

Abstract
A series of nine C‐functionalized cationic diazaoxatriangulene (DAOTA) dyes have been successfully synthesized and fully characterized, including X‐ray structural analysis of four derivatives. The introduction of electron‐withdrawing or ‐donating functions enables the tuning of both electro‐ and photochemical properties with, for instance, two consecutive (reversible) reