

Abstract
Aims: Lipoprotein glomerulopathy (LPG) is a rare inherited renal disease. Several apolipoprotein E (apoE) mutations have been reported to be related to LPG. Herein, we report a case of a LPG patient with a novel apoE mutation.
Methods: A 45-year-old Chinese female was diagnosed as LPG by renal biopsy. APOE gene was sequenced. Clinical and genetic studies were conducted.
Results: The patient presented with nephrotic syndrome and hypertension. A fasting lipid panel showed mild hyperlipidemia and elevated serum apoE (5.6 mg/dL). Renal biopsy revealed typical LPG lesions with whorled, mesh-like material in dilated glomerular capillary lumens that stained positive for Sudan III and apoE. apoE gene analysis revealed a T-to-C point mutation at amino acid 173 that caused a substitution of a proline residue for a leucine residue, which has not been reported previously. We named this mutation apoE Chengdu (c.518T> C, p.L173P). Two of five of the family members carried this mutation, including the patient's brother who was receiving hemodialysis, and her sister, whose urine protein levels were normal. All mutation carriers were heterozygotes with the apoE genotype ε3/ε3. This mutation was not found among 200 of the local people. Fenofibrate treatment for one year induced clinical improvement.
Conclusions: ApoE Chengdu (p.L173P) is a novel mutation causing LPG. This case supports the hypothesis that the substitution of proline in or near the LDL receptor-binding area contributes to the development of LPG. The detailed mechanism of action of this variant remains to be elucidated.
Keywords: Apolipoprotein E, ApoE Chengdu, Lipoprotein glomerulopathy
Introduction
Lipoprotein glomerulopathy (LPG) is a rare autosomal dominant renal disease with incomplete penetrance that has mainly been reported in eastern Asia1, 2). LPG patients often present with nephrotic-range proteinuria, hypertriglyceridemia and elevated apolipoprotein E (apoE) levels. Massive lipoprotein thrombi in the lumens of dilated glomerular capillaries are the typical pathological manifestation of LPG3). Renal biopsy is crucial for the LPG diagnosis.
To date, several apoE mutations have been reported to be related to LPG, including apoE Sendai4) and apoE Kyoto5). However, DNA from 17 patients with LPG were reported to have no apoE variants6). Herein, we present a case of a Chinese LPG patient with a novel apoE mutation (apoE Chengdu). This mutation causes the substitution of a leucine residue with a proline residue at position 173 of the apoE protein. The clinicopathological features and treatment are also discussed.
Methods
Participants
In this study, we recruited five family members of the LPG patient and 200 local elderly people. Written informed consent was obtained from all participants prior to genetic testing. This study was approved by the ethics committee of the West China Hospital and was in accordance with the Declaration of Helsinki.
ApoE Mutation and Genotype Detection
To verify the connection between apoE mutation and lipoprotein glomerulopathy, the coding region of the APOE gene was sequenced. Genomic DNA was extracted from peripheral blood leukocytes collected from five family members using a DNA extraction kit (Tiangen, China). All of the family members were screened for mutations in the APOE coding sequence. Four primer pairs (Table 1) were designed with Primer Premier 5.0 to amplify exons 2, 3, 4 and the adjacent intron/exon boundaries of APOE (reference sequence GenBank accession no. NM_000041.3). The polymerase chain reaction (PCR) products were purified and then sequenced on an ABI377A DNA sequencer (Applied Biosystems, Foster City, CA, USA). Genotype was confirmed by restriction fragment length polymorphism (RFLP) analysis using enzyme HhaI7).
