 2). The mosaic aneuploidies confirmed the diagnosis of MVA.
Figure 2.
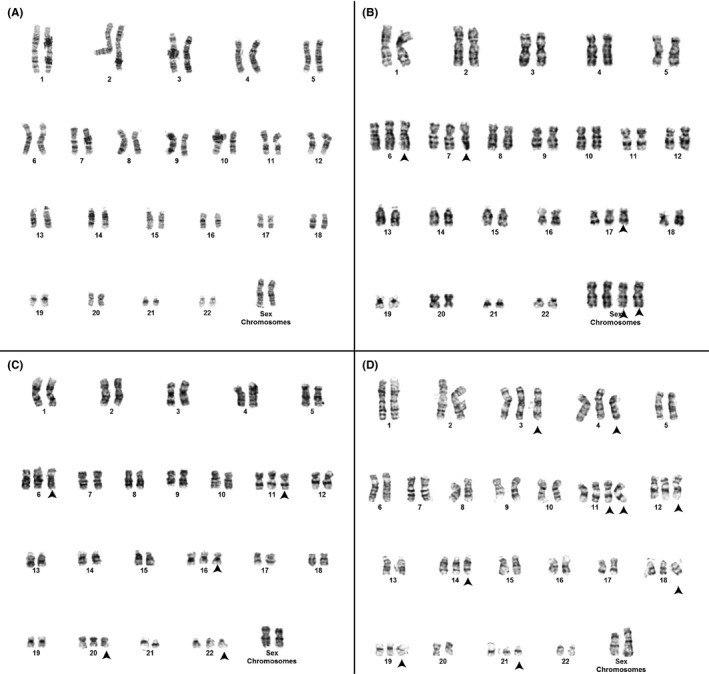
Chromosome results indicate mosaic variegated aneuploidy syndrome. The karyotype of 17 of 22 cells was 46,XX (A). Various aneuploidies were observed in the remaining five cells, three of which are pictured (B‐D). These include 51,XXXX,+6,+7,+17 (B), 51,XX,+6,+11,+16,+20,+22 (C), 55,XX,+3,+4,+11,+11,+12,+14,+18,+19,+21 (D). The aneuploidies are indicated with an arrowhead
4. DISCUSSION
Only 5 cases of MVA caused by CEP57 mutations have been reported in the literature.2, 3 Interestingly, MVA caused by homozygous mutations in BUB1B or TRIP13 is associated with an increased risk of cancer, but it is unknown if MVA caused by mutations in CEP57 is associated with increased cancer risks.1, 2, 3, 7, 10 Had this been known, growth hormone might have not been prescribed. Consistent with prior reports of MVA caused by mutations in CEP57, this patient has growth retardation, IUGR, ID, rhizomelic limb shortening, and skull anomalies (temporal bossing and triangular face) (Table 1).2, 3 This patient also has precocious puberty and acanthosis nigricans, which may be previously undescribed features of MVA or may be unrelated to this syndrome. Additional case reports of patients with MVA caused by mutations in CEP57 are needed to further define the phenotypic spectrum.
Table 1.
Review of the phenotype of published cases of CEP57 mutations
| Previous reports | Present report | Total | |
|---|---|---|---|
| Reference | Snape et al 2011, Pinson et al 2014 | This Case Report | |
| Age | 3 wk‐15 y | 12 y 7 mo | 3 wk‐15 y |
| Sex | 3 Males, 2 Females | 1 Female | 3 Males, 3 Females |
| Ethnicity | Mexican, Caucasian, Moroccan | Pakistani | |
| Growth retardation | 5/5 | + | 6/6 |
| IUGR | 3/5 | + | 4/6 |
| Mild intellectual disability | 2/4 | + | 3/4 |
| Rhizomelic shortening | 3/5 | + | 4/6 |
| Skull anomalies | 3/3 | + | 4/4 |
| Precocious puberty | Not reported | + | |
| Acanthosis nigricans | Not reported | + | |
| Cancer | 0/5 | ‐ | 0/6 |
IUGR, Intrauterine Growth Restriction.
