

Abstract
Objective
To evaluate the efficacy and safety of atacicept, an antagonist of B lymphocyte stimulator/APRIL–mediated B cell activation, in patients with systemic lupus erythematosus (SLE).
Methods
ADDRESS II is a 24‐week, multicenter, randomized, double‐blind, placebo‐controlled, parallel‐arm, phase IIb study evaluating the safety and efficacy of atacicept in patients with SLE (ClinicalTrials.gov identifier NCT01972568). Patients with active, autoantibody‐positive SLE receiving standard therapy were randomized (1:1:1) to receive atacicept (75 mg or 150 mg) or placebo for 24 weeks. The primary end point was the SLE responder index 4 (SRI‐4) at week 24.
Results
The intent‐to‐treat (ITT) population included 306 patients. There was a trend toward an improved SRI‐4 response rate with atacicept 75 mg (57.8%; adjusted odds ratio [OR] 1.78, P = 0.045) and 150 mg (53.8%; adjusted OR 1.56, P = 0.121) at week 24 as compared with placebo (44.0%) (primary analysis; using the screening visit as baseline). In a prespecified sensitivity analysis using study day 1 as baseline, a significantly larger proportion of patients receiving atacicept 75 mg and 150 mg achieved an SRI‐4 response at week 24 compared with placebo. In predefined subpopulations with high levels of disease activity (HDA) at baseline, serologically active disease, or both, statistically significant improvements in the SRI‐4 and SRI‐6 response rates were seen with atacicept versus placebo. A severe risk of disease flare was reduced with atacicept therapy in both the ITT and the HDA populations. The risks of serious adverse events and serious or severe infection were not increased with atacicept as compared with placebo.
Conclusion
Atacicept treatment showed evidence of efficacy in SLE, particularly in HDA and serologically active patients. Reductions in disease activity and severe flare were observed with atacicept treatment, with an acceptable safety profile.
Systemic lupus erythematosus (SLE) is a multisystem autoimmune disease with a fluctuating disease course characterized by sporadic, unpredictable disease flares 1, 2, 3. Standard treatments include antimalarial, corticosteroid, and immunosuppressive drugs 4. Despite improved understanding of the disease process, there remains a significant unmet need for new treatment because of the continued high risk of death and progressive organ damage 5, 6, 7. The long‐term burden of disease symptoms and toxic effects of immunosuppressive therapies also significantly affects quality of life 8, 9, 10.
Elevated serum levels of the cytokines B lymphocyte stimulator (BLyS) and APRIL in SLE patients correlate with both disease activity 11, 12 and autoantibody production 13, 14, 15, 16. These factors are therefore promising targets for new investigational therapies. The BLyS inhibitor belimumab has demonstrated efficacy and safety in phase III studies in SLE 17, 18 and is approved for treating patients with active disease. Efficacy of atacicept, the dual APRIL/BLyS inhibitor, was suggested by the APRIL SLE study, which also confirmed its biologic activity in reducing total B cell, plasma cell, and serum immunoglobulin levels in SLE patients 19, 20.
We report herein the findings of ADDRESS II, a randomized, placebo‐controlled phase IIb study of weekly doses of atacicept (75 or 150 mg) versus placebo in patients with active, autoantibody‐positive SLE receiving standard therapy (ClinicalTrials.gov identifier NCT01972568).
Patients and Methods
Study design. In this 24‐week, multicenter, randomized, double‐blind, placebo‐controlled, parallel‐arm, phase IIb study, patients with SLE receiving standard therapy were assigned to once‐weekly subcutaneous injections of placebo or atacicept, 75 or 150 mg. The study included a screening period of up to 4 weeks, a treatment period of 24 weeks (reported herein), and a safety follow‐up period of 24 weeks (see Supplementary Figure 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40360/abstract). A long‐term extension was offered as part of a separate protocol (see Supplementary Figure 2, available at http://onlinelibrary.wiley.com/doi/10.1002/art.40360/abstract). The data reported herein were generated by the ADDRESS II study investigators in Latin America, Asia, South Africa, Europe, UK, and the US.
Prednisone‐equivalent corticosteroid dosages could be adjusted during screening (up to 40 mg/day) but had to be ≤30 mg/day and no more than the dosage at the screening visit by week 4, except that patients receiving <7.5 mg/day at screening could be taking as much as 7.5 mg/day at week 4. Dosage tapering was encouraged during weeks 5–16. One corticosteroid rescue with ≤30 mg/day was allowed, but the dosage had to be reduced to the dosage at week 4 within 7 days. The dosage at week 16 remained stable during weeks 17–24.
Single immunosuppressive or immunomodulatory drugs and/or an antimalarial drug were permitted. We excluded patients who had received treatment with other investigational agents within the previous 3 months, within a period of 5 half‐lives of that drug from the screening visit, or per the washout requirement from the previous protocol, whichever was longest. Patients who had received belimumab (or other anti‐BLyS therapy), rituximab, ocrelizumab, or other B cell–directed biologic drug within 1 year before the screening visit were excluded. Background therapy had to remain stable during the screening and treatment periods. Use of nonpermitted medicines or therapies required discontinuation of atacicept and was considered a treatment failure (for details, see Supplementary Text 1, available at http://onlinelibrary.wiley.com/doi/10.1002/art.40360/abstract.)
Baseline was defined as the screening visit for assessments of disease activity and as treatment day 1 for all other assessments. Interim safety and disease activity data were regularly monitored by an independent Data Safety Monitoring Board.
Eligibility criteria. Eligible patients were age ≥18 years with at least moderately active SLE, as defined by an SLE Disease Activity Index 2000 (SLEDAI‐2K) score of ≥6, met at least 4 of the American College of Rheumatology revised classification criteria for SLE 21, 22, had a disease duration of ≥6 months, and were positive for antinuclear antibody (titer ≥1:80 on HEp‐2 cell substrate) and/or anti–double‐stranded DNA (anti‐dsDNA) antibody (≥30 IU/ml) at screening. Up‐to‐date vaccinations against Streptococcus pneumoniae and influenza virus were required (could be given during screening). Patients with severe glomerulonephritis (urine protein‐to‐creatinine ratio >2.0 mg/mg and/or estimated glomerular filtration rate <40 ml/minute/1.73 m2) and those with major central nervous system manifestations were excluded. (For further eligibility criteria, see Supplementary Text 2, available at http://onlinelibrary.wiley.com/doi/10.1002/art.40360/abstract.)
All patients provided written informed consent. The study was performed in accordance with the Declaration of Helsinki, the International Conference on Harmonisation Note for Guidance on Good Clinical Practice (ICH Topic E6, 1996), and applicable regulatory requirements. All study sites received approval for the study from their local ethics board.
Randomization. An interactive web response system was used to randomize the patients 1:1:1 to either of 3 study arms. Randomization was stratified according to the SLEDAI‐2K total score (6–9 versus ≥10), race (black/African American versus other), and use of mycophenolate mofetil at screening.
Assessment of end points. The primary end point was the SLE Responder Index 4 (SRI‐4) 17, 18 without clinically significant use of nonpermitted medication or treatment at week 24 compared with the screening visit. The SRI‐4 is a composite end point that includes SLEDAI‐2K score ≥4 point reduction; <10% increase in physician's global assessment; no new organ with British Isles Lupus Assessment Group (BILAG) [2004] A (severe) disease, and no more than 1 new BILAG B (moderate) organ score.
Key secondary end points were corticosteroid dosage reduction and patient's global assessment of change at 24 weeks. (For complete details, see Supplementary Text 3, available at http://onlinelibrary.wiley.com/doi/10.1002/art.40360/abstract.) Other disease activity end points were severe flares, as assessed by the Safety of Estrogens in Lupus Erythematosus National Assessment (SELENA) version of the SLEDAI (the SELENA–SLEDAI flare index [SFI]) or by a new BILAG A (severe) manifestation 23. An evaluation of the SRI‐6 in the predefined high disease activity (HDA) subpopulation (SLEDAI‐2K score ≥10 at screening) was performed.
Assessment of biomarkers. Biomarker assessments included serum concentrations of IgG, IgM, and IgA, complement C3 and C4 (measured using Tina‐Quant complement C4 and C3c tests; Roche Diagnostics), and anti‐dsDNA antibodies (measured by enzyme‐linked immunosorbent assay; Phadia).
Assessment of safety. Safety was assessed according to the findings on the physical examination, vital signs measurements, electrocardiograms, and clinical laboratory tests. Reports of adverse events (AEs) and serious AEs were also evaluated.
Statistical analysis. Assuming a 30% response rate with placebo and a 2‐sided alpha value of 0.05, a total of 93 patients per arm would provide 80% power to detect a 20% absolute difference in the proportion of patients achieving an SRI‐4 response for each of the 2 active drug groups versus placebo. With the 1:1:1 randomization ratio, the planned total sample size was therefore 279.
Data analysis was planned for the following populations: intent‐to‐treat (ITT; all randomized patients), modified ITT (all randomized patients who received at least 1 dose of study medication, whether atacicept or placebo), safety (all randomized patients who received at least 1 dose of study medication, whether atacicept or placebo, and were analyzed according to actual treatment received), and HDA (a subgroup of the modified ITT population with a SLEDAI‐2K score of ≥10 at screening). The primary efficacy analysis was performed using the modified ITT population. Step‐down sequential testing was used to control for multiplicity in testing the 2 atacicept doses. The atacicept 150‐mg arm was to be first compared with placebo (primary analysis; 2‐sided α = 0.05) and, if statistically significant, the atacicept 75‐mg arm was compared with placebo. Prespecified sensitivity analyses for the primary end point were conducted as follows: 1) using treatment day 1 as baseline (rather than the screening visit), 2) in the HDA subpopulation, and 3) in patient subgroups with serologically active disease (anti‐dsDNA antibody level ≥15 IU/ml and low levels of complement [C3 <0.9 gm/liter and/or C4 <0.1 gm/liter]).
Analyses of key secondary end points (corticosteroid dosage reduction and patient's global assessment of change at week 24) were performed in a hierarchical manner (first, the 150‐mg dose versus placebo, then, the 75‐mg dose versus placebo) to control for overall Type I error (2‐sided α = 0.05) but became exploratory if the primary end point was not met for either atacicept dose. All other secondary end points were analyzed descriptively using appropriate summary statistics.
All treatment effect tests were conducted at a 2‐sided alpha level of 0.05. P values and 2‐sided 95% confidence intervals (95% CIs) are presented where applicable. Binary end points were analyzed using logistic regression, adjusted for randomization stratification factors. Continuous end points were analyzed using analysis of covariance, adjusted for the baseline value and randomization stratification factors. Time to severe SLE flares was defined according to the BILAG and SFI flare indices separately, and analyzed using a Cox proportional hazard regression model adjusted for baseline stratification factors. Patients not experiencing severe flare were censored at time of last treatment.
Results
Study population. A total of 306 patients were randomized to receive either placebo (n = 100), atacicept 75 mg (n = 102), or atacicept 150 mg (n = 104). Disposition of the study patients is shown in Supplementary Figure 3 (available at http://onlinelibrary.wiley.com/doi/10.1002/art.40360/abstract). All randomized patients (n = 306, the ITT population) received at least 1 dose of atacicept or placebo and were included in the modified ITT and safety analyses. Forty‐four patients (14.4%) discontinued treatment prematurely after randomization (16.0% of the placebo group, 15.7% of the atacicept 75‐mg group, and 11.5% of the atacicept 150‐mg group). The main reasons for discontinuation were AEs (16 patients [36.4%]) or the patient's decision to withdraw (n = 16 patients [36.4%]). One patient was lost to follow‐up.
The demographic features, disease characteristics and severity, and background medications at baseline were similar between groups, except that more patients in the atacicept 150‐mg group than in the 75‐mg group had BILAG 2004 1A or 2B scores at screening (Table 1). BILAG A and B organ system manifestations were mainly mucocutaneous and musculoskeletal (Supplementary Table 1, available at http://onlinelibrary.wiley.com/doi/10.1002/art.40360/abstract), and the most common SLEDAI‐2K disease manifestations included arthritis, rash, and low levels of complement (Supplementary Table 2, available at http://onlinelibrary.wiley.com/doi/10.1002/art.40360/abstract). In the ITT population, 84 patients (27.5%) had serologically active disease.
Table 1.
Demographic features and clinical characteristics at screening (intent‐to‐treat population)a
| Placebo (n = 100) | Atacicept | ||
|---|---|---|---|
| 75 mg (n = 102) | 150 mg (n = 104) | ||
| Age, mean ± SD years | 40 ± 13.0 | 37 ± 11.2 | 39 ± 11.6 |
| Sex, no. (%) | |||
| Female | 90 (90) | 93 (91.2) | 97 (93.3) |
| Male | 10 (10) | 9 (8.8) | 7 (6.7) |
| Race, no. (%) | |||
| White | 78 (78.0) | 72 (70.6) | 66 (63.5) |
| Black/African American | 5 (5.0) | 6 (5.9) | 9 (8.7) |
| Asian | 7 (7.0) | 15 (14.7) | 14 (13.5) |
| Native American or Alaska Native | 4 (4.0) | 3 (2.9) | 4 (3.8) |
| Native Hawaiian or other Pacific Islander | 1 (1.0) | 0 (0.0) | 0 (0.0) |
| Other | 5 (5.0) | 6 (5.9) | 11 (10.6) |
| Hispanic or Latino ethnicity, no. (%) | 57 (57.0) | 51 (50.0) | 45 (43.3) |
| Geographic region | |||
| Europe | 26 (26.0) | 23 (22.5) | 30 (28.8) |
| Asia | 5 (5.0) | 12 (11.8) | 10 (9.6) |
| North America | 22 (22.0) | 21 (20.6) | 20 (19.2) |
| Central and South America | 47 (47.0) | 46 (45.1) | 44 (42.3) |
| Disease duration, mean ± SD years | 6.79 ± 7.648 | 6.77 ± 6.854 | 6.93 ± 6.954 |
| SLEDAI‐2K | |||
| Mean ± SD score | 10 ± 2.8 | 10 ± 3.3 | 10 ± 3.0 |
| No. (%) with score of ≥10 | 52 (52.0) | 54 (52.9) | 51 (49.0) |
| Physician's global assessment score, mean ± SD | 1.50 ± 0.452 | 1.42 ± 0.532 | 1.46 ± 0.460 |
| BILAG 2004 1A or 2B score, no. (%) | 60 (60.0) | 57 (55.9) | 72 (69.2) |
| Serologically active diseaseb | 29 (29.0) | 29 (28.4) | 26 (25.0) |
| Medications | |||
| Corticosteroid (prednisone equivalent) | |||
| Mean ± SD dose, mg/day | 9.40 ± 7.503 | 10.18 ± 8.898 | 9.41 ± 7.417 |
| No. (%) taking >7.5 mg/day | 54 (54.0) | 56 (54.9) | 55 (52.9) |
| Antimalarial drug, no. (%) | 78 (78.0) | 75 (73.5) | 80 (76.9) |
| Immunosuppressive drug, no. (%) | |||
| Azathioprine | 20 (20.0) | 20 (19.6) | 21 (20.2) |
| Methotrexate | 18 (18.0) | 12 (11.8) | 13 (12.5) |
| Mycophenolate mofetil | 16 (16.0) | 16 (15.7) | 18 (17.3) |
| Otherc | 0 (0.0) | 1 (1.0) | 3 (2.9) |
| Serum biomarkers | |||
| ANA titer ≥1:80, no. (%) | 96 (96.0) | 99 (97.1) | 98 (94.2) |
| Anti‐dsDNA ≥15 IU/ml, no. (%) | 47 (47.0) | 51 (50.0) | 49 (47.1) |
| Complement, no. (%) under LLN | |||
| C3 <0.9 gm/liter | 32 (32.0) | 36 (35.3) | 33 (31.7) |
| C4 <0.1 gm/liter | 19 (19.0) | 16 (15.7) | 21 (20.2) |
| IgG, mean ± SD gm/liter | 14.2 ± 4.64 | 13.9 ± 4.66 | 15.0 ± 5.52 |
The intent‐to‐treat population consisted of all patients randomized into the study who received at least 1 dose of study medication. SLEDAI‐2K = Systemic Lupus Erythematosus Disease Activity Index 2000; BILAG = British Isles Lupus Assessment Group; ANA = antinuclear antibody; LLN = lower limit of normal.
Anti–double‐stranded DNA (anti‐dsDNA) antibody positivity (≥15 IU/ml) and low levels of complement (<0.9 gm/liter of C3 and/or <0.1 gm/liter of C4).
Other immunosuppressive drugs were cyclosporine and leflunomide.
The HDA subpopulation included 158 patients (51.6%). Baseline BILAG A and B organ system and SLEDAI‐2K manifestations were mainly balanced across treatment arms, although more patients in the atacicept 150‐mg group than the 75‐mg group experienced BILAG B mucocutaneous (76.5% versus 58.2%) and musculoskeletal (66.7% versus 56.4%) manifestations (Supplementary Table 1). Other clinical features of SLE in the HDA subpopulation were comparable to those in the ITT population, except for the following SLEDAI‐2K manifestations, which were more frequent in HDA patients: low complement levels (50.0% versus 35.6%), anti‐dsDNA antibody positivity (67.7% versus 48.0%), and proteinuria (14.6% versus 8.2%) (Supplementary Tables 1 and 2). Daily doses of corticosteroids at screening were similar in the ITT and HDA populations (Supplementary Table 3, available at http://onlinelibrary.wiley.com/doi/10.1002/art.40360/abstract).
SRI responses. SRI‐4 response. In the primary efficacy analysis, there was a trend toward an improved SRI‐4 response rate at week 24 with atacicept 75 mg (57.8%; adjusted odds ratio [OR] 1.78 [95% CI 1.01–3.12], P = 0.045) and atacicept 150 mg (53.8%; adjusted OR 1.56 [95% CI 0.89–2.72], P = 0.121) versus placebo (44.0%) (Table 2). Differences in treatment response versus placebo were observed from around week 16 (Figure 1A).
Table 2.