Table 1. Primer sequences and PCR product sizes.
| Exon | Primer sequence (5′→3′) | PCR product size (bp) |
|---|---|---|
| Exon2 | 1F: TTGCAGATAATGCAACAAGGC 1R: GTCAAATCGCTGTTCAGAGCC |
324 |
| Exon3 | 2F: ATGGCTCCAAAGAAGCATTTG 2R: GCAGAATGAAACCTGGACCTG |
374 |
| Exon4 | 3F: GTCTCCTTCTCTCGGCCTCT 3R: ACCTGCTCCTTCACCTCGT |
662 |
| Exon4 | 4F: GCCGATGACCTGCAGAAG 4R: GCTGGGGCTTAGAGGAAATC |
628 |
After identifying the specific mutation site, we searched the apoE Chengdu (p.L173P) mutation among 200 local elderly people. A 692-bp segment of the apoE gene including the mutation in codon 173 (reference sequence GenBank accession no. NM_000041.3) was amplified by PCR using the oligonucleotide primers E4F1 (GCCTACAAATCGGAACTGGA) and E4R1 (TTCGGCGTTCAGTGATTGTC). A total of 10 pmol of each primer and 2 µL of genomic DNA were added to a 50-microliter PCR system that consisted of 10 µL of 5× prime STAR GXL Buffer, 4 µL of a dNTP mixture (2.5 mM each), 1 µL of the prime STAR GXL DNA polymerase, and 31 µL of sterilized distilled water. The amplification conditions were as follows: denaturation at 98 °C for 10 s; annealing at 55°C for 15 s; and polymerization at 68°C for 42 s with 32 cycles. The PCR products were purified and sequenced directly.
Clinicopathological Examination
All family members underwent routine laboratory testing including measurement of serum albumin, serum creatinine, 24-h proteinuria, the urinary protein/creatinine ratio (g/g), urine sediment, and a lipid profile analysis including the serum total cholesterol (TC), total triglyceride (TG), low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C), and apoE levels (using single radial immunodiffusion methods; Sekisui Medical Co. Tokyo, Japan).
Formalin-fixed, 2-µm-thick, paraffin-embedded sections of renal biopsy tissues were stained with hematoxylin and eosin, periodic acid-Schiff (PAS), periodic acid-silver methenamine (PASM), Masson's trichrome, and Sudan III. Immunofluorescent examinations (including apoE) and electron microscopy were also performed.
Case Presentation
A 48-year-old Chinese woman was admitted to our hospital in April 2016. Two years prior to admission she noticed edema of her lower extremities. The patient's blood pressure was 160/104 mmHg. Laboratory evaluation revealed the following: urinary protein 3.8 g/24 h; serum creatinine 0.85 mg/dL; serum albumin 3.5 g/dL; TG 224 mg/dL; TC 307 mg/dL; and LDL-C 193 mg/dL. She was diagnosed with nephrotic syndrome. Oral prednisone at a dose of 30 mg per day was initiated for six months and then tapered. Candesartan at 4 mg per day and amlodipine at 5 mg per day were used to maintain the patient's blood pressure below 140/90 mmHg. Atorvastatin was used to control hyperlipidemia. The patient's proteinuria did not improve with this regimen.
Upon admission, the patient's height was 150 cm, and weight was 61 kg (body mass index 27.1 kg/m2). Her blood pressure was 146/103 mmHg. She did not show any corneal opacity or xanthoma. The laboratory evaluation revealed a serum creatinine level of 0.76 mg/dL, urea nitrogen of 16 mg/dL, serum albumin of 3.74 g/dL, TG of 135.4 mg/dL, TC of 198.1 mg/dL, LDL-C of 86.85 mg/dL, HDL-C of 87.08 mg/dL, serum apoE of 5.6 mg/dL, urinary protein of 3.74 g/24 h, and urinary sediment with 11 red blood cells per high-power field. The serum anti-nuclear antibody (ANA), double-stranded DNA (ds-DNA), anti-neutrophil cytoplasmic antibody (ANCA), anti-glomerular basement membrane (anti-GBM) antibody, and rheumatoid factor tests were all negative. The serum C3 and C4 levels were 102 and 25.7 mg/dL, respectively. The hepatitis B virus (HBV) and hepatitis C virus (HCV) tests were negative. Ultrasonographic evaluation revealed that both kidneys were a normal size.
