

Abstract
Ischemic cardiovascular disease is a major cause of morbidity and mortality worldwide and thrombus formation on disrupted atherosclerotic plaques is considered to trigger its onset. Although the activation of platelets and coagulation pathways has been investigated intensively, the mechanisms of thrombus formation on disrupted plaques have not been understood in detail. Platelets are thought to play a central role in the formation of arterial thrombus because of rapid flow conditions; however, thrombus that develops on disrupted plaques consistently includes large amounts of fibrin in addition to aggregated platelets. While, thrombus does not always become large enough to completely occlude the vascular lumen, indicating that the propagation of thrombus is also critical for the onset of cardiovascular events. Various factors, such as vascular wall thrombogenicity, altered blood flow and imbalanced blood hemostasis, modulate thrombus formation and propagation on disrupted plaques. Pathological findings derived from humans and experimental animal models of atherothrombosis have identified important factors that affect thrombus formation and propagation, namely platelets, extrinsic and intrinsic coagulation factors, proinflammatory factors, plaque hypoxia and blood flow alteration. These findings might provide insight into the mechanisms of thrombus formation and propagation on disrupted plaques that lead to the onset of cardiovascular events.
Keywords: Atherothrombosis, Platelet, Tissue factor, Blood flow, Factor XI
Introduction
Thrombus formation on disrupted atherosclerotic plaques is generally considered as being a trigger for atherothrombosis, namely the onset of cardiovascular events. Since thrombus does not always become large enough to completely occlude vessels with subsequent acute symptomatic events, thrombus propagation is also critical to the onset of clinical events1). Thrombus formation is modulated by factors, including the thrombogenicity of plaques and blood, local hemorheology and proinflammatory factors. Although the activation of platelets and coagulation pathways has been intensively investigated, the mechanisms involved in thrombogenesis or thrombus propagation at plaque disruption sites remain obscure.
Many studies of experimental animals have determined the molecular mechanisms of thrombus formation. However, the mechanisms of thrombus formation and propagation in atherosclerotic vessels remain vague because thrombi are induced in the “normal” arteries of many experimental animal models via chemical or physical damage. Since atherothrombosis develops on disrupted atherosclerotic plaque, vascular wall components and associated blood flow alterations are essential factors for thrombogenesis.
This article examines the mechanisms of thrombus formation and propagation on disrupted atherosclerotic plaques from pathological aspects, including recently identified factors.
Pathology of Plaque Disruption
The underlying mechanisms of the onset of cardiovascular events comprise plaque rupture and plaque erosion (Fig. 1).
Fig. 1.
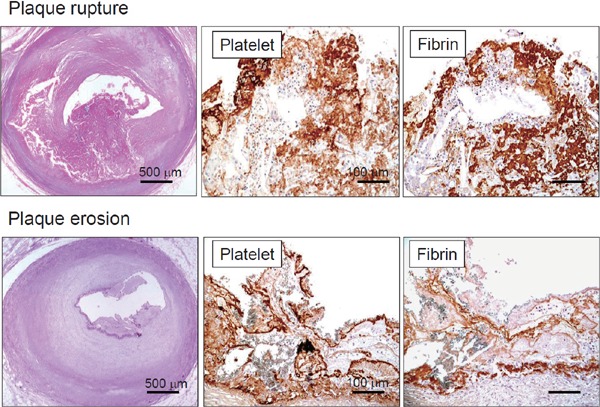
Microphotographs of coronary plaque rupture and erosion with thrombi
Ruptured plaque comprises large necrotic core and disrupted thin fibrous cap accompanied by thrombus formation. Eroded plaque is fibrous and rich in smooth muscle cells, without visible atheromatous component. Both types of thrombi comprise platelets and fibrin (Ref. 8, with permission).
Plaque rupture is the most common cause of acute coronary syndrome (ACS), and it is induced by fibrous cap disruption that allows blood to contact a highly thrombogenic necrotized core, resulting in thrombus formation. It is likely rupture occurs in plaques with a large necrotic core, fibrous caps that are usually < 65 µm thick, and the massive infiltration of macrophages, T lymphocytes and occasional smooth muscle cells (SMC)2, 3). Accumulating evidence supports the notion that inflammation plays a key role in the pathogenesis of plaque rupture. Macrophages mainly with the M1 phenotype4) overexpress proteolytic enzymes in plaques. In addition, activated T lymphocytes and macrophages secrete interferon γ, which inhibits collagen synthesis and induces SMC apoptosis5). These findings show that inflammatory pathways significantly participate in plaque destabilization.
Plaque erosion is responsible for 22%–44% of ACS6), which is characterized by an absence of endothelium or intimal injury without reaching lipid core. The morphological characteristics include an abundance of SMC and proteoglycan matrix, especially versican and hyaluronan, whereas the necrotic core is small or absent and macrophages and T cells are scant2, 3). Patients with plaque erosion are younger, and unlike those with plaque rupture, males do not predominate,6). Mechanisms that might contribute to erosion include endothelial dysfunction, hemodynamic forces, hypervasoconstriction and sub-endothelial matrix modification; however, details remain unclear6). A contribution of Toll-like receptor 2 and neutrophil activation has recently been reported7).
Pathology of Thrombus on Disrupted Plaques
Arterial thrombi are thought to mainly comprise aggregated platelets because of rapid flow conditions. However, thrombi that develop on disrupted plaques consist of not only aggregated platelets, but also large amounts of fibrin in addition to other types of blood cells (Fig. 1)8–10). In addition, the proportions of fibrin and platelets differs between coronary thrombi on ruptured and eroded plaques, which are fibrin- and rather platelet-rich, respectively8). These morphological features indicate that the coagulation pathway is activated as well as the platelets at the sites of plaque disruption even under rapid flow conditions, and that thrombogenetic mechanisms differ between plaque rupture and erosion.
Platelet Activation on Plaques
Platelet activation at the sites of disrupted plaques is an initial step in the formation of thrombus. Activated platelets release adenosine diphosphate (ADP), serotonin and thromboxane A2, which promote platelet activation further. This self-amplifying process results in thrombus propagation. In addition, released adenosine triphosphate (ATP) from erythrocytes and leukocytes can also activate platelets. Because ADP and ATP play key roles in platelet aggregation11), their metabolisms in the bloodstream are important for the regulation of platelet activation and recruitment.
1). Ecto-Nucleoside Triphosphate Diphosphohydrolase
Ecto-nucleoside triphosphate diphosphohydrolase (E-NTPDase, CD39) is a membrane-bound enzyme that rapidly hydrolyzes both ADP and ATP to AMP and thereby inhibits platelet activation and thrombus formation12, 13). Endothelial E-NTPDase expression is down-regulated under conditions of inflammation and disrupted blood flow14, 15). Hatakeyama et al.16) showed that E-NTPDase was also expressed in vascular SMC and down-regulated in atherosclerotic lesions, being more significantly decreased in patients with unstable, than stable angina. These findings suggest that E-NTP-Dase expression by SMC is also reduced by inflammatory and/or oxidative stress. The local expression of E-NTPDase by gene transfer in the injured arterial walls of rats significantly suppresses platelet aggregation and occlusive thrombus formation17). These lines of evidence suggest that reduced E-NTPDase activity in atherosclerotic lesions largely promotes thrombus formation on disrupted plaques.
