

Abstract
This open‐label, single‐sequence study in healthy subjects investigated the effects of steady‐state carbamazepine on the pharmacokinetic (PK) profile of a single 2‐mg dose of fingolimod. In period 1, a single oral dose of fingolimod 2 mg (day 1) was followed by PK and safety assessments up to 36 days. In period 2, carbamazepine was administered in flexible, up‐titrated doses (600 mg twice daily maximum) for 49 days. Fingolimod was administered on day 35, followed by a study completion evaluation (day 71). The PK analysis included 23 of 26 of the enrolled subjects (88.5%). Coadministration of fingolimod at steady‐state carbamazepine concentrations resulted in increased fingolimod CL/F by 67% through the induction of CYP3A4, a cytochrome with negligible involvement in fingolimod clearance in an uninduced state. Fingolimod Cmax was reduced by 18% and AUCinf by 40%, as was T1/2 (106 vs 163 hours). A similar trend was observed for fingolimod‐P. Models linking fingolimod‐P blood concentrations to lymphocyte count or annual relapse rate suggest that such a decrease would have a low impact on the treatment effect. However, in the absence of efficacy data of fingolimod at doses lower than the therapeutic dose, their coadministration should be used with caution.
Keywords: fingolimod, carbamazepine, drug‐drug interaction, healthy subjects, pharmacokinetics
Fingolimod (FTY720; Gilenya, Novartis Pharma AG) 0.5 mg once daily is a first‐in‐class sphingosine 1‐phosphate receptor modulator approved in several countries (first‐line therapy: United States and Switzerland) as an oral disease‐modifying therapy for relapsing‐remitting multiple sclerosis (RRMS).1, 2, 3 The efficacy and safety profiles of fingolimod have been well characterized in a large clinical program in RRMS.4, 5, 6, 7, 8 In vivo, fingolimod is phosphorylated by the enzyme sphingosine‐1‐kinase to form the biologically active metabolite, fingolimod phosphate (fingolimod‐P), which is dephosphorylated back to the inactive form, fingolimod, before further metabolism.9, 10, 11 The pharmacokinetic (PK) profiles of fingolimod and fingolimod‐P have been extensively investigated in healthy subjects, renal transplant recipients, and patients with multiple sclerosis (MS) and are identical across the populations.12, 13, 14, 15, 16 Fingolimod exhibits high oral bioavailability (>90%),17 and its absorption is unaffected by dietary intake.15 It has a large volume of distribution (1200‐1700 L) and relatively low clearance (6‐8 L/h).13 Owing to a long half‐life (6‐9 days), steady‐state PK for fingolimod and fingolimod‐P is typically attained within 1 to 2 months of daily dosing, with steady‐state levels approximately 10‐fold greater than with the initial dose. Fingolimod and fingolimod‐P show dose‐proportional exposure after a single dose and at steady‐state over a wide dose range, and the PK is time independent.13 Moreover, both fingolimod and fingolimod‐P exhibited low to moderate intersubject variability in PK parameters (approximately 30%).13 In vitro studies have shown that fingolimod and fingolimod‐P have little or no capacity to inhibit or induce the activity of the main cytochrome P450 enzymes or transporters (Center for Drug Evaluation and Research [CDER] I, pages 54‐55).12 Well‐designed clinical studies showed that fingolimod has no effect on oral contraceptives (ethinylestradiol and levonorgestrel) exposure18 or cyclosporine levels.19
Fingolimod undergoes extensive metabolism through several pathways in humans, including reversible phosphorylation to form active fingolimod‐P, ω‐hydroxylation followed by rapid further oxidation, and conjugation with endogenous fatty acids to form ceramide analogues.10, 11 The cytochrome P450 (CYP) 4F subfamily, in particular CYP4F2, constitutes the major enzymes responsible for the ω‐hydroxylation of fingolimod, the primary elimination pathway of fingolimod in vivo.10 Other CYP enzymes, in particular, CYP3A4, have no or little contribution to fingolimod metabolism.10 Earlier, we reported that coadministration of ketoconazole and fingolimod results in increased fingolimod and fingolimod‐P exposure by 1.7‐fold through the inhibition of CYP4F2.20 However, despite the negligible role of CYP3A4 in fingolimod metabolism, evidence from in vitro experiments and a population PK analysis in patients with MS suggest a potential of drug‐drug interactions between strong CYP3A4 inducers and fingolimod when administered together.
Carbamazepine, a frequently prescribed treatment for neuropathic pain in patients with MS,21, 22 is a US Food and Drug Administration (FDA)‐recommended inductory probe for the CYP3A4 enzyme.23, 24 Thus, the present study was conducted to assess the potential effects of carbamazepine on the PK profile of fingolimod in healthy subjects. In addition, this study evaluated the safety and tolerability of simultaneous coadministration of these 2 drugs. The study methodology supported the FDA recommendations to fully explore the maximal dose range of carbamazepine in drug‐drug interaction studies of this nature.
Methods
Subjects
Eligible subjects included healthy men and women of non‐child bearing potential, aged between 18 and 50 years. Male subjects with a female partner of childbearing potential were advised to use 2 effective/highly effective methods of contraception for the entire duration of the study up to the completion visit. The key exclusion criteria were: smokers (urine cotinine ≥ 500 ng/mL); use of any prescription drugs or herbal supplements within 4 weeks prior to the first dosing and/or over‐the‐counter medication or dietary supplements (including vitamins) within 2 weeks prior to the first dosing; use of any investigational product at the time of enrollment or within 5 half‐lives of enrollment or within 30 days, whichever was longer; and a history of epilepsy and/or multiple and recurring allergies or allergy to the investigational compound/compound class. Subjects with ancestry across broad regions of Asia (including Chinese and South Asian Indians) were also excluded because of the increased risks of carbamazepine hypersensitivity, Steven‐Johnson syndrome, and toxic epidermal necrolysis.25, 26, 27
The study was conducted at Buffalo Clinical Research Center (Buffalo, New York) in accordance with the International Council for Harmonization Guidelines for Good Clinical Practice and the principles of the Declaration of Helsinki. The study protocol was approved by the institutional review board of the study site (IntegReview, Inc. Ethical Review Board, Lamar Blvd. Austin, Texas). All study subjects provided written informed consent before any study‐related procedure was performed.