Pathological Findings
A renal biopsy was performed. Among the eight glomeruli sampled for light microscopy analysis, two were globally sclerotic and two were segmentally sclerotic with synechia. The glomeruli were diffusely enlarged due to greatly dilated capillaries filled with whorled, mesh-like material in the glomerular capillary lumens (Figs. 1A, B, and C). The mesh-like material stained positive with Sudan III and apoE (Figs. 1D and E). Foam cells were absent in the glomeruli. A moderate increase in mesangial cells and the matrix was present and accompanied by segmental mesangial interposition and duplication of the glomerular basement membranes. Tubular atrophy and interstitial fibrosis affected approximately 10% of the cortex sampled. No obvious vascular lesions were observed. Immunofluorescence staining was negative for IgG, IgA, IgM, C3, C4, and C1q. Electron microscopy (Fig. 1F) revealed numerous lipid granules deposited in the dilated glomerular capillary lumens without evidence of macrophage infiltration.
Fig. 1.
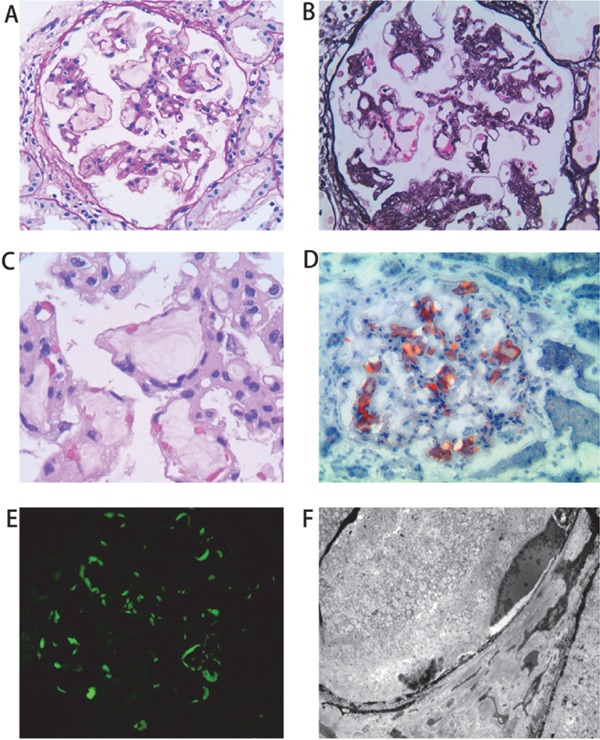
Pathological findings of renal biopsy specimens from LPG patient
Panel A shows a glomerulus with the presence of a pale-stained accumulation of amorphous material within the dilated lumens of the glomerular capillaries (PAS staining, ×200). Panel B shows intraglomerular lipoprotein thrombi and a moderate increase in the mesangial matrix (PASM staining, ×200). Panel C shows magnified glomerular capillaries containing lamellate thrombuslike depositions (PAS staining, ×400). Panel D shows that the mesh-like material in the glomerular capillaries is positive by Sudan III staining (frozen section, ×200). Immunofluorescence reveals that apoE is present primarily in the capillary lumen (panel E, frozen section, ×200). Electron microscopy shows numerous lipid granules deposited in the dilated glomerular capillary lumens (panel F, ×6000).
Familial Evaluation
The patient had three brothers and one sister (Fig. 2). One of her brothers (II-2) was diagnosed with uremia and had been on hemodialysis. Four years previously he had gone to hospital complaining of headache for two months. Physical examination showed elevated blood pressure 160/95 mmHg. Serum creatinine was 15.8 mg/dL. Urine protein was 2+. Ultrasonography did not show evidence of urinary tract obstruction. He did not undergo a renal biopsy. His lipid profile was TG of 257 mg/dL, TC of 231 mg/dL, LDL-C of 131 mg/dL, HDL-C of 47 mg/dL, and serum apoE of 3.2 mg/dL. Other family members had no evidence of proteinuria or renal dysfunction. Her sister (II-1) had mild hypertriglyceridemia and serum apoE of 4.9 mg/dL. The other two brothers (II-3, II-5) had mild hypercholesterolemia. The detailed lipid profiles of this family are listed in Table 2.
Fig. 2.
