

Abstract
Ribociclib (KISQALI), a cyclin‐dependent kinase 4/6 inhibitor approved for the first‐line treatment of HR+/HER2– advanced breast cancer with an aromatase inhibitor, is administered with no restrictions on concomitant gastric pH‐elevating agents or food intake. The influence of proton pump inhibitors (PPIs) on ribociclib bioavailability was assessed using 1) biorelevant media solubility, 2) physiologically based pharmacokinetic (PBPK) modeling, 3) noncompartmental analysis (NCA) of clinical trial data, and 4) population PK (PopPK) analysis. This multipronged approach indicated no effect of gastric pH changes on ribociclib PK and served as a platform for supporting ribociclib labeling language, stating no impact of gastric pH‐altering agents on the absorption of ribociclib, without a dedicated drug–drug interaction trial. The bioequivalence of ribociclib exposure with or without a high‐fat meal was demonstrated in a clinical trial. Lack of restrictions on ribociclib dosing may facilitate better patient compliance and therefore clinical benefit.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Oral anticancer therapies vary widely in terms of permitted concomitant medication and food intake. Ribociclib, an orally bioavailable selective CDK4/6 inhibitor, is currently administered without regard to concomitant PPI use or food intake.
WHAT QUESTION DID THE STUDY ADDRESS?
☑ Does elevated gastric pH affect ribociclib absorption? Does food intake affect ribociclib bioavailability?
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ PBPK simulations and PopPK and NCA analyses revealed no effect of PPI use on ribociclib bioavailability. The PK parameters for ribociclib observed in healthy volunteers are not affected by food intake. Dedicated clinical pharmacology studies evaluating the effect of PPIs may not be needed with proper use and interpretation of advanced modeling and simulation techniques in combination with available clinical PK data.
HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE
☑ Ribociclib may be administered with no regard to food intake and concomitant PPI use. Ease of administration may improve patient adherence. Utilization of this multipronged platform may lead to waiving of dedicated clinical pharmacology trials.
Ribociclib (KISQALI) is an orally bioavailable, selective, small‐molecule inhibitor of cyclin‐dependent kinase 4/6 (CDK4/6).1 Preclinical studies of ribociclib demonstrated cell cycle arrest and tumor growth inhibition in both in vitro and in vivo nonclinical models for a variety of retinoblastoma protein‐positive (Rb+) solid tumor types.2, 3 Ribociclib received breakthrough therapy designation and was approved for use in combination with an aromatase inhibitor as an initial endocrine‐based therapy for the treatment of postmenopausal women with hormone receptor‐positive (HR+), human epidermal growth factor receptor‐2–negative (HER2–) advanced breast cancer,4, 5 after significant improvement in progression‐free survival was observed with ribociclib plus letrozole vs. letrozole alone in the phase III MONALEESA‐2 trial.6 Per the label, ribociclib can be taken with or without a meal, and with no restriction on concomitant use of gastric pH‐elevating agents such as proton pump inhibitors (PPIs), H2‐blocking agents, and other gastric acid‐reducing agents (ARAs).4, 5
Approximately 20–33% of all patients with cancer are treated with ARAs, most frequently with PPIs.7 PPIs elevate gastric pH by inhibiting hydrogen potassium (H,K)‐ATPase‐mediated gastric acid secretion and are administered to treat preexisting conditions and gastric damage resulting from anticancer therapies and cancer‐related stress.8, 9 The widespread use of long‐term ARA therapy increases the potential for interactions with oral anticancer therapies with pH‐dependent solubility.10, 11 Similarly, food intake may dramatically alter drug bioavailability through changes in drug dissolution kinetics, increased gastric acid secretion, and delayed gastric emptying.12
The recent increase in targeted oral anticancer therapies offers a convenient and flexible method of treatment for patients. The impact of concomitant drug use (e.g., PPIs) or meal timing on these compounds can be evaluated using in vitro and clinical data.13 However, early evaluation of targeted agents in the context of oncology drug development is often limited by the need to make rapid decisions based on a small number of patients. Therefore, a multipronged approach, using preclinical data and modeling and simulation approaches as well as clinical pharmacokinetics (PK) data from early‐phase trials, could be useful to evaluate drug exposure and tolerability.
Here, the results of in silico and clinical investigations into the influence of gastric pH changes and food intake on ribociclib PK are presented. In vitro and clinical data were used to build and qualify a physiologically based pharmacokinetic (PBPK) model of ribociclib and to assess the effect of changes in gastric pH on ribociclib PK. The effect of food intake on ribociclib PK was evaluated in a two‐period crossover clinical study in healthy volunteers. PK data from several clinical trials in patients with cancer were used to examine the effects of concomitant PPI use on ribociclib bioavailability using noncompartmental analysis (NCA) and population PK (PopPK) analysis. Together, these data supported the label, which indicates that ribociclib absorption is not affected by concomitant administration of food or gastric pH‐altering agents.
RESULTS
Full descriptions of clinical studies included in these analyses are in Supplemental Table S1.
Ribociclib drug substance solubility
Ribociclib succinate salt is a weak base (pKa 5.5, 8.6). In aqueous media, ribociclib solubility decreased with increasing pH (range, 2.0–7.5). Ribociclib exhibited high solubility in acidic media, acting as a buffering agent and driving the pH toward 5, with solubility up to 200 mg/mL. The maximum concentration for assessing solubility was set to 2.4 mg/mL, which corresponded to the maximum clinical dose (600 mg) in the stomach at a volume of 250 mL. Solubility in fed‐state simulated intestinal fluid (FeSSIF, pH 5.0) and fasted‐state simulated intestinal fluid (FaSSIF, pH 6.5) was similar. Solubility in biorelevant media (FeSSIF and FaSSIF) was found to be similar to that observed in buffered media at a lower pH (range, 2.0–4.5) (Table 1). These data served as the basis for further studies of ribociclib bioavailability (Figure 1 a).
Table 1.
Ribociclib solubility vs. pH
| pH | Buffer | Solubility, mg/mL | Bile salt concentration |
|---|---|---|---|
| 2.0 | HCl/KCl | >2.4a | — |
| 4.5 | Acetate | >2.4a | — |
| 6.8 | Phosphate | 0.8 | — |
| 7.5 | Phosphate | 0.3 | — |
| 6.5 | FaSSiF‐V1b | >2.4 | 3 mM |
| 5.0 | FeSSiF‐V1c | >2.2 | 15 mM |
Equates to 600 mg (clinical dose) of free base in 250 mL solution (volume represents typical quantity of water taken during drug administration).
