

Summary
CD4 T cells contribute to protection against pathogens through numerous mechanisms. Incorporating the goal of memory CD4 T‐cell generation into vaccine strategies therefore offers a powerful approach to improve their efficacy, especially in situations where humoral responses alone cannot confer long‐term immunity. These threats include viruses such as influenza that mutate coat proteins to avoid neutralizing antibodies, but that are targeted by T cells that recognize more conserved protein epitopes shared by different strains. A major barrier in the design of such vaccines is that the mechanisms controlling the efficiency with which memory cells form remain incompletely understood. Here, we discuss recent insights into fate decisions controlling memory generation. We focus on the importance of three general cues: interleukin‐2, antigen and co‐stimulatory interactions. It is increasingly clear that these signals have a powerful influence on the capacity of CD4 T cells to form memory during two distinct phases of the immune response. First, through ‘programming’ that occurs during initial priming, and second, through ‘checkpoints’ that operate later during the effector stage. These findings indicate that novel vaccine strategies must seek to optimize cognate interactions, during which interleukin‐2‐, antigen‐ and co‐stimulation‐dependent signals are tightly linked, well beyond initial antigen encounter to induce robust memory CD4 T cells.
Keywords: CD4 T cell, memory, vaccination
Abbreviations
- APC
antigen presenting cell
- IAV
influenza A virus
- ICOS
inducible T‐cell co‐stimulatory
- IFN‐γ
interferon‐γ
- IL‐2
interleukin‐2
- MHC‐II
major histocompatibility complex class II
- TCR
T‐cell receptor
- Th17
T helper type 17
- TRAIL
tumour necrosis factor‐related apoptosis‐inducing ligand
- TRM
tissue‐resident memory
Introduction
Vaccines that aim to generate high titres of protective antibody can fail to provide long‐lived protection against threats with variable surface protein expression. Such pathogens include RNA viruses like influenza A virus (IAV) that can rapidly mutate coat proteins that are the targets for neutralizing antibodies due to immune pressure and error‐prone viral replication machinery. In the case of IAV, annual vaccination can provide protection when the vaccine formulation contains targets that accurately reflect the season's major circulating strains. However, the estimated vaccine effectiveness over the past decade has only peaked at 60% and dropped below 20% during the 2014–2015 season, in which the H3N2 component of the vaccine did not match the circulating virus.1 Furthermore, the neutralizing antibodies elicited by seasonal vaccination are not protective against unexpected pandemic strains that can arise from animal reservoirs.
Current vaccines, including those for IAV, do not induce robust T‐cell responses.2, 3, 4 Developing strategies to induce strong T‐cell memory could improve protection against not only IAV, but also other global health threats including human immunodeficiency virus,5 tuberculosis,6 malaria7 and leishmaniasis8 against which effective vaccination is currently absent. In the case of IAV, this strategy is supported by human studies correlating more robust CD49 and CD810 T‐cell responses with improved clinical outcomes, and decades of studies in animal models.11 Furthermore, T cells recognize epitopes of IAV proteins that are remarkably conserved across strains.12 For example, priming with seasonal IAV strains can generate protective CD4 T cells against pandemic IAV isolates.13 Promoting T‐cell memory could therefore provide a platform for ‘universal’ protection that is less‐dependent on global IAV tracking, prediction of seasonal strains to be targeted, and annual vaccine reformulation.
CD4 T cells directly contribute to pathogen clearance through many mechanisms.14 In the setting of IAV infection, CD4 T cells promote viral clearance through the production of T helper type 17 (Th17) ‐associated cytokines,15, 16 and through perforin‐dependent killing of infected cells.17 These effector mechanisms are in addition to helper functions that improve B‐cell antibody production and cytolytic CD8 T‐cell responses.18 Interestingly, a protective role for the signature ‘Th1’ cytokine interferon‐γ (IFN‐γ) by CD4 T cells during primary IAV infection has been difficult to demonstrate. Indeed, protection mediated by the transfer of activated Th1‐polarized, but IFNγ‐deficient effector populations recognizing IAV to unprimed mice causes reduced weight loss and earlier recovery than is seen following transfer of an equal number of wild‐type cells.17, 19 This suggests that CD4 T‐cell‐derived IFN‐γ can directly contribute to immunopathology. Nevertheless, IFN‐γ produced by CD4 T cells is required for optimal clearance of many other intracellular pathogens.14
Memory CD4 T cells can bring to bear additional protective functions compared with naive cells.11, 18, 20, 21, 22, 23 These memory‐specific mechanisms include faster and more robust cytokine production compared with naive cells,24 enhanced extrafollicular and follicular helper activity that accelerate antibody production,24, 25 and the rapid activation of dendritic cells at the site of infection leading to a ‘jumpstart’ of protective innate immune responses.26, 27 Surprisingly, we and others have also identified a protective role for IFN‐γ produced by memory CD4 T cells during recall responses against IAV.20, 28 The reason for the emergent role for IFN‐γ during secondary CD4 T‐cell responses is unclear but might reflect the more rapid production or greater magnitude of IFN‐γ produced by memory versus naive CD4 T cells.24
A central impediment to the incorporation of T cells into vaccine strategies is that key parameters regulating how memory T cells form are not fully understood. Here, we discuss the impact of three general signals received by CD4 T cells during cognate interactions with antigen‐presenting cells (APC): (i) stimulation through the T‐cell receptor (TCR), (ii) interleukin‐2 (IL‐2), and (iii) co‐stimulation. Recent observations demonstrate that these same signals regulate memory development at multiple time‐points during the T‐cell response. Our discussion will be centred on memory generated in response to acute stimuli rather than during chronic antigen/pathogen exposure in which the line between ‘memory’ and ‘effector’ is more difficult to define. We will also focus exclusively on CD4 T cells. Although many signals regulating memory impact CD4 and CD8 T cells similarly, important differences also distinguish these pathways,29 and excellent reviews have recently concentrated on CD8 T‐cell memory.30, 31, 32
When is memory fate decided?
Perhaps the clearest evidence of uncertainty regarding how memory CD4 T‐cell formation operates are the many models proposed. The model backed by a preponderance of experimental evidence suggests that most memory cells arise from activated effector cells,33, 34 but that the capacity to form memory diminishes as effectors reach an increasingly differentiated, terminal state.35 Indeed, most CD4 T‐cell effectors die through apoptosis and other mechanisms during the resolution of an immune response, leaving behind only a small population that survives long‐term. The transition from an activated effector to a resting memory cell can be quite rapid: acquisition of memory‐associated phenotypic and functional attributes requires only 3 days.36 This transition is largely default in that it requires no discernible instructional signal to CD4 T cells beyond the removal of antigen and inflammatory cytokines.36, 37 However, it appears not to be an entirely stochastic process. In certain settings, effectors can be phenotypically categorized into populations with a greater and lesser potential to survive long term.38, 39 The control over this divergence in fate is not completely understood, but asymmetric division following activation of CD4 T cells has been observed to correlate with distinct cell fates of daughter cells.40, 41 This indicates that, as has been documented for CD8 T cells,42, 43 critical events regulating memory potential may occur within the first few cell divisions following CD4 T‐cell activation.