Study Design
This was an open‐label, single‐sequence, 2‐period study that comprised a 14‐day screening period, 2 treatment periods, and a study completion evaluation 71 days after initiation of period 2 (Figure 1). In period 1 (37 days), eligible subjects underwent baseline evaluation on the day prior to fingolimod dosing (day ‐1). Subjects received a single 2‐mg oral dose of fingolimod under fasting conditions on day 1 and stayed in the clinic until the morning of day 3, followed by PK and safety assessments performed up to 36 days (864 hours) after dosing. In period 2 (71 days, starting between 1 and 10 days after the end of period 1), prior to carbamazepine dosing (day ‐1), subjects underwent baseline evaluations again. Subsequently, subjects received carbamazepine (carbamazepine 100/200/400 mg extended‐release tablets; Tegretol‐XR; Novartis) twice daily for 49 days in flexible, incremental, up‐titrated doses. The administration of each titration dose (starting from 100 mg twice daily) could vary from 3 to 7 days depending on the individual subject's tolerance. For example, if carbamazepine was well tolerated during dose titration, the dose was up‐titrated by 100 mg twice daily every 3 days until the maximum dose (600 mg twice daily) was achieved on day 16. If carbamazepine was poorly tolerated during dose titration, the dose was up‐titrated by 100 mg twice daily every 7 days until the maximum dose (400 mg twice daily) was achieved on day 21. The dose administered on day 21 was to range between 400 and 600 mg twice daily. This maximally achieved dose was to be maintained for the next 28 days (days 21‐49). However, flexibility was provided in the protocol, wherein this dose could be decreased to a minimum of at least 300 mg twice daily. The dose was not to be increased again during this phase. In period 2, fingolimod was administered on day 35. The study completion evaluation was performed on day 71 (±2 days) of period 2.
Figure 1.
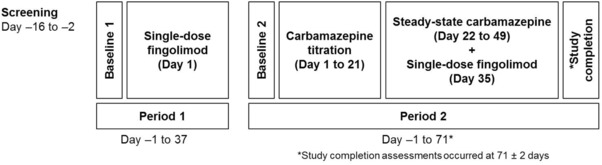
Study design.
Pharmacokinetic Assessments
In period 1, fingolimod and fingolimod‐P PK blood sampling was performed up to 36 days (864 hours) at the following time points: predose and 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 16, 24, 36, 48, 72, 96, 168, 240, 336, 504, 672, and 864 hours after a single 2‐mg fingolimod dose. In period 2, predose carbamazepine PK blood sampling was performed in the morning on days 1, 15, 18, 21, 24, 27, 30, 33, 34, 35, 36, 37, 38, 42, 45, and 49. Following the coadministration of carbamazepine and fingolimod on the morning of day 35, fingolimod and fingolimod‐P PK blood sampling was performed up to the study completion visit on day 71 of period 2 at the same post–fingolimod dose time points as in period 1.
Blood Collection and Processing
Blood samples were collected and processed as per the standard protocol (for details, please refer to supplementary information). The concentrations of fingolimod and fingolimod‐P were measured in whole blood using validated liquid chromatography/tandem mass spectrometry (m/z fingolimod, 308.50/255.50; m/z fingolimod‐P, 386.27/78.89). The lower limits of quantification (LLOQ) for fingolimod and fingolimod‐P were 0.080 ng/mL and 0.100 ng/mL, respectively, using 100 μL of blood; the upper limits of quantification (ULOQ) were 16.0 ng/mL and 20.0 ng/mL, respectively. The method was specific for fingolimod and fingolimod‐P, and no interference was observed. The assay performance during the study was evaluated using 4 quality control (QC) samples for fingolimod (0.24, 1.2, 6, and 12 ng/mL). The accuracy (% bias) and the precision (% coefficient of variation [%CV]) of mean values for the QC samples ranged from 1.7% to 5.8% and from 4.4% to 8.1%, respectively. The assay performance for fingolimod‐P was also evaluated using 4 QC samples (0.3, 1.5, 7.5, and 15 ng/mL). The accuracy (% bias) of mean values ranged from 2.7% to 5.3%, whereas the precision (%CV) ranged from 3.7% to 8.2% (data on file; Novartis Pharma AG, Basel, Switzerland). Carbamazepine concentrations in plasma were determined by a validated liquid chromatography/tandem mass spectrometry method using electrospray ionization as the ionization technique (m/z carbamazepine, 237.1/194.1). Full details are provided in the supplementary information. The LLOQ and ULOQ for carbamazepine were 0.10 and 25.0 μg/mL, respectively, using 50 μL of plasma. The method was specific for carbamazepine, and no interference was observed. The assay performance during the study was evaluated using 4 QC samples: 0.25, 1.25, 12.5, and 20 μg/mL. The accuracy (% bias) of the mean values ranged from 1.6% to 6.8%, whereas the precision (%CV) ranged from 2.7% to 3.5% (data on file; Novartis Pharma AG, Basel, Switzerland).
Fingolimod and Fingolimod‐P Pharmacokinetics
All PK parameters were derived from blood concentration‐time curves after fingolimod administration on day 1 of period 1 and on day 35 of period 2 using non‐compartmental method(s) in Phoenix WinNonlin, version 6.2. Computed PK parameters included apparent systemic clearance from blood (CL/F), apparent volume of distribution during the terminal phase (Vz/F), maximum blood concentration (Cmax), area under curve (AUC) from time zero to the sampling time of the last measurable concentration (AUClast), AUC from time zero extrapolated to infinity (AUCinf), time to reach maximum (peak) blood concentration (Tmax), lag time between drug intake and the first quantifiable blood concentration (Tlag), and apparent terminal half‐life (T1/2). There was no fixed maximum limit in the percentage of AUC extrapolated to infinity triggering the exclusion of an AUCinf value from the analysis; despite a sampling period of more than 35 days and a low LLOQ, it was known by experience that the values could be as high as 45%, still associated with a reliable estimation of AUCinf. T1/2 was computed with concentrations measured at times known to be in the terminal phase, providing the regression line going through them had a minimum r 2 value as defined by Novartis internal PK guidelines.
Carbamazepine Pharmacokinetics
No PK parameter was computed for carbamazepine. Only predose plasma concentrations were assessed.
Safety Assessments
Safety assessments comprised recording of all adverse events (AEs) and serious adverse events. Assessments included regular monitoring of laboratory values (hematology, blood chemistry, and urine analysis) and regular assessments of vital signs, physical examinations, and standard 12‐lead electrocardiograms (ECGs). The safety and tolerability of carbamazepine were assessed based on incidence of AEs and standard clinical laboratory data.
Sample‐Size Calculations
Sample‐size calculations were based on a potential decrease of 20% in the AUC of fingolimod. The estimate of within‐subject standard deviation (SD) for Cmax and AUClast on the log scale in healthy subjects was 0.10 and 0.17, respectively (data on file; Novartis Pharma AG, Basel, Switzerland). The calculations assumed a within‐subject SD of 0.2 to be more conservative. When the sample size was 16, a 2‐sided 90% confidence interval (CI) for the difference of 2 means of a log‐transformed PK parameter had an interval that extended no more than 0.216 from the observed difference in means with a coverage probability of 90%. When back‐transformed onto the original scale, this equated to a 90%CI for the test versus reference ratio of the PK parameter extending no more than 0.81 to 1.24, when the observed ratio of test versus reference was 1.00. Allowing for a dropout rate of 40%, the total number of subjects randomized to obtain 16 evaluable subjects was 26. The sample‐size calculations assumed that the PK parameter data were log‐normal.
Statistical Analysis
All subjects who received at least 1 dose of the study drug were included in the safety analysis set. The PK analysis set included all subjects with data for at least 1 primary PK variable (Cmax, AUClast, AUCinf) for fingolimod or fingolimod‐P in at least 1 period and no major protocol deviations that would have any impact on PK data. Descriptive statistics were presented for all PK parameters, which included arithmetic mean, SD, CV, geometric mean, CV of geometric mean, median, and minimum and maximum. Individual fingolimod and fingolimod‐P concentrations below the LLOQ were set to zero for statistical and graphic purposes. A linear mixed‐effects model was used to analyze the log‐transformed (natural logarithm) primary PK parameters of fingolimod and fingolimod‐P with treatment as a fixed factor and subject as a random factor. The estimated differences between the PK parameters for carbamazepine‐fingolimod coadministration versus fingolimod alone as well as the corresponding 90%CIs were calculated. These estimates (90%CIs) were back‐transformed to obtain an estimate and a 90%CI for the ratio of geometric means for the PK parameter (for both fingolimod alone and carbamazepine‐fingolimod coadministration). Descriptive statistics were used for safety assessments by treatment and visit. All analyses were performed using SAS 9.2 (SAS Institute, Cary, North Carolina).
Results
Subject Disposition and Demographics
Of the 26 enrolled subjects, 20 (76.9%) completed the study. The main reasons for discontinuation included AEs (n = 3), withdrawal of consent (n = 2), and abnormal test procedure results (n = 1). All subjects were male of either Caucasian (n = 11) or African American (n = 15) origin. The mean ± SD age of the study population was 30.9 ± 7.3 years (range, 19‐44 years). The mean body mass index was 25.65 ± 2.7 kg/m2 (range, 20.8–29.8 kg/m2).
Study Drug Exposure
In period 1, all 26 subjects received a single dose of fingolimod 2 mg on day 1. In period 2, 23 of 26 subjects received carbamazepine, as 3 subjects discontinued the study prior to entering period 2 and did not receive any dose of carbamazepine. Subsequently, 3 more subjects discontinued prior to day 35 of period 2. The remaining 20 subjects received a single dose of fingolimod 2 mg along with a morning dose of carbamazepine on day 35 of period 2. During the titration period, all subjects who completed the study received carbamazepine doses of 600 mg twice daily, the maximum dose, from day 16. All subjects but 1 (dose reduced to 400 mg twice daily from day 42) remained on this dose until the end of the carbamazepine administration (day 49).
Pharmacokinetics
The PK analysis data set comprised 23 subjects for period 1, and 19 for period 2 (on top of the 6 subjects who discontinued the study, 1 subject with a protocol deviation was excluded). The PK parameters for fingolimod and fingolimod‐P in the absence (period 1) and presence (period 2) of carbamazepine are presented in Table 1. The mean blood concentration profiles of fingolimod and fingolimod‐P in periods 1 and 2 are provided in Figure 2.
Table 1.
Fingolimod and Fingolimod‐P Pharmacokinetics in the Absence (Period 1) and Presence (Period 2) of Carbamazepine in Healthy Subjects (Period 1, n = 23; Period 2, n = 19)
| Fingolimod | Fingolimod‐P | |||
|---|---|---|---|---|
| PK Parameters | Fingolimod Alone (Period 1) | Fingolimod + Carbamazepine (Period 2) | Fingolimod Alone (Period 1) | Fingolimod + Carbamazepine (Period 2) |
| Tlag a (h) | 0.00 | 0.50 | 1.00 | 1.00 |
| (0.00–0.50) | (0.00–0.50) | (0.50–1.50) | (0.50–1.50) | |
| Tmax a (h) | 12.00 | 12.00 | 6.52 | 6.00 |
| (6.00–36.00) | (8.00–36.00) | (6.00–12.00) | (6.00–12.00) | |
| Cmax b (ng/mL) | 1.38 ± 0.203 | 1.15 ± 0.185 | 1.68 ± 0.365 | 1.38 ± 0.292 |
| (1.37, 15.4) | (1.14, 16.3) | (1.64, 22.5) | (1.35, 22.8) | |
| AUClast b (ng·h/mL) | 249 ± 90 | 155 ± 47.7 | 91.4 ± 31.0 | 55.0 ± 18.2 |
| (232, 42) | (148, 31.7) | (85.8, 39.7) | (51.9, 37.3) | |
| AUCinf b (ng·h/mL) | 283 ± 100 | 173 ± 51.5 | 125 ± 37.9c | 84.7 ± 15.4d |
| (264, 41.5) | (166, 30.8) | (120, 30.8) | (83.5, 17.7) | |
| CL/Fb (L/h) | 8.24 ± 3.91 | 12.6 ± 3.78 | ||
| (7.57, 41.5) | (12.1, 30.8) | |||
| Vz /Fb (L) | 1700 ± 315 | 1810 ± 346 | ||
| (1670, 18.9) | (1780, 17.5) | |||
| T1/2 b (h) | 163 ± 56.3 | 106 ± 25.2 | 154 ± 63.2c | 106 ± 27.4d |
| (153, 37.2) | (102, 26.7) | (143, 40.2) | (102, 26.6) | |
| Tlast a (h) | 504 | 336 | 240 | 168 |
| (240–864) | (168–504) | (96–504) | (96–240) | |
AUC, area under curve; AUCinf, AUC from time zero extrapolated to infinity; AUClast, AUC from time zero to the sampling time of the last measurable concentration; CL/F, apparent systemic clearance from blood; Cmax, maximum blood concentration; T1/2, apparent terminal half‐life; Tlag, lag time between drug intake and the first quantifiable blood concentration; Tlast, time of the last quantifiable drug blood concentration; Tmax, time to reach maximum (peak) blood concentration; Vz/F, apparent volume of distribution during the terminal phase.
Median (minimum‐maximum).
Arithmetic mean ± standard deviation (geometric mean, % geometric mean coefficient of variation).
n = 20.
n = 13 as T1/2 could not be estimated reliably in all subjects.
Figure 2.
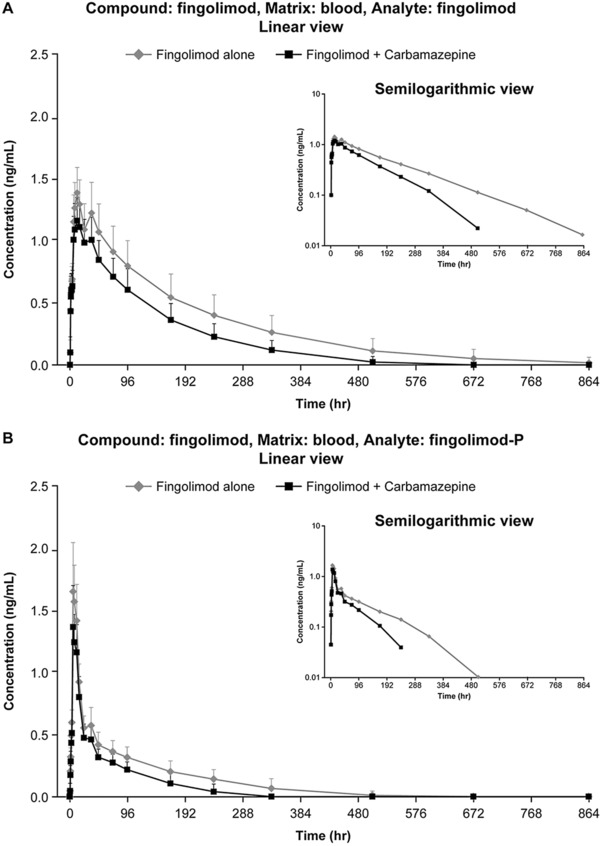
Mean ± SD blood concentration profiles of fingolimod (A) and fingolimod‐P (B) in periods 1 and 2. Error bars represent standard deviation (SD).
Fingolimod
In both periods, after a median lag time smaller than 0.5 hours (range, 0‐0.5 hours), fingolimod blood concentrations slowly increased to Cmax, occurring at a time ranging from 6 to 36 hours postdose with a median of 12 hours. In many subjects, fingolimod blood concentrations remained in a broad plateau region (from 12 to 36 hours) and slowly declined thereafter in a monoexponential manner, with a geometric mean T1/2 of 153 hours in period 1 and 102 hours in period 2. The last measurable sample was earlier in period 2 than in period 1 in 17 of 19 subjects (and equal in 1 of 19 subjects).
The geometric mean CL/F of fingolimod increased in the presence of carbamazepine at steady‐state, whereas Vz/F remained nearly unchanged, resulting in a shorter T1/2 and a decreased Cmax and AUC. In the presence of carbamazepine, on average, fingolimod Cmax was reduced by 18%, AUClast by 39%, and AUCinf by 40%; the corresponding geometric mean ratios (carbamazepine‐fingolimod coadministration over fingolimod alone) and 90%CIs were 0.82 (0.78‐0.85), 0.61 (0.55‐0.67), and 0.60 (0.54‐0.66); see Table 2. The variability, as measured by the %CV geometric mean, was low for Cmax in both periods (period 1, 15.4%; period 2, 16.3%) and moderate to high for AUClast (period 1, 42%; period 2, 31.7%) and AUCinf (period 1, 41.5%; period 2, 30.8%).
Table 2.
Geometric Mean Ratio (Carbamazepine‐Fingolimod Coadministration Versus Fingolimod Alone) and 90% Confidence Interval for PK Parameters (PK Analysis Set)
| Adjusted Geometric Meana | Geometric Mean Ratioa | |||||
|---|---|---|---|---|---|---|
| Analyte | PK Parameter | Fingolimod+ Carbamazepine | Fingolimod Alone | (Fingolimod + Carbamazepine/Fingolimod Alone) | Lower 90%CI | Upper 90%CI |
| Fingolimod | Cmax (ng/mL) | 1.12 | 1.37 | 0.82 | 0.78 | 0.85 |
| AUClast (ng·h/mL) | 141 | 232 | 0.61 | 0.55 | 0.67 | |
| AUCinf (ng·h/mL) | 158 | 264 | 0.60 | 0.54 | 0.66 | |
| Fingolimod‐P | Cmax (ng/mL) | 1.34 | 1.64 | 0.82 | 0.74 | 0.90 |
| AUClast (ng·h/mL) | 49.7 | 85.8 | 0.58 | 0.53 | 0.64 | |
| AUCinf (ng·h/mL) | 74.3 | 121 | 0.62 | 0.55 | 0.69 | |
AUC, area under the curve; AUCinf, AUC from time zero extrapolated to infinity; AUClast, AUC from time zero to the sampling time of the last measurable concentration; CL, confidence level; Cmax, maximum blood concentration; PK, pharmacokinetics.
Back‐transformed from log scale. The log‐transformed PK parameter data were analyzed using a linear mixed model with treatment as a fixed factor and subject as a random factor.
Fingolimod‐P
In both periods, after a median lag time of 1 hour (range, 0.5‐1.5 hours), longer than that observed for fingolimod, fingolimod‐P blood concentrations sharply increased to Cmax, occurring at a time ranging from 6 to 12 hours postdose with a median of 6‐6.52 hours. Fingolimod‐P blood concentrations declined rapidly in a first phase and then much slowly in a second phase, with T1/2 comparable to that of fingolimod (geometric mean of 143 hours in period 1 and 102 hours in period 2). As for fingolimod, the last measurable fingolimod‐P concentration was recorded earlier in the presence of carbamazepine in most subjects.
Fingolimod‐P exposure parameters were reduced in a range similar to fingolimod. On average, Cmax was reduced by 18%, AUClast by 42%, and AUCinf by 38%; the corresponding geometric mean ratios and 90%CIs were 0.82 (0.74‐0.90), 0.58 (0.53‐0.64), and 0.62 (0.55‐0.69); see Table 2.
Cmax variability was low to moderate in both periods for fingolimod‐P (period 1, 22.5%; period 2, 22.8%), whereas the variability in AUClast (period 1, 39.7%; period 2, 37.3%) and AUCinf (period 1, 30.8%; period 2, 17.7%) was low to high.
Carbamazepine
In most subjects, the highest predose plasma concentrations of carbamazepine were reached on day 18 or day 21. Thereafter, the concentrations fluctuated from each day to the next, with an overall tendency to decline until day 30 or day 33, when steady‐state was likely to be reached, as there was no further decline (Figure 3). A majority of the subjects were exposed to a predose plasma carbamazepine concentration of 7‐10 μg/mL from day 21 to day 49 (for details, please refer to the table in the supplementary information). The fingolimod 2‐mg single dose had no marked effects on the predose concentrations of carbamazepine.