FaSSiF = Fasted‐state simulated intestinal fluid (pH = 6.5, sodium taurocholate 3 mM, lecithin 0.75 mM, sodium chloride 105.9 mM, monobasic sodium phosphate 28.4 nM, sodium hydroxide 8.7 mM, pancreatin 10 mg/mL).
FeSSiF = Fed‐state simulated intestinal fluid (pH = 5.0, sodium taurocholate 15 nM, lecithin 3.75 nM, sodium chloride 203.2 nM, acetic acid 144.1 mM, sodium hydroxide 101 mM, pancreatin 40 mg/mL).
Figure 1.
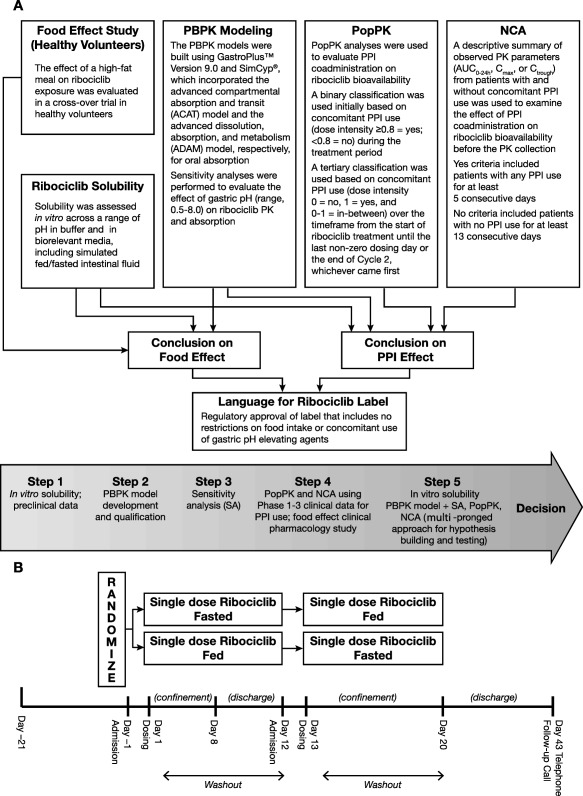
Study design and rationale. (a) Multipronged approach used to assess ribociclib bioavailability. (b) Design of the food effect study in healthy volunteers (Study A2301).
PBPK modeling and sensitivity analysis of ribociclib PK
The GastroPlus advanced compartmental absorption and transit (ACAT) model14 predicted high absorption for ribociclib given at 600 mg (fraction of a dose absorbed ∼90%), with the majority of absorption occurring in the small intestine. The established model adequately simulated the PK profile of ribociclib in fasted healthy subjects at 600 mg (Study A2111) using the Johnson dissolution model15 (Figure 2 a). This model was then qualified using PK profiles from patients with cancer (Study X2101) (Figure 2 b) and from healthy subjects (Study A2103; data not shown). Additionally, the simulations from the alternate dissolution model (Takano et al.16) matched well with the observed data and the Johnson dissolution model (data not shown), further indicating that pH does not impact the dissolution of ribociclib. A sensitivity analysis (Simcyp v13) using an advanced dissolution, absorption, and metabolism (ADAM) model demonstrated that varying stomach pH (range, 0.5–8.0) did not influence ribociclib absorption (Figure 3 a) or its PK profile (Figure 2 c). A sensitivity analysis examining the effects of varying gastric pH (range, 1.5–8.0) on ribociclib absorption predicted results similar to the GastroPlus ACAT model (Figure 3 b). These models support the lack of effect of changes in gastric pH on ribociclib absorption.
Figure 2.
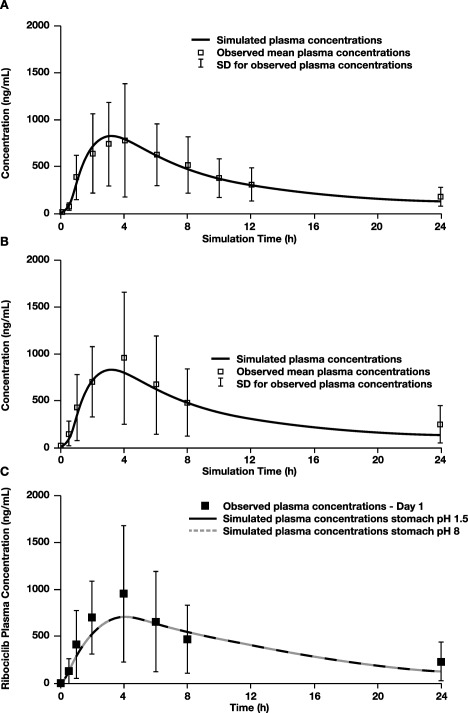
Simulated vs. observed mean human PK profiles following oral 600‐mg dose of ribociclib using PBPK models. Simulations were performed using (a) GastroPlus ACAT model with dissolution data from varying pH levels, using PK values from healthy volunteers in Study A2111; (b) GastroPlus ACAT model using observed PK values from patients in Study X2101; (c) Simcyp ADAM model at gastric pH 1.5 and 8.0, using data from patients with cancer in Study X2101. ACAT, advanced compartmental absorption and transit; ADAM, advanced dissolution, absorption, and metabolism; AUC, area under the drug concentration–time curve; Cmax, maximum concentration; F, oral bioavailability; Fa, fraction of dose absorbed; FDP, fraction of dose to the portal vein; N/A, not available; PBPK, physiologically based pharmacokinetics; PK, pharmacokinetics; SD, standard deviation; Tmax, time to reach Cmax.
Figure 3.

Sensitivity analysis of varying stomach pH on ribociclib absorption in PBPK models. Simulations were performed using qualified models with (a) Simcyp ADAM model and (b) GastroPlus ACAT model with dissolution data from varying pH levels.