Many factors control the extent of T‐cell contraction and the efficiency of memory generation, but their impacts are often context dependent.44, 45 In general, signals delivered to CD4 T cells at two distinct phases of the immune response affect the quantity and the quality of the memory cells formed. Early events during activation can ‘programme’ the memory capacity of effector cells, but signals that act on effector cells at defined ‘checkpoints’ later during immune responses regulate the efficiency with which memory is ultimately formed (Figure 1). We will not exhaustively discuss the myriad of variables found to affect this process. Instead, we will review how IL‐2, antigen and co‐stimulation influence memory CD4 T‐cell fate. Interestingly, these same signals can act both during programming and again at later checkpoints to regulate memory potential. Their importance throughout the immune response indicates the critical role that continuing cognate interactions play in shaping CD4 T‐cell memory, with important implications for vaccine design.
Figure 1.
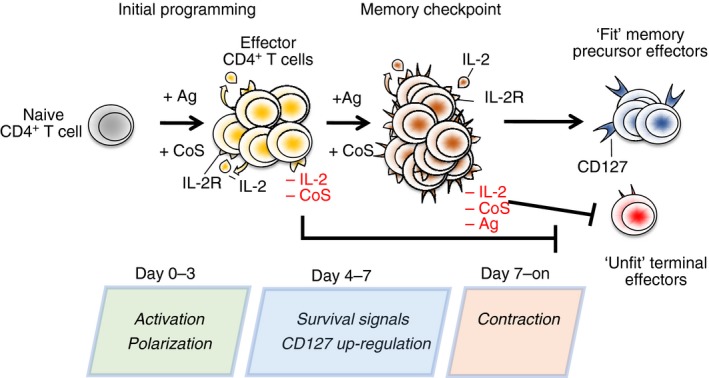
Many key fate decisions are made within the first 2 days or so following CD4 T‐cell priming including T helper type 1 (Th1)/Th2 polarization and the degree to which the cells can form memory. Such early decisions (programming) are especially key in experimental models and immunization protocols using non‐replicating antigen in the absence of adjuvants that facilitate prolonged presentation, which often restrict cognate interactions to within the ‘initial programming’ window. Antigen (Ag), Co‐stimulation (Cos) and interleukin‐2 (IL‐2) are critical signals in programming memory potential. In contrast, replicating pathogens present CD4 T cells with the chance to reencounter antigen during the effector phase of the immune response. Ag‐, IL‐2‐, and Cos‐dependent signals delivered to effector cells during a defined ‘memory checkpoint’ in these situations can modulate the realization of memory fate programmed during initial priming. In the absence of proper signalling during programming or during the memory checkpoint, effector cells form terminal populations with limited capacity to give rise to memory.
Signals that impact memory delivered during initial priming
We will first review findings that either focus on variables impacting initial CD4 T‐cell priming on memory formation, or in which an impact on the priming phase is largely presumed to be responsible for the outcome. It is important to note that some of the experimental models used in studies that will be discussed cannot preclude the possibility that molecules of interest could affect the responding cells not only during priming, but throughout the response (i.e. at the later memory checkpoint). This is especially relevant for models employing animals knocked out for specific molecules or animals treated throughout the period of an immune response to inhibit signalling by specific factors.
Interleukin‐2
Using peptide and APC to active naive CD4 T cells in vitro, IL‐2 was shown to play a decisive role in promoting the capacity of the cells to survive long term. When IL‐2‐deficient (Il2 −/−) effectors were generated with and without exogenous IL‐2, the cells not exposed to IL‐2 rapidly died when transferred to adoptive hosts whereas those activated in the presence of IL‐2 formed durable memory.46 Survival of cells primed with or without IL‐2 was, however, equal when transferred to lymphopenic hosts. Interleukin‐2 is therefore not essential for memory development per se, but instead programmes the fitness of cells to compete for entry into and subsequent survival within a tightly regulated memory pool.46
Interleukin‐2 signals during priming up‐regulate expression of CD127, the α‐chain of the IL‐7 receptor, on effector cells.47 As IL‐7 is required for long‐term memory CD4 T‐cell survival,48, 49 effectors with increased CD127 are better equipped to compete for limiting levels of IL‐7 in T‐cell replete mice.47 This explains why Il2 −/− cells survive well in lymphopenic hosts, where competition for IL‐7 imposed by other T cells is absent. It is interesting that while IL‐2 programmes memory potential through this mechanism, it is also well known to prime CD4 and CD8 T cells for apoptotic death in certain situations.50 It is likely that the level of IL‐2 and the exact state of T‐cell activation together dictate whether IL‐2 delivers a pro‐survival, or pro‐death signal.51 The complex interplay of these two variables presents a challenge for incorporating IL‐2‐based signalling into vaccines with the aim of promoting memory.
Although IL‐7 signals are necessary for long‐term memory CD4 T‐cell survival, we stress that improved access to IL‐7 by effectors during contraction does not appear to impact the size of the memory population formed. In experiments using cells constitutively expressing higher or normal amounts of CD127, the degree of contraction and the number of pathogen‐specific memory CD4 T cells generated after infection was equivalent.52 Similarly, neutralizing IL‐7 in vivo during the contraction phase following priming with vaccinia virus,53 and blocking IL‐7 receptor during the contraction phase following priming with IAV39 had no impact on memory cells recovered. Furthermore, in studies defining a crucial role for the anti‐apoptotic molecule Bcl‐6 in CD4 T‐cell memory generation, no differences in IL‐7 receptor were detected between wild‐type and Bcl6 −/− effectors following peptide immunization, but virtually no memory cells formed from the latter.54 Hence, the relative expression level of IL‐7 receptor on effector CD4 T cells alone is not a definitive marker of memory fate.
Antigen
The strength of the antigen stimulus during priming can be a major determinant of whether T cells, both CD4+ 55 and CD8+,56 form memory precursor cells versus terminal effectors. This may at least in part relate to increased IL‐2 production by T cells stimulated with higher affinity or more antigen,36 through the mechanism discussed above. Indeed, using a Listeria infection model, effector cells that failed to form memory due to lower functional avidity for priming antigen produced reduced IL‐2 versus effectors with higher avidity that formed memory.57 Experiments in which the duration of antigen presentation could be controlled found memory induction to require greater than 24 hr of antigen stimulation whereas less time was nevertheless sufficient to induce robust activation.58 This supports a model in which the stability of cognate interactions during priming, influenced by the amount and duration of antigen, has a profound impact in programming memory potential.59 A similar conclusion was made tracking TCR transgenic cells responding against pathogens that provided more (efficient memory) or less (virtually no memory) antigen.57 However, other studies find that the duration of antigen presentation beyond 24 hr does not play a role in determining CD4 T‐cell fate.60 In addition, too much stimulation during priming can be detrimental in priming CD4 T cells61 and in promoting memory generation62 and function.63 Somewhat at odds to results stressing the importance of relatively fine differences in TCR‐dependent signals, a recent study reported that most CD4 T‐cell clones able to enter the response upon Listeria infection form memory to at least some degree.64 The exact nature of antigen‐dependent signalling required to promote optimal memory programming during priming are therefore yet to be fully defined.
A related variable found to impact memory formation is the number of CD4 T cells able to respond. In a model employing adoptive transfer of TCR transgenic CD4 T cells and challenge with a virus recognized by the TCR, increasing donor cells beyond a critical threshold virtually abolished memory formation.65 Higher precursor frequencies probably do not affect the ability of cells to access peptide‐bearing APC, but instead appear to impair the longer‐term interactions needed to programme memory development.66
These studies also stress the importance of designing experiments using TCR transgenic cells to mirror as much as possible the response of endogenous CD4 T‐cell populations. In addition to the number of responding cells impacting response potential, larger populations of monoclonal CD4 T cells have been shown to display altered response kinetics compared with smaller populations of polyclonal cells.67 The timing of critical signals involved in priming and, as will be discussed, during the memory checkpoint, may therefore be substantially impacted by precursor frequency.
Co‐stimulation
Co‐stimulatory signals delivered by APC are critical for activation and maximal expansion of CD4 T cells. As such, co‐stimulation on one level can be viewed to simply maximize the effector population, some of which will be precursors for memory. But certain co‐stimulatory signals seem to programme CD4 T cells with an enhanced capacity to survive long term. Both CD28‐ and CD40‐dependent signals during priming can promote optimal efficiency with which effectors form memory following peptide stimulation.68 Inducible T‐cell co‐stimulatory (ICOS; CD278) ‐dependent signalling has also been found to improve memory primed by bacterial challenge, even though ICOS‐ligand‐deficient and ICOS‐ligand‐sufficient T cells generate effector populations of similar magnitude.69 The requirement for co‐stimulation may be related to maximizing IL‐2 production. Indeed, using protein immunization, OX‐40‐dependent interactions were found to promote optimal memory, correlating with enhanced IL‐2 production by the CD4 T cells.70 CD27‐dependent signals during priming may be especially important for memory formed by CD4 T cells expressing lower affinity TCRs.71 Whether or not different co‐stimulatory molecule interactions direct or favour the generation of distinct kinds of memory cells is not clear. However, signalling through OX‐40 (CD134) expressed by T cells may be especially important in generating ‘effector’ versus ‘central’ memory cells.72 This suggests the possibility that novel vaccines could shape distinct subsets to optimize protection in a pathogen‐specific manner by modulating the expression of single or multiple co‐stimulatory markers on APC.
Control of CD4 T‐cell memory during the effector phase of immune responses
It is increasingly clear that signals received by CD4 T‐cell effectors well after their activation can have a profound impact on memory fate. The relative emphasis on the contribution of early versus later‐acting signals in controlling memory can be influenced by the experimental system employed. For example, non‐replicating antigens given without adjuvants that provide sustained presentation, and many in vitro models, focus analysis on variables that act during the first couple of days following priming. This is because functional antigen presentation in such experiments beyond the first few days is absent or extremely limited.73 As mentioned earlier, it is also difficult to ascribe relative importance to ‘programming’ versus later ‘checkpoints’ in models where opportunities for continuing antigen encounter exist without careful differentiation of these stages. Below, we will discuss the growing body of evidence supporting the argument that the same signals that influence memory potential during priming again act to regulate memory fate at the effector phase of CD4 T‐cell responses.
Interleukin‐2
We identified a critical need for autocrine IL‐2 signals well past priming to generate virtually all CD4 T‐cell memory following an IAV challenge.39 Although we confirmed the need for naive CD4 T cells to receive IL‐2 during activation to be primed for memory fate (programming), without a second IL‐2 signal several days later, the IL‐2‐primed cells did not survive contraction.39 The second IL‐2 signal is required during a restricted window between 5 and 7 days after infection at which time the CD4 T cells are highly activated effectors. A similar requirement for IL‐2 signals after priming to promote maximal CD4 T‐cell memory is seen following lymphocytic choriomeningitis virus challenge,74 indicating that late‐acting memory checkpoints have key roles in settings other than IAV infection.
Interleukin‐2 rescues memory generation during the memory checkpoint by at least two distinct mechanisms (Figure 2). First, IL‐2 up‐regulates sustained CD127 expression on effector cells. This action of IL‐2 presumably operates through the same mechanisms as described earlier by which IL‐2 during priming increases CD127. Increasing amounts of exogenous IL‐2 given to mice from days 4 to 6 post‐infection in the form of IL‐2: anti‐IL‐2 antibody complexes (that enhance the longevity and potency of IL‐2 in vivo 75) directly correlate with the degree of CD127 up‐regulation, and with the number of memory cells recovered. This indicates that the limiting factor restricting memory is often the availability of IL‐2 and not the number of effector cells inherently able to form memory (i.e. properly ‘programmed’ and not terminally differentiated). Furthermore, as the major pathway of IL‐2 signalling in T cells is autocrine following TCR triggering, this suggests that another crucial bottleneck on memory generation under physiological conditions is the ability of effector cells to engage in cognate interactions that result in autocrine IL‐2 signals.
Figure 2.
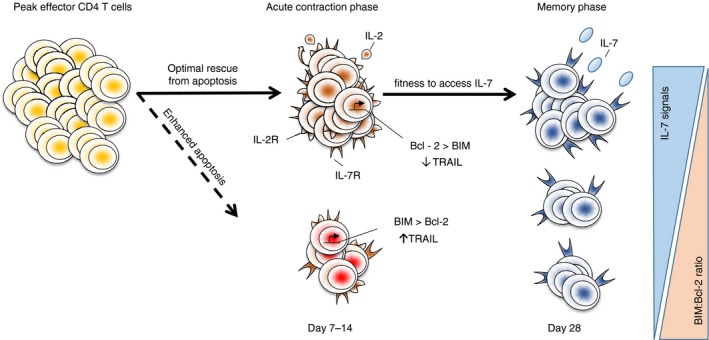
The efficiency of memory generation is impacted by two major mechanisms. First, the degree of acute contraction following the peak of effector CD4 T‐cell responses determines the number of effector cells able to serve as precursors for long‐lived memory cells. Interleukin‐2 (IL‐2) signals during the memory checkpoint increase Bcl‐2 and reduce Bim and tumour necrosis factor‐related apoptosis‐inducing ligand (TRAIL) expression which protects effector cells from apoptotic death leading to a larger (solid line versus dotted line) population of effectors with potential to form memory. Cognate interactions also up‐regulate IL‐7 receptor on effector cells. Although not impacting the degree of acute apoptosis, CD4 T cells able to outcompete other T cells for IL‐7 signals following contraction gain a fitness advantage to survive long‐term. The resulting number of long‐lived memory cells formed following an immune response therefore depends both on how well effector cells survive contraction and on their relative fitness to compete for IL‐7. Both of these processes are impacted by Antigen (Ag), Co‐stimulation (Cos) and IL‐2‐dependent signals during the memory checkpoint.
The second, unique mechanism by which IL‐2 acts at the memory checkpoint is through preventing acute effector cell apoptosis (Figure 2). Interleukin‐2 signalling from days 4 to 6 post‐infection up‐regulates the expression of the anti‐apoptotic protein Bcl‐2 and down‐regulates the pro‐apoptotic molecule Bim throughout the contraction phase following IAV infection.39 Late IL‐2 administration also increases Bcl‐2 in effector T cells responding to lymphocytic choriomeningitis virus.74 Other molecules regulated by IL‐2 probably also contribute to preventing acute apoptosis. For example, we observed that IL‐2 signals during the memory checkpoint reduced tumour necrosis factor‐related apoptosis‐inducing ligand (TRAIL; CD253) expression on effectors,39 and TRAIL‐dependent apoptosis of Th1‐polarized cells during pathogen responses has been demonstrated.76, 77 A recent study in a malaria model also identified a critical role for IL‐2 during the effector phase of the CD4 T‐cell response in preventing terminal differentiation.78 It is likely that other cytokines acting on effector cells also impact memory formation. For example, transforming growth factor‐β can synergize with IL‐2 to promote the survival of activated CD4 T cells, at least in vitro.79 Similarly, IL‐15 can support effector cell survival in culture,80 although whether IL‐15 can synergize with IL‐2 in this regard is not clear.
Antigen
By blocking major histocompatibility complex class II (MHC‐II) ‐dependent interactions in mice responding to IAV only during the memory checkpoint phase of the IAV response we observed a sharp reduction in the magnitude of CD4 memory formed.39 One explanation for reduced memory development in mice where MHC‐II‐dependent interactions are blocked during the effector phase of the CD4 T‐cell response is that autocrine IL‐2 signalling is much reduced. However, TCR stimulation also impacts several aspects of the memory programme that are independent of IL‐2 signalling.81 Importantly, these experiments used a small number of TCR transgenic donor cells that closely mirrored the response kinetics, and patterns of antigen encounter of endogenous CD4 T cells responding against IAV detected by tetramer analysis.81 This suggests that polyclonal CD4 T cells bearing TCRs with different affinities share a similar need for antigen recognition to form optimal memory during the effector phase. However, given that response kinetics of individual clones can vary based on their relative frequency, it is possible that the precise timing of antigen‐dependent interactions that define the memory checkpoint are asynchronous within endogenous effector populations.
The cognate interactions engaged in by effector cells during the memory checkpoint also appear to induce optimal functional potential of the memory cells generated. In these experiments, IAV‐specific CD4 T effectors were isolated from infected mice and cultured briefly with dendritic cells in the presence or absence of cognate peptide. Equal numbers of effectors were then transferred to new hosts and analysed 1 week later. The effectors cultured with peptide gave rise to memory cells with enhanced capacity for IFN‐γ production and were able to produce multiple cytokines compared with effectors cultured without cognate antigen.81 The ability of Th1 cells to co‐produce multiple cytokines is a commonly used indicator of their improved protective potential.82 Late antigen encounter therefore impacts both quantitative and qualitative aspects of memory formation.
A requirement for antigen beyond priming to generate optimal CD4 T‐cell memory has also been seen after Salmonella infection. Using an antibiotic to rapidly clear bacteria from primed mice, researchers found that treatment initiated before the second week of infection severely limited the generation of protective IFN‐γ‐secreting memory cells whereas bacterial clearance after this time‐point had no effect on memory induction.83 Initiation of antibiotic treatment before the second week post‐infection did not, however, impact the expansion of CD4 T cells.83 A similar decrease in CD4 T‐cell memory and protective immunity has been reported in a murine model of Chlamydia infection using antibiotics to cut short the natural course of infection.84
Although antibiotic treatment in these studies probably does not result in the immediate elimination of antigen presentation, it may reduce antigen levels below a critical threshold and/or severely shorten its duration compared with natural infection.85 Additionally, antibiotic treatment may alter the context of antigen presentation by reducing infection‐associated inflammation and co‐stimulatory molecule expression on APC that may be needed to promote memory (see next section). Further studies are required to investigate the precise relationship between antibacterial and antiviral drug treatment on memory CD4 T‐cell generation, and the mechanisms impacted.
Together, these findings lead to a teleological model in which the continuing presence of antigen into the effector phase of the CD4 T‐cell response is central in determining the extent of memory formation. It makes sense that little or no memory should be formed against antigens that are present for only a few days and then rapidly cleared. Such antigens do not pose a strong need for recall as their removal by elements of the innate immune response occurs before the generation of strong T‐cell responses. On the other hand, antigens that persist, and especially those that increase over the course of several days, probably reflect an encounter with a replicating microorganism that is not efficiently contained by innate immunity alone. In such cases, recall responses by T cells would be highly beneficial to reduce the severity of infection should the pathogen be encountered again.
Co‐stimulation
We found CD70‐dependent signalling through CD27 expressed on effector CD4 T cells to maximize IAV‐primed memory. CD27 stimulation enhanced IL‐2 production from effectors, suggesting that this is a major mechanism by which this pathway acts to promote effector survival.39 This was confirmed by restoration of memory in mice treated with blocking antibodies against CD70 together with administration of IL‐2 complexes from days 4 to 6 post‐infection, circumventing the need for autocrine IL‐2 production induced by CD27‐dependent signalling during the memory checkpoint. Interestingly, CD70+ APC detected in mice peak during the 5‐ to 7‐day period following IAV priming that coincides with the memory checkpoint phase of the response.39 The number of CD70+ APC present during this timeframe may impose a critical limitation on the number of memory cells generated. An appealing strategy to improve vaccine‐induced CD4 T‐cell memory is to target the amplification of those APC able to deliver pro‐memory signals to antigen‐specific effector cells, or to directly modulate co‐stimulatory molecule expression by APC to alleviate bottlenecks constricting memory formation. Further research is needed to determine whether other co‐stimulatory pathways found to impact memory during priming (see above) also have roles during the effector phase to maximize memory fate.
It is highly likely that other pathways beyond those summarized here that act during the effector phase of immune responses also impact the efficiency of memory generation. Studies assessing the ability of effector CD4 T cells isolated 6–8 days after peptide priming to form memory in adoptive hosts found a strong pro‐memory impact of Toll‐like receptor ligand administration at the time of transfer.86 A direct impact of the adjuvant on the T cells was observed, resulting in enhanced ability to produce IL‐2. This indicates that pathogen‐associated molecular patterns sensed by effector CD4 T cells can contribute during the memory checkpoint to promote short‐term effector survival, and ultimately improved memory formation.
Distinct regulation of tissue‐resident memory establishment?