2). C-Type Lectin-Like Receptor 2
Platelet activation is mediated by platelet-specific receptors including those for glycoprotein (GP) Ib, GPVI, GPIIb/IIIa, protease activated receptors (PAR) 1, PAR4, and P2Y.18). C-type lectin-like receptor 2 (CLEC-2) is a novel receptor for the platelet-activating, snake-venom protein, rhodocytin. CLEC-2 elicits powerful platelet activation signals in conjunction with Src, Syk kinases, and phospholipase Cγ2, like the collagen receptor GPVI/FcRγ-chain complex19). Therefore, endogenous ligands for CLEC-2 in vessel walls contribute to thrombosis and hemostasis. Podoplanin has recently become recognized as an endogenous ligand for CLEC-219). Hatakeyama et al.20) showed that podoplanin was expressed in human advanced, but not early atherosclerotic lesions (diffuse intimal thickening), that primarily consist of SMC. Inoue et al.21) showed that SMC stimulate platelets through binding CLEC-2 and identified S100A13 as one of its ligands. S100A13 is a Ca2+-binding protein that belongs to the S100 protein family, and it is implicated in inflammation, angiogenesis and tumor development22, 23). S100A13 is expressed on the surface of SMC under oxidative stress, but not on that of normal SMC21). Increased amounts of CLEC-2 ligands, podoplanin and S100A13 in advanced atherosclerotic plaques access platelet activation and promote thrombus formation.
Coagulation Factors on Thrombus Formation
The coagulation pathway also plays a critical role in thrombus formation. The binding of plasma factor VII/VIIa (FVII/FVIIa) to tissue factor (TF) initiates this pathway, and TF/FVIIa complexes of the extrinsic pathway initiate blood coagulation by activating both FX and FIX. The FVIIIa/FIXa complexes of the intrinsic pathway provide an alternative route to the generation of FXa, which is part of the prothrombinase complex (FVa/FXa) that activates prothrombin to thrombin that subsequently plays a central role in the coagulation protease cascade24). In addition, activated platelets enhance the coagulation cascade by providing a negatively charged phospholipid surface for the assembly of prothrombinase complexes and by binding FXI via the GPIb receptor25).
TF triggers the extrinsic pathway, and it is widely distributed in non-uniform tissues of the brain, lungs, heart, kidneys and other organs26). Adventitial fibroblasts in normal arteries, as well as macrophages and SMC in atherosclerotic lesions express TF, large amounts of which locate in the extracellular matrix of advanced lesions in humans27, 28). These pathological findings indicate that TF expression in atherosclerotic plaques plays a major procoagulant role in thrombus formation after plaque disruption. Mallat et al.29) reported that 97% of the total procoagulant activity extracted from atherosclerotic plaques was due to TF. In addition, TF circulates in a blood microparticle (MP)-associated form, and clinical studies have identified increased amounts of TF antigen circulating in the plasma of patients with cardiovascular disease (CAD)30, 31). However, Hisada et al.32) reported that the activity of the MP-associated form of TF did not significantly increase in the cardiovascular disease patients; therefore, plaques would be the major source of TF that drives atherothrombosis.
Rabbit Model of Atherothrombosis
We developed an animal model that expressed TF in vascular lesions to understand the role of TF in plaques on thrombus formation33–35).
Plaques were created in rabbit arteries injured using a balloon catheter and, then, the rabbits were fed with either a conventional or a cholesterol diet to developing SMC- or macrophage-rich plaques, respectively. The expression and activities of TF increased in both types of plaques, with more being localized in the macrophage-rich types35); this mimics the situation in human atherosclerotic plaques.
Large thrombi produced on injured plaques in this model comprised a mixture of platelets and fibrin, whereas small thrombi that developed on the injured normal artery were rich in platelets (Fig. 2)35). The intravenous injection of a recombinant TF pathway inhibitor (TFPI) or TFPI gene transfection into injured vessel walls significantly reduced thrombus size, suggesting that thrombus formation on arterial lesion largely depends on the TF-dependent coagulation pathway34, 36, 37).
Fig. 2.
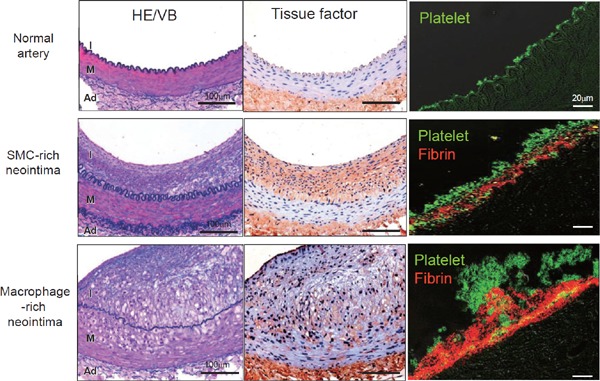
Immunohistochemical microphotographs of tissue factor and thrombus in rabbit normal and atherosclerotic femoral arteries
(Left column) Representative microphotographs of normal femoral artery and of femoral arteries at 3 weeks after balloon injury of conventional (SMC-rich neotima) or 0.5% cholesterol diet (Macrophage-rich neointima). (Middle column) Tissue factor is expressed in SMC- and macrophage-rich neointima, and in adventitia. (Right column) Thrombus 15 minutes after balloon injury on normal artery comprises only small aggregated platelets, whereas that on neointima comprises a mixture of platelets and fibrin. Thrombus on macrophage-rich neointima is much larger. Abbreviations: Ad, adventitia; HE/VB, hematoxylin and eosin/Victoria blue; I, intima; M, media (modified from Ref. 35, with permission).
Tissue Factor Expression in Plaques
The expression of TF in vascular cells is induced by many factors, such as proinflammatory cytokines (interleukin-1, tumor necrosis factor-a), bacterial endotoxin and modified low density lipoprotein38).
1). C-Reactive Protein
C-reactive protein (CRP) is an inflammatory acute-phase reactant that has emerged as a powerful predictor of CAD39–41). This protein is present in all stages of atherosclerotic plaques from the early to advanced lesions, and more prevalent in thrombotic, than non-thrombotic plaques42, 43), implicating CRP in atherothrombogenesis. Because CRP functions in rabbits but not in mice as an acute-phase reactant during inflammation in the same way that it does in humans44), Koike et al.45) established a line of transgenic (Tg) rabbits that overexpressed human CRP (hCRP). Matsuda et al.46) found that hCRP enhanced vascular wall thrombogenicity via increased TF expression in SMC and promoted thrombus formation after plaque disruption in this model.
2). Plaque Hypoxia
Hypoxia affects the biological functions of vascular cells via the regulation of metabolism, inflammation, angiogenesis and several other processes47). Hypoxia signaling can modulate tissue remodeling or the severity of cardiovascular disorders. Hypoxia inducible factor-1α (HIF-1α) and HIF-2α belong to a group of transcription factors that mediate most of the cellular responses to hypoxia at the transcriptional level48). Sluimer et al.49) demonstrated that hypoxia was present in the center of an advanced human carotid plaque by pimonidazole hydrochloride, a hypoxic marker, and also that the hypoxic area, HIF and macrophages were correlated with intra-plaque angiogenesis. Plaque hypoxia has been detected in animal models of atherosclerosis50, 51). Leppänen et al.52) revealed that even though the rabbit normal aorta was not hypoxic, the cores of plaques > 500 µm thick were hypoxic, and they were characterized by ATP depletion, low glucose, low glycogen and high lactate values. These findings suggest that limited oxygen diffusion capacity due to increased plaque thickness is a potent determinant of the onset of hypoxia within atherosclerotic lesions. Matsuura et al.50) also detected hypoxic areas located predominantly within lipid-laden macrophage-rich plaque in a rabbit model of atherosclerosis. These hypoxic areas positively correlated with the number of HIF-1α and NF-κB p65-immunopositive nuclei and TF-immunopositivity. They also found hypoxic conditions increased TF and plasminogen activator inhibitor (PAI)-1 expression and procoagulant activity in atherosclerotic plaque tissues and cultured macrophages. These results suggest that hypoxia itself is an important determinant of TF expression. O'Rourke et al.53) reported that hypoxia can induce TF mRNA in HIF-1α-deficient HeLa cells, suggesting that hypoxia could also increase TF independently of HIF-1α.