Effect of food on ribociclib bioavailability
Twenty‐four healthy volunteers were randomized 1:1 to receive 600 mg ribociclib under fasted conditions or after a high‐fat, high‐calorie meal, followed by the alternative condition after a washout period (Figure 1 b). Demographics and baseline characteristics are summarized in Supplemental Table S2. Plasma concentrations for ribociclib were similar regardless of food intake (Figure 4). Pharmacokinetic parameters for ribociclib were comparable between conditions, with geometric mean maximum concentration (Cmax) 792 vs. 790 ng/mL, geometric mean area under the plasma concentration–time curve from time zero to infinity (AUCinf) 14,300 vs. 15,000 h·ng/mL, and median time to reach maximum concentration (Tmax) 3.0 vs. 4.0 h (fasted vs. fed states, respectively). Geometric mean ratios for PK parameters (Cmax and AUCinf) were ∼1.0, with 90% confidence interval (CI) ranges of 0.898–1.11 for Cmax and 1.01–1.12 for AUCinf (Table 2). No effect of food intake on ribociclib was observed for any PK parameters analyzed.
Figure 4.
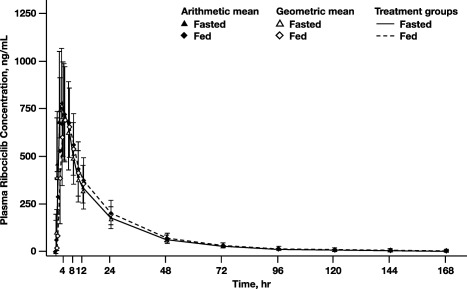
Ribociclib PK concentration under fasted and fed conditions (A2103).
Table 2.
Single‐dose ribociclib (600 mg) PK parameters in healthy volunteers (A2103)
|
Cmax, ng/mL |
Tmax, h |
AUCinf, h·ng/mL |
T1/2, h |
||
|---|---|---|---|---|---|
|
Fasted state (n = 23) |
n | 23 | 23 | 23 | 23 |
|
Geo‐mean (CV% Geo‐mean) |
792 (35.0) |
N/A | 14,300 (32.3) |
32.0 (18.6) |
|
| Adjusted Geo‐mean | 790 | 3 | 14,100 | N/A | |
|
Fed state (n = 24) |
n | 24 | 24 | 24 | 24 |
|
Geo‐mean (CV% Geo‐mean) |
790 (33.2) |
N/A |
15,000 (29.2) |
33.6 (24.6) |
|
| Adjusted Geo‐mean | 790 | 4 | 15,000 | N/A | |
|
Fed:fasted GMRa (90% CI) |
1.00 (0.898, 1.11) |
1 (–2, 8) |
1.06 (1.01, 1.12) |
||
AUCinf, area under the plasma concentration‐time curve from time zero to infinity; Cmax, maximum concentration; CV, coefficient of variation; Geo‐mean, geometric mean; GMR, geometric mean ratio; T1/2, elimination half‐life; Tmax, time to reach maximum concentration.
Adjusted geometric mean is calculated based on a mixed model adjusted for random effects and other covariates.
GMR is the ratio of the adjusted geometric mean for Cmax and AUCinf and the difference of the medians for Tmax.
CV% geo‐mean = sqrt (exp (variance for log transformed data) – 1) · 100
The values for the treatment comparison include the geo‐mean ratio with the lower and upper limits of treatment comparison (90% CI) in parentheses for each parameter.
PopPK analysis
The final model adequately described PK data. Prediction‐ and residual‐based diagnostic plots are shown in Supplemental Figures S1 and S2, respectively. Retaining body weight (BW) in the final model mitigated the trend between BW and PK parameters (Supplemental Figure S3). Visual predictive check (VPC) simulations confirmed that the model could reproduce the data on which it was developed (Supplemental Figure S4). Parameter estimates of the final model are listed in Supplemental Table S3. Satisfactory model performance supported use of the PopPK model for evaluation of covariate effects, including PPI use.
Among the 208 patients with cancer included in the analysis, 52 patients had concomitant PPI use by the binary (Yes/No) classification system. PPI use was determined to be a statistically insignificant variable on ribociclib PK, with relative bioavailability with PPI use estimated to be 0.95 (95% CI, 0.82–1.09; %RSE = 7.04) relative to the reference state (no concomitant PPI use). PPI use was therefore removed from the final PopPK model. With a tertiary classification system (Yes/No/In‐between), 145 patients were categorized as having no PPI use (PPI dose intensity = 0, reference state), 41 patients were categorized as having concomitant PPI use (PPI dose intensity = 1), and 15 patients were categorized as in between (PPI dose intensity greater than 0 and less than 1). The effect of concomitant PPI use, as defined by the tertiary classification system, was determined to be 0.96 (95% CI, 0.80–1.11; %RSE = 8.3). Given the proximity of the covariate effect point estimate to 1 and that the CI included the null value of 1 for both binary and tertiary classification, the analysis indicated that concomitant PPI use did not affect ribociclib exposure.
Noncompartmental analysis of clinical data
The same studies used in the PopPK analysis were used for the NCA approach. Of 164 total patients with evaluable steady‐state PK parameters, 41 were categorized as having concomitant PPI use. Across studies, the geometric means for steady‐state PK parameters (AUC from time zero to 24 h (AUC0‐24h), Cmax, and trough concentration (Ctrough)) were similar regardless of concomitant PPI use (Table 3), demonstrating that concomitant PPI use did not have a clinically significant effect on ribociclib PK.
Table 3.
Steady‐state ribociclib (600 mg) PK parameters by PPI use
| Study no. | PK parameter | n | PPI use |
Geometric mean (geometric coefficient of variation, %) |
|---|---|---|---|---|
| X2107 | AUC0‐24h, h·ng/mL | 8 | Yes | 24,700 (30.6) |
| 10 | No | 21,100 (57.2) | ||
| Cmax, ng/mL | 10 | Yes | 1,780 (34.6) | |
| 13 | No | 1,620 (53.2) | ||
| X2101 | AUC0‐24h, h·ng/mL | 12 | Yes | 25,900 (79.1) |
| 46 | No | 23,700 (61.3) | ||
| Cmax, ng/mL | 13 | Yes | 2,050 (74.7) | |
| 48 | No | 1,870 (60.3) | ||
| X1101 | AUC0‐24h, h·ng/mL | 2 | Yes | 42,600 (28.7) |
| 6 | No | 55,100 (68.6) | ||
| Cmax, ng/mL | 2 | Yes | 2,700 (53.0) | |
| 6 | No | 3,500 (65.8) | ||
| A2301 | Ctrough, ng/mL | 8 | Yes | 587 (55.8) |
| 36 | No | 711 (72.9) |
AUC0‐24h, area under the concentration‐time curve from time zero to 24 h; Cmax, maximum concentration; Ctrough, trough concentration; PK, pharmacokinetics; PPI, proton pump inhibitor.
DISCUSSION
Ribociclib is a weak base with a pH‐dependent solubility profile over the range found in the human gastrointestinal tract. Despite the fact that ribociclib solubility decreased with increasing pH, solubility in biorelevant media was maintained, and no difference was observed between fed‐state vs. fasted‐state biorelevant media. As weakly basic compounds are known to have pH‐dependent solubility,11 and simple buffered media may not accurately represent gut solubility, it is essential to determine compound solubility in biorelevant media (FaSSIF and FeSSIF) early in the discovery process. These data suggest that ribociclib absorption is unlikely to be affected by changes in gastric pH that occur following food intake due to stimulation of gastric acid secretion or following concomitant use of ARAs such as PPIs.17
PBPK modeling is a useful tool that combines drug‐specific information, such as compound physiochemical properties and human physiologic information, to predict the influence of factors such as gastric pH and food intake on drug absorption, distribution, metabolism, and excretion.18 While there is some debate on the accuracy of predictions from available absorption models,19 increasing evidence has demonstrated their applicability to clinical scenarios.20, 21, 22 Indeed, a number of recent works have confirmed their validity,23, 24, 25 and regulatory authorities are more frequently accepting PBPK modeling to assess potential drug–drug interactions (DDIs) and drug exposure in special patient populations in clinical drug development, with draft guidance available from the US Food and Drug Administration (FDA) and European Medicines Agency (EMA).26, 27, 28, 29, 30, 31 However, the potential for these simulations to impact labeling recommendations for gastric pH‐elevating agents remains largely untapped.32
PBPK simulations using in vitro data and combined with data from clinical studies and PopPK approaches can provide a platform to assess clinically relevant drug interaction scenarios in the target population. In a recent review of approvals of oral oncology targeted agents, 15 of 44 exhibited pH‐dependent solubility,33 underscoring the need to assess for changes in their bioavailability with changes in pH. PBPK modeling has been used to investigate food effect and pH‐dependent DDIs for several agents, including alectinib, a basic anaplastic lymphoma kinase (ALK) inhibitor.34, 35 Additionally, the use of biorelevant media was found to accurately predict food effects on the absorption of celecoxib, a poorly soluble nonsteroidal antiinflammatory drug.36 The lack of effect of gastric pH‐altering agents on panobinostat absorption was evaluated solely on PBPK modeling and directly impacted the labeling language.37 The long‐term impact of PPIs on the PK and efficacy of ceritinib was evaluated in fulfillment of a postmarketing requirement based on available clinical data and PopPK modeling approaches.38 Our approach used the in vitro solubility and PBPK modeling results as hypothesis‐generating data that allowed phase II/III clinical trials to proceed without restriction on PPI use, permitting further NCA and PopPK analyses. This multipronged approach demonstrates the utility of PBPK modeling in combination with clinical data and PopPK analysis in lieu of dedicated phase I trials in healthy volunteers, which can help circumvent additional postmarketing evaluation through registration trials, wherein efficacy and safety could be established with food or concomitant ARA use.
The in vitro solubility data also provided a foundation for the hypothesis that food may not affect ribociclib absorption, further supported by PBPK models for ribociclib, which were constructed and validated with clinical data from healthy volunteers and patients with cancer. Sensitivity analyses based on PBPK models of ribociclib absorption built into both the GastroPlus and Simcyp software packages showed that exposure was independent of gastric pH over the physiologic range (pH range, 1.0–8.0),39 consistent with the lack of impact of pH on the in vitro solubility of ribociclib in the biorelevant media. This hypothesis was then tested and confirmed by the food effect study in healthy volunteers, which showed that ribociclib PK parameters were similar in the fed or fasted state. This evaluation early in development allowed administration of ribociclib in clinical trials without regard to meals.
Examination of clinical data from patients with cancer receiving ribociclib and concomitant PPIs in several studies revealed no difference in observed PK parameters. Additionally, neither food nor PPI coadministration was restricted in MONALEESA‐2 (Study A2301), wherein ribociclib in combination with letrozole was demonstrated to be clinically effective and tolerable as a first‐line therapy for patients with HR+, HER2– advanced breast cancer.6 Concomitant PPI use was identified as a statistically insignificant and clinically unimportant covariate in ribociclib bioavailability using a PopPK approach. The use of a binary system (Yes/No) was complemented with a more conservative tertiary system (Yes = 1 / No = 0 / In between), and both consistently showed that PPIs did not have a clinical impact on ribociclib PK. Furthermore, the long duration of suppression of gastric secretion by PPIs, in addition to dose intensity of ≥0.8, represents an adequate assessment of drug interactions due to PPI use, regardless of the timing of PPI administration relative to ribociclib dosing. Taken together, these results suggest that gastric pH‐elevating agents and food intake do not affect the rate or extent of ribociclib absorption and, as such, ribociclib may be administered without regard to meals or concomitant use of ARAs (Figure 1 a).
The clinical data evaluated here via PopPK and NCA analyses indicated no effect of PPIs on ribociclib exposure, and similarly no effect is expected from other ARAs (e.g., H2‐blocking agents and antacids). Although both PPIs and H2‐blocking agents have demonstrated dose‐related suppression of gastric acid secretion, they differ in onset and duration of effect. H2‐blocking agents have rapid onset of action (<1 h) and short duration of activity (<12 h), while PPIs have a delayed onset of action but a prolonged duration of activity (3–5 days) due to irreversible inhibition of proton pumps as a result of covalent bonding with the drug.40 The FDA has commonly requested DDI study sponsors to evaluate the effect of gastric pH‐elevating agents, but recent requests have included a gated approach wherein the effect of changes in gastric pH on drug exposure are first examined using PPIs.33, 41 As PPIs are used as a first assessment in this gated approach, they may be considered a “worst‐case scenario” in the evaluation of gastric pH changes on drug exposure, as a result of their longer duration of activity compared with H2‐blocking agents and antacids.33, 41 If the coadministration of PPIs results in a large effect on drug exposure, the effect of H2‐blocking agents and antacids should then be evaluated. As PPI use was not found to have an effect on ribociclib exposure, no effect is expected from H2‐blocking agents or antacids.