Specialized subsets of CD4+ and CD8+ memory T cells persist at sites of infection without systemic recirculation.87 In many cases tissue‐resident memory (TRM) CD4 T cells have been found to make critical contributions to pathogen clearance. Examples include in mice challenged with IAV after intranasal vaccination, in animals primed and re‐challenged with Bordetella pertussis or with oral Listeria challenge, and after vaccination and rechallenge with Francisella tularensis.88, 89, 90, 91, 92 Their distinct impact is due in large part to location, allowing TRM cells to respond before conventional memory cells in lymphoid tissues are stimulated and traffic in large numbers to inflamed sites, a process than can take up to 1 week.24
It is not well understood when during the immune response TRM cell differentiation occurs. It is reasonable to assume that decisions governing TRM cell fate take place only after effector CD4 T cells traffic to sites of infection to engage in responses against pathogens. It is also plausible that unique factors regulate this pathway versus the signals critical for the formation of conventional CD4 T‐cell memory. In support of this hypothesis, we found memory cells in the lung were reduced in the absence of IL‐2 only by about twofold compared with the almost complete ablation of memory cells in lymphoid organs.93 We furthermore found that all of the IL‐2‐independent memory cells fit TRM cell criteria and were indistinguishable from TRM cells formed in mice where IL‐2 signalling was not blocked.93 We found a similar number of lung memory cells in mice in which we blocked MHC‐II‐dependent antigen‐presentation from days 4 to 6 post‐infection,39 suggesting that TRM cells can develop in the absence of interactions with APC during the memory checkpoint that are required to form most other memory cells.
These observations suggest that the formation of some TRM cells may be directly guided by inflammatory signals received by effector cells in infected tissues independently of cognate interactions during which IL‐2, antigen and co‐stimulatory signals are delivered (Figure 3). In support of this hypothesis, we found that blocking IL‐15 signalling from 1 to 7 days post‐infection eliminated the IL‐2‐independent TRM cell subset.93 Interleukin‐15, which is not produced by CD4 T cells, is only detected in the lungs during IAV challenge and peaks during the same 4‐ to 7‐day post‐infection window in which late IL‐2 acts on effectors to facilitate memory formation.93 Interleukin‐15 signalling to CD4 T‐cell effectors therefore appears to constitute a tissue‐derived signal, generated in response to infection, that is restricted in promoting local memory. Indeed, equal populations of memory CD4 T cells are seen in secondary lymphoid organs of wild‐type and IL15 −/− mice primed by IAV, indicating that circulating memory cell formation is largely independent of IL‐15. Future work is needed to determine if IL‐15, or other factors, play similar roles in other tissues during infection to direct CD4+ TRM cell formation. Fully elucidating this pathway may be valuable for vaccine strategies as a means to specifically target TRM cell formation versus the IL‐2‐dependent mechanism that can support the formation of both circulating and local memory pools.
Figure 3.
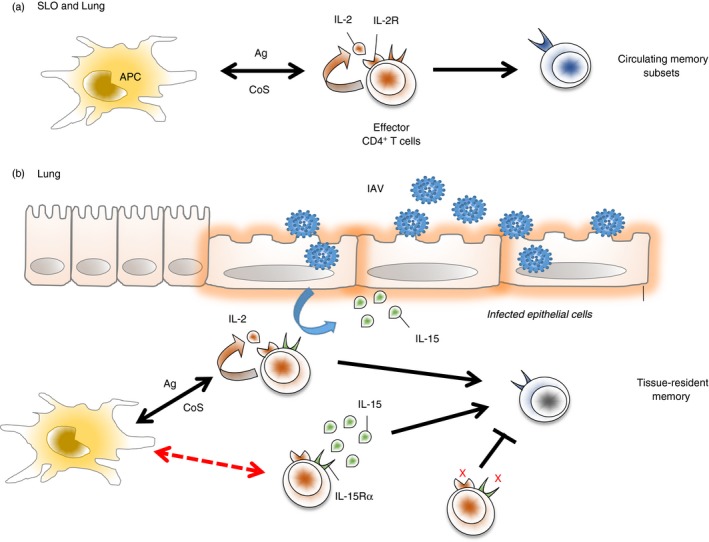
Tissue‐resident memory primed by influenza A virus (IAV) is supported by two independent pathways acting at the memory ‘checkpoint’. (a) Antigen (Ag), interleukin‐2 (IL‐2), and Co‐stimulation (CoS) ‐dependent signals support the generation of circulating memory cells in secondary lymphoid organs and in the lung (the site of infection). (b) These same signals can also support the generation of a cohort of tissue‐resident memory cells in the lung. An additional IL‐2‐independent, IL‐15‐dependent mechanism can also support robust tissue‐resident memory formation. IL‐15 is an inflammatory cytokine induced by IAV infection that is restricted mainly to the lung. This pathway appears not to require cognate interactions during the memory checkpoint (indicated by the red dashed line). In the absence of IL‐15 and IL‐2 signals, effector CD4 T cells fail to survive the contraction phase, generating virtually no memory either in the lung or systemically.
Summary and future directions
There has been tremendous progress towards understanding the basic mechanisms governing T‐cell memory generation. For example, studies revealing the importance of the metabolic94 and transcriptional95 state of effectors in facilitating memory development have provided novel mechanistic clarity into how fate decisions are governed as well as targets for intervention. One of the pressing challenges facing the field though is to incorporate these insights into improved vaccination strategies that are able to harness the protective potential of T‐cell memory. Perhaps the most direct means towards this goal is for vaccines to mimic as much as possible the signals and environments induced by actual infection. Indeed, the focus on developing novel adjuvant regimens to optimize lymphocyte priming reflects this approach. However, in most cases, and especially with non‐replicating vaccine platforms, the priming phase is the only aspect of the T‐cell response that is considered and targeted. This has tended to place an emphasis on improving memory T‐cell generation through manipulating variables during the initial ‘programming’ of T‐cell responses.
On the other hand, a growing body of experimental evidence indicates that to maximize their ability to prime T‐cell memory, vaccines must also aim to replicate key aspects of the environments and signals induced by infection in which activated T‐cell effectors respond. It is at this later phase of the immune response against replicating pathogens that the realization of memory fate, and in at least some cases the maximal functional and protective potential of memory cells, can be powerfully influenced by antigen, IL‐2 and co‐stimulatory signals. Further characterization of the cognate interactions that effector CD4 T cells engage in with APC, as well as the influence of inflammatory signals at the site of infection, will continue to provide novel insight into how CD4 T‐cell memory can be optimized through innovative vaccine approaches.
Conflict of interest
The authors declare no financial or commercial conflicts of interest.
Acknowledgements
We thank Dr Tara Strutt for critical review and expert graphic design. This work was supported by the American Heart Association grant 14SDG18600020 (to KKM), and by the University of Central Florida.