SRI responder rates at week 24a
| Population | Response rate, no. (%) | Atacicept 75 mg versus placebo response | Atacicept 150 mg versus placebo response | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Placebo | Atacicept 75 mg | Atacicept 150 mg | Treatment effect size, % | Adjusted OR (95% CI) | P | Treatment effect size, % | Adjusted OR (95% CI) | P | |
| ITT populationb | |||||||||
| No. of patients | 100 | 102 | 104 | ||||||
| SRI‐4 score | 44 (44.0) | 59 (57.8) | 56 (53.8) | 13.8 | 1.78 (1.01–3.12) | 0.045 | 9.8 | 1.56 (0.89–2.72) | 0.121 |
| SRI‐6 score | 30 (30.0) | 32 (31.4) | 38 (36.5) | 1.4 | 1.08 (0.59–1.98) | 0.810 | 6.5 | 1.44 (0.79–2.62) | 0.230 |
| No. serologically active | 29 | 29 | 26 | ||||||
| SRI‐4 score | 7 (24.1) | 18 (62.1) | 16 (61.5) | 37.9 | 5.96 (1.85–19.15) | 0.003 | 37.4 | 7.49 (2.12–26.44) | 0.002 |
| SRI‐6 score | 4 (13.8) | 13 (44.8) | 12 (46.2) | 31.0 | 5.48 (1.49–20.13) | 0.010 | 32.4 | 6.45 (1.66–25.06) | 0.007 |
| HDA populationc | |||||||||
| No. of patients | 52 | 55 | 51 | ||||||
| SRI‐4 score | 22 (42.3) | 33 (60.0) | 32 (62.7) | 17.7 | 2.11 (0.97–4.59) | 0.060 | 20.4 | 2.44 (1.09–5.44) | 0.029 |
| SRI‐6 score | 15 (28.8) | 24 (43.6) | 28 (54.9) | 14.8 | 1.98 (0.88–4.46) | 0.098 | 26.1 | 3.31 (1.44–7.61) | 0.005 |
| No. serologically active | 24 | 25 | 20 | ||||||
| SRI‐4 score | 6 (25.0) | 16 (64.0) | 13 (65.0) | 39.0 | 5.97 (1.70–21.02) | 0.005 | 40.0 | 7.72 (1.88–31.67) | 0.005 |
| SRI‐6 score | 4 (16.7) | 12 (48.0) | 11 (55.0) | 31.3 | 4.88 (1.28–18.64) | 0.020 | 38.3 | 7.31 (1.71–31.28) | 0.007 |
The Systemic Lupus Erythematosus Responder Index 4 (SRI‐4) score represents a ≥4‐point reduction, and the SRI‐6 score represents a ≥6‐point reduction, in the score on the Safety of Estrogens in Lupus Erythematosus National Assessment (SELENA) version of the SLEDAI, with no new BILAG domain score or no more than 1 new BILAG B domain score, and no deterioration from baseline of >0.3 points in the physician's global assessment. Serologically active was defined as the anti‐dsDNA antibody positivity (≥15 IU/ml) and low levels of complement (<0.9 gm/liter of C3 and/or <0.1 gm/liter of C4). Adjusted odds ratios (ORs), 95% confidence intervals (95% CIs), and P values were estimated from a logistic regression model and adjusted for prespecified covariates. See Table 1 for other definitions.
The intent‐to‐treat (ITT) population consisted of all patients randomized into the study who received at least 1 dose of study medication.
The high disease activity (HDA) population consisted of all patients with a SLEDAI‐2K score of ≥10 at screening.
Figure 1.

Effect of atacicept on the disease response of patients with systemic lupus erythematosus (SLE), as determined by the SLE Responder Index 4 (SRI‐4). A, Proportion of SRI‐4 responders in the intent‐to‐treat (ITT) population. B, Proportion of SRI‐4 responders in the high disease activity (HDA) subpopulation. Values at the right are the effect size (Δ) for the indicated treatment groups. * = P < 0.05 versus placebo. D1= treatment day 1.
Since the primary end point was not met, all other analyses are considered exploratory. In a prespecified sensitivity analysis using day 1 as baseline, both atacicept doses improved SRI‐4 response rates at week 24 in the ITT population (P < 0.05 for each comparison) (Supplementary Figure 4, available at http://onlinelibrary.wiley.com/doi/10.1002/art.40360/abstract). The serologically active patient subgroup achieved significantly higher SRI‐4 response rates with both 75 mg (62.1% [adjusted OR 5.96 (95% CI 1.85–19.15)], P = 0.003) and 150 mg (61.5% [adjusted OR 7.49 (95% CI 2.12–26.44)], P = 0.002) of atacicept versus placebo (24.1%) at week 24 (Table 2). Furthermore, dose‐dependent improvements in SRI‐4 response rates were seen in the HDA subpopulation, with atacicept 150 mg improvement (62.7%) being significantly higher than placebo (42.3%) at week 24 (adjusted OR 2.44 [95% CI 1.09–5.44], P = 0.029) (Table 2). Improvement in treatment response was observed from week 4, and this increased to the end of treatment (Figure 1B).
SRI‐6 response. In the HDA subpopulation, an SRI‐6 response at week 24 occurred more frequently with atacicept 150 mg (54.9% [adjusted OR 3.31 (95% CI 1.44–7.61)], P = 0.005) versus placebo (28.8%) (Table 2). Separation of treatment effects was observed as early as week 8 and attained a significant difference by week 16 (Figure 2B). The effect was not significant at week 24 in the ITT population (Figure 2A). The treatment effect size was pronounced in the patients with serologically active disease, both in the ITT population and in the HDA population (Table 2 and Figures 2C and D). Similarly, a subgroup of patients within the HDA subpopulation who were positive for anti‐dsDNA antibody, had low complement levels, or both at baseline achieved a 30.6% increase in the SRI‐6 response rate with atacicept 150 mg treatment versus placebo.
Figure 2.

Effect of atacicept on the disease response of patients with systemic lupus erythematosus (SLE), as determined by the SLE Responder Index 6 (SRI‐6). A, Proportion of SRI‐6 responders in the intent‐to‐treat (ITT) population. B, Proportion of SRI‐6 responders in the high disease activity (HDA) subpopulation. C, Proportion of SRI‐6 responders in the serologically active subgroup (anti–double‐stranded DNA [anti‐dsDNA] antibody positive [≥15 IU/ml] and low levels of complement) of the ITT population. D, Proportion of SRI‐6 responders in the serologically active subgroup (anti‐dsDNA antibody positive and low levels of complement) of the HDA subpopulation. * = P < 0.05 versus placebo. D1 = treatment day 1.