Evaluation of food effect and concomitant PPI use on the PK of a compound early in the drug development process can have a major impact on the drug approval process. This evaluation process can include measuring solubility and dissolution in a biorelevant medium in the discovery space, use of PBPK modeling with in vitro and limited PK data to simulate compound absorption and bioavailability in the translational space, and PopPK modeling and/or leveraging clinical data from the early phase I/II clinical trial space. Upon establishing the impact of food and/or PPI use on drug exposure early during clinical development, late‐stage clinical trials could include appropriate recommendations with respect to the expected impact of food or PPI use on PK, exposure, safety, and efficacy. Also, the lack of PPI and food effect on drug exposure may facilitate greater patient compliance and clinical benefit by reducing restrictions on meal timing and concomitant drug use, which may be especially important in special populations such as elderly patients with achlorhydria. Later‐stage clinical trials can then include these processes with greater confidence that such factors will not affect drug safety or efficacy.
The multipronged approach used in this study serves as an effective platform to assess the effect of PPI use on ribociclib bioavailability without a dedicated PPI trial, and was used to support the labeling language. Reducing the need for dedicated clinical trials in combination with PPIs and other commonly used medications, particularly for oncology drugs with established efficacy in randomized controlled trials, reinforces the effectiveness of a combined approach using advanced modeling and simulation techniques in combination with clinical data. This comes at a time when regulatory agencies are increasing their acceptance of this approach.
METHODS
This work evaluated the effect of food and ARAs on ribociclib PK. The evaluation of food effect was based on 1) in vitro solubility and 2) single‐dose food effect study. The evaluation PPI effect was based on 1) in vitro solubility, 2) PBPK modeling, 3) NCA of patient PK data, and 4) PopPK analyses. A schematic representation of the analyses is shown in Figure 1 a.
In vitro solubility
Ribociclib succinate salt (∼763 mg) in crystalline form (corresponding to ∼600 mg equivalent free form and equal to the 600‐mg clinical dose) was added to 250 mL of aqueous buffered solution (50 mM; pH range, 2.0–7.5) at 37 ± 1°C. Biorelevant media (FaSSIF v1 and FeSSIF v1) were prepared by diluting ready‐to‐use powder sourced out from http://Biorelevant.com. The estimated volume (250 mL) was derived from bioequivalent study protocols in which a drug product was administered to a fasting volunteer with a glass of water. The suspensions were stirred with a magnetic bar, liquid fractions were separated after 24 h, and concentrations were determined by high‐performance liquid chromatography. The pH and chemical purity of the solutions were verified, and no significant chemical degradation or significant pH shifts were observed compared with the initial solution. Each experiment was repeated in triplicate and mean concentration values were reported.
PBPK model development and simulation
PBPK modeling was used to evaluate the effect of changes in gastric pH on ribociclib absorption. With physicochemical properties (e.g., reference solubility, pKa, mean precipitation time) and dissolution data for ribociclib, a two‐compartment model coupled to the ACAT model was built using GastroPlus v. 9.0 with default fasted gut physiology. The ACAT model (Supplemental Table S4) was fitted using PK data from healthy volunteers in Study A2111 and was qualified using PK data from patients in Study X2101 and from healthy volunteers in Study A2103 receiving a single oral dose (600 mg) of ribociclib. The initial ACAT model included the Johnson dissolution model (the default in GastroPlus),15 and predicted rapid dissolution of ribociclib with no precipitation during transit through the gut. In addition, an alternate dissolution model (Takano et al.16) in GastroPlus was used to simulate PK profiles for ribociclib at various gastric pH levels (1.0, 2.0, 4.5, and 6.8) with a default stomach transit time (0.1 h).
A previously established qualified model of ribociclib kinetics using Simcyp was modified to a mechanistic ADAM model42, 43 to include ribociclib solubility data at varying pH. The model was validated using PK data from patients receiving a single oral dose (600 mg) of ribociclib. The gastric pH in the model was modified to allow evaluation of values ranging from pH 1.5 to 8.0 during PK simulations. Sensitivity analyses were conducted in both GastroPlus and Simcyp models to evaluate the impact of changing pH from 1.5 to 8.0 on the absorption of ribociclib.
The impact of changes in gastric pH on the absorption of ribociclib observed from the PBPK models was used to generate a hypothesis for the food effect clinical study.
Food effect study
Two phase I studies, A2103 and A2111, evaluated the effect of food on ribociclib PK using the film‐coated tablet and drug‐in‐capsule formulations, respectively. The design and results of A2103 are discussed in this report because of its relevance to the marketed formulation (film‐coated tablets). Study A2103 was a phase I, open‐label, randomized, 2‐period, 2‐sequence crossover study that examined the impact of a high‐fat, high‐calorie meal on ribociclib absorption in healthy volunteers. Men and postmenopausal women between 18 and 55 years of age, with laboratory values in the normal range and normal cardiac function, were eligible for enrollment. Subjects taking any concomitant medication, using caffeine or tobacco products, or those with abnormal electrocardiogram (ECG) values were excluded from the study. Subjects were randomized 1:1 to receive a single dose of ribociclib (600 mg) in fed or fasted conditions, and then received a second dose of ribociclib under the alternate condition after a washout period of 12 days (Figure 1 b). Blood samples were collected for determination of plasma ribociclib concentration at predose and at 0.5, 1, 2, 3, 4, 6, 8, 10, 12, and 24 h postdose, and then daily through the end of the treatment period. Subjects were followed for 30 days after the last dose. Safety was assessed by recording of adverse events. This study was conducted according to the ethical principles of the Declaration of Helsinki. All subjects provided written informed consent before enrollment.