Figure 3.
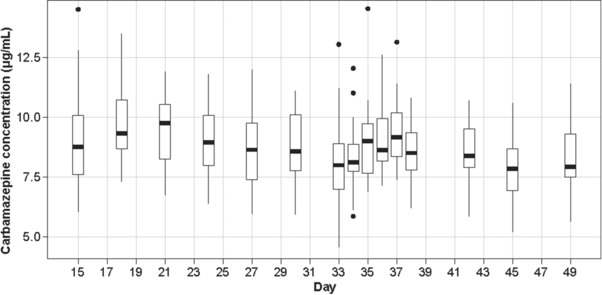
Predose plasma concentration (μg/mL) of carbamazepine during the study (days 15 to 49). The box‐and‐whiskers plot shows the medians, quartiles, and ranges for predose plasma concentration of carbamazepine for each subject during the study. The bottom and top of the boxes represent the 25th and 75th percentiles, respectively. The horizontal lines in the middle of the boxes represent the median. The top whiskers extend from the 75th percentile up to the largest value no farther than 1.5 times the interquartile range (75th‐25th percentile). The bottom whiskers extend from the 25th percentile to the smallest value at most 1.5 times the interquartile range. The individual points represent outliers that go beyond the whiskers.
Safety
Overall, fingolimod was well tolerated when administered as a single 2‐mg dose alone and on day 35, once steady‐state carbamazepine concentrations were achieved. The incidence of AEs was highest during the carbamazepine titration phase (69.6%), followed by the carbamazepine‐fingolimod coadministration phase (30%) and the fingolimod‐alone phase (23.1%). The most commonly reported AEs included somnolence, dizziness, fatigue, and nausea. All AEs reported in the study were mild to moderate (n = 5) in severity, and there were no serious or severe AEs (Table 3). Of the 3 AEs (increased blood triglycerides, AE of neck mass, and increased hepatic enzymes) that led to study discontinuation, only the increase in hepatic enzymes was suspected to be related to the study drug. Carbamazepine‐related AEs such as dizziness, nausea, and headache were reported in 7 subjects (30.4%), 2 subjects (8.7%), and 2 subjects (8.7%), respectively, during the titration phase and were also reported in 2 subjects (10%) during the carbamazepine‐fingolimod coadministration phase. Nausea and headache were reported in 1 subject (3.8%) when fingolimod was administered alone in period 1. Clinically significant ECG abnormalities were reported in 7 subjects and included sinus tachycardia (at baseline of period 1, n = 1), sinus bradycardia (at screening and end of study [EOS], n = 1), right bundle branch block (at EOS, n = 1), atrial fibrillation (at baseline of period 1, n = 1), sinus rhythm, nonspecific ST/T‐wave changes (at screening, n = 1; at baseline of period 2, n = 2), and sinus rhythm, first‐degree atrioventricular block (at screening, at baseline of period 1, and EOS, n = 1). None of these ECG abnormalities were reported as AEs or led to study discontinuation. In addition, in most of the subjects, these abnormalities resolved by the EOS visit. No clinically significant changes were observed in vital signs, hematology, or laboratory values.
Table 3.
Incidence of AEs
| Incidence of AEs | Fingolimod Alone, n = 26 | Carbamazepine Alone (Titration and Top Dose), n = 23 | Fingolimod + Carbamazepine Combination, n = 20 |
|---|---|---|---|
| Subjects with AE(s) | 6 (23.1) | 16 (69.6) | 6 (30.0) |
| Nervous system disorders | 1 (3.8) | 14 (60.9) | 2 (10.0) |
| Gastrointestinal disorders | 3 (11.5) | 6 (26.1) | 2 (10.0) |
| General disorders and administration‐site condition | 0 (0.0) | 3 (13.0) | 3 (15.0) |
| Skin and subcutaneous tissue disorders | 0 (0.0) | 5 (21.7) | 0 (0.0) |
| Investigations | 1 (3.8) | 3 (13.0) | 0 (0.0) |
| Metabolism and nutrition disorders | 0 (0.0) | 4 (17.4) | 0 (0.0) |
| Musculoskeletal and connective tissue disorders | 0 (0.0) | 4 (17.4) | 0 (0.0) |
| Eye disorders | 0 (0.0) | 1 (4.3) | 1 (5.0) |
| Infections and infestations | 1 (3.8) | 0 (0.0) | 1 (5.0) |
| Injury, poisoning, and procedural complications | 0 (0.0) | 1 (4.3) | 0 (0.0) |
| Psychiatric disorders | 0 (0.0) | 1 (4.3) | 0 (0.0) |
| Renal and urinary disorders | 0 (0.0) | 1 (4.3) | 0 (0.0) |
AE, adverse event.
Discussion
Fingolimod is primarily metabolized via human CYP4F2 and possibly other enzymes of the CYP4F subfamily. Other CYP enzymes, in particular CYP3A4, have a negligible contribution to fingolimod metabolism.10
Prior to this clinical study, in vitro experiments were conducted to assess the impact of known CYP inducers on fingolimod metabolism (CDER I, page 103).12 Primary human hepatocytes were treated with the strong CYP inducers rifampicin, phenytoin, phenobarbital, or carbamazepine or vehicle for 72 hours. Thereafter, the rate of fingolimod metabolism (formation of metabolites by the CYP4F ω‐hydroxylation), the rate of marker activities for common CYPs and CYP4F, and mRNA levels of these CYP enzymes were determined in the hepatocytes. The results showed that the inducer‐treated hepatocytes metabolized fingolimod at a 1.3‐ to 4.1‐fold higher rate than with vehicle control, whereas the mRNA of CYP4F enzymes was essentially unaffected (increase < 1.4‐fold for CYP4F2 and <1.6‐fold for CYP4F3B). In contrast, the activity of CYP3A and the mRNA of CYP3A4 were strongly induced (up to 19‐fold and up to 180‐fold, respectively). The ω‐hydroxylation of leukotriene B4 (LTB4) was increased by the 4 inducers between 1.6‐ and 3.7‐fold. The latter reaction was originally thought to be a specific marker for the activity of CYP4F enzymes (CDER I, page 103),12 but was later discovered to also be catalyzed, to a lower extent, by other CYP enzymes, particularly CYP3A4 (data on file; Novartis Pharma AG, Basel, Switzerland). Across the different inducer treatments, the induction pattern of fingolimod metabolism correlated well with LTB4 ω‐hydroxylation and CYP3A activity. Activities of CYP2B6, CYP2C8, CYP2C9, and CYP2C19 were increased as expected by the inducers and correlated poorly with the increase in fingolimod metabolism. Together, these data suggest that the small increase in fingolimod metabolism on treatment of the hepatocytes by the potent inducers was a result of CYP3A4 induction. This evidence shows that although in the uninduced state this cytochrome is not involved to a relevant extent in the primary step of fingolimod metabolism, strong induction can lead to a notable contribution of this enzyme. Hence, a potential drug‐drug interaction may exist between CYP3A4 inducers and fingolimod when administered together.