Modified SRI‐4 and SRI‐6 response rates excluding anti‐dsDNA antibodies and complement levels. Although the treatment effect size (∆) for atacicept versus placebo was lower after these SLEDAI serologic parameters were excluded from the calculation of improvement in the SLEDAI‐2K score, differences between atacicept 150 mg and placebo were still apparent in the HDA subpopulation. For the modified SRI‐4, the values ranged from Δ16.5% in HDA patients to Δ23.2% in HDA patients who had anti‐dsDNA antibodies or low complement levels at screening. For the modified SRI‐6, the values ranged from Δ18.2% in HDA patients to Δ21.8% in HDA patients who had anti‐dsDNA antibodies or low complement levels at screening (Supplementary Table 4, available at http://onlinelibrary.wiley.com/doi/10.1002/art.40360/abstract).
Severe disease flares. In the ITT population, the incidence of severe disease flares was reduced with atacicept 75 mg according to the incidence of new BILAG A manifestation (Figure 3A) and with atacicept 150 mg according to the SFI (2.9% versus 14.0% with placebo; hazard ratio (HR) 0.18 [95% CI 0.05–0.62], P = 0.002). The impact on disease flare was more pronounced in the HDA subpopulation: both atacicept doses led to reductions in severe flares according to both the incidence of a new BILAG A manifestation (Figure 3B) and the SFI (with 150 mg, HR 0.19 [95% CI 0.05–0.68], P = 0.004; with 75 mg, HR 0.33 [95% CI 0.12–0.94], P = 0.029) versus placebo (25.0%).
Figure 3.
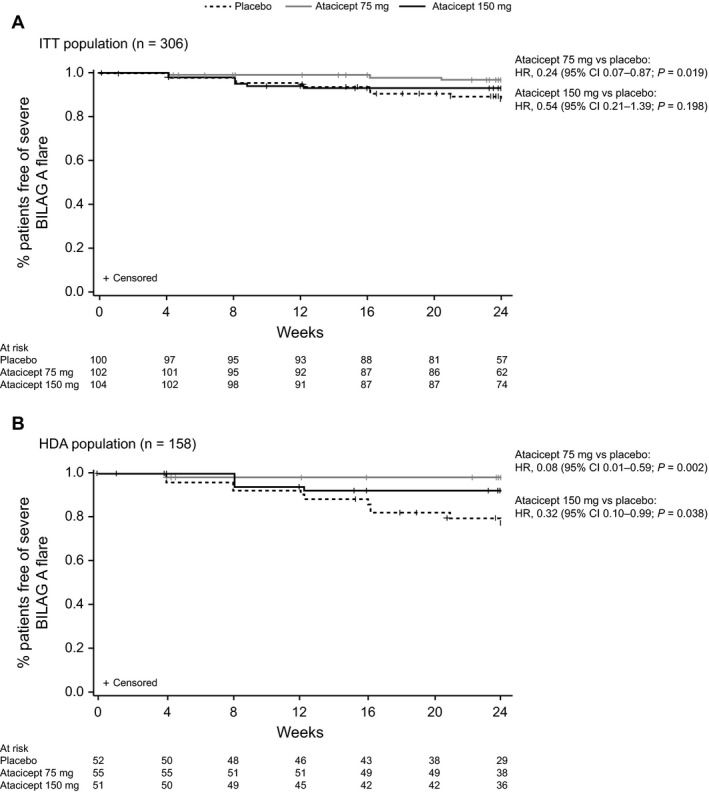
Kaplan‐Meier analysis of time to first severe flare according to scores on the British Isles Lupus Assessment Group (BILAG) A grade manifestations. A, Intent‐to‐treat (ITT) population. B, High disease activity (HDA) subpopulation. Numbers across the bottom of the x‐axes are the numbers of patients at the indicated time points. HR = hazard ratio; 95% CI = 95% confidence interval.
Findings of key secondary end points. Atacicept 75 mg or 150 mg did not significantly increase the proportion of patients achieving a corticosteroid dosage reduction to ≤7.5 mg/day at week 24 versus placebo (17.9%, 11.3%, and 18.9%, respectively) in patients whose corticosteroid dosage was ≥10 mg/day at screening. Similarly, with atacicept versus placebo, no difference was observed in the proportion of patients reporting the following 7 categories of change in the patient's global assessment: very much improved, much improved, minimally improved, no change, minimally worse, much worse, or very much worse at week 24 since beginning the treatment (data not shown).
Levels of biomarkers. In patients with low levels of serum complement C3 (n = 101) or C4 (n = 56) at baseline, atacicept at either dosage led to a steady increase in serum C3 or C4 levels, respectively, from week 4 until week 24, but in the placebo group, the levels remained similar to baseline throughout the treatment period. At week 24, the median percentage increase in the serum C3 levels compared with baseline (treatment day 1) was 5.3% with atacicept 75 mg and 22.1% with atacicept 150 mg versus 1.5% with placebo. The median percentage increase in the serum C4 level was 64.5% with atacicept 75 mg and 128.6% with atacicept 150 mg (Figures 4A and B). In patients with a low C3 and/or C4 level at baseline (n = 109), a normalized C3 and C4 level at week 24 was achieved in 52.6% and 30.6% taking atacicept 150 mg and 75 mg, respectively, versus 17.1% taking placebo.
Figure 4.

Changes in serum biomarkers over time. The median percentage change from baseline over 24 weeks is shown for A, serum complement C3 levels (C3‐low patients), B, serum complement C4 levels (C4‐low patients), C, anti–double‐stranded DNA (anti‐dsDNA) antibody levels (anti‐dsDNA antibody–positive patients), and D, serum IgG levels.
In patients with anti‐dsDNA antibodies at baseline (n = 147), both atacicept dosages reduced anti‐dsDNA antibody levels over time. At week 24, the median percentage change versus baseline was –23.6% with atacicept 75 mg and –28.2% with atacicept 150 mg. Anti‐dsDNA antibody levels increased by a median of 16.0% with placebo treatment (Figure 4C). Treatment with atacicept 150 mg and 75 mg increased the likelihood of not having a positive anti‐dsDNA antibody result at week 24 as compared with placebo (30.6%, 19.6%, and 2.1%, respectively).
