

Key Clinical Message
We describe the prenatal and postnatal course of an infant with a large 19p deletion. Cases such as ours will improve the knowledge of specific gene functions for every medical specialist. The goal is to allow for a more rapid diagnosis, accurate prognosis and to decrease the likelihood of complications.
Keywords: 19p deletion, hiatal hernia, hypotonia, intrauterine growth restriction, neonate, pyloric stenosis, small for gestational age
1. INTRODUCTION
Sixteen cases with a deletion in the short arm of chromosome 19 are described in detail in the literature. Patients presented with neonatal hypotonia and developmental delay. Other findings varied significantly. We describe the perinatal course of an infant with a 3.7‐megabase deletion in the short arm of chromosome 19.
Deletions in the short arm of chromosome 19 are exceedingly rare.1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11While reported cases of chromosome 19p deletions have varying sizes and breakpoints and a wide range of phenotypes, all have developmental delays. We present the clinical course of a male newborn with a large deletion in the short arm of chromosome 19.
2. CASE REPORT
Twin B of a spontaneous dichorionic and diamniotic twin pregnancy of a 30‐year‐old multigravida was found to have symmetrical intrauterine growth restriction (IUGR) at 25‐week gestation (estimated fetal weight (EFW) <3rd percentile, intertwin weight discordance 28%), with abnormal Dopplers indicating increased placental resistance and cranial sparing with cephalization of blood flow. Also noted were mild right lateral cerebral ventriculomegaly, small stomach, and enlarged gallbladder. Aneuploidy screening results were low‐risk, with cell‐free DNA negative for trisomy 21, 18, and 13. Amniocentesis was declined. Maternal viral serology for cytomegalovirus, toxoplasmosis, and parvovirus was negative for recent infection. From 27 weeks onward, persistently absent end‐diastolic flow of the umbilical artery was noted. Ventriculomegaly worsened to moderate range at 28 and 33 weeks, with bitemporal skull narrowing and strawberry‐shaped skull appearance noted at 31 weeks (Figure 1). Anhydramnios for Twin B was noted at 36‐week gestation. As diagnosis of aneuploidy would affect postnatal management, bladder aspiration of Twin B was performed, but cell culture was without growth. At 38 weeks, the EFW for Twin B was 1559 g, with twin discordance of 48%.
Figure 1.
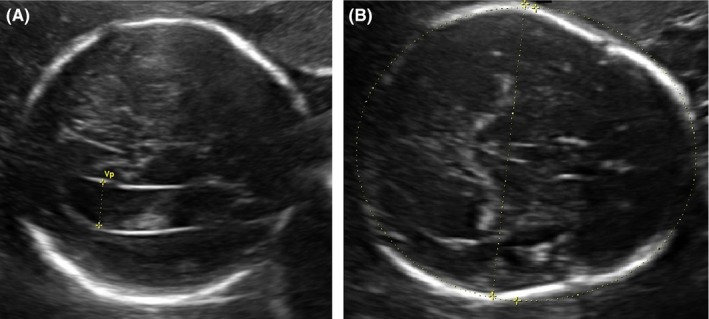
Select prenatal ultrasound images representative of (A) right lateral ventriculomegaly (12.4 mm), and (B) strawberry‐shaped head
The patient was born at 38 weeks and 3 days gestation, via scheduled repeat cesarean delivery. Apgar scores were 5, 7, and 8 at 1, 5, and 10 minutes of life, respectively. He was small for gestational age (SGA) with birthweight 1133 g (Z‐score: −5.49), birth length 39.4 cm (Z‐score: −4.43), and head circumference 29.5 cm (Z‐score: −3.23). Physical examination was significant for small facies with underdeveloped chin, relative macrocephaly, overlapping cranial sutures, mild crumpling of the ears, wide‐spaced nipples, second and fifth fingers of the right hand overlapping the third and fourth fingers, and a small sacral hemangioma (Figure 2). The patient developed hypoglycemia and hypophosphatemia, likely secondary to his IUGR/SGA status, which responded to modifications in intravenous fluids. He tolerated full enteral feeds by the second week of life, but due to poor oromotor skills, he was fully nasogastric tube‐dependent. Patient developed self‐resolving leukopenia. Thrombocytopenia, reaching a nadir on day of life (DOL) 2, responded to 1 platelet transfusion. Mild self‐resolving anemia was noted with a high reticulocyte count. Second anemia nadir at fourth week of life was treated with epoetin alfa, with normalization of erythrocyte indices. From DOL 5 onward, patient had a prolonged need for respiratory support, attributed to chronic microaspirations, with a waxing‐and‐waning degree of respiratory distress, which was subsequently attributed to a hiatal hernia. Gastrointestinal contrast study showed approximately half the stomach within the thoracic cavity, along with an incidental finding of inguinal hernia.