PopPK analysis
The effect of PPIs was evaluated with a PopPK model developed using PK data pooled from patients with cancer from three clinical trials (X1101, X2101, and X2107; N = 208). Details of the PopPK analyses are available elsewhere.44, 45 Briefly, the model development followed the typical stepwise procedure from base model to full covariate model and to final model. The model was built in NONMEM v. 7.3 (ICON Development Solutions, Ellicott City, MD) using the first‐order conditional estimation method. The base model featured a two‐compartment model structure with delayed zero‐order oral absorption, linear intercompartmental distribution, and linear clearance from the central compartment with dose as a structural covariate to account for nonlinearity of ribociclib PK. A list of covariates for evaluation was predefined based on general physiologic and pharmacologic understanding. Simultaneous incorporation of the covariates into the base model yielded a full covariate model. Evaluation of the covariate effects was based on prespecified criteria that took both statistical significance and clinical importance into account. Covariates that were not statistically significant (i.e., 95% CI of a parameter estimated to include the null value) and/or not clinically important (i.e., 95% CI of a covariate effect being entirely within ±20% from the reference value) were removed simultaneously to create the final model. In the final model, dose and BW were retained. However, BW‐based dose adjustment was not warranted because the variation in steady‐state exposure of ribociclib (600 mg q.d.) simulated using the final model, which included BW, was judged to be not clinically relevant. Model stability was assessed throughout the development process by testing three separate sets of initial estimates for fixed‐effect parameters. Model performance was assessed according to the success of convergence, plausibility of parameter estimates, and diagnostic plots. The final model was further evaluated using VPC and validated externally against PK data from study A2301 (MONALEESA‐2).
The effect of PPIs on ribociclib PK was incorporated into the full model as a covariate on the relative bioavailability, wherein PPI dose intensity was calculated as the ratio of number of days the patient had taken the medication in a time window over the total number of days in that time window. Concomitant PPI use was categorized using two classification systems. A binary classification based on PPI dose intensity (≥0.8 = yes; <0.8 = no) during the treatment period (from treatment initiation to either the last nonzero dosing date or the last PK assessment date with concentration value above lower limit of quantification, whichever came first) was included in the initial analysis. A tertiary classification was also used based on PPI dose intensity grouped into three categories: no PPIs (dose intensity = 0), with PPIs (dose intensity = 1), and in between (dose intensity greater than 0 and less than 1) over the timeframe from the start of ribociclib treatment until the last nonzero dosing day or the end of Cycle 2, whichever came first. The numbers of patients in the no and yes categories were sufficient for evaluation of the covariate effect of PPI.
Noncompartmental analysis of PK data in patients
The effect of PPIs was evaluated using observed PK data from patients with cancer (X2101, X2107, X1101, and A2301). Pharmacokinetic parameters (Cmax, AUC0‐24h, Ctrough) were derived from plasma concentration–time profiles using NCA (Phoenix; Pharsight, Mountain View, CA) and descriptively summarized. Concomitant PPI use was defined as PPI use for at least 5 consecutive days prior to the PK assessment, while no PPI use was defined as no PPI use for at least 13 consecutive days prior to the PK assessment.
AUTHOR CONTRIBUTIONS
T.S.S., S.D., Y.L., M.L., S.Y., A.G., M.M.‐Z., K.U., F.H., M.M., C.G., and M.E. wrote the article; T.S.S., S.D., and M.E. designed the research; T.S.S., S.D., Y.L., M.L., S.Y., A.G., M.M.‐Z., K.U., F.H., and M.E. performed the research; T.S.S., Y.L., M.L., S.Y., A.G., and M.M.‐Z. analyzed the data; K.U. and F.H. contributed new reagents/analytical tools.
CONFLICT OF INTEREST
T.S.S., Y.L., S.Y., M.M., C.G., and M.E. are employees of Novartis Pharmaceuticals Corporation; S.D. was an employee of Novartis Pharmaceuticals Corporation at the time this study was conducted and is currently a consultant for Novartis Pharmaceuticals Corporation; and M.L., A.G., M.M.‐Z., K.U., and F.H. are employees of Novartis Pharma AG.
Supporting information
Table S1.—Clinical Studies Included in Evaluations of Ribociclib Pharmacokinetic Parameter.
Table S2.—Patient Demographics and Baseline Characteristics (Study A2103).
Table S3.—Final PopPK Model Parameter Estimates. ALAG1, absorption lag; D1, time for absorption; CL, apparent clearance; Q, intercompartmental clearance; V1, volume of central compartment; V2, volume of peripheral compartment. All parameters in the form of covariate (Dose or BW)∼PK parameter (CL, Q, or V2) are parameters representing covariate effects; all ω2 represent interindividual variations, and the σ2 represent the residual error, both expressed as variance; RSE: relative standard error, calculated using SE/Estimate×100%; shrinkage reported by NONMEM v7.3.
Table S4.—Input Parameters for Ribociclib ACAT Model in GastroPlus™ Software.
Figure S1.—Observations (DV) vs. population (PRED) and individual prediction (IPRED) from final PopPK model. SS: steady state; SS 0–24h represents exposure time points within the 24 h interval after previous dose at steady state; Non‐SS represents all other data points at non‐steady state included in the analysis dataset.
Figure S2.—Residual‐based diagnostic plots for final PopPK model. CWRES, conditional weighted residuals.
Figure S3.—Retention of covariates in final PopPK model. (A) mitigated parameter‐covariate relationships identified from base model (B).
Figure S4.—Body weight stratified visual predictive check (VPC) on Day 21 after 600‐mg daily dosing using final PopPK model.
ACKNOWLEDGMENTS
We thank Tycho Heimbach for reviewing this article and providing expertise. Financial support for medical editorial assistance was provided by Novartis Pharmaceuticals. We thank Elise Blankenship, PhD, ProEd Communications, Inc., for medical editorial assistance with this article. Previous Publication: Data were included in an abstract submitted to ESMO 2017. Funding provided by Novartis.
References
- 1. Kim, S. et al LEE011: an orally bioavailable, selective small molecule inhibitor of CDK4/6—reactivating Rb in cancer. Presented at: AACR‐NCI‐EORTC International Conference on Molecular Targets and Cancer Therapeutics; October 19–23, 2013; Boston, MA. Abstract B264.