Non-Hemostatic Functions for TF
Accumulating evidence shows that TF/FVIIa complexes not only initiate the coagulation pathway, but also affect various non-hemostatic biological agents by activating proteinase-activated receptors (PARs)54, 55). Many cell types express these receptors, which play important roles in cardiovascular physiology and pathophysiology, and in the systems of other many organs55). The TF/VIIa complex, FXa and thrombin are potent PAR activators54). Therefore, TF/VIIa complex directly or indirectly activates PARs (Fig. 3), which mediate contraction, migration, proliferation, hypertrophy, and production of the extracellular matrix of SMC, thereby contributing to the development of vascular lesions and the pathophysiology of vascular diseases such as atherosclerosis56–59). Furthermore, the cytoplasmic domain of TF serves as a regulator for angiogenesis and SMC migration/proliferation60, 61). Therefore, TF expressed in plaques contributes to not only thrombus formation after plaque disruption, but also contributes to the development of plaque.
Fig. 3.
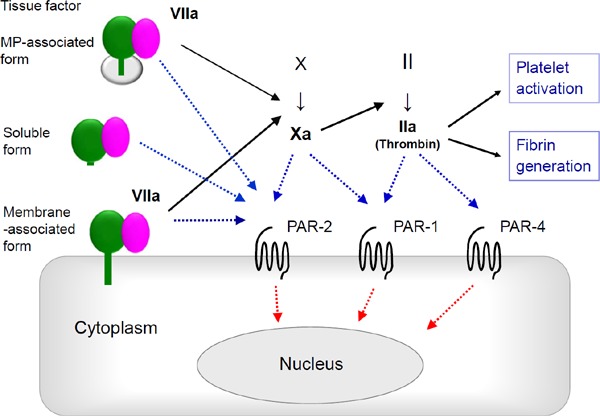
Tissue factor-dependent coagulation pathway and PARs
Membrane- and microparticle (MP)-associated TF binding to factor VIIa triggers coagulation pathway, whereas soluble form of TF with factor VIIa does not. Downstream coagulation factors activate proteinase-activated receptors (PARs) that play other non-coagulative biological roles.
Factors That Contribute to Thrombus Propagation
Thrombus formation on disrupted plaques is a fundamental process in the onset of acute cardiovascular events. However, it does not always lead to complete thrombotic occlusion with subsequent acute symptomatic events1). Autopsy studies of patients who died of non-cardiac causes have found a 4%–10% incidence of coronary plaque disruption with fresh non-occlusive thrombi62–64). Clinical studies using invasive vascular imaging techniques have revealed a much higher incidence of asymptomatic coronary plaque disruption, and multiple plaque disruption is a frequent complication in patients with ACS65–67). Disrupted plaques at various stages of healing are also occasionally found post-mortem in patients with or without ACS64, 68). Furthermore, several pathologic studies of aspirated coronary material have shown that plaque disruption and thrombus formation occur significantly earlier than symptom onset, coronary thrombi are days or weeks old in many patients, and the presence of an older thrombus is an independent predictor of mid or long-term mortality in patients with acute myocardial infarction (MI)69–71). This evidence indicates that coronary plaque disruption with thrombosis is rather prevalent and that many lesions might not be associated with acute symptomatic events. Therefore, other factors affecting thrombus propagation must contribute to the onset of acute coronary events.
1). Blood Flow Changes and VWF-ADAMTS13 Axis
Blood flow is a key modulator of thrombosis. Stenosis or irregular vessel geometries in advanced atherosclerotic arteries might induce disturbed blood flow (i.e., flow separation, recirculation, and reattachment of forward flow) that are thought to favor thrombus propagation72, 73). In addition, plaque disruption and coronary intervention can induce distal embolisms and vasoconstriction, which reduce or disrupt coronary blood flow at sites of plaque disruption74–76). Regions with low wall shear stress, particularly oscillatory or reversed shear stress that can occur at bifurcations or at sites distal to stenotic lesions, are likely associated with the recruitment of platelets and coagulation factors73). Reduced blood flow promoted fibrin-rich mural thrombi on atherosclerotic lesions in our animal model77). Disrupted blood flow induced by luminal stenosis can induce plaque disruption (erosion), and computational flow simulation suggests that increased wall shear stress, turbulence kinetic energy and blood pressure significantly contribute to the onset of plaque erosion78). The combination of changes in blood flow and the increased thrombogenicity of vascular walls is crucial for thrombus formation and propagation79) (Fig. 4).
Fig. 4.
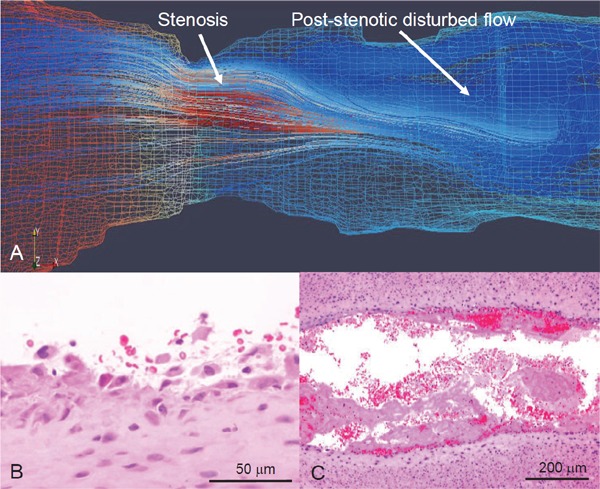
Computational flow simulation and microphotographs of erosive injury of rabbit stenotic femoral artery with SMC-rich plaque
Rabbit femoral arteries 3 weeks after balloon injury are constricted using a vascular occluder. (A) Representative computational reconstructed image and flow simulation in Reynolds-Averaged Navier-Stokes model. Red and blue mesh indicates high and low pressure, respectively, on wall. Flow velocity in this model increases at stenosis and decreases at post-stenotic portion, resulting in disrupted flow. (B, C) Representative microphotographs of erosive injury and thrombus formation. Neointimal endothelial cells and SMCs are broadly detached at post-stenotic portion 15 minute after vascular stenosis (B) (Ref. 79, with permission). Large mural thrombi are formed at the portion 60 minute after vascular stenosis (C).