References
- 1. Zimmerman RK, Nowalk MP, Chung J, Jackson ML, Jackson LA, Petrie JG et al 2014–2015 influenza vaccine effectiveness in the United States by vaccine type. Clin Infect Dis 2016; 63:1564–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hoft DF, Babusis E, Worku S, Spencer CT, Lottenbach K, Truscott SM et al Live and inactivated influenza vaccines induce similar humoral responses, but only live vaccines induce diverse T‐cell responses in young children. J Infect Dis 2011; 204:845–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bodewes R, Fraaij PL, Geelhoed‐Mieras MM, van Baalen CA, Tiddens HA, van Rossum AM et al Annual vaccination against influenza virus hampers development of virus‐specific CD8+ T cell immunity in children. J Virol 2011; 85:11995–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hoft DF, Lottenbach KR, Blazevic A, Turan A, Blevins TP, Pacatte TP et al Comparisons of the humoral and cellular immune responses induced by live attenuated influenza vaccine and inactivated influenza vaccine in adults. Clin Vaccine Immunol 2017; 24:e00414–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Streeck H, D'Souza MP, Littman DR, Crotty S. Harnessing CD4+ T cell responses in HIV vaccine development. Nat Med 2013; 19:143–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lewinsohn DA, Lewinsohn DM, Scriba TJ. Polyfunctional CD4+ T cells as targets for tuberculosis vaccination. Front Immunol 2017; 8:1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Doll KL, Harty JT. Correlates of protective immunity following whole sporozoite vaccination against malaria. Immunol Res 2014; 59:166–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Glennie ND, Scott P. Memory T cells in cutaneous leishmaniasis. Cell Immunol 2016; 309:50–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wilkinson TM, Li CK, Chui CS, Huang AK, Perkins M, Liebner JC et al Preexisting influenza‐specific CD4+ T cells correlate with disease protection against influenza challenge in humans. Nat Med 2012; 18:274–80. [DOI] [PubMed] [Google Scholar]
- 10. Sridhar S, Begom S, Bermingham A, Hoschler K, Adamson W, Carman W et al Cellular immune correlates of protection against symptomatic pandemic influenza. Nat Med 2013; 19:1305–12. [DOI] [PubMed] [Google Scholar]
- 11. Zens KD, Farber DL. Memory CD4 T cells in influenza. Curr Top Microbiol Immunol 2015; 386:399–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Powell TJ, Strutt T, Reome J, Hollenbaugh JA, Roberts AD, Woodland DL et al Priming with cold‐adapted influenza A does not prevent infection but elicits long‐lived protection against supralethal challenge with heterosubtypic virus. J Immunol 2007; 178:1030–8. [DOI] [PubMed] [Google Scholar]
- 13. Alam S, Sant AJ. Infection with seasonal influenza virus elicits CD4 T cells specific for genetically conserved epitopes that can be rapidly mobilized for protective immunity to pandemic H1N1 influenza virus. J Virol 2011; 85:13310–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Swain SL, McKinstry KK, Strutt TM. Expanding roles for CD4+ T cells in immunity to viruses. Nat Rev Immunol 2012; 12:136–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McKinstry KK, Strutt TM, Buck A, Curtis JD, Dibble JP, Huston G et al IL‐10 deficiency unleashes an influenza‐specific Th17 response and enhances survival against high‐dose challenge. J Immunol 2009; 182:7353–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eliasson DG, Omokanye A, Schon K, Wenzel UA, Bernasconi V, Bemark M et al M2e‐tetramer‐specific memory CD4 T cells are broadly protective against influenza infection. Mucosal Immunol 2017; 11:273–89. [DOI] [PubMed] [Google Scholar]
- 17. Brown DM, Lee S, Garcia‐Hernandez Mde L, Swain SL. Multifunctional CD4 cells expressing gamma interferon and perforin mediate protection against lethal influenza virus infection. J Virol 2012; 86:6792–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McKinstry KK, Dutton RW, Swain SL, Strutt TM. Memory CD4 T cell‐mediated immunity against influenza A virus: more than a little helpful. Arch Immunol Ther Exp (Warsz) 2013; 61:341–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brown DM, Dilzer AM, Meents DL, Swain SL. CD4 T cell‐mediated protection from lethal influenza: perforin and antibody‐mediated mechanisms give a one‐two punch. J Immunol 2006; 177:2888–98. [DOI] [PubMed] [Google Scholar]
- 20. Teijaro JR, Verhoeven D, Page CA, Turner D, Farber DL. Memory CD4 T cells direct protective responses to influenza virus in the lungs through helper‐independent mechanisms. J Virol 2010; 84:9217–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McKinstry KK, Strutt TM, Swain SL. Hallmarks of CD4 T cell immunity against influenza. J Intern Med 2011; 269:507–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. DiPiazza A, Richards KA, Knowlden ZA, Nayak JL, Sant AJ. The role of CD4 T cell memory in generating protective immunity to novel and potentially pandemic strains of influenza. Front Immunol 2016; 7:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. La Gruta NL, Turner SJ. T cell mediated immunity to influenza: mechanisms of viral control. Trends Immunol 2014; 35:396–402. [DOI] [PubMed] [Google Scholar]
- 24. Strutt TM, McKinstry KK, Kuang Y, Bradley LM, Swain SL. Memory CD4+ T‐cell‐mediated protection depends on secondary effectors that are distinct from and superior to primary effectors. Proc Natl Acad Sci USA 2012; 109:E2551–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. MacLeod MK, David A, McKee AS, Crawford F, Kappler JW, Marrack P. Memory CD4 T cells that express CXCR5 provide accelerated help to B cells. J Immunol 2011; 186:2889–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Strutt TM, McKinstry KK, Dibble JP, Winchell C, Kuang Y, Curtis JD et al Memory CD4+ T cells induce innate responses independently of pathogen. Nat Med 2010; 16:558–64, 1p following 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chapman TJ, Lambert K, Topham DJ. Rapid reactivation of extralymphoid CD4 T cells during secondary infection. PLoS ONE 2011; 6:e20493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McKinstry KK, Strutt TM, Kuang Y, Brown DM, Sell S, Dutton RW et al Memory CD4+ T cells protect against influenza through multiple synergizing mechanisms. J Clin Invest 2012; 122:2847–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Seder RA, Ahmed R. Similarities and differences in CD4+ and CD8+ effector and memory T cell generation. Nat Immunol 2003; 4:835–42. [DOI] [PubMed] [Google Scholar]
- 30. Kurtulus S, Tripathi P, Hildeman DA. Protecting and rescuing the effectors: roles of differentiation and survival in the control of memory T cell development. Front Immunol 2012; 3:404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Valbon SF, Condotta SA, Richer MJ. Regulation of effector and memory CD8 T cell function by inflammatory cytokines. Cytokine 2015; 82:16–23. [DOI] [PubMed] [Google Scholar]
- 32. Gerritsen B, Pandit A. The memory of a killer T cell: models of CD8+ T cell differentiation. Immunol Cell Biol 2016; 94:236–41. [DOI] [PubMed] [Google Scholar]