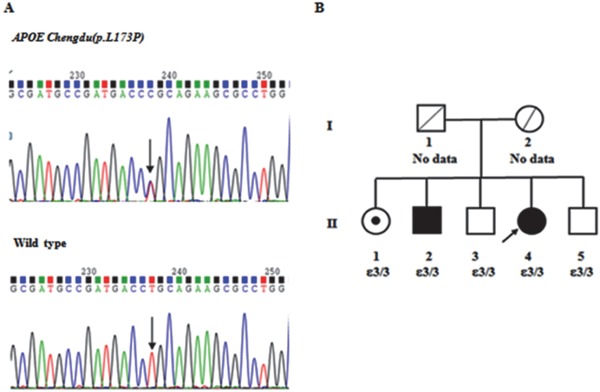
Mutation detection of apoE and family pedigree of LPG patient
Panel A shows a partial sequence of APOE exon 4 from the patient (upper) and a healthy control (lower) and the position of the apoE Chengdu mutation (arrow). Panel B shows the family pedigree of the LPG patient. The proband (II-4) is indicated by an arrow. A square represents a male, a circle represents a female, blank symbols represent unaffected family members, and black symbols represent patients heterozygous for apoE Chengdu (p.L173P). II-1 (dotted symbol) is an asymptomatic carrier of the mutation. II-2 is on hemodialysis with the same mutation. I-1 and I-2 died ten years ago. The causes of death are unknown.
Table 2. Lipid profiles and apolipoprotein E levels of the lipoprotein glomerulopathy pedigree with apoE Chengdu.
| Subject | Gender | Age years | TG mg/dL | TC mg/dL | HDL-C mg/dL | LDL-C mg/dL | ApoE mg/dL | ApoA1 g/L | ApoB100 g/L | ApoA1/ApoB100 |
|---|---|---|---|---|---|---|---|---|---|---|
| Carrier | ||||||||||
| II-4 | Female | 48 | 224 | 307 | 56 | 193 | 5.6 | 1.8 | 0.72 | 2.34 |
| II-1 | Female | 58 | 299 | 168 | 28 | 53 | 4.9 | 1.41 | 0.96 | 1.47 |
| II-2 | Male | 54 | 257 | 231 | 47 | 131 | 3.2 | 1.51 | 1.01 | 1.50 |
| Non-carrier | ||||||||||
| II-3 | Male | 51 | 100 | 277 | 48 | 195 | 4.0 | 1.31 | 1.67 | 0.78 |
| II-5 | Male | 46 | 68 | 247 | 65 | 162 | 4.0 | 1.48 | 1.25 | 1.18 |
Note: Patient II-2 was on hemodialysis, patient II-4 had LPG, and patient II-1was an asymptomatic carrier.
Laboratory reference range: TG (triglycerides): 25.7–162 mg/dL, TC (total cholesterol): 108.4–220.6 mg/dL, HDL-C (high-densitylipoprotein cholesterol): > 34.8 mg/dL, apoE (apolipoprotein E): 2.7–4.5 mg/dL, LDL-C (low-densitylipoprotein cholesterol): < 154.4 mg/dL, apoA1 (apolipoprotein A1): 1.04–2.25 g/L, apoB100 (apolipoprotein B100): 0.60–1.33 g/L.
DNA Sequence Analysis, APOE Genotype, and Pedigree Study
The APOE gene analysis of the patient (II-4) revealed a T-to-C nucleotide substitution at codon 173 of the apoE gene that caused the substitution of a leucine residue with a proline residue (Fig. 2A). No other apoE mutation was identified in this patient. Familial evaluation (Fig. 2B) showed that two additional family members (II-1, II-2) carried the same variant. Subject II-2 might have had LPG although he did not undergo a renal biopsy because he suffered from uremia and showed hypertriglyceridemia. Subject II-1 was an asymptomatic carrier. All of these carriers were heterozygous for this novel missense mutation and had the same apoE genotype (ε3/ε3). This variant was not identified among the 200 local elderly people surveyed. Search in the Exome Sequencing Project, 1000 Genomes Project, and Exome Aggregation Consortium revealed that this mutation was a novel apoE variant. We named this mutation apoE Chengdu (c.518T > C, p.L173P). The pedigree study showed that LPG secondary to apoE Chengdu is an autosomal dominant trait with incomplete penetrance.
Clinical Course
After the LPG diagnosis, the treatment regimen for this patient included 200 mg of fenofibrate and 62.5 mg of hyzaar per day. Three months later, the patient's urinary protein/creatinine ratio was markedly decreased to 0.93 g/g. One year after fenofibrate treatment, her urine protein/creatinine ratio had decreased to 0.58 g/g, TGs had decreased to 55 mg/dL, apoE level had decreased to 4.3 mg/dL, and serum albumin level had returned to the normal range. Additionally, stable renal functions were obtained. The patient showed good tolerance to fenofibrate treatment (Fig. 3). Myalgia and rhabdomyolysis were not reported, and her serum creatine kinase remained within the normal range.
Fig. 3.
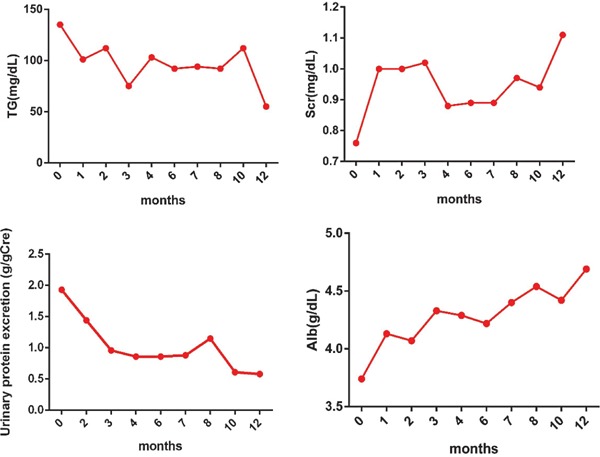
Laboratory data for LPG patient before and after fenofibrate treatment
Discussion
Human apoE is a 34-kDa protein with 299 amino acids. apoE mediates the tissue uptake of triglyceriderich lipoproteins through both the low-density lipoprotein receptor (LDLr) and LDL receptor-related protein (LRP) pathways8). Variants of apoE that are defective in receptor binding capacity may result in type III hyperlipoproteinemia (HLP) or LPG9). The apoE gene (APOE) is located at 19q13.2 and has four exons and three introns. To date, 16 apoE variants10) have been reported to be related to LPG, including apoE Chengdu. Among them, 12 variants are located in the LDL receptor-binding region (residues p. 158–168) or nearby areas (Fig. 4). Interestingly, the substitution of proline has been reported in 5/12 variants in this hotspot region, including apoE Sendai (p.R163P)4), apoE Chicago (p.R165P)11), apoE Guangzhou (p.R168P)12), apoE Osaka/Kurashiki (p.R176P)13, 14), and apoE Chengdu (p.L173P). apoE is highly helical and has a labile tertiary structure. Proline acts as a structural breaker in the middle of the α-helix of the protein, which is not located in or near the LDL receptor-binding region. Georgiadou et al. investigated the structural and conformational integrity of apoE Sendai, apoE Chicago and apoE Osaka/Kurashiki, which all shared this proline substitution15). All three variants had a significantly reduced helical content, which exposed a larger portion of the hydrophobic surface to the solvent and exhibited generalized unfolding of the N-terminal domain, which resulted in a more molten, globule-like structure. These changes led to a defective binding capacity for the LDL receptor and enhanced protein and lipoprotein aggregation, which contribute to the pathogenesis of hyperlipoproteinemia and the formation of lipoprotein thrombi15). apoE Chengdu is also a proline substitution near the LDL receptor-binding area. Thus, apoE Chengdu may lead to LPG in a manner similar to other apoE variants with proline substitutions. Notably, the substitution of proline in apoE was all for arginine in the LPG cases reported previously. apoE Chengdu is the substitution of proline for leucine. It provides the evidence that the substitution of proline plays an important role in the development of LPG, irrespective of the original amino acid (arginine or leucine).
Fig. 4.
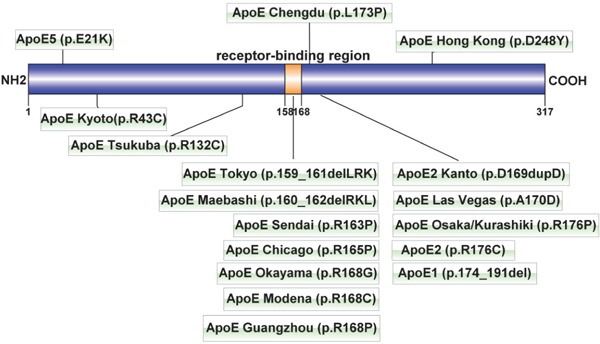
LPG-related apoE mutations
The orange portion (p. 158–168) represents the LDL receptor-binding region of apoE. The mutations that caused lipoprotein glomerulopathy were concentrated in this region. The apoE Kanto (p.D169dupD) mutation is an unpublished case, which was cited in a review24).
Clinically, LPG often presents as moderate to severe proteinuria and hypertriglyceridemia with elevated serum apoE levels2). The hyperlipidemia of LPG is usually attributed to reduced binding capacity of mutated apoE with the LDL receptor9, 16), just like type III HLP caused by APOE2 homozygosity. However, normolipidemic or mildly hyperlipidemic cases of LPG have also been reported, regardless of whether the apoE mutations are located within or distant from the LDL receptor-binding area17–19). Accordingly, it suggests that factors different to those related to hyperlipidemia may be involved in the development of LPG. Murano et al. reported that triglyceride-rich lipoproteins possessing apoE Kyoto showed increased binding to endothelial cells20). Sam et al. showed that serum apoE from a LPG patient with the apoE Chicago mutation could strongly bind to glomerular capillary walls compared with control11). These studies suggest that mutated apoE exhibits enhanced binding to glomerular capillaries, which might be a part of the pathogenic mechanism of LPG.
In this study, carriers of mutated apoE showed normally or mildly elevated serum apoE levels. Actually, LPG patients with normal serum apoE levels have been reported previously13, 14, 17), although these levels in LPG are usually two or three times higher than the normal range. But the mechanism for why these cases did not show elevated serum apoE levels is unclear.
Many cases in our previous work and others have showed that intensive lipid-lowering treatment including fibrate for under 100 mg/dL of serum triglyceride could significantly reduce proteinuria and achieve clinical remission1, 13, 17, 21–23). Notably, normolipidemic or mildly hyperlipidemic LPG patients also benefit from fibrate treatment17, 22, 23). Therefore, strict control of serum triglyceride levels in patients with LPG may be necessary to induce a decline in apoE variants sufficient to achieve clinical remission. In our study, the proband (II-4) presented mild hypertriglyceridemia but pathologic examinations revealed apoE deposition in glomerular capillary lumina, and clinical remission was achieved with fibrate treatment accompanying serum triglycerides reduction to 55 mg/dL. This supports the idea that intraglomerular accumulation of mutant apoE may play a more crucial role for the pathogenesis of LPG than may the serum apoE level, and fibrate treatment should be effective even in normolipidemic or mildly hyperlipidemic LPG patients.
In summary, we presented the novel apoE variant apoE Chengdu (p.L173P) in a Chinese LPG patient with mild dyslipidemia. This case supports the hypothesis that the substitution of proline in or near the LDL receptor-binding area contributes to the development of LPG. Fibrate treatment could be effective to reduce proteinuria in LPG patients with mild hyperlipidemia as well as severe hyperlipidemia.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (grant number81470916) and the Chengdu Municipal Bureau of Science and Technology (grant number 2014-HM01-00320-SF).
Conflict of Interest Statement
The authors declare that they have no conflict of interest.