In a population PK evaluation, the effect of carbamazepine on fingolimod and fingolimod‐P predose concentrations (as markers of total blood exposure) was explored (CDER I, pages 60‐61).12 The data from more than 1200 multiple sclerosis patients from the 2 phase 3 clinical studies (FREEDOMS and FREEDOMS II) were used. The fingolimod and fingolimod‐P concentrations measured in samples from patients receiving carbamazepine were compared with those from samples drawn from patients who were not receiving carbamazepine (the total number of fingolimod‐P samples was approximately 7000). Based on more than 100 samples, carbamazepine had little effect (<30% decrease) on fingolimod‐P concentration. Therefore, only a moderate effect of carbamazepine administered at the maximum therapeutic dose of 600 mg twice daily could be expected (data on file; Novartis Pharma AG, Basel, Switzerland).
Carbamazepine is a potent inducer of CYP isoenzymes that are involved in the metabolism of several drugs (CYP1A2, CYP2C9, CYP2C19, and CYP3A4), leading to the possibility of significant drug‐drug interactions when coadministered.24 Carbamazepine is frequently prescribed in patients with MS to alleviate neuropathic pain and could therefore alter the PK profile of several drugs when coadministered, thereby affecting their clinical efficacy.21 Thus, this study was conducted to assess the potential effects of multiple carbamazepine doses (at steady‐state concentrations) on the PK profile of fingolimod in healthy subjects.
There is some evidence in the literature that carbamazepine induction is dose‐dependent and that the maximum effect may be reached at 500 mg daily28 or 600 mg daily.29 In our study, doses of 1200 mg daily (600 mg twice daily) were administered, on request from the FDA, to fully explore the induction effect of the highest therapeutic dose. The study design allowed flexibility with the dosing‐escalation scheme to enable subjects to be up‐titrated more conservatively and continue in the study only if they were able to tolerate the dose of 300 mg twice daily from day 21 onward. All subjects who completed the study received carbamazepine doses of 600 mg twice daily, the maximum dose, from day 16, suggesting the robustness of the dose‐escalation scheme. All subjects but 1 (dose reduced to 400 mg twice daily from day 42) remained on this dose until the end of the carbamazepine administration (day 49). Interestingly, the carbamazepine predose concentrations (Figure 3) reached a maximum value on day 18 or 21 in all subjects (ie, 3‐5 days after the start of the 600‐mg dose administration) and then tended to slightly decrease to day 33 (for details, please refer to the table in the supplementary information). This suggests that induction may not have reached full capacity before that day. However, caution needs to be applied to any conclusion based on these observations, as they rely on predose concentrations that are quite variable between the subjects and from one day to the next within the subjects.
The elimination half‐life of carbamazepine is approximately 35‐40 hours with initial dosing, which can be reduced to 10‐20 hours following full induction after multiple dosing.30 However, it is known that the time to reach full induction on repeated administration and the time for deinduction after treatment discontinuation are linked to the cytochrome turnover time, measured with a difficult‐to‐estimate half‐life.31 Yang et al31 highlighted that there are marked discrepancies in the literature on the CYP3A4 half‐life, with average values ranging between 70 and 140 hours (using in vivo methods) and individual values even more variable. Magnusson et al32 evaluated that the induction and deinduction half‐life for carbamazepine is approximately 3 days. This value is consistent with the findings from Xu et al,29 suggesting that carbamazepine near maximal induction is reached after 10 days of treatment. In their simulations using rifampicin as a strong inducer, Baneyx et al33 found that a hepatic CYP3A4 half‐life of 3 days was appropriate to describe the induction and deinduction process in their model and that a 2‐week period would be sufficient to allow the CYP3A4 levels to return to baseline. However, Reitman et al34 showed in a clinical study that the half‐life for deinduction is in the range of 8 days and that a 2‐week rifampin discontinuation is not sufficient to completely reverse its inductive effect.
Given these uncertainties, the design of our study was conservative. The study comprised 2 periods in a single‐sequence, open‐label design. Although a randomized crossover study would have been a more rigorous study design (absence of period effect), we wanted to ensure that subjects were naïve to CYP3A4 inducer in the period in which fingolimod was administered in the absence of carbamazepine. Starting the study with the carbamazepine period would have required a long washout at the end of the carbamazepine treatment, of at least 5 times the mean deinduction half‐life value determined by Reitman et al (ensuring more than 95% elimination),34 that is, 40 days, which would make the study more challenging to conduct. The single‐sequence approach with fingolimod administered in period 1 eliminated any risk of CYP3A4 induction carryover. The duration of the sampling periods after fingolimod administration in both periods (37 days) in our design provided sufficient time to ensure enough washout of fingolimod and fingolimod‐P blood concentrations to accurately characterize the AUCinf of both analytes.
This study design also ensured that carbamazepine concentrations at the highest tolerated dose reached steady‐state at the time of fingolimod administration in period 2 (day 35) and that full induction of the enzyme was well approached before the administration of fingolimod. Furthermore, carbamazepine administration was continued until day 49 (ie, for 14 days after fingolimod administration) to maintain full induction over approximately 2 half‐lives of fingolimod and fingolimod‐P. To our knowledge, this is the longest duration for which carbamazepine has been coadministered in any drug‐drug interaction study in healthy subjects. A majority of subjects were exposed to predose plasma carbamazepine concentrations consistent with values reported in the literature.35
In this study, fingolimod was administered at a dose of 2 mg (ie, 4 times greater than the approved dose of 0.5 mg) to allow appropriate assessment of the PK profiles of both analytes in the event of a 50% decrease in exposure in the presence of carbamazepine. A single dose of fingolimod, as high as 40 mg, has been shown to be well tolerated in healthy subjects. The blood concentration‐time profiles of the 2 analytes and estimated fingolimod CL/F and Vz/F in period 1 (ie, in the absence of carbamazepine) are consistent with previous observations.13 As the PK of fingolimod and fingolimod‐P are dose‐ and time independent (ie, clearance and volume of distribution are identical over a wide dose range and do not change on repeated administration), the effects of carbamazepine on this 2‐mg single‐dose exposure of these 2 analytes were assumed to be identical to those at steady‐state concentrations.