- 2. Rader, J. et al Dual CDK4/CDK6 inhibition induces cell‐cycle arrest and senescence in neuroblastoma. Clin. Cancer Res. 19, 6173–6182 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang, Y.X. et al Antiproliferative effects of CDK4/6 inhibition in CDK4‐amplified human liposarcoma in vitro and in vivo. Mol. Cancer Ther. 13, 2184–2193 (2014). [DOI] [PubMed] [Google Scholar]
- 4. Kisqali (ribociclib) [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2017. [Google Scholar]
- 5. Kisqali 200 mg film‐coated tablets [summary of product characteristics]. Camberley, Surrey, UK: Novartis Pharmaceuticals UK; 2017. [Google Scholar]
- 6. Hortobagyi, G.N. et al Ribociclib as first‐line therapy for HR‐positive, advanced breast cancer. N. Engl. J. Med. 375, 1738–1748 (2016). [DOI] [PubMed] [Google Scholar]
- 7. Smelick, G.S. et al Prevalence of acid‐reducing agents (ARA) in cancer populations and ARA drug‐drug interaction potential for molecular targeted agents in clinical development. Mol. Pharm. 10, 4055–4062 (2013). [DOI] [PubMed] [Google Scholar]
- 8. Numico, G. , Fusco, V. , Franco, P. & Roila, F. Proton pump inhibitors in cancer patients: how useful they are? A review of the most common indications for their use. Crit. Rev. Oncol. Hematol. 111, 144–151 (2017). [DOI] [PubMed] [Google Scholar]
- 9. Shin, J.M. & Sachs, G. Pharmacology of proton pump inhibitors. Curr. Gastroenterol. Rep. 10, 528–534 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wedemeyer, R.S. & Blume, H. Pharmacokinetic drug interaction profiles of proton pump inhibitors: an update. Drug Saf. 37, 201–211 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. van Leeuwen, R.W.F. et al Tyrosine kinase inhibitors and proton pump inhibitors: an evaluation of treatment options. Clin. Pharmacokinet. 56, 683–688 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Charman, W.N. , Porter, C.J. , Mithani, S. & Dressman, J.B. Physiochemical and physiological mechanisms for the effects of food on drug absorption: the role of lipids and pH. J. Pharm. Sci. 86, 269–282 (1997). [DOI] [PubMed] [Google Scholar]
- 13. Singh, B.N. & Malhotra, B.K. Effects of food on the clinical pharmacokinetics of anticancer agents: underlying mechanisms and implications for oral chemotherapy. Clin. Pharmacokinet. 43, 1127–1156 (2004). [DOI] [PubMed] [Google Scholar]
- 14. Agoram, B. , Woltosz, W.S. & Bolger, M.B. Predicting the impact of physiological and biochemical processes on oral drug bioavailability. Adv. Drug Deliv. Rev. 50 Suppl 1, S41–67 (2001). [DOI] [PubMed] [Google Scholar]
- 15. Hintz, R.J. & Johnson, K.C. The effect of particle size distribution on dissolution rate and oral absorption. Int. J. Pharm. 51, 9–17 (1989). [Google Scholar]
- 16. Takano, R. et al Oral absorption of poorly water‐soluble drugs: computer simulation of fraction absorbed in humans from a miniscale dissolution test. Pharm. Res. 23, 1144–1156 (2006). [DOI] [PubMed] [Google Scholar]
- 17. Brooks, F.P. Effect of diet on gastric secretion. Am. J. Clin. Nutr. 42, 1006–1019 (1985). [DOI] [PubMed] [Google Scholar]
- 18. Sager, J.E. , Yu, J. , Ragueneau‐Majlessi, I. & Isoherranen, N. Physiologically based pharmacokinetic (PBPK) modeling and simulation approaches: a systematic review of published models, applications, and model verification. Drug Metab. Dispos. 43, 1823–1837 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Darwich, A.S. et al IMI — Oral biopharmaceutics tools project — Evaluation of bottom‐up PBPK prediction success part 3: Identifying gaps in system parameters by analysing In Silico performance across different compound classes. Eur. J. Pharm. Sci. 96, 626–642 (2017). [DOI] [PubMed] [Google Scholar]
- 20. Suarez‐Sharp, S. , Li, M. , Duan, J. , Shah, H. & Seo, P. Regulatory experience with in vivo in vitro correlations (IVIVC) in new drug applications. AAPS J. 18, 1379–1390 (2016). [DOI] [PubMed] [Google Scholar]
- 21. Kostewicz, E.S. et al PBPK models for the prediction of in vivo performance of oral dosage forms. Eur. J. Pharm. Sci. 57, 300–321 (2014). [DOI] [PubMed] [Google Scholar]
- 22. Lin, L. & Wong, H. Predicting oral drug absorption: mini review on physiologically‐based pharmacokinetic models. Pharmaceutics. 9, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Poulin, P. & Theil, F.P. Prediction of pharmacokinetics prior to in vivo studies. 1. Mechanism‐based prediction of volume of distribution. J. Pharm. Sci. 91, 129–156 (2002). [DOI] [PubMed] [Google Scholar]
- 24. Margolskee, A. et al IMI — Oral biopharmaceutics tools project — Evaluation of bottom‐up PBPK prediction success part 2: An introduction to the simulation exercise and overview of results. Eur. J. Pharm. Sci. 96, 610–625 (2017). [DOI] [PubMed] [Google Scholar]
- 25. Sjogren, E. , Thorn, H. & Tannergren, C. In silico modeling of gastrointestinal drug absorption: predictive performance of three physiologically based absorption models. Mol. Pharm. 13, 1763–1778 (2016). [DOI] [PubMed] [Google Scholar]
- 26. Wagner, C. et al Application of physiologically based pharmacokinetic (PBPK) modeling to support dose selection: report of an FDA public workshop on PBPK. CPT Pharmacometrics Syst. Pharmacol. 4, 226–230 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sinha, V. , Zhao, P. , Huang, S.M. & Zineh, I. Physiologically based pharmacokinetic modeling: from regulatory science to regulatory policy. Clin. Pharmacol. Ther. 95, 478–480 (2014). [DOI] [PubMed] [Google Scholar]
- 28. Rowland, M. , Lesko, L.J. & Rostami‐Hodjegan, A. Physiologically based pharmacokinetics is impacting drug development and regulatory decision making. CPT Pharmacometrics Syst. Pharmacol. 4, 313–315 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. US Food and Drug Administration Center for Drug Evaluation and Research . Physiologically Based Pharmacokinetic Analyses—Format and Content. Guidance for Industry. <https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM531207.pdf> Published December 2016. Accessed June 27, 2017.