Figure 2.
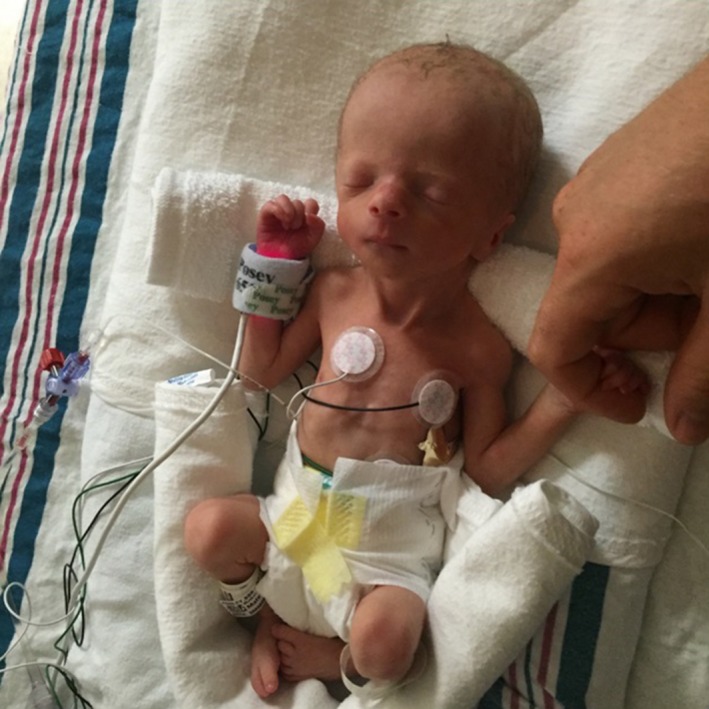
Photograph of the patient. Note the relative macrocephaly, downslanting palpebral fissures, low‐set ears, and mild micrognathia. The second finger overlaps the third finger, and the fifth overlaps the fourth finger. The nipples are wide‐spaced (left not visible)
During the patient's hospital course, extensive workup was completed as part of evaluation for the chromosome 19p deletion (see Section 3). Initial head ultrasound showed a small hyperechogenic focus inferior to the third ventricle. Noncontrast brain MRI performed on DOL 23 showed normal findings. Follow‐up head ultrasound performed on DOL 50 showed resolution of the previously described echogenic focus, with a new finding of mild dilation of frontal horns of lateral ventricles. Spinal ultrasound was significant for a low lying conus and a tethered spinal cord. Whole spine MRI confirmed this finding, additionally showing 6 lumbar vertebrae. Abdominal and focused renal ultrasounds were normal. Echocardiogram was significant for suspected interrupted inferior vena cava with hemiazygos continuation to the left superior vena cava draining to the coronary sinus; no right superior vena cava was noted. Suspected cervical aortic arch was present with mild tortuosity of transverse arch without evidence of coarctation. Normal pulmonary venous drainage and a patent foramen ovale with left‐to‐right shunt were also seen. Ophthalmologic examination performed on DOL 13 showed a thin iris with rugged pupils. Mild stippling of the left retina with irregularly shaped optic disk was also noted.
On DOL 53, laparoscopy was performed to repair the hiatal hernia. Intraoperative diagnoses were: type I paraesophageal hernia with almost the entire stomach located within the chest, and a significant pyloric stenosis. The infant underwent paraesophageal hernia repair, Nissen fundoplication, G‐tube placement, and pyloromyotomy. On postoperative day four, the patient developed a firm abdomen and respiratory compromise. Abdominal X‐ray revealed intestinal pneumatosis and presence of portal vein gas. Emergent laparotomy found a 3 mm gastric perforation and necrotic small bowel. Unfortunately, the patient passed away intraoperatively. Cause of death was shock secondary to gastric perforation, peritonitis, and bowel necrosis.