At week 24, serum IgG levels were reduced from baseline by ~30% and 25% with atacicept 150 and 75 mg, respectively (Figure 4D and Supplementary Figure 5, available at http://onlinelibrary.wiley.com/doi/10.1002/art.40360/abstract). There were no incidents of severe hypogammaglobulinemia (serum IgG <3 gm/liter). The median percentage reduction in the serum levels of IgA (~50% with 150 mg and ~45% with 75 mg) and IgM (~70% with 150 mg and ~60% with 75 mg) compared with baseline were of greater magnitude than the median percentage reduction in the serum level of IgG (Supplementary Figure 6, available at http://onlinelibrary.wiley.com/doi/10.1002/art.40360/abstract).
Safety. Rates of treatment‐emergent AEs (TEAEs) were higher with atacicept 75 mg and 150 mg versus placebo (81.4%, 80.8%, and 72.0%, respectively). The most commonly reported AEs across treatment arms were injection site reactions, injection site pain, urinary tract infections, upper respiratory tract infections, and diarrhea (Supplementary Table 5, available at http://onlinelibrary.wiley.com/doi/10.1002/art.40360/abstract). Incidence rates of TEAEs were similar with atacicept 75 and 150 mg and placebo, except for upper respiratory tract infections (9.8%, 12.5%, and 3.0%, respectively) and diarrhea (6.9%, 11.5%, and 5.0%), which were more common with atacicept, and urinary tract infections (11.8%, 11.5%, and 17.0%), which were more common with placebo. Other commonly reported infections with atacicept 75 and 150 mg included nasopharyngitis (4.9% and 6.7%), bronchitis (1.0% and 3.8%), and influenza (2.9% and 2.9%). Pneumonia occurred in 1 patient (1.0%) in each atacicept group. There were fewer serious TEAEs with atacicept 75 and 150 mg than with placebo (5.8%, 8.8%, and 12.0%, respectively). One patient treated with atacicept 150 mg (1%) reported a serious infection, compared with 5.9% treated with atacicept 75 mg and 5.0% treated with placebo.
The incidence of TEAEs leading to treatment discontinuation was comparable between study arms. Severe treatment‐emergent injection site reactions were infrequent, occurring in 1 patient (1.0%) receiving atacicept 75 mg and 1 patient (1.0%) receiving placebo. There were no notable differences in clinical laboratory parameters (i.e., hematology, liver function tests) between either of the atacicept groups and placebo. No deaths occurred during the study. Immunogenicity was assessed predose and postdose: 7 patients in the atacicept 75‐mg group and 1 patient in the atacicept 150‐mg group had measurable antibodies to atacicept at week 24.
Discussion
ADDRESS II compared the efficacy and safety of 2 doses of atacicept (75 and 150 mg) with placebo in patients with active, autoantibody‐positive SLE receiving standard treatment. The primary end point of an SRI‐4 response at week 24 was not met in the ITT population, although there was a trend toward increased response rates with atacicept. A more robust increase in the SRI‐4 response rates for both atacicept doses was observed in the sensitivity analysis in which day 1 was used as baseline. The treatment effect size according to the SRI‐4 in the ITT population observed with atacicept 150 mg versus placebo (∆9.8% [adjusted OR 1.56] and ∆14.8% [adjusted OR 1.96] in the primary and sensitivity analyses, respectively) was similar to that observed with the highest dose of belimumab (10 mg/kg) in the 52‐week BLISS‐52 and BLISS‐76 studies (∆14.0% and ∆9.7%, respectively) 17, 18. Similar to the much larger BLISS studies, this trial was conducted on a background of standard care and included some adjustments in the steroid dosage, which protects the safety of the sickest subpopulations, but supports high SRI‐4 response rates in the placebo group among patients with moderate disease, a tradeoff that may limit the effect size if there is a ceiling of response rates for targeted treatments in a heterogeneous disease.
We analyzed response in the stratified subpopulation of patients with HDA (SLEDAI‐2K of ≥10 at baseline). Major treatment effects were observed with atacicept 150 mg versus placebo according to both the SRI‐4 (∆20.4%), a measure of clinically meaningful response, and the SRI‐6 (∆26.1%), an end point that requires a greater response. Even with the greater threshold for an SRI‐6 response in patients with severe disease, the response rate with atacicept 150 mg remained high (54.9%). These findings are consistent with the findings of subgroup analyses in blisibimod and belimumab studies that demonstrated an increased treatment effect in patients with HDA 24, 25. These data suggest that when the target population is patients with HDA, there is a greater likelihood of discriminating an effective treatment from placebo, and this may have implications for future trial designs.
Serologic activity also defined a subpopulation with increased responsiveness to belimumab 18, 24. We observed increased treatment effect sizes for SRI‐4 and SRI‐6 with atacicept in patients with serologically active disease, both in the ITT and HDA populations, consistent with the roles played by BLyS in B cell proliferation and maturation, autoantibody production, and thus, disease pathogenesis 16, 26, 27, 28. Since serologic normalization can lead to a potential 4‐point improvement in the SLEDAI score (potentially accounting for achievement of an SRI‐4 response without clinical improvement), the extent to which serologic effects contributed to SRI response rates in these populations was examined by excluding serologic data from the SRI assessment. Despite reductions in treatment effect sizes, response differences were still pronounced, confirming that atacicept treatment led to both clinical and serologic improvements.
Consistent with the post hoc analysis of the APRIL‐SLE study, in which flare rates were reduced with atacicept 150 mg 29, we observed reductions in the incidence of new, severe BILAG A scores and SFI severe flares with atacicept versus placebo in both the ITT population and the HDA subpopulation. Atacicept has therefore demonstrated consistent flare reduction in 2 studies.
The biomarker results were comparable with those previously observed in studies of atacicept, including the APRIL‐SLE study 29, 30, 31. Reduced serum immunoglobulin and anti‐dsDNA autoantibody levels and increased complement levels were apparent soon after administration of atacicept and continued to the end of treatment. The magnitude of changes appeared to be greater than those reported for agents targeting BLyS alone 17, 18, 25, 32. This is consistent with the role of APRIL in plasma cell survival 33, 34, antibody production, and potentially, associated complement consumption.