- 30. European Medicines Agency Committee for Medicinal Products for Human Use . Guideline on the qualification and reporting of physiologically based pharmacokinetic (PBPK) modelling and simulation. EMA/CHMP/458101/2016. <http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2016/07/WC500211315.pdf> Published July 21, 2016. Accessed June 27, 2017.
- 31. Bloomer, J.C. et al Identification and characterisation of a salt form of Danirixin with reduced pharmacokinetic variability in patient populations. Eur. J. Pharm. Biopharm. 117, 224–231 (2017). [DOI] [PubMed] [Google Scholar]
- 32. Yoshida, K. , Budha, N. & Jin, J.Y. Impact of physiologically based pharmacokinetic models on regulatory reviews and product labels: frequent utilization in the field of oncology. Clin. Pharmacol. Ther. 101, 597–602 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Howard, D.R. Practical considerations for clinical pharmacology in drug development: a survey of 44 FDA oncology approvals In: Pharmacokinetics in Drug Development, Vol. 4 (ed. Bonate P.L.) 237–301 (Springer International Publishing, Switzerland, 2016). [Google Scholar]
- 34. Parrott, N.J. , Yu, L.J. , Takano, R. , Nakamura, M. & Morcos, P.N. Physiologically based absorption modeling to explore the impact of food and gastric pH changes on the pharmacokinetics of alectinib. AAPS J. 18, 1464–1474 (2016). [DOI] [PubMed] [Google Scholar]
- 35. Zhu, A.Z. , Ho, M.D. , Gemski, C.K. , Chuang, B.C. , Liao, M. & Xia, C.Q. Utilizing in vitro dissolution‐permeation chamber for the quantitative prediction of pH‐dependent drug‐drug interactions with acid‐reducing agents: a comparison with physiologically based pharmacokinetic modeling. AAPS J. 18, 1512–1523 (2016). [DOI] [PubMed] [Google Scholar]
- 36. Shono, Y. et al Prediction of food effects on the absorption of celecoxib based on biorelevant dissolution testing coupled with physiologically based pharmacokinetic modeling. Eur. J. Pharm. Biopharm. 73, 107–114 (2009). [DOI] [PubMed] [Google Scholar]
- 37. Farydak (pabinostat) [package insert]. East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2016. [Google Scholar]
- 38. Lau, Y.Y. et al Assessment of drug‐drug interaction potential between ceritinib and proton pump inhibitors in healthy subjects and in patients with ALK‐positive non‐small cell lung cancer. Cancer Chemother. Pharmacol. 79, 1119–1128 (2017). [DOI] [PubMed] [Google Scholar]
- 39. Hens, B. et al Exploring gastrointestinal variables affecting drug and formulation behavior: methodologies, challenges and opportunities. Int. J. Pharm. 519, 79–97 (2017). [DOI] [PubMed] [Google Scholar]
- 40. US Food and Drug Administration Center for Drug Evaluation and Research . Pharmacodynamic aspects of H2‐blockers versus proton pump inhibitors. <https://www.fda.gov/ohrms/dockets/ac/00/backgrd/3650b1b_tab_03.pdf>. Accessed September 12, 2017.
- 41. Zhang, L. , Wu, F. , Lee, S.C. , Zhao, H. & Zhang, L. pH‐dependent drug‐drug interactions for weak base drugs: potential implications for new drug development. Clin. Pharmacol. Ther. 96, 266–277 (2014). [DOI] [PubMed] [Google Scholar]
- 42. Darwich, A.S. , Neuhoff, S. , Jamei, M. & Rostami‐Hodjegan, A. Interplay of metabolism and transport in determining oral drug absorption and gut wall metabolism: a simulation assessment using the “Advanced Dissolution, Absorption, Metabolism (ADAM)” model. Curr. Drug Metab. 11, 716–729 (2010). [DOI] [PubMed] [Google Scholar]
- 43. Jamei, M. et al Population‐based mechanistic prediction of oral drug absorption. AAPS J. 11, 225–237 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. US Food and Drug Administration, Center for Drug Evaluation and Research . NDA/BLA Multi‐disciplinary Review and Evaluation 209,092: Kisqali. August 29, 2016.
- 45. Lu, Y. , Huang, P.‐H. , Zieba, R. , Samant, T.S. , Dhuria, S. & Elmeliegy, M. Ribociclib (KISQALI®) population pharmacokinetics (PopPK) and exposure‐response (E‐R) analysis of neutropenia. Presented at: American Conference on Pharmacometrics (ACoP); October 15–18, 2017; Fort Lauderdale, FL.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1.—Clinical Studies Included in Evaluations of Ribociclib Pharmacokinetic Parameter.
Table S2.—Patient Demographics and Baseline Characteristics (Study A2103).
Table S3.—Final PopPK Model Parameter Estimates. ALAG1, absorption lag; D1, time for absorption; CL, apparent clearance; Q, intercompartmental clearance; V1, volume of central compartment; V2, volume of peripheral compartment. All parameters in the form of covariate (Dose or BW)∼PK parameter (CL, Q, or V2) are parameters representing covariate effects; all ω2 represent interindividual variations, and the σ2 represent the residual error, both expressed as variance; RSE: relative standard error, calculated using SE/Estimate×100%; shrinkage reported by NONMEM v7.3.
Table S4.—Input Parameters for Ribociclib ACAT Model in GastroPlus™ Software.
Figure S1.—Observations (DV) vs. population (PRED) and individual prediction (IPRED) from final PopPK model. SS: steady state; SS 0–24h represents exposure time points within the 24 h interval after previous dose at steady state; Non‐SS represents all other data points at non‐steady state included in the analysis dataset.
Figure S2.—Residual‐based diagnostic plots for final PopPK model. CWRES, conditional weighted residuals.
Figure S3.—Retention of covariates in final PopPK model. (A) mitigated parameter‐covariate relationships identified from base model (B).
Figure S4.—Body weight stratified visual predictive check (VPC) on Day 21 after 600‐mg daily dosing using final PopPK model.