3. METHODS AND RESULTS
Prenatal screening included First Trimester Screen (Eurofins NTD, NY) and cell‐free DNA screen (MaterniT21, Sequenom, CA). Maternal viral serologies were performed at Quest Diagnostics, VA. Prenatal fluorescent in situ hybridization (FISH) was performed by Integrated Genetics, LabCorp, NC (probes: nuc ish Xcen (DXZ1x1), Ycen (DYZ3x1), 13q14 (IRB1x1), 18cent (D18Z1x2), 21q22.13‐q22.2 (D2S342/D211S341/D21S259)x2). All prenatal imaging performed by a registered diagnostic medical sonographer with Voluson E8 (GE Healthcare, WI) and reviewed by a board certified maternal fetal medicine specialist. Neonatal FISH screen showed a normal male chromosome complement, suggesting no numerical abnormality of chromosome 13, 18, 21 or sex chromosomes. Subsequent karyotype analysis showed a normal male karyotype (46, XY). High‐resolution chromosomal microarray showed a 3.7 Mb interstitial deletion of the short arm of chromosome 19, bands p13.2 p13.12 (11 836 342‐15 505 009)x1 (Quest Diagnostics Nichols Institute, Chantilly, VA).
4. DISCUSSION
We present a newborn with multiple anomalies (Table 1) and a large 3.7‐Mb interstitial deletion of the short arm of chromosome 19, bands p13.2 p13.12 (11 836 342‐15 505 009)x1, which accounts for approximately 5.8% of the total length of chromosome 19 and contains at least 111 genes, including 60 OMIM‐annotated genes. There have been a total of thirteen patients with a deletion in the short arm of chromosome 19, that partially or fully overlapped with the deletion seen in our patient (Figure 3), reported in the literature with a detailed prenatal and neonatal course. Additionally, DECIPHER database provides a list of lifetime phenotypic changes for up to 41 patients with an overlapping deletion, 10 of which were classified as having “definitive or likely pathogenic deletions”.
Table 1.
Clinical, laboratory and imaging findings seen in our patient
| Prenatal findings |
| Severe intrauterine growth restriction with abnormal umbilical artery and middle cerebral artery Dopplers |
| Dilated gallbladder |
| Small stomach |
| Bitemporal narrowing/strawberry‐shaped skull |
| Unilateral moderate ventriculomegaly |
| Anhydramnios in the third trimester |
| Newborn physical examination |
| Small for gestational age |
| Small facies with micrognathia |
| Relative macrocephaly |
| Overlapping cranial sutures |
| Downslanting palpebral fissures |
| Low set ears |
| Mild crumpling of the ears bilaterally |
| Wide‐spaced nipples |
| Second and fifth fingers of the right hand overlapped the third and fourth fingers, respectively |
| Hypoglycemia, hypophosphatemia |
| Hematologic anomalies |
| Thrombocytopenia, transitory (25 000 per microliter) |
| Leukopenia, transitory (3660 per microliter) |
| Anemia |
| Neurologic anomalies |
| Hyperechogenic focus inferior to the third ventricle, resolved by 2 months of age |
| Mild dilatation of frontal horns of lateral ventricles, noted at 2 months of age |
| Low lying conus medullaris, tethered spinal cord |
| Thin iris with rugged pupils, with mild left retinal stippling and irregularly shaped optic disk |
| Musculoskeletal anomalies |
| Six lumbar vertebrae |
| Cardiovascular anomalies |
| Interrupted inferior vena cava |
| Hemiazygos continuation to left superior vena cava, draining into the coronary sinus |
| Absent right superior vena cava |
| Suspected cervical aortic arch |
| Gastrointestinal anomalies |
| Paraesophageal hernia, type I |
| Pyloric stenosis |
| Inguinal hernia, bilateral |
Figure 3.