References
- 1). Hu Z, Huang S, Wu Y, Liu Y, Liu X, Su D, Tao Y, Fu P, Zhang X, Peng Z, Zhang S, Yang Y: Hereditary features, treatment, and prognosis of the lipoprotein glomerulopathy in patients with the APOE Kyoto mutation. Kidney Int, 2014; 85: 416-424 [DOI] [PubMed] [Google Scholar]
- 2). Saito T, Matsunaga A, Oikawa S: Impact of lipoprotein glomerulopathy on the relationship between lipids and renal diseases. Am J Kidney Dis, 2006; 47: 199-211 [DOI] [PubMed] [Google Scholar]
- 3). Saito T, Sato H, Kudo K, Oikawa S, Shibata T, Hara Y, Yoshinaga K, Sakaguchi H: Lipoprotein glomerulopathy: glomerular lipoprotein thrombi in a patient with hyperlipoproteinemia. Am J Kidney Dis, 1989; 13: 148-153 [DOI] [PubMed] [Google Scholar]
- 4). Oikawa S, Matsunaga A, Saito T, Sato H, Seki T, Hoshi K, Hayasaka K, Kotake H, Midorikawa H, Sekikawa A, Hara S, Abe K, Toyota T, Jingami H, Nakamura H, Sasaki J: Apolipoprotein E Sendai (arginine 145-->proline): a new variant associated with lipoprotein glomerulopathy. J Am Soc Nephrol, 1997; 8: 820-823 [DOI] [PubMed] [Google Scholar]
- 5). Matsunaga A, Sasaki J, Komatsu T, Kanatsu K, Tsuji E, Moriyama K, Koga T, Arakawa K, Oikawa S, Saito T, Kita T, Doi T: A novel apolipoprotein E mutation, E2 (Arg25Cys), in lipoprotein glomerulopathy. Kidney Int, 1999; 56: 421-427 [DOI] [PubMed] [Google Scholar]
- 6). Zhang B, Liu ZH, Zeng CH, Zheng JM, Chen HP, Li LS: Clinicopathological and genetic characteristics in Chinese patients with lipoprotein glomerulopathy. J Nephrol, 2008; 21: 110-117 [PubMed] [Google Scholar]
- 7). Zhong L, Xie YZ, Cao TT, Wang Z, Wang T, Li X, Shen RC, Xu H, Bu G, Chen XF: A rapid and cost-effective method for genotyping apolipoprotein E gene polymorphism. Mol Neurodegener, 2016; 11: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8). Rall SC, Jr., Weisgraber KH, Mahley RW: Human apolipoprotein E. The complete amino acid sequence. The Journal of biological chemistry, 1982; 257: 4171-4178 [PubMed] [Google Scholar]
- 9). Hoffmann M, Scharnagl H, Panagiotou E, Banghard W, Wieland H, Marz W: Diminished LDL receptor and high heparin binding of apolipoprotein E2 Sendai associated with lipoprotein glomerulopathy. J Am Soc Nephrol, 2001; 12: 524-530 [DOI] [PubMed] [Google Scholar]
- 10). Saito T, Matsunaga A, Ito K, Nakashima H: Topics in lipoprotein glomerulopathy: an overview. Clin Exp Nephrol, 2014; 18: 214-217 [DOI] [PubMed] [Google Scholar]
- 11). Sam R, Wu H, Yue L, Mazzone T, Schwartz MM, Arruda JA, Dunea G, Singh AK: Lipoprotein glomerulopathy: a new apolipoprotein E mutation with enhanced glomerular binding. Am J Kidney Dis, 2006; 47: 539-548 [DOI] [PubMed] [Google Scholar]
- 12). Luo B, Huang F, Liu Q, Li X, Chen W, Zhou SF, Yu X: Identification of apolipoprotein E Guangzhou (arginine 150 proline), a new variant associated with lipoprotein glomerulopathy. Am J Nephrol, 2008; 28: 347-353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13). Tokura T, Itano S, Kobayashi S, Kuwabara A, Fujimoto S, Horike H, Satoh M, Komai N, Tomita N, Matsunaga A, Saito T, Sasaki T, Kashihara N: A novel mutation ApoE2 Kurashiki (R158P) in a patient with lipoprotein glomerulopathy. J Atheroscler Thromb, 2011; 18: 536-541 [DOI] [PubMed] [Google Scholar]