Blood flow velocity significantly affects the function of von Willebrand factor (VWF), a large multimeric glycoprotein that is synthesized and stored by endothelial cells and megakaryocytes. The VWF multimers released upon stimulation are rich in ultra-large (UL) forms that are hyperactive in binding platelet GP Ib and can induce platelet aggregation80, 81). Released UL-VWF multimers are rapidly cleaved by the plasma protease, a disintegrin and metalloprotease with a thrombospondin type 1 motif 13 (ADAMTS-13), to smaller and less active forms under flow conditions82). An extreme deficiency of ADAMTS-13 activity results in increased circulating levels of UL-VWF multimers that correlate with the onset of thrombotic thrombocytopenic purpura (TTP)83). Shida et al.84) reported that ADAMTS-13 cleaved VWF and down-regulated mural thrombus growth at the site of ongoing thrombus generation under high shear rate conditions. This evidence suggests that ADAMTS-13 prevents thrombus propagation and thrombotic occlusion by regulating the size of VWF multimers under high shear rate conditions. Clinical studies have found decreased ADAMTS-13 activity or a higher ratio of VWF/ADAMTS-13 in patients with acute MI85–87). We reported that ADAMTS-13 closely localized with VWF in coronary thrombi from patients with ACS, and that reducing ADAMTS-13 activity enhanced thrombus formation88). ADAMTS-13 activity is decreased during aging, in systemic inflammation, and in focal vascular inflammation89–91).
2). Factor XI in the Intrinsic Coagulation Pathway
The intrinsic coagulation pathway is essential for thrombus stabilization and propagation. This pathway is activated when blood contacts negatively charged substances or artificial surfaces and induces the conversion of FXII into activated FXII, which then cleaves prekallikrein to generate active kallikrein, and activate FXI. Factor XI is also activated by thrombin and is essential for further thrombin generation during thrombus formation92). It also causes the inhibition of fibrinolysis that is dependent on thrombin activatable fibrinolysis inhibitor (TAFI)93). These lines of evidence indicate that FXI-mediated thrombin activation and inhibited fibrinolysis contribute to thrombus propagation.
On the other hand, FXI is generally considered to be less important in normal hemostasis, because patients with an FXI deficiency have a mild or absent bleeding tendency and a mild trauma-induced bleeding disorder94, 95). However, recent findings indicate that FXI is activated during blood coagulation, and that even small amounts of FXI can induce thrombus growth by generating thrombin and by protecting thrombi from fibrinolysis via TAFI. Therefore, FXI apparently plays a critical role in thrombus stability and propagation. Clinical studies have associated high levels of FXI with the onset of venous thrombosis and ischemic stroke96–98), whereas patients with a congenital FXI deficiency appear to be protected from these diseases99–101). We showed that inhibiting FXI activity suppressed thrombus propagation without prolonging bleeding time in a rabbit model of arterial and venous thrombosis102–104). Other clinical and experimental animal studies have also shown that lowering plasma FXI activity reduces thrombus formation while minimally affecting bleeding time105, 106).
These findings together indicate that plasma FXI plays a pivotal role in thrombus propagation and that FXI is a potential therapeutic target for preventing thrombosis without the adverse effect of bleeding.
Conclusion
Atherosclerotic plaques are in a prothrombotic state. The downregulation of E-NTPDase as well as nitric oxide and prostaglandin I2 in plaque endothelial cells promotes platelet activation, and increased podoplanin and S100A13 expression also activates platelets via CLEC-2. Furthermore, high levels of TF are expressed in plaques under conditions of inflammation, oxidative stress and hypoxia that together largely promote thrombus formation at sites of plaque disruption. Intrinsic coagulation factors and changes in blood flow also play critical roles in thrombus propagation, leading to clinical manifestations (Fig. 5).
Fig. 5.
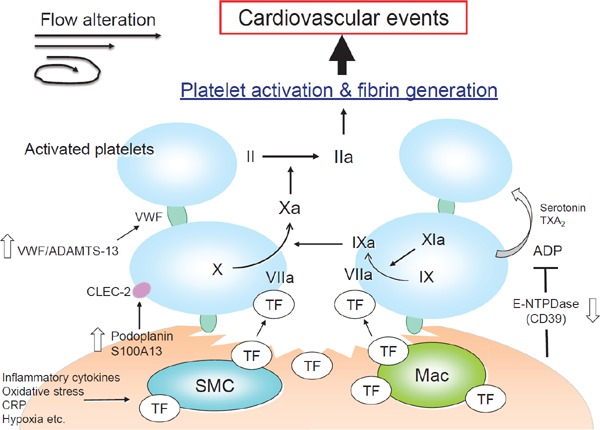
Activation of platelets and coagulation pathway at disrupted atherosclerotic plaque
ADAMTS-13, a disintegrin and metalloprotease with a thrombospondin type 1 motif 13; ADP, adenosine diphosphate; CLEC-2, c-type lectin-like receptor 2; CRP, c-reactive protein; E-NTPDase, ecto-nucleoside triphosphate diphosphohydrolase; Mac, macrophage; SMC, smooth muscle cell; TF, tissue factor; TXA2, thromboxane A2; VWF, von Willebrand factor.
Acknowledgments
This work was supported in part by Grants-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan (20390102, 23390084, 16H05163), an Intramural Research Fund (25-4-3) for Cardiovascular Diseases from the National Cerebral and Cardiovascular Center, and for Clinical Research from Miyazaki University Hospital.
Conflict of Interest
None declared
References
- 1). Falk E, Shah PK, Fuster V. Coronary plaque disruption. Circulation. 1995; 92: 657-671 [DOI] [PubMed] [Google Scholar]
- 2). Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000; 20: 1262-1275 [DOI] [PubMed] [Google Scholar]
- 3). Shah PK. Mechanisms of plaque vulnerability and rupture. J Am Coll Cardiol. 2003; 41: 15S-22S [DOI] [PubMed] [Google Scholar]
- 4). Komohara Y, Fujiwara Y, Ohnishi K, Shiraishi D, Takeya M. Contribution of macrophage polarization to metabolic diseases. J Atheroscler Thromb. 2016; 23: 10-17 [DOI] [PubMed] [Google Scholar]
- 5). Libby P. Mechanisms of acute coronary syndromes and their implications for therapy. N Engl J Med. 2013; 368: 2004-2013 [DOI] [PubMed] [Google Scholar]
- 6). White SJ, Newby AC, Johnson TW. Endothelial erosion of plaques as a substrate for coronary thrombosis. Thromb Haemost. 2016; 115: 509-519 [DOI] [PubMed] [Google Scholar]
- 7). Quillard T, Araújo HA, Franck G, Shvartz E, Sukhova G, Libby P. TLR2 and neutrophils potentiate endothelial stress, apoptosis and detachment: implications for superficial erosion. Eur Heart J. 2015; 36: 1394-1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8). Sato Y, Hatakeyama K, Yamashita A, Marutsuka K, Sumiyoshi A, Asada Y. Proportion of fibrin and platelets differs in thrombi on ruptured and eroded coronary atherosclerotic plaques in humans. Heart. 2005; 91: 526-530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9). Yamashita A, Sumi T, Goto S, Hoshiba Y, Nishihira K, Kawamoto R, Hatakeyama K, Date H, Imamura T, Ogawa H, Asada Y. Detection of von Willebrand factor and tissue factor in platelets-fibrin rich coronary thrombi in acute myocardial infarction. Am J Cardiol. 2006; 97: 26-28 [DOI] [PubMed] [Google Scholar]
- 10). Hoshiba Y, Hatakeyama K, Tanabe T, Asada Y, Goto S. Co-localization of von Willebrand factor with platelet thrombi, tissue factor and platelets with fibrin, and consistent presence of inflammatory cells in coronary thrombi obtained by an aspiration device from patients with acute myocardial infarction. J Thromb Haemost. 2006; 4: 114-120 [DOI] [PubMed] [Google Scholar]
- 11). Gachet C, Leon C, Hechler B. The platelet P2 receptors in arterial thrombosis. Blood Cells Mol Dis. 2006; 36: 223-227 [DOI] [PubMed] [Google Scholar]
- 12). Robson SC, Wu Y, Sun X, Knosalla C, Dwyer K, Enjyoji K. Ectonucleotidases of CD39 family modulate vascular inflammation and thrombosis in transplantation. Semin Thromb Hemost 2005; 31: 217-233 [DOI] [PubMed] [Google Scholar]
- 13). Marcus AJ, Broekman MJ, Drosopoulos JH, Olson KE, Islam N, Pinsky DJ, Levi R. Role of CD39 (NTPDase-1) in thromboregulation, cerebroprotection, and cardioprotection. Semin Thromb Hemost. 2005; 31: 234-246 [DOI] [PubMed] [Google Scholar]
- 14). Robson SC, Kaczmarek E, Siegel JB, Candinas D, Koziak K, Millan M, Hancock WW, Bach FH. Loss of ATP diphosphohydrolase activity with endothelial cell activation J Exp Med. 1997; 185: 153-163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15). Kanthi Y, Hyman MC, Liao H, Baek AE, Visovatti SH, Sutton NR, Goonewardena SN, Neral MK, Jo H, Pinsky DJ. Flow-dependent expression of ectonucleotide tri(di)phosphohydrolase-1 and suppression of atherosclerosis. J Clin Invest. 2015; 125: 3027-3036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16). Hatakeyama K, Hao H, Imamura T, Ishikawa T, Shibata Y, Fujimura Y, Eto T, Asada Y. Relation of CD39 to plaque instability and thrombus formation in directional atherectomy specimens from patients with stable and unstable angina pectoris. Am J Cardiol. 2005; 95: 632-625 [DOI] [PubMed] [Google Scholar]
- 17). Furukoji E, Matsumoto M, Yamashita A, Yagi H, Sakurai Y, Marutsuka K, Hatakeyama K, Morishita K, Fujimura Y, Tamura S, Asada Y. Adenovirus-mediated transfer of human placental ectonucleoside triphosphate diphosphohydrolase to vascular smooth muscle cells suppresses platelet aggregation in vitro and arterial thrombus formation in vivo. Circulation. 2005; 111: 808-815 [DOI] [PubMed] [Google Scholar]
- 18). Jamasbi J, Ayabe K, Goto S, Nieswandt B, Peter K, Siess W. Platelet receptors as therapeutic targets: Past, present and future. Thromb Haemost. 2017; 117: 1249-1257 [DOI] [PubMed] [Google Scholar]
- 19). Suzuki-Inoue K, Inoue O, Ozaki Y. Novel platelet activation receptor CLEC-2: from discovery to prospects. J Thromb Haemost. 2011; 9 Suppl 1: 44-55 [DOI] [PubMed] [Google Scholar]
- 20). Hatakeyama K, Kaneko MK, Kato Y, Ishikawa T, Nishihira K, Tsujimoto Y, Shibata Y, Ozaki Y, Asada Y. Podoplanin expression in advanced atherosclerotic lesions of human aortas. Thromb Res. 2012; 129: e70-76 [DOI] [PubMed] [Google Scholar]
- 21). Inoue O, Hokamura K, Shirai T, Osada M, Tsukiji N, Hatakeyama K, Umemura K, Asada Y, Suzuki-Inoue K, Ozaki Y. Vascular smooth muscle cells stimulate platelets and facilitate thrombus formation through platelet CLEC-2: Implications in atherothrombosis. PLoS One. 2015; 10: e0139357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22). Massi D, Landriscina M, Piscazzi A, Cosci E, Kirov A, Paglierani M, Di Serio C, Mourmouras V, Fumagalli S, Biagioli M, Prudovsky I, Miracco C, Santucci M, Marchionni N, Tarantini F. S100A13 is a new angiogenic marker in human melanoma. Mod Pathol. 2010; 23: 804-813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23). Pierce A, Barron N, Linehan R, Ryan E, O'Driscoll L, Daly C, Clynes M. Identification of a novel, functional role for S100A13 in invasive lung cancer cell lines. Eur J Cancer. 2008; 44: 151-159 [DOI] [PubMed] [Google Scholar]
- 24). Grant MA, Aird WC. Molecular evolution of the vertebrate blood coagulation system. Hemostasis and Thrombosis, 6th ed, Marder VJ, Arid WC, Bennett JS, Schulman S, White GC., II Lippincott Williams & Wilkins, 2013, pp11-25 [Google Scholar]
- 25). Baglia FA, Badellino KO, Li CQ, Lopez JA, Walsh PN. Factor XI binding to the platelet glycoprotein I b-IX-V complex promotes factor XI activation by thrombin. J Biol Chem 2002; 277: 1662-1668 [DOI] [PubMed] [Google Scholar]
- 26). Drake TA, Morrissey JH, Edgington TS. Selective cellular expression of tissue factor in human tissues. Implications for disorders of hemostasis and thrombosis. Am J Pathol. 1989; 134: 1087-1097 [PMC free article] [PubMed] [Google Scholar]
- 27). Wilcox JN, Smith KM, Schwartz SM, Gordon D. Localization of tissue factor in the normal vessel wall and in the atherosclerotic plaque. Proc Natl Acad Sci U S A. 1989; 86: 2839-2843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28). Hatakeyama K, Asada Y, Marutsuka K, Sato Y, Kamikubo Y, Sumiyoshi A. Localization and activity of tissue factor in human aortic atherosclerotic lesions. Atherosclerosis. 1997; 133: 213-219 [DOI] [PubMed] [Google Scholar]
- 29). Mallat Z, Hugel B, Ohan J, Leseche G, Freyssinet JM, Tedgui A: Shed membrane microparticles with procoagulant potential in human atherosclerotic plaques: a role for apoptosis in plaque thrombogenicity. Circulation, 1999; 99: 348-353 [DOI] [PubMed] [Google Scholar]
- 30). Nomura S. Microparticle and atherothrombotic diseases. J Atheroscler Thromb. 2016; 23: 1-9 [DOI] [PubMed] [Google Scholar]
- 31). Owens AP, 3rd, Mackman N. Microparticles in hemostasis and thrombosis. Circ Res. 2011; 108: 1284-1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32). Hisada Y, Alexander W, Kasthuri R, Voorhees P, Mobarrez F, Taylor A, McNamara C, Wallen H, Witkowski M, Key NS, Rauch U, Mackman N. Measurement of microparticle tissue factor activity in clinical samples: A summary of two tissue factor-dependent FXa generation assays. Thromb Res. 2016; 139: 90-97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33). Hatakeyama K, Asada Y, Marutsuka K, Kataoka H, Sato Y, Sumiyoshi A. Expression of tissue factor in the rabbit aorta after balloon injury. Atherosclerosis. 1998; 139: 265-271 [DOI] [PubMed] [Google Scholar]