This report highlights an important lesson for genetic testing. Karyotype was normal (46,XX) despite the presence of mosaic aneuploidies in a subset of cells. For a chromosome gain to be considered clonal, it must be observed in at least two cells per ISCN guidelines.11 This patient could have been diagnosed sooner had mosaic aneuploidies been detected and reported in her first karyotype. Laboratory and Clinical Geneticists should be aware that MVA should be considered if multiple mosaic aneuploidies are observed.
AUTHOR CONTRIBUTIONS
DB: analyzed WES and wrote manuscript. SE: evaluated the patient, wrote the clinical description, and edited the manuscript. AD: analyzed WES, edited, and approved manuscript.
CONFLICT OF INTEREST
The authors have no conflict of interests to disclose.
Brightman DS, Ejaz S, Dauber A. Mosaic variegated aneuploidy syndrome caused by a CEP57 mutation diagnosed by whole exome sequencing. Clin Case Rep. 2018;6:1531–1534. 10.1002/ccr3.1655
Diana S. Brightman and Sehar Ejaz contributed equally to this work.
REFERENCES
- 1. Hanks S, Coleman K, Reid S, et al. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat Genet. 2004;36:1159‐1161. [DOI] [PubMed] [Google Scholar]
- 2. Snape K, Hanks S, Ruark E, et al. Mutations in CEP57 cause mosaic variegated aneuploidy syndrome. Nat Genet. 2011;43:527‐529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pinson L, Mannini L, Willems M, et al. CEP57 mutation in a girl with mosaic variegated aneuploidy syndrome. Am J Med Genet A. 2014;164A:177‐181. [DOI] [PubMed] [Google Scholar]
- 4. He R, Wu Q, Zhou H, Huang N, Chen J, Teng J. Cep57 protein is required for cytokinesis by facilitating central spindle microtubule organization. J Biol Chem. 2013;288:14384‐14390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chan GK, Jablonski SA, Sudakin V, Hittle JC, Yen TJ. Human BUBR1 is a mitotic checkpoint kinase that monitors CENP‐E functions at kinetochores and binds the cyclosome/APC. J Cell Biol. 1999;146:941‐954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Emanuele MJ, Stukenberg PT. Xenopus Cep57 is a novel kinetochore component involved in microtubule attachment. Cell. 2007;130:893‐905. [DOI] [PubMed] [Google Scholar]
- 7. Yost S, de Wolf B, Hanks S, et al. Biallelic TRIP13 mutations predispose to Wilms tumor and chromosome missegregation. Nat Genet. 2017;49:1148‐1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Garcia‐Castillo H, Vasquez‐Velasquez AI, Rivera H, Barros‐Nunez P. Clinical and genetic heterogeneity in patients with mosaic variegated aneuploidy: delineation of clinical subtypes. Am J Med Genet A. 2008;146A:1687‐1695. [DOI] [PubMed] [Google Scholar]
- 9. Micale MA, Schran D, Emch S, Kurczynski TW, Rahman N, Van Dyke DL. Mosaic variegated aneuploidy without microcephaly: implications for cytogenetic diagnosis. Am J Med Genet A. 2007;143a:1890‐1893. [DOI] [PubMed] [Google Scholar]
- 10. Rio Frio T, Lavoie J, Hamel N, et al. Homozygous BUB1B mutation and susceptibility to gastrointestinal neoplasia. N Engl J Med. 2010;363:2628‐2637. [DOI] [PubMed] [Google Scholar]
- 11. McGowan‐Jordan JS, Simons A, Schmid M, International Standing Committee on Human Cytogenomic Nomenclature . ISCN: an international system for human cytogenomic nomenclature. New York: Karger; 2016. [Google Scholar]