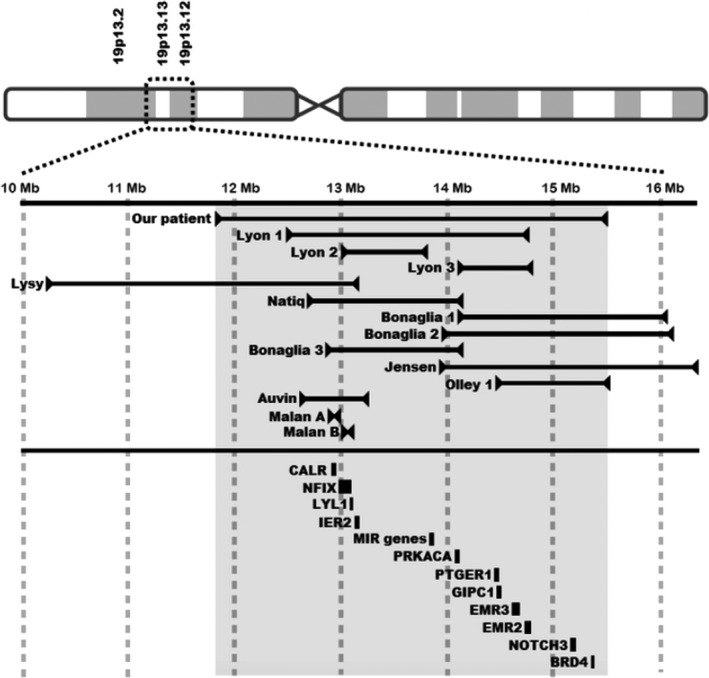
Graphic representation of sites of 19p deletion in our patient, and previously reported patients by distance in megabases (Mb) from the centromere. The shaded area corresponds to the extent of the deletion seen in our patient. OMIM‐annotated genes, pertinent to the findings seen in our patient, and their positions relative to the deletion are shown in the bottom part of the graph. The width of the bar next to the gene corresponds with the size of the gene. Hg19 positions of the genes: CALR (19:12,938,599‐12,944,489), NFIX (19:12,995,511‐13,098,795), LYL1 (19:13,099,027‐13,103,159), MIR genes (19:13,836,286‐ 13,874,807), PRKACA (19:14,091,687‐14,117,746), PTGER1 (19:14,472,465‐14,475,361), GIPC1 (19:14,477,758‐14,496,148), EMR3 (19:14,600,174‐14,674,917), EMR2 (19:14,732,696‐14,778,540), NOTCH (19:15,159,632‐15,200,980), BRD4 (19:15,236,835‐15,332,542). IER2 (GenBank accession no. NM_004907)
The patients described in detailed case reports all had varying degree of neonatal hypotonia. In addition, 2 patients4, 10 had IUGR with head sparing, one10 had neonatal seizures, one4 had severe microcephaly, while two4, 6 presented with facial dysmorphism. Lysy et al and Lyon et al described patients with large 19p deletions and craniosynostosis. The authors raised the possibility that DAND5 and CALR genes might be involved in the pathogenesis of the neurocranial abnormality. As the same 2 genes were affected in our patient, it is unlikely that they alone would be responsible for the craniosynostosis. Additionally, the patient with a 1 Mb‐deletion in Lyon et al's report had several phenotypic features similar to our patient: antenatal onset of ventriculomegaly, postnatally noted dysmorphisms (low‐set ears and micrognathia), and bilateral optic dysplasia. The authors also raised a potential role of CACNA1A and CC2D1A genes in epilepsy and developmental delay, respectively. It is possible our patient would have developed these features, but given his early demise the full phenotype was not able to develop. Malan et al described 2 patients with a deletion of NFIX gene, who presented with drastically different phenotypes. One patient presented with Sotos‐like overgrowth syndrome, while the other presented with Marshall‐Smith syndrome (MSS). The difference was postulated to be a result of differing effects of nonsense‐mediated mRNA decay of the NFIX gene product. Many physical findings of our patient overlap with those seen in MSS: underdeveloped/small face, retrognathia, failure to thrive, respiratory distress, and pyloric stenosis. It is interesting to note that our patient's deletion affected the microRNA genes (MIR24‐2, MIR27A, MIR23A, MIR181C, MIR181D, and MIR639), potentially contributing to the MSS‐like phenotype. NFIX mutations have also been associated with pathologic thinning of the intestinal wall and diminished blood supply.12 Olley et al showed that haploinsufficiency of BRD4, either as part of a larger deletion or as a single gene mutation, is the likely genetic mechanism of Cornelia de Lange (CdL)‐like syndrome. Our patient had several features that would fall under either major or secondary characteristics of CdL syndrome, including: small for gestational age, microcephaly, low‐set ears, clinodactyly of the fifth finger and gastrointestinal abnormalities. It is interesting to note that of the patients described in the DECIPHER database, ones with deletions overlapping with the telomeric side of our patient's deletion had macrocephaly and optic nerve hypoplasia, while the ones overlapping with the centromeric side tended to have microcephaly and low‐set ears.