The safety profile of atacicept was acceptable. Compared with placebo, there was no increase in the overall frequency of serious AEs or in the subset of serious infections associated with active treatment. Reassuringly, there were no cases of severe hypogammaglobulinemia and no deaths. The atacicept 150 mg treatment arm of the APRIL‐SLE study was prematurely terminated as a cautionary measure, as recommended by the Independent Data Monitoring Committee, following 2 deaths from pulmonary infections complicated by pulmonary alveolar hemorrhage. Although most large clinical trials of new therapy in SLE report similar small numbers of deaths 17, 18, 24, 25, 32, several additional risk‐mitigation measures were established for the ADDRESS II study in response to these fatal infection outcomes. These included requirements for medical monitor review of the patients’ screening data to confirm eligibility as well as up‐to‐date vaccinations against pneumococcus and seasonal influenza (could be administered during the screening period up to 2 weeks prior to randomization). These measures may have minimized the risk of serious infections in this study population.
Potential limitations of the study include the low proportion of black/African American patients enrolled and the 24‐week duration. Future studies will be needed to evaluate responses in patients from populations with renal and/or central nervous system disease and populations known to be at high risk of developing lupus and experiencing poor outcomes. More prolonged atacicept treatment may have provided more reliable efficacy discrimination, and it will be important to assess the safety of longer‐term treatment with atacicept. Completers of this 24‐week study were offered enrollment in a long‐term extension study, the results of which will be reported as soon as they are available.
In summary, although this phase IIb study did not meet its primary end point, there was robust discrimination between atacicept and placebo treatments in multiple end points, particularly in subpopulations with serologic activity and/or high levels of disease activity. There was no increase in the risk of serious AEs, including serious infections, in patients treated with atacicept. These results support further clinical evaluation of atacicept in SLE.
Author Contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Merrill had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Merrill, Fraser, Isenberg.
Acquisition of data
Merrill, Kao, Fraser.
Analysis and interpretation of data
Merrill, Wallace, Wax, Kao, Fraser, Chang, Isenberg.
Role of the Study Sponsor
EMD Serono, Inc., a division of Merck KGaA (Darmstadt, Germany), was involved in the study design, the collection, analysis, and interpretation of the data, and the writing of the manuscript. EMD Serono (including EMD Serono authors) and all other authors approved the final version of the manuscript and were involved in the final decision to submit the manuscript for publication. Writing assistance was provided by the Bioscript Group (Macclesfield, UK, a scientific communications group) and was directed and supported by EMD Serono, Inc.
Supporting information
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Supinfo
Acknowledgments
The authors thank the patients and their families, as well as the ADDRESS II study team, for their participation.
ClinicalTrials.gov identifier: NCT01972568.
Supported by EMD Serono, Inc., a division of Merck KGaA, Darmstadt, Germany.
Dr. Merrill has received consulting fees from EMD Serono, Anthera Pharmaceuticals, GlaxoSmithKline, and Eli Lilly (less than $10,000 each). Dr. Wallace has received consulting fees from EMD Serono (less than $10,000). Drs. Wax, Kao, Fraser, and Chang have long‐term incentives from EMD Serono. Dr. Isenberg has received consulting fees from EMD Serono (less than $10,000); fees provided to Dr. Isenberg are passed on to a local arthritis charity.
[The copyright line for this article was changed on August 4, 2018 after original online publication.]
References
- 1. Rekvig OP, Van der Vlag J. The pathogenesis and diagnosis of systemic lupus erythematosus: still not resolved. Semin Immunopathol 2014;36:301–11. [DOI] [PubMed] [Google Scholar]
- 2. Bertsias G, Cervera T, Boumpas D. Systemic lupus erythematosus: pathogenesis and clinical features. 2012. URL: http://www.eular.org/myuploaddata/files/sample%20chapter20_mod%2017.pdf.
- 3. Lisnevskaia L, Murphy G, Isenberg D. Systemic lupus erythematosus. Lancet 2014;384:1878–88. [DOI] [PubMed] [Google Scholar]
- 4. Bertsias G, Ioannidis JP, Boletis J, Bombardieri S, Cervera R, Dostal C, et al. EULAR recommendations for the management of systemic lupus erythematosus: report of a task force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics. Ann Rheum Dis 2008;67:195–205. [DOI] [PubMed] [Google Scholar]
- 5. Bernatsky S, Boivin JF, Joseph L, Manzi S, Ginzler E, Gladman DD, et al. Mortality in systemic lupus erythematosus. Arthritis Rheum 2006;54:2550–7. [DOI] [PubMed] [Google Scholar]
- 6. Cervera R, Khamashta MA, Font J, Sebastiani GD, Gil A, Lavilla P, et al. Morbidity and mortality in systemic lupus erythematosus during a 10‐year period: a comparison of early and late manifestations in a cohort of 1,000 patients. Medicine (Baltimore) 2003;82:299–308. [DOI] [PubMed] [Google Scholar]
- 7. Lateef A, Petri M. Unmet medical needs in systemic lupus erythematosus. Arthritis Res Ther 2012;14 Suppl 4:S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schmeding A, Schneider M. Fatigue, health‐related quality of life and other patient‐reported outcomes in systemic lupus erythematosus. Best Pract Res Clin Rheumatol 2013;27:363–75. [DOI] [PubMed] [Google Scholar]
- 9. Gordon C, Isenberg D, Lerstrom K, Norton Y, Nikai E, Pushparajah DS, et al. The substantial burden of systemic lupus erythematosus on the productivity and careers of patients: a European patient‐driven online survey. Rheumatology (Oxford) 2013;52:2292–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Thamer M, Hernan MA, Zhang Y, Cotter D, Petri M. Prednisone, lupus activity, and permanent organ damage. J Rheumatol 2009;36:560–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Salazar‐Camarena DC, Ortiz‐Lazareno PC, Cruz A, Oregon‐Romero E, Machado‐Contreras JR, Munoz‐Valle JF, et al. Association of BAFF, APRIL serum levels, BAFF‐R, TACI and BCMA expression on peripheral B‐cell subsets with clinical manifestations in systemic lupus erythematosus. Lupus 2016;25:582–92. [DOI] [PubMed] [Google Scholar]