- 34). Asada Y, Hara S, Tsuneyoshi A, Hatakeyama K, Kisanuki A, Marutsuka K, Sato Y, Kamikubo Y, Sumiyoshi A. Fibrin-rich and platelet-rich thrombus formation on neointima: recombinant tissue factor pathway inhibitor prevents fibrin formation and neointimal development following repeated balloon injury of rabbit aorta. Thromb Haemost. 1998; 80: 506-511 [PubMed] [Google Scholar]
- 35). Yamashita A, Matsuda S, Matsumoto T, Moriguchi-Goto S, Takahashi M, Sugita C, Sumi T, Imamura T, Shima M, Kitamura K, Asada Y. Thrombin generation by intimal tissue factor contributes to thrombus formation on macrophage-rich neointima but not normal intima of hyperlipidemic rabbits. Atherosclerosis. 2009; 206: 418-426 [DOI] [PubMed] [Google Scholar]
- 36). Nishida T, Ueno H, Atsuchi N, Kawano R, Asada Y, Nakahara Y, Kamikubo Yi, Takeshita A, Yasui H. Adenovirus-mediated local expression of human tissue factor pathway inhibitor eliminates shear stress-induced recurrent thrombosis in the injured carotid artery of the rabbit. Circ Res. 1999; 84: 1446-1452 [DOI] [PubMed] [Google Scholar]
- 37). Atsuchi N, Nishida T, Marutsuka K, Asada Y, Kamikubo Y, Takeshita A, Ueno H. Combination of a brief irrigation with tissue factor pathway inhibitor (TFPI) and adenovirus-mediated local TFPI gene transfer additively reduces neointima formation in balloon-injured rabbit carotid arteries. Circulation. 2001; 103: 570-575 [DOI] [PubMed] [Google Scholar]
- 38). Bode M, Mackman N. Regulation of tissue factor gene expression in monocytes and endothelial cells: Thromboxane A2 as a new layer. Vascul Pharmacol. 2014; 62: 57-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39). Ridker PM. C-reactive protein and the prediction of cardiovascular events among those at intermediate risk: moving an inflammatory hypothesis toward consensus. J Am Coll Cardiol. 2007; 49: 2129-2138 [DOI] [PubMed] [Google Scholar]
- 40). Emerging Risk Factors Collaboration. Kaptoge S, Di Angelantonio E, Lowe G, Pepys MB, Thompson SG, Collins R, Danesh J. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta-analysis. Lancet. 2010; 375: 132-140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41). Kitagawa K, Hosomi N, Nagai Y, Kagimura T, Ohtsuki T, Origasa H, Minematsu K, Uchiyama S, Nakamura M, Matsumoto M, J-STARS Investigators Reduction in high-sensitivity C-reactive protein levels in patients with ischemic stroke by statin treatment: Hs-CRP Sub-Study in J-STARS. J Atheroscler Thromb. 2017; 24: 1039-1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42). Sun H, Koike T, Ichikawa T, Hatakeyama K, Shiomi M, Zhang B, Kitajima S, Morimoto M, Watanabe T, Asada Y, Chen YE, Fan J. C-reactive protein in atherosclerotic lesions: its origin and pathophysiological significance. Am J Pathol, 2005; 167: 1139-1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43). Ishikawa T, Hatakeyama K, Imamura T, Date H, Shibata Y, Hikichi Y, Asada Y, Eto T. Involvement of C-reactive protein obtained by directional coronary atherectomy in plaque instability and developing restenosis in patients with stable or unstable angina pectoris. Am J Cardiol. 2003; 91: 287-292 [DOI] [PubMed] [Google Scholar]
- 44). Torzewski J. C-reactive protein and atherogenesis: new insights from established animal models. Am J Pathol. 2005; 167: 923-925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45). Koike T, Kitajima S, Yu Y, Nishijima K, Zhang J, Ozaki Y, Morimoto M, Watanabe T, Bhakdi S, Asada Y, Chen YE, Fan J. Human C-reactive protein does not promote atherosclerosis in transgenic rabbits. Circulation. 2009; 120: 2088-2094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46). Matsuda S, Yamashita A, Sato Y, Kitajima S, Koike T, Sugita C, Moriguchi-Goto S, Hatakeyama K, Takahashi M, Koshimoto C, Matsuura Y, Iwakiri T, Chen YE, Fan J, Asada Y. Human C-reactive protein enhances thrombus formation after neointimal balloon injury in transgenic rabbits. J Thromb Haemost. 2011; 9: 201-208 [DOI] [PubMed] [Google Scholar]
- 47). Semenza GL. Oxygen homeostasis. Wiley Interdiscip Rev Syst Biol Med 2010; 2: 336-361 [DOI] [PubMed] [Google Scholar]
- 48). Abe H, Semba H, Takeda N. The Roles of Hypoxia Signaling in the Pathogenesis of Cardiovascular Diseases. J Atheroscler Thromb. 2017; 24: 884-894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49). Sluimer JC, Gasc JM, van Wanroij JL, Kisters N, Groeneweg M, Sollewijn Gelpke MD, Cleutjens JP, van den Akker LH, Corvol P, Wouters BG, Daemen MJ, Bijnens AP. Hypoxia, hypoxia-inducible transcription factor, and macrophages in human atherosclerotic plaques are correlated with intraplaques angiogenesis. J Am Coll Cardiol. 2008; 51: 1258-1265 [DOI] [PubMed] [Google Scholar]
- 50). Matsuura Y, Yamashita A, Iwakiri T, Sugita C, Okuyama N, Kitamura K, Asada Y. Vascular wall hypoxia promotes arterial thrombus formation via augmentation of vascular thrombogenicity. Thromb Haemost. 2015; 114: 158-172 [DOI] [PubMed] [Google Scholar]
- 51). Nie X, Randolph GJ, Elvington A, Bandara N, Zheleznyak A, Gropler RJ, Woodard PK, Lapi SE. Imaging of hypoxia in mouse atherosclerotic plaques with (64) Cu-ATSM. Nucl Med Biol. 2016; 43: 534-542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52). Leppänen O, Björnheden T, Evaldsson M, Borén J, Wiklund O, Levin M. ATPdepletion in macrophages in the core of advanced rabbit atherosclerotic plaques in vivo. Atherosclerosis. 2006; 188: 323-330 [DOI] [PubMed] [Google Scholar]
- 53). O'Rourke JF, Pugh CW, Bartlett SM, Ratcliffe PJ. Identification of hypoxically inducible mRNAs in HeLa cells using differential-display PCR. Role of hypoxia-inducible factor-1. Eur J Biochem. 1996; 241: 403-410 [DOI] [PubMed] [Google Scholar]
- 54). Mackman N. Role of tissue factor in hemostasis, thrombosis, and vascular development. Arterioscler Thromb Vasc Biol. 2004; 24: 1015-1022 [DOI] [PubMed] [Google Scholar]
- 55). Isermann B. Homeostatic effects of coagulation protease-dependent signaling and protease activated receptors. J Thromb Haemost. 2017; 15: 1273-1284 [DOI] [PubMed] [Google Scholar]
- 56). Sato Y, Asada Y, Marutsuka K, Hatakeyama K, Kamikubo Y, Sumiyoshi A. Tissue factor pathway inhibitor inhibits aortic smooth muscle cell migration induced by tissue factor/factor VIIa complex. Thromb Haemost. 1997; 78: 1138-1141 [PubMed] [Google Scholar]