LYL1 and IER2 gene products are involved in angiogenesis, capillary formation, and vessel stabilization.13, 14 We speculate that the gastrointestinal tract in a patient with deletions of these genes would be more prone to tissue injury in a setting of peritonitis or surgery, as seen in our patient. Other potentially clinically significant genes were affected (Figure 3). CALR codes for calreticulin, which acts as a general recognition signal on apoptotic cells, thus having a crucial role in resolution of inflammation.15 EMR2 and EMR3 genes are involved in potentiating inflammatory responses.16 Deletion of these 3 genes could have led to poor healing and increased risk of gastric perforation in a setting of surgical manipulation. Steroid production is increased with increase in activity level of PRKACA‐coded protein kinase17 and modulated at the hypothalamic level through prostaglandin E2 receptor, coded by PTGER1.18 Our patient did not exhibit any hemodynamic instability that would prompt us to consider initiation of steroid replacement therapy. Nonetheless, we would recommend that with future patients who have a deletion of one or both of these genes, measurement of cortisol level be considered. It is interesting to note that despite chronic microaspiration and a remarkable number of desaturation episodes, our patient never developed any signs of pulmonary hypertension. Animal models showed that NOTCH3‐null mice did not develop pulmonary hypertension in a setting of hypoxic stimulation.19 It is possible that the deletion of NOTCH3 gene might have protected our patient from developing clinically significant pulmonary hypertension. GIPC1 codes for synectin, a protein crucial for cell surface receptor expression.20 Synectin‐null mice have abnormal pattern of arterial formation and branching, with hypoplastic dorsal aorta and stunted intersomitic vessels.21 Our patient's echocardiogram showed a cervical aortic arch with transverse arch tortuosity, a finding consistent with the animal models. Major vascular anomalies noted in our patient affected the large systemic veins. None of the genes deleted in our patient have previously been related to such phenotypes.
In this report, we describe the prenatal and postnatal courses of an infant with a large 19p13 deletion and address possible clinical correlation to the deletion of specific genes. Cases such as our will further improve our knowledge of specific gene functions.
CONFLICT OF INTERESTS
Authors have no conflict of interests to declare.
AUTHORSHIP
MC: involved in postnatal management of the patient, literature review of the 19p deletions, preclinical and clinical studies of genes involved by the deletion, writing the manuscript. JR: provided prenatal genetic counseling to the patient and coordinated prenatal aneuploidy screening and testing during the pregnancy. RSH: aided in the interpretation of the genetic anomaly, review of microarray, genes and gene location involved, and revisions of the manuscript. RD: involved in prenatal management of the mother, providing the antenatal course, and interpretation of the ultrasound findings. KK: involved in postnatal management of the patient, providing the postnatal clinical course. JT: involved in prenatal management of the mother, providing the antenatal course and interpretation of the ultrasound findings, edits, and revisions of the manuscript.