- 33. Lohning M, Hegazy AN, Pinschewer DD, Busse D, Lang KS, Hofer T et al Long‐lived virus‐reactive memory T cells generated from purified cytokine‐secreting T helper type 1 and type 2 effectors. J Exp Med 2008; 205:53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Harrington LE, Janowski KM, Oliver JR, Zajac AJ, Weaver CT. Memory CD4 T cells emerge from effector T‐cell progenitors. Nature 2008; 452:356–60. [DOI] [PubMed] [Google Scholar]
- 35. Wu CY, Kirman JR, Rotte MJ, Davey DF, Perfetto SP, Rhee EG et al Distinct lineages of TH1 cells have differential capacities for memory cell generation in vivo . Nat Immunol 2002; 3:852–8. [DOI] [PubMed] [Google Scholar]
- 36. McKinstry KK, Golech S, Lee WH, Huston G, Weng NP, Swain SL. Rapid default transition of CD4 T cell effectors to functional memory cells. J Exp Med 2007; 204:2199–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Harbertson J, Biederman E, Bennett KE, Kondrack RM, Bradley LM. Withdrawal of stimulation may initiate the transition of effector to memory CD4 cells. J Immunol 2002; 168:1095–102. [DOI] [PubMed] [Google Scholar]
- 38. Marshall HD, Chandele A, Jung YW, Meng H, Poholek AC, Parish IA et al Differential expression of Ly6C and T‐bet distinguish effector and memory Th1 CD4+ cell properties during viral infection. Immunity 2011; 35:633–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McKinstry KK, Strutt TM, Bautista B, Zhang W, Kuang Y, Cooper AM et al Effector CD4 T‐cell transition to memory requires late cognate interactions that induce autocrine IL‐2. Nat Commun 2014; 5:5377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chang JT, Palanivel VR, Kinjyo I, Schambach F, Intlekofer AM, Banerjee A et al Asymmetric T lymphocyte division in the initiation of adaptive immune responses. Science 2007; 315:1687–91. [DOI] [PubMed] [Google Scholar]
- 41. Nish SA, Zens KD, Kratchmarov R, Lin WW, Adams WC, Chen YH et al CD4+ T cell effector commitment coupled to self‐renewal by asymmetric cell divisions. J Exp Med 2017; 214:39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Arsenio J, Metz PJ, Chang JT. Asymmetric cell division in T lymphocyte fate diversification. Trends Immunol 2015; 36:670–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reiner SL, Adams WC. Lymphocyte fate specification as a deterministic but highly plastic process. Nat Rev Immunol 2014; 14:699–704. [DOI] [PubMed] [Google Scholar]
- 44. McKinstry KK, Strutt TM, Swain SL. Regulation of CD4+ T‐cell contraction during pathogen challenge. Immunol Rev 2010; 236:110–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McKinstry KK, Strutt TM, Swain SL. The effector to memory transition of CD4 T cells. Immunol Res 2008; 40:114–27. [DOI] [PubMed] [Google Scholar]
- 46. Dooms H, Kahn E, Knoechel B, Abbas AK. IL‐2 induces a competitive survival advantage in T lymphocytes. J Immunol 2004; 172:5973–9. [DOI] [PubMed] [Google Scholar]
- 47. Dooms H, Wolslegel K, Lin P, Abbas AK. Interleukin‐2 enhances CD4+ T cell memory by promoting the generation of IL‐7Rα‐expressing cells. J Exp Med 2007; 204:547–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kondrack RM, Harbertson J, Tan JT, McBreen ME, Surh CD, Bradley LM. Interleukin 7 regulates the survival and generation of memory CD4 cells. J Exp Med 2003; 198:1797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li J, Huston G, Swain SL. IL‐7 promotes the transition of CD4 effectors to persistent memory cells. J Exp Med 2003; 198:1807–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lenardo MJ. Interleukin‐2 programs mouse αβ T lymphocytes for apoptosis. Nature 1991; 353:858–61. [DOI] [PubMed] [Google Scholar]
- 51. Bachmann MF, Oxenius A. Interleukin 2: from immunostimulation to immunoregulation and back again. EMBO Rep 2007; 8:1142–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Haring JS, Jing X, Bollenbacher‐Reilley J, Xue HH, Leonard WJ, Harty JT. Constitutive expression of IL‐7 receptor α does not support increased expansion or prevent contraction of antigen‐specific CD4 or CD8 T cells following Listeria monocytogenes infection. J Immunol 2008; 180:2855–62. [DOI] [PubMed] [Google Scholar]
- 53. Tripathi P, Mitchell TC, Finkelman F, Hildeman DA. Cutting Edge: limiting amounts of IL‐7 do not control contraction of CD4+ T cell responses. J Immunol 2007; 178:4027–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ichii H, Sakamoto A, Arima M, Hatano M, Kuroda Y, Tokuhisa T. Bcl6 is essential for the generation of long‐term memory CD4+ T cells. Int Immunol 2007; 19:427–33. [DOI] [PubMed] [Google Scholar]
- 55. Keck S, Schmaler M, Ganter S, Wyss L, Oberle S, Huseby ES et al Antigen affinity and antigen dose exert distinct influences on CD4 T‐cell differentiation. Proc Natl Acad Sci USA 2014; 111:14852–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gett AV, Sallusto F, Lanzavecchia A, Geginat J. T cell fitness determined by signal strength. Nat Immunol 2003; 4:355–60. [DOI] [PubMed] [Google Scholar]
- 57. Williams MA, Ravkov EV, Bevan MJ. Rapid culling of the CD4+ T cell repertoire in the transition from effector to memory. Immunity 2008; 28:533–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Obst R, van Santen HM, Melamed R, Kamphorst AO, Benoist C, Mathis D. Sustained antigen presentation can promote an immunogenic T cell response, like dendritic cell activation. Proc Natl Acad Sci USA 2007; 104:15460–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kim C, Wilson T, Fischer KF, Williams MA. Sustained interactions between T cell receptors and antigens promote the differentiation of CD4+ memory T cells. Immunity 2013; 39:508–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Corbin GA, Harty JT. Duration of infection and antigen display have minimal influence on the kinetics of the CD4+ T cell response to Listeria monocytogenes infection. J Immunol 2004; 173:5679–87. [DOI] [PubMed] [Google Scholar]
- 61. Jelley‐Gibbs DM, Dibble JP, Filipson S, Haynes L, Kemp RA, Swain SL. Repeated stimulation of CD4 effector T cells can limit their protective function. J Exp Med 2005; 201:1101–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Blair DA, Turner DL, Bose TO, Pham QM, Bouchard KR, Williams KJ et al Duration of antigen availability influences the expansion and memory differentiation of T cells. J Immunol 2011; 187:2310–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Han S, Asoyan A, Rabenstein H, Nakano N, Obst R. Role of antigen persistence and dose for CD4+ T‐cell exhaustion and recovery. Proc Natl Acad Sci USA 2010; 107:20453–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tubo NJ, Fife BT, Pagan AJ, Kotov DI, Goldberg MF, Jenkins MK. Most microbe‐specific naive CD4+ T cells produce memory cells during infection. Science 2016; 351:511–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Blair DA, Lefrancois L. Increased competition for antigen during priming negatively impacts the generation of memory CD4 T cells. Proc Natl Acad Sci USA 2007; 104:15045–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Garcia Z, Pradelli E, Celli S, Beuneu H, Simon A, Bousso P. Competition for antigen determines the stability of T cell‐dendritic cell interactions during clonal expansion. Proc Natl Acad Sci USA 2007; 104:4553–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Foulds KE, Shen H. Clonal competition inhibits the proliferation and differentiation of adoptively transferred TCR transgenic CD4 T cells in response to infection. J Immunol 2006; 176:3037–43. [DOI] [PubMed] [Google Scholar]