The study showed that steady‐state dosing of carbamazepine at 600 mg twice daily increased fingolimod CL/F by 67%, with a mean decrease of approximately 20% in Cmax and approximately 40% in AUCinf and AUClast. These results are in line with the observations made in the in vitro induction experiments and the population PK study.12 An effect of a similar extent was observed on fingolimod‐P Cmax, AUClast, and AUCinf. This was expected, as the phosphorylation/dephosphorylation reactions involved in fingolimod‐P formation and metabolism are not catalyzed by any cytochrome. Therefore, the dynamic equilibrium between the 2 analytes was not altered by carbamazepine, and the changes in fingolimod exposure are directly reflected in fingolimod‐P. Fingolimod apparent volume of distribution was nearly unchanged between the 2 periods. This suggests that the bioavailability of the drug was minimally affected by carbamazepine administration and that the changes in exposure are mainly a result of changes in the clearance. Although not powered for such an analysis, the magnitude of the carbamazepine impact on fingolimod and fingolimod‐P exposure parameters did not seem to differ between the Caucasian and African American subjects.
The impact of decreased exposure in fingolimod‐P, as observed in this study, on treatment efficacy in MS patients receiving the approved 0.5‐mg dose is currently unknown, as there is no clinical experience with lower doses. The relationship between fingolimod‐P exposure and lymphocyte count (a pharmacodynamic marker of fingolimod effect) has been modeled using data from the MS patients in the pivotal studies. The decrease in lymphocyte count with increasing fingolimod‐P exposure is described well by an ordinary Imax function, with an estimated maximum reduction, compared with baseline, of approximately 80% (CDER II, page 85).36 Per the model, the mean fingolimod‐P exposure of a 0.5 mg dose corresponds to 88% of this maximum decrease and that of an exposure 40% smaller corresponds to approximately 80% of the maximum reduction. A model describing fingolimod‐P exposure versus annualized relapse rate (ARR) suggests that the ARR for a decreased exposure of 40% is approximately 0.22 versus 0.2 for a 0.5 mg dose (CDER II, page 87).36 Although these models tend to show that a 40% reduction in fingolimod‐P exposure has a low impact on the treatment effect, it is not possible to evaluate the true clinical response without a proper, well‐designed clinical study. Given this uncertainty, the coadministration of fingolimod and carbamazepine should be done with caution.
In our study, fingolimod was well tolerated in healthy subjects when administered as a single 2‐mg dose alone and at steady‐state carbamazepine concentrations. Moreover, the safety findings in this study were in line with previously known profiles of individual drugs.
Conclusion
Overall, carbamazepine administration at the maximal therapeutic dose of 600 mg twice daily through the induction of CYP3A4, a cytochrome with negligible involvement in fingolimod clearance in an uninduced state, decreased fingolimod and fingolimod‐P exposure by approximately 40% in healthy subjects. Models linking fingolimod‐P blood concentrations to lymphocyte count or annual relapse rate suggest that such a decrease would have a low impact on the treatment effect. However, in the absence of efficacy data of fingolimod at doses lower than the therapeutic dose, the coadministration of fingolimod and carbamazepine should be used with caution.
Supporting information
Supporting information
Acknowledgments
The authors acknowledge Rahul Birari and Vimal Kumar Muthyala (Medical Communications, Novartis Healthcare Pvt. Ltd., Hyderabad, India) for providing medical writing support, which encompassed preparation of the first draft, formatting, referencing, preparing tables and figures, incorporating authors’ revisions, and submission, all under the direction of the authors; and Brigitte Weisshaar (Medical Communications, Novartis Pharma AG, Basel, Switzerland) for editorial assistance and manuscript coordination. Complying with the guidelines of the International Committee of Medical Journal Editors, all authors have significantly contributed to the study and have been thoroughly involved in the critical review of the article for important intellectual content.
Declaration of Conflicting Interests
Authors Olivier David, Rhett Behrje, Pal Parasar, Hisanori Hara, and Robert Schmouder were employed by Novartis during the conduct of the study and are still employed by Novartis outside the submitted work. Author Christian Lates is a subcontractor of the Buffalo Clinical Research Center, which was contracted by Novartis Pharma AG for the conduct of this study.
Funding
This study was funded by Novartis Pharma AG, Basel.
References
- 1. Brinkmann V, Billich A, Baumruker T, et al. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov. 2010;9(11):883–897. [DOI] [PubMed] [Google Scholar]
- 2. Chun J, Hartung HP. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin Neuropharmacol. 2010;33(2):91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kappos L, Antel J, Comi G, et al. Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med. 2006;355(11):1124–1140. [DOI] [PubMed] [Google Scholar]
- 4. Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):402–415. [DOI] [PubMed] [Google Scholar]
- 5. Comi G, O'Connor P, Montalban X, et al. Phase II study of oral fingolimod (FTY720) in multiple sclerosis: 3‐year results. Mult Scler. 2010;16(2):197–207. [DOI] [PubMed] [Google Scholar]
- 6. Kappos L, Cohen J, Collins W, et al. Fingolimod in relapsing multiple sclerosis: an integrated analysis of safety findings. Mult Scler Relat Disord. 2014;3(4):494–504. [DOI] [PubMed] [Google Scholar]
- 7. Kappos L, Radue EW, O'Connor P, et al. A placebo‐controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362(5):387–401. [DOI] [PubMed] [Google Scholar]
- 8. Khatri B, Barkhof F, Comi G, et al. Comparison of fingolimod with interferon beta‐1a in relapsing‐remitting multiple sclerosis: a randomised extension of the TRANSFORMS study. Lancet Neurol. 2011;10(6):520–529. [DOI] [PubMed] [Google Scholar]
- 9. Albert R, Hinterding K, Brinkmann V, et al. Novel immunomodulator FTY720 is phosphorylated in rats and humans to form a single stereoisomer. Identification, chemical proof, and biological characterization of the biologically active species and its enantiomer. J Med Chem. 2005;48(16):5373–5377. [DOI] [PubMed] [Google Scholar]
- 10. Jin Y, Zollinger M, Borell H, Zimmerlin A, Patten CJ. CYP4F enzymes are responsible for the elimination of fingolimod (FTY720), a novel treatment of relapsing multiple sclerosis. Drug Metab Dispos. 2011;39(2):191–198. [DOI] [PubMed] [Google Scholar]
- 11. Zollinger M, Gschwind HP, Jin Y, Sayer C, Zecri F, Hartmann S. Absorption and disposition of the sphingosine 1‐phosphate receptor modulator fingolimod (FTY720) in healthy volunteers: a case of xenobiotic biotransformation following endogenous metabolic pathways. Drug Metab Dispos. 2011;39(2):199–207. [DOI] [PubMed] [Google Scholar]
- 12. Center for Drug Evaluation and Research [CDER]. Application number 22‐527. Clinical Pharmacology and Biopharmaceutical Reviews. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2010/022527Orig1s000clinpharmr.pdf. Accessed February 8, 2018.