- 57). Marutsuka K, Hatakeyama K, Sato Y, Yamashita A, Sumiyoshi A, Asada Y. Protease-activated receptor 2 (PAR2) mediates vascular smooth muscle cell migration induced by tissue factor/factor VIIa complex. Thromb Res. 2002; 107: 271-276 [DOI] [PubMed] [Google Scholar]
- 58). Hirano K. The roles of proteinase-activated receptors in the vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol. 2007; 27: 27-36 [DOI] [PubMed] [Google Scholar]
- 59). Alberelli MA, De Candia E. Functional role of protease activated receptors in vascular biology. Vascul Pharmacol. 2014; 62: 72-81 [DOI] [PubMed] [Google Scholar]
- 60). Belting M, Dorrell M, Sandgren S, Aguilar E, Ahamed J, Dorfleutner A, Carmeliet P, Mueller B, Friedlander M, Ruf W. Regulation of angiogenesis by tissue factor cytoplasmic domain signaling. Nat Med. 2004; 10: 502-509 [DOI] [PubMed] [Google Scholar]
- 61). Otta I, Michaelis C, Schuermann M, Steppich B, Seitz I, Dewerchin M, Zohlnhofer D, Wessely R, Rudelius M, Schoming A, Carmeliet P. Vascular remodeling in mice lacking the cytoplasmic domain of tissue factor. Circ Res. 2005; 97: 293-298 [DOI] [PubMed] [Google Scholar]
- 62). Davies MJ, Bland JM, Hangartner JR, Angelini A, Thomas AC. Factors influencing the presence or absence of acute coronary artery thrombi in sudden ischaemic death. Eur Heart J 1989; 10: 203-208 [DOI] [PubMed] [Google Scholar]
- 63). Arbustini E, Grasso M, Diegoli M, Morbini P, Aguzzi A, Fasani R, et al. Coronary thrombosis in non-cardiac death. Coron Artery Dis 1993; 4: 751-759 [DOI] [PubMed] [Google Scholar]
- 64). Sato Y, Hatakeyama K, Marutsuka K, Asada Y. Incidence of asymptomatic coronary thrombosis and plaque disruption: comparison of non-cardiac and cardiac deaths among autopsy cases. Thromb Res. 2009; 124: 19-23 [DOI] [PubMed] [Google Scholar]
- 65). Rioufol G, Finet G, Ginon I, Andre-Fouet X, Rossi R, Vialle E, Desjoyaux E, Convert G, Huret JF, Tabib A. Multiple atherosclerotic plaque rupture in acute coronary syndrome: a three-vessel intravascular ultrasound study. Circulation 2002; 106: 804-808 [DOI] [PubMed] [Google Scholar]
- 66). Kotani J, Mintz GS, Castagna MT, Pinnow E, Berzingi CO, Bui AB, Pichard AD, Satler LF, Suddath WO, Waksman R, Laird JR, Jr, Kent KM, Weissman NJ. Intravascular ultrasound analysis of infarct-related and non-infarct-related arteries in patients who presented with an acute myocardial infarction. Circulation 2003; 107: 2889-2893 [DOI] [PubMed] [Google Scholar]
- 67). Shimamura K, Ino Y, Kubo T, Nishiguchi T, Tanimoto T, Ozaki Y, Satogami K, Orii M, Shiono Y, Komukai K, Yamano T, Matsuo Y, Kitabata H, Yamaguchi T, Hirata K, Tanaka A, Imanishi T, Akasaka T. Difference of ruptured plaque morphology between asymptomatic coronary artery disease and non-ST elevation acute coronary syndrome patients: an optical coherence tomography study. Atherosclerosis. 2014; 235: 532-537 [DOI] [PubMed] [Google Scholar]
- 68). Burke AP, Kolodgie FD, Farb A, Weber DK, Malcom GT, Smialek J, Virmani R. Healed plaque ruptures and sudden coronary death: evidence that subclinical rupture has a role in plaque progression. Circulation 2001; 103: 934-940 [DOI] [PubMed] [Google Scholar]
- 69). Rittersma SZ, van der Wal AC, Koch KT, Piek JJ, Henriques JP, Mulder KJ, Ploegmakers JP, Meesterman M, de Winter RJ. Plaque instability frequently occurs days or weeks before occlusive coronary thrombosis: a pathological thrombectomy study in primary percutaneous coronary intervention. Circulation. 2005; 111: 1160-1165 [DOI] [PubMed] [Google Scholar]
- 70). Kramer MC, van der Wal AC, Koch KT, Ploegmakers JP, van der Schaaf RJ, Henriques JP, Baan J, Jr, Rittersma SZ, Vis MM, Piek JJ, Tijssen JG, de Winter RJ. Presence of older thrombus is an independent predictor of longterm mortality in patients with ST-elevation myocardial infarction treated with thrombus aspiration during primary percutaneous coronary intervention. Circulation. 2008; 118: 1810-1816 [DOI] [PubMed] [Google Scholar]
- 71). Nishihira K, Shibata Y, Yamashita A, Kuriyama N, Asada Y. Relationship between thrombus age in aspirated coronary material and mid-term major adverse cardiac and cerebrovascular events in patients with acute myocardial infarction. Atherosclerosis. 2018; 268: 138-144 [DOI] [PubMed] [Google Scholar]
- 72). Chatzizisis YS, Coskun UA, Jonas M, Edelman ER, Feldman CL, Stone PH. Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling. J Am Coll Cardiol. 2007; 49: 2379-2393 [DOI] [PubMed] [Google Scholar]
- 73). Hathcock JJ. Flow effects on coagulation and thrombosis. Arterioscler Thromb Vasc Biol. 2006; 26: 1729-1737 [DOI] [PubMed] [Google Scholar]
- 74). Marzilli M, Sambuceti G, Fedele S, L'Abbate A. Coronary microcirculatory vasoconstriction during ischemia in patients with unstable angina. Am J Coll Cardiol 2000; 35: 327-334 [DOI] [PubMed] [Google Scholar]
- 75). Heusch G, Kleinbongard P, Böse D, Levkau B, Haude M, Schulz R, Erbel R. Coronary microembolization: from bedside to bench and back to bedside. Circulation. 2009; 120: 1822-1836 [DOI] [PubMed] [Google Scholar]
- 76). Teramoto R, Sakata K, Miwa K, Matsubara T, Yasuda T, Inoue M, Okada H, Kanaya H, Kawashiri MA, Yamagishi M, Hayashi K. Impact of Distal protection with filter-type device on long-term outcome after percutaneous coronary intervention for acute myocardial infarction: clinical results with Filtrap®. J Atheroscler Thromb. 2016; 23: 1313-1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77). Yamashita A, Furukoji E, Marutsuka K, Hatakeyama K, Yamamoto H, Tamura S, Ikeda Y, Sumiyoshi A, Asada Y. Increased vascular wall thrombogenicity combined with reduced blood flow promotes occlusive thrombus formation in rabbit femoral artery. Arterioscler Thromb Vasc Biol. 2004; 24: 2420-2424 [DOI] [PubMed] [Google Scholar]
- 78). Sameshima N, Yamashita A, Sato S, Matsuda S, Matsuura Y, Asada Y. The value of wall shear stress, turbulence kinetic energy and blood pressure gradient are associated with atherosclerotic plaque erosion in rabbits. J Atheroscler Thromb. 2014; 21: 831-838 [DOI] [PubMed] [Google Scholar]
- 79). Sumi T, Yamashita A, Matsuda S, Goto S, Nishihira K, Furukoji E, Sugimura H, Kawahara H, Imamura T, Kitamura K, Tamura S, Asada Y. Disturbed blood flow induces erosive injury to smooth muscle cell-rich neointima and promotes thrombus formation in rabbit femoral arteries. J Thromb Haemost. 2010; 8: 1394-1402 [DOI] [PubMed] [Google Scholar]