- 68. Dooms H, Abbas AK. Control of CD4+ T‐cell memory by cytokines and costimulators. Immunol Rev 2006; 211:23–38. [DOI] [PubMed] [Google Scholar]
- 69. Marriott CL, Carlesso G, Herbst R, Withers DR. ICOS is required for the generation of both central and effector CD4+ memory T‐cell populations following acute bacterial infection. Eur J Immunol 2015; 45:1706–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gramaglia I, Jember A, Pippig SD, Weinberg AD, Killeen N, Croft M. The OX40 costimulatory receptor determines the development of CD4 memory by regulating primary clonal expansion. J Immunol 2000; 165:3043–50. [DOI] [PubMed] [Google Scholar]
- 71. Baumgartner CK, Yagita H, Malherbe LP. A TCR affinity threshold regulates memory CD4 T cell differentiation following vaccination. J Immunol 2012; 189:2309–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Soroosh P, Ine S, Sugamura K, Ishii N. Differential requirements for OX40 signals on generation of effector and central memory CD4+ T cells. J Immunol 2007; 179:5014–23. [DOI] [PubMed] [Google Scholar]
- 73. Jelley‐Gibbs DM, Lepak NM, Yen M, Swain SL. Two distinct stages in the transition from naive CD4 T cells to effectors, early antigen‐dependent and late cytokine‐driven expansion and differentiation. J Immunol 2000; 165:5017–26. [DOI] [PubMed] [Google Scholar]
- 74. Blattman JN, Grayson JM, Wherry EJ, Kaech SM, Smith KA, Ahmed R. Therapeutic use of IL‐2 to enhance antiviral T‐cell responses in vivo . Nat Med 2003; 9:540–7. [DOI] [PubMed] [Google Scholar]
- 75. Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody‐cytokine immune complexes. Science 2006; 311:1924–7. [DOI] [PubMed] [Google Scholar]
- 76. Li X, McKinstry KK, Swain SL, Dalton DK. IFN‐γ acts directly on activated CD4+ T cells during mycobacterial infection to promote apoptosis by inducing components of the intracellular apoptosis machinery and by inducing extracellular proapoptotic signals. J Immunol 2007; 179:939–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Herbeuval JP, Grivel JC, Boasso A, Hardy AW, Chougnet C, Dolan MJ et al CD4+ T‐cell death induced by infectious and noninfectious HIV‐1: role of type 1 interferon‐dependent, TRAIL/DR5‐mediated apoptosis. Blood 2005; 106:3524–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Opata MM, Carpio VH, Ibitokou SA, Dillon BE, Obiero JM, Stephens R. Early effector cells survive the contraction phase in malaria infection and generate both central and effector memory T cells. J Immunol 2015; 194:5346–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zhang X, Giangreco L, Broome HE, Dargan CM, Swain SL. Control of CD4 effector fate: transforming growth factor β1 and interleukin 2 synergize to prevent apoptosis and promote effector expansion. J Exp Med 1995; 182:699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Dooms H, Desmedt M, Vancaeneghem S, Rottiers P, Goossens V, Fiers W et al Quiescence‐inducing and antiapoptotic activities of IL‐15 enhance secondary CD4+ T cell responsiveness to antigen. J Immunol 1998; 161:2141–50. [PubMed] [Google Scholar]
- 81. Bautista BL, Devarajan P, McKinstry KK, Strutt TM, Vong AM, Jones MC et al Short‐lived antigen recognition but not viral infection at a defined checkpoint programs effector CD4 T cells to become protective memory. J Immunol 2016; 197:3936–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Darrah PA, Patel DT, De Luca PM, Lindsay RW, Davey DF, Flynn BJ et al Multifunctional TH1 cells define a correlate of vaccine‐mediated protection against Leishmania major . Nat Med 2007; 13:843–50. [DOI] [PubMed] [Google Scholar]
- 83. Griffin AJ, McSorley SJ. Generation of Salmonella‐specific Th1 cells requires sustained antigen stimulation. Vaccine 2011; 29:2697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Su H, Morrison R, Messer R, Whitmire W, Hughes S, Caldwell HD. The effect of doxycycline treatment on the development of protective immunity in a murine model of chlamydial genital infection. J Infect Dis 1999; 180:1252–8. [DOI] [PubMed] [Google Scholar]
- 85. Benoun JM, Labuda JC, McSorley SJ. Collateral damage: detrimental effect of antibiotics on the development of protective immune memory. MBio 2016; 7:e01520–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Guloglu FB, Ellis JS, Wan X, Dhakal M, Hoeman CM, Cascio JA et al Antigen‐free adjuvant assists late effector CD4 T cells to transit to memory in lymphopenic hosts. J Immunol 2013; 191:1126–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Mueller SN, Mackay LK. Tissue‐resident memory T cells: local specialists in immune defence. Nat Rev Immunol 2015; 16:79–89. [DOI] [PubMed] [Google Scholar]
- 88. Zens KD, Chen JK, Farber DL. Vaccine‐generated lung tissue‐resident memory T cells provide heterosubtypic protection to influenza infection. JCI Insight 2016; 1:e85832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wilk MM, Misiak A, McManus RM, Allen AC, Lynch MA, Mills KHG. Lung CD4 tissue‐resident memory T cells mediate adaptive immunity induced by previous infection of mice with Bordetella pertussis . J Immunol 2017; 199:233–43. [DOI] [PubMed] [Google Scholar]
- 90. Christensen D, Mortensen R, Rosenkrands I, Dietrich J, Andersen P. Vaccine‐induced Th17 cells are established as resident memory cells in the lung and promote local IgA responses. Mucosal Immunol 2017; 10:260–70. [DOI] [PubMed] [Google Scholar]
- 91. Romagnoli PA, Fu HH, Qiu Z, Khairallah C, Pham QM, Puddington L et al Differentiation of distinct long‐lived memory CD4 T cells in intestinal tissues after oral Listeria monocytogenes infection. Mucosal Immunol 2017; 10:520–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Roberts LM, Wehrly TD, Crane DD, Bosio CM. Expansion and retention of pulmonary CD4+ T cells after prime boost vaccination correlates with improved longevity and strength of immunity against tularemia. Vaccine 2017; 35:2575–81. [DOI] [PubMed] [Google Scholar]
- 93. Strutt TM, Dhume K, Finn CM, Hwang JH, Castonguay C, Swain SL et al IL‐15 supports the generation of protective lung‐resident memory CD4 T cells. Mucosal Immunol 2017; doi: 10.1038/mi.2017.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. van der Windt GJ, Pearce EL. Metabolic switching and fuel choice during T‐cell differentiation and memory development. Immunol Rev 2012; 249:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Kaech SM, Cui W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat Rev Immunol 2012; 12:749–61. [DOI] [PMC free article] [PubMed] [Google Scholar]