Culjat M, Razak J, Saadeh‐Haddad R, Driggers R, Kamholz K, Timofeev J. Perinatal findings in a patient with a novel large chromosome 19p deletion. Clin Case Rep. 2018;6:1525–1530. 10.1002/ccr3.1615
REFERENCES
- 1. Archer HL, Gupta S, Enoch S, et al. Distinct phenotype associated with a cryptic subtelomeric deletion of 19p13.3‐pter. Am J Med Genet A. 2005;136:38‐44. [DOI] [PubMed] [Google Scholar]
- 2. Aten E, den Hollander N, Ruivenkamp C, et al. Split hand‐foot malformation, tetralogy of Fallot, mental retardation and a 1 Mb 19p deletion‐evidence for further heterogeneity? Am J Med Genet A. 2009;149A:975‐981. [DOI] [PubMed] [Google Scholar]
- 3. Auvin S, Holder‐Espinasse M, Lamblin MD, Andrieux J. Array‐CGH detection of a de novo 0.7‐Mb deletion in 19p13.13 including CACNA1A associated with mental retardation and epilepsy with infantile spasms. Epilepsia. 2009;50:2501‐2503. [DOI] [PubMed] [Google Scholar]
- 4. Bonaglia MC, Marelli S, Novara F, et al. Genotype‐phenotype relationship in three cases with overlapping 19p13.12 microdeletions. Eur J Hum Genet. 2010;18:1302‐1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hurgoiu V, Suciu S. Occurrence of 19p‐ in an infant with multiple dysmorphic features. Ann Genet. 1984;27:56‐57. [PubMed] [Google Scholar]
- 6. Jensen DR, Martin DM, Gebarski S, et al. A novel chromosome 19p13.12 deletion in a child with multiple congenital anomalies. Am J Med Genet A. 2009;149A:396‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lyon SM, Waggoner D, Halbach S, Thorland EC, Khorasani L, Reid RR. Syndromic craniosynostosis associated with microdeletion of chromosome 19p13.12‐19p13.2. Genes Dis. 2015;2:347‐352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lysy PA, Ravoet M, Wustefeld S, et al. A new case of syndromic craniosynostosis with cryptic 19p13.2‐p13.13 deletion. Am J Med Genet A. 2009;149A:2564‐2568. [DOI] [PubMed] [Google Scholar]
- 9. Malan V, Rajan D, Thomas S, et al. Distinct effects of allelic NFIX mutations on nonsense‐mediated mRNA decay engender either a Sotos‐like or a Marshall‐Smith syndrome. Am J Hum Genet. 2010;87:189‐198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Natiq A, Elalaoui SC, Miesch S, et al. A new case of de novo 19p13.2p13.12 deletion in a girl with overgrowth and severe developmental delay. Mol Cytogenet. 2014;7:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Olley G, Ansari M, Bengani H, et al. BRD4 interacts with NIPBL and BRD4 is mutated in a Cornelia de Lange‐like syndrome. Nat Genet. 2018;50:329‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Driller K, Pagenstecher A, Uhl M, et al. Nuclear factor I X deficiency causes brain malformation and severe skeletal defects. Mol Cell Biol. 2007;27:3855‐3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Giroux S, Kaushik AL, Capron C, et al. lyl‐1 and tal‐1/scl, two genes encoding closely related bHLH transcription factors, display highly overlapping expression patterns during cardiovascular and hematopoietic ontogeny. Gene Expr Patterns. 2007;7:215‐226. [DOI] [PubMed] [Google Scholar]
- 14. Wu W, Zhang X, Lv H, et al. Identification of immediate early response protein 2 as a regulator of angiogenesis through the modulation of endothelial cell motility and adhesion. Int J Mol Med. 2015;36:1104‐1110. [DOI] [PubMed] [Google Scholar]
- 15. Gardai SJ, McPhillips KA, Frasch SC, et al. Cell‐surface calreticulin initiates clearance of viable or apoptotic cells through trans‐activation of LRP on the phagocyte. Cell. 2005;123:321‐334. [DOI] [PubMed] [Google Scholar]
- 16. Yona S, Lin HH, Dri P, et al. Ligation of the adhesion‐GPCR EMR2 regulates human neutrophil function. FASEB J. 2008;22:741‐751. [DOI] [PubMed] [Google Scholar]
- 17. Beuschlein F, Fassnacht M, Assié G, et al. Constitutive activation of PKA catalytic subunit in adrenal Cushing's syndrome. N Engl J Med. 2014;370:1019‐1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Matsuoka Y, Furuyashiki T, Bito H, et al. Impaired adrenocorticotropic hormone response to bacterial endotoxin in mice deficient in prostaglandin E receptor EP1 and EP3 subtypes. Proc Natl Acad Sci USA. 2003;100:4132‐4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li X, Zhang X, Leathers R, et al. Notch 3 signaling promotes the development of pulmonary arterial hypertension. Nat Med. 2009;15:1289‐1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee NY, Ray B, How T, Blobe GC. Endoglin promotes transforming growth factor beta‐mediated Smad 1/5/8 signaling and inhibits endothelial cell migration through its association with GIPC. J Biol Chem. 2008;283:32527‐33233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chittenden TW, Claes F, Lanahan AA, et al. Selective regulation of arterial branching morphogenesis by synectin. Dev Cell. 2006;10:783‐795. [DOI] [PubMed] [Google Scholar]