- 13. David OJ, Kovarik JM, Schmouder RL. Clinical pharmacokinetics of fingolimod. Clin Pharmacokinet. 2012;51(1):15–28. [DOI] [PubMed] [Google Scholar]
- 14. Kovarik JM, Schmouder R, Barilla D, Riviere GJ, Wang Y, Hunt T. Multiple‐dose FTY720: tolerability, pharmacokinetics, and lymphocyte responses in healthy subjects. J Clin Pharmacol. 2004;44(5):532–537. [DOI] [PubMed] [Google Scholar]
- 15. Kovarik JM, Schmouder R, Barilla D, Wang Y, Kraus G. Single‐dose FTY720 pharmacokinetics, food effect, and pharmacological responses in healthy subjects. Br J Clin Pharmacol. 2004;57(5):586–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kovarik JM, Slade A, Voss B, et al. Ethnic sensitivity study of fingolimod in white and Asian subjects. Int J Clin Pharmacol Ther. 2007;45(2):98–109. [DOI] [PubMed] [Google Scholar]
- 17. Kovarik JM, Hartmann S, Bartlett M, et al. Oral‐intravenous crossover study of fingolimod pharmacokinetics, lymphocyte responses and cardiac effects. Biopharm Drug Dispos. 2007;28(2):97–104. [DOI] [PubMed] [Google Scholar]
- 18. David OJ, Ocwieja M, Meiser K, et al. Pharmacokinetics of fingolimod (FTY720) and a combined oral contraceptive coadministered in healthy women: drug‐drug interaction study results. Int J Clin Pharmacol Ther. 2012;50(8):540–544. [DOI] [PubMed] [Google Scholar]
- 19. Kovarik JM, Schmouder RL, Barilla D, et al. FTY720 and cyclosporine: evaluation for a pharmacokinetic interaction. Ann Pharmacother. 2004;38(7‐8):1153–1158. [DOI] [PubMed] [Google Scholar]
- 20. Kovarik JM, Dole K, Riviere GJ, et al. Ketoconazole increases fingolimod blood levels in a drug interaction via CYP4F2 inhibition. J Clin Pharmacol. 2009;49(2):212–218. [DOI] [PubMed] [Google Scholar]
- 21. Solaro C, Uccelli MM. Management of pain in multiple sclerosis: a pharmacological approach. Nat Rev Neurol. 2011;7(9):519–527. [DOI] [PubMed] [Google Scholar]
- 22. Zakrzewska JM, Linskey ME. Trigeminal neuralgia. BMJ. 2014;348:g474. [DOI] [PubMed] [Google Scholar]
- 23. U.S. Food and Drug Administration: Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers. https://www.fda.gov/Drugs/DevelopmentApprovalProcess/DevelopmentResources/DrugInteractionsLabeling/ucm093664.htm. Accessed February 8, 2018.
- 24. Spina E, Pisani F, Perucca E. Clinically significant pharmacokinetic drug interactions with carbamazepine. An update. Clin Pharmacokinet. 1996;31(3):198–214. [DOI] [PubMed] [Google Scholar]
- 25. Hung SI, Chung WH, Jee SH, et al. Genetic susceptibility to carbamazepine‐induced cutaneous adverse drug reactions. Pharmacogenet Genomics. 2006;16(4):297‐306. [DOI] [PubMed] [Google Scholar]
- 26. Man CB, Kwan P, Baum L, et al. Association between HLA‐B*1502 allele and antiepileptic drug‐induced cutaneous reactions in Han Chinese. Epilepsia. 2007;48(5):1015–1018. [DOI] [PubMed] [Google Scholar]
- 27. Lonjou C, Thomas L, Borot N, et al. A marker for Stevens‐Johnson syndrome …: ethnicity matters. Pharmacogenomics J. 2006;6(4):265–268. [DOI] [PubMed] [Google Scholar]
- 28. Scheyer RD, Cramer JA, Mattson RH. A pharmacodynamic approach to the estimate of carbamazepine autoinduction. J Pharm Sci. 1994;83(4):491–494. [DOI] [PubMed] [Google Scholar]
- 29. Xu Y, Zhou Y, Hayashi M, Shou M, Skiles GL. Simulation of clinical drug‐drug interactions from hepatocyte CYP3A4 induction data and its potential utility in trial designs. Drug Metab Dispos. 2011;39(7):1139–1148. [DOI] [PubMed] [Google Scholar]
- 30. Bertilsson L. Clinical pharmacokinetics of carbamazepine. Clin Pharmacokinet. 1978;3(2):128–143. [DOI] [PubMed] [Google Scholar]
- 31. Yang J, Liao M, Shou M, et al. Cytochrome p450 turnover: regulation of synthesis and degradation, methods for determining rates, and implications for the prediction of drug interactions. Curr Drug Metab. 2008;9(5):384–394. [DOI] [PubMed] [Google Scholar]
- 32. Magnusson MO, Dahl ML, Cederberg J, Karlsson MO, Sandstrom R. Pharmacodynamics of carbamazepine‐mediated induction of CYP3A4, CYP1A2, and Pgp as assessed by probe substrates midazolam, caffeine, and digoxin. Clin Pharmacol Ther. 2008;84(1):52–62. [DOI] [PubMed] [Google Scholar]
- 33. Baneyx G, Parrott N, Meille C, Iliadis A, Lave T. Physiologically based pharmacokinetic modeling of CYP3A4 induction by rifampicin in human: influence of time between substrate and inducer administration. Eur J Pharm Sci. 2014;56:1–15. [DOI] [PubMed] [Google Scholar]
- 34. Reitman ML, Chu X, Cai X, et al. Rifampin's acute inhibitory and chronic inductive drug interactions: experimental and model‐based approaches to drug‐drug interaction trial design. Clin Pharmacol Ther. 2011;89(2):234–242. [DOI] [PubMed] [Google Scholar]
- 35. Kudriakova TB, Sirota LA, Rozova GI, Gorkov VA. Autoinduction and steady‐state pharmacokinetics of carbamazepine and its major metabolites. Br J Clin Pharmacol. 1992;33(6):611–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. CDER application number 22‐527. Medical Reviews. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2010/022527Orig1s000medr.pdf. Accessed February 8, 2018.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information