- 80). Franchini M, Lippi G. Von Willebrand factor and thrombosis. Ann Hematol. 2006; 85: 415-423 [DOI] [PubMed] [Google Scholar]
- 81). Shiozaki S, Takagi S, Goto S. Prediction of molecular interaction between platelet glycoprotein Ibα and von Willebrand factor using molecular dynamics simulations. J Atheroscler Thromb. 2016; 23: 455-464 [DOI] [PubMed] [Google Scholar]
- 82). Levy GG, Motto DG, Ginsburg D. ADAMTS13 turns 3. Blood. 2005; 106: 11-17 [DOI] [PubMed] [Google Scholar]
- 83). Moake JL. Thrombotic microangiopathies. N Engl J Med. 2002; 347: 589-600 [DOI] [PubMed] [Google Scholar]
- 84). Shida Y, Nishio K, Sugimoto M, Mizuno T, Hamada M, Kato S, Matsumoto M, Okuchi K, Fujimura Y, Yoshioka A. Functional imaging of shear-dependent activity of ADAMTS13 in regulating mural thrombus growth under whole blood flow conditions. Blood. 2008; 111: 1295-1298 [DOI] [PubMed] [Google Scholar]
- 85). Kaikita K, Soejima K, Matsukawa M, Nakagaki T, Ogawa H. Reduced von Willebrand factor-cleaving protease (ADAMTS13) activity in acute myocardial infarction. J Thromb Haemost. 2006; 4: 2490-2493 [DOI] [PubMed] [Google Scholar]
- 86). Horii M, Uemura S, Uemura M, Matsumoto M, Ishizashi H, Imagawa K, Iwama H, Takeda Y, Kawata H, Nakajima T, Fujimura Y, Saito Y. Acute myocardial infarction as a systemic prothrombotic condition evidenced by increased von Willebrand factor protein over ADAMTS13 activity in coronary and systemic circulation. Heart Vessels. 2008; 23: 301-307 [DOI] [PubMed] [Google Scholar]
- 87). Maino A, Siegerink B, Lotta LA, Crawley JT, le Cessie S, Leebeek FW, Lane DA, Lowe GD, Peyvandi F, Rosendaal FR. Plasma ADAMTS-13 levels and the risk of myocardial infarction: an individual patient data meta-analysis. J Thromb Haemost. 2015; 13: 1396-1404 [DOI] [PubMed] [Google Scholar]
- 88). Moriguchi-Goto S, Yamashita A, Tamura N, Soejima K, Takahashi M, Nakagaki T, Goto S, Asada Y. ADAMTS-13 attenuates thrombus formation on type I collagen surface and disrupted plaques under flow conditions. Atherosclerosis. 2009; 203: 409-416 [DOI] [PubMed] [Google Scholar]
- 89). Mannucci PM, Canciani MT, Forza I, Lussana F, Lattuada A, Rossi E. Changes in health and disease of the metalloprotease that cleaves von Willebrand factor. Blood. 2001; 98: 2730-2735 [DOI] [PubMed] [Google Scholar]
- 90). Reiter RA, Varadi K, Turecek PL, Jilma B, Knöbl P. Changes in ADAMTS13 (von-Willebrand-factor-cleaving protease) activity after induced release of von Willebrand factor during acute systemic inflammation. Thromb Haemost. 2005; 93: 554-558 [DOI] [PubMed] [Google Scholar]
- 91). Gandhi C, Khan MM, Lentz SR, Chauhan AK. ADAMTS13 reduces vascular inflammation and the development of early atherosclerosis in mice. Blood. 2012; 119: 2385-2391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92). von dem Borne PAK, Meijers JCM, Bouma BN. Feedback activation of factor XI by thrombin in plasma results in additional formation of thrombin that protects fibrin clots from fibrinolysis. Blood. 1995; 86: 3035-3042 [PubMed] [Google Scholar]
- 93). von dem Borne PAK, Bajzar L, Meijers JCM, Nesheim ME, Bouma BN. Thrombin-mediated activation of factor XI results in a thrombin activatable fibrinolysis inhibitor-dependent inhibition of fibrinolysis. J Clin Invest. 1997; 99: 2323-2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94). Bolton-Maggs PH. Factor XI deficiency and its management. Haemophilia. 2000; 6: 100-109 [DOI] [PubMed] [Google Scholar]
- 95). Schmaier AH. Physiologic activities of the contact activation system. Thromb Res. 2014; 133 Suppl 1: S41-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96). Meijers JC, Tekelenburg WL, Bouma BN, Bertina RM, Rosendaal FR. High levels of coagulation factor XI as a risk factor for venous thrombosis. N Engl J Med. 2000; 342: 696-701 [DOI] [PubMed] [Google Scholar]
- 97). Berliner JI, Rybicki AC, Kaplan RC, Monrad ES, Freeman R, Billett HH. Elevated levels of factor XI are associated with cardiovascular disease in women. Thromb Res. 2002; 107: 55-60 [DOI] [PubMed] [Google Scholar]
- 98). Yang DT, Flanders MM, Kim H, Rodgers GM. Elevated factor XI activity levels are associated with an increased odds ratio for cerebrovascular events. Am J Clin Pathol. 2006; 126: 411-415 [DOI] [PubMed] [Google Scholar]
- 99). Salomon O, Steinberg DM, Zucker M, Varon D, Zivelin A, Seligsohn U. Patients with severe factor XI deficiency have a reduced incidence of deep-vein thrombosis. Thromb Haemost. 2011; 105: 269-273 [DOI] [PubMed] [Google Scholar]
- 100). Salomon O, Steinberg DM, Koren-Morag N, Tanne D, Seligsohn U. Reduced incidence of ischemic stroke in patients with severe factor XI deficiency. Blood. 2008; 111: 4113-4117 [DOI] [PubMed] [Google Scholar]
- 101). Preis M, Hirsch J, Kotler A, Zoabi A, Stein N, Rennert G, Saliba W. Factor XI deficiency is associated with lower risk for cardiovascular and venous thromboembolism events. Blood. 2017; 129: 1210-1215 [DOI] [PubMed] [Google Scholar]
- 102). Yamashita A, Nishihira K, Kitazawa T, Yoshihashi K, Soeda T, Esaki K, Imamura T, Hattori K, Asada Y. Factor XI contributes to thrombus propagation on injured neointima of the rabbit iliac artery. J Thromb Haemost. 2006; 4: 1496-1501 [DOI] [PubMed] [Google Scholar]
- 103). Takahashi M, Yamashita A, Moriguchi-Goto S, Sugita C, Matsumoto T, Matsuda S, Sato Y, Kitazawa T, Hattori K, Shima M, Asada Y. Inhibition of factor XI reduces thrombus formation in rabbit jugular vein under endothelial denudation and/or blood stasis. Thromb Res. 2010; 125: 464-470 [DOI] [PubMed] [Google Scholar]
- 104). Sugita C, Yamashita A, Matsuura Y, Iwakiri T, Okuyama N, Matsuda S, Matsumoto T, Inoue O, Harada A, Kitazawa T, Hattori K, Shima M, Asada Y. Elevated plasma factor VIII enhances venous thrombus formation in rabbits: contribution of factor XI, von Willebrand factor and tissue factor. Thromb Haemost. 2013; 110: 62-75 [DOI] [PubMed] [Google Scholar]
- 105). Gailani D, Bane CE, Gruber A. Factor XI and contact activation as targets for antithrombotic therapy. J Thromb Haemost. 2015; 13: 1383-1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106). Büller HR, Bethune C, Bhanot S, Gailani D, Monia BP, Raskob GE, Segers, Verhamme P, Weitz JI, FXI-ASO TKA Investigators Factor XI antisense oligonucleotide for prevention of venous thrombosis. N Engl J Med. 2015; 372: 232-240 [DOI] [PMC free article] [PubMed] [Google Scholar]