- 14). Mitani A, Ishigami M, Watase K, Minakata T, Yamamura T: A novel apolipoprotein E mutation, ApoE Osaka (Arg158 Pro), in a dyslipidemic patient with lipoprotein glomerulopathy. J Atheroscler Thromb, 2011; 18: 531-535 [DOI] [PubMed] [Google Scholar]
- 15). Georgiadou D, Stamatakis K, Efthimiadou EK, Kordas G, Gantz D, Chroni A, Stratikos E: Thermodynamic and structural destabilization of apoE3 by hereditary mutations associated with the development of lipoprotein glomerulopathy. J Lipid Res, 2013; 54: 164-176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16). Ishigaki Y, Oikawa S, Suzuki T, Usui S, Magoori K, Kim DH, Suzuki H, Sasaki J, Sasano H, Okazaki M, Toyota T, Saito T, Yamamoto TT: Virus-mediated transduction of apolipoprotein E (ApoE)-sendai develops lipoprotein glomerulopathy in ApoE-deficient mice. The Journal of biological chemistry, 2000; 275: 31269-31273 [DOI] [PubMed] [Google Scholar]
- 17). Hagiwara M, Yamagata K, Matsunaga T, Arakawa Y, Usui J, Shimizu Y, Aita K, Nagata M, Koyama A, Zhang B, Mastunaga A, Saku K, Saito T: A novel apolipoprotein E mutation, ApoE Tsukuba (Arg 114 Cys), in lipoprotein glomerulopathy. Nephrol Dial Transplant, 2008; 23: 381-384 [DOI] [PubMed] [Google Scholar]
- 18). Konishi K, Saruta T, Kuramochi S, Oikawa S, Saito T, Han H, Matsunaga A, Sasaki J: Association of a novel 3-amino acid deletion mutation of apolipoprotein E (Apo E Tokyo) with lipoprotein glomerulopathy. Nephron, 1999; 83: 214-218 [DOI] [PubMed] [Google Scholar]
- 19). Saito T, Sato H, Oikawa S, Kudo K, Kurihara I, Nakayama K, Abe K, Yoshinaga K, Sakaguchi H: Lipoprotein glomerulopathy. Report of a normolipidemic case and review of the literature. Am J Nephrol, 1993; 13: 64-68 [DOI] [PubMed] [Google Scholar]
- 20). Murano T, Matsumura R, Misawa Y, Ozaki H, Miyashita Y, Yoshida S, Sueioshi M, Sugiyama T, Shirai K: Interaction of endothelial cells and triglyceride-rich lipoproteins with apolipoprotein E (Arg-->Cys) from a patient with lipoprotein glomerulopathy. Metabolism, 2002; 51: 201-205 [DOI] [PubMed] [Google Scholar]
- 21). Matsunaga A, Furuyama M, Hashimoto T, Toyoda K, Ogino D, Hayasaka K: Improvement of nephrotic syndrome by intensive lipid-lowering therapy in a patient with lipoprotein glomerulopathy. Clin Exp Nephrol, 2009; 13: 659-662 [DOI] [PubMed] [Google Scholar]
- 22). Kinomura M, Sugiyama H, Saito T, Matsunaga A, Sada K-e, Kanzaki M, Takazawa Y, Maeshima Y, Yanai H, Makino H: A novel variant apolipoprotein E Okayama in a patient with lipoprotein glomerulopathy. Nephrol Dial Transplant, 2008; 23: 751-756 [DOI] [PubMed] [Google Scholar]
- 23). Arai T, Yamashita S, Yamane M, Manabe N, Matsuzaki T, Kiriyama K, Kanayama Y, Himeno S, Matsuzawa Y: Disappearance of intraglomerular lipoprotein thrombi and marked improvement of nephrotic syndrome by bezafibrate treatment in a patient with lipoprotein glomerulopathy. Atherosclerosis, 2003; 169: 293-299 [DOI] [PubMed] [Google Scholar]
- 24). Matsunaga A, Saito T: Apolipoprotein E mutations: a comparison between lipoprotein glomerulopathy and type III hyperlipoproteinemia. Clin Exp Nephrol, 2014; 18: 220-224 [DOI] [PubMed] [Google Scholar]