- 12. Hegazy M, Darwish H, Darweesh H, El‐Shehaby A, Emad Y. Raised serum level of APRIL in patients with systemic lupus erythematosus: correlations with disease activity indices. Clin Immunol 2010;135:118–24. [DOI] [PubMed] [Google Scholar]
- 13. McCarthy EM, Lee RZ, Ni Gabhann J, Smith S, Cunnane G, Doran MF, et al. Elevated B lymphocyte stimulator levels are associated with increased damage in an Irish systemic lupus erythematosus cohort. Rheumatology (Oxford) 2013;52:1279–84. [DOI] [PubMed] [Google Scholar]
- 14. Zhang J, Roschke V, Baker KP, Wang Z, Alarcon GS, Fessler BJ, et al. Cutting edge: a role for B lymphocyte stimulator in systemic lupus erythematosus. J Immunol 2001;166:6–10. [DOI] [PubMed] [Google Scholar]
- 15. Stohl W, Metyas S, Tan SM, Cheema GS, Oamar B, Roschke V, et al. Inverse association between circulating APRIL levels and serological and clinical disease activity in patients with systemic lupus erythematosus. Ann Rheum Dis 2004;63:1096–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Koyama T, Tsukamoto H, Miyagi Y, Himeji D, Otsuka J, Miyagawa H, et al. Raised serum APRIL levels in patients with systemic lupus erythematosus. Ann Rheum Dis 2005;64:1065–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Navarra SV, Guzmán RM, Gallacher AE, Hall S, Levy RA, Jimenez RE, et al, for the BLISS‐52 Study Group . Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo‐controlled, phase 3 trial. Lancet 2011;377:721–31. [DOI] [PubMed] [Google Scholar]
- 18. Furie R, Petri M, Zamani O, Cervera R, Wallace DJ, Tegzová D, et al. A phase III, randomized, placebo‐controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum 2011;63:3918–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dall'Era M, Chakravarty E, Wallace D, Genovese M, Weisman M, Kavanaugh A, et al. Reduced B lymphocyte and immunoglobulin levels after atacicept treatment in patients with systemic lupus erythematosus: results of a multicenter, phase Ib, double‐blind, placebo‐controlled, dose‐escalating trial. Arthritis Rheum 2007;56:4142–50. [DOI] [PubMed] [Google Scholar]
- 20. Pena‐Rossi C, Nasonov E, Stanislav M, Yakusevich V, Ershova O, Lomareva N, et al. An exploratory dose‐escalating study investigating the safety, tolerability, pharmacokinetics and pharmacodynamics of intravenous atacicept in patients with systemic lupus erythematosus. Lupus 2009;18:547–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1982;25:1271–7. [DOI] [PubMed] [Google Scholar]
- 22. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1997;40:1725. [DOI] [PubMed] [Google Scholar]
- 23. Isenberg DA, Rahman A, Allen E, Farewell V, Akil M, Bruce IN, et al. BILAG 2004: development and initial validation of an updated version of the British Isles Lupus Assessment Group's disease activity index for patients with systemic lupus erythematosus. Rheumatology (Oxford) 2005;44:902–6. [DOI] [PubMed] [Google Scholar]
- 24. Van Vollenhoven RF, Petri MA, Cervera R, Roth DA, Ji BN, Kleoudis CS, et al. Belimumab in the treatment of systemic lupus erythematosus: high disease activity predictors of response. Ann Rheum Dis 2012;71:1343–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Furie RA, Leon G, Thomas M, Petri MA, Chu AD, Hislop C, et al. A phase 2, randomised, placebo‐controlled clinical trial of blisibimod, an inhibitor of B cell activating factor, in patients with moderate‐to‐severe systemic lupus erythematosus, the PEARL‐SC study. Ann Rheum Dis 2015;74:1667–75. [DOI] [PubMed] [Google Scholar]
- 26. Ota M, Duong BH, Torkamani A, Doyle CM, Gavin AL, Ota T, et al. Regulation of the B cell receptor repertoire and self‐reactivity by BAFF. J Immunol 2010;185:4128–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yu G, Boone T, Delaney J, Hawkins N, Kelley M, Ramakrishnan M, et al. APRIL and TALL‐I and receptors BCMA and TACI: system for regulating humoral immunity. Nat Immunol 2000;1:252–6. [DOI] [PubMed] [Google Scholar]
- 28. Ju S, Zhang D, Wang Y, Ni H, Kong X, Zhong R. Correlation of the expression levels of BLyS and its receptors mRNA in patients with systemic lupus erythematosus. Clin Biochem 2006;39:1131–7. [DOI] [PubMed] [Google Scholar]
- 29. Isenberg D, Gordon C, Licu D, Copt S, Rossi CP, Wofsy D. Efficacy and safety of atacicept for prevention of flares in patients with moderate‐to‐severe systemic lupus erythematosus (SLE): 52‐week data (APRIL‐SLE randomised trial). Ann Rheum Dis 2015;74:2006–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Van Vollenhoven RF, Kinnman N, Vincent E, Wax S, Bathon J. Atacicept in patients with rheumatoid arthritis and an inadequate response to methotrexate: results of a phase II, randomized, placebo‐controlled trial. Arthritis Rheum 2011;63:1782–92. [DOI] [PubMed] [Google Scholar]
- 31. Van Vollenhoven RF, Wax S, Li Y, Tak PP. Safety and efficacy of atacicept in combination with rituximab for reducing the signs and symptoms of rheumatoid arthritis: a phase II, randomized, double‐blind, placebo‐controlled pilot trial. Arthritis Rheumatol 2015;67:2828–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Isenberg DA, Petri M, Kalunian K, Tanaka Y, Urowitz MB, Hoffman RW, et al. Efficacy and safety of subcutaneous tabalumab in patients with systemic lupus erythematosus: results from ILLUMINATE‐1, a 52‐week, phase III, multicentre, randomised, double‐blind, placebo‐controlled study. Ann Rheum Dis 2016;75:323–31. [DOI] [PubMed] [Google Scholar]
- 33. Benson MJ, Dillon SR, Castigli E, Geha RS, Xu S, Lam KP, et al. Cutting edge: the dependence of plasma cells and independence of memory B cells on BAFF and APRIL. J Immunol 2008;180:3655–9. [DOI] [PubMed] [Google Scholar]
- 34. Belnoue E, Pihlgren M, McGaha TL, Tougne C, Rochat AF, Bossen C, et al. APRIL is critical for plasmablast survival in the bone marrow and poorly expressed by early‐life bone marrow stromal cells. Blood 2008;111:2755–64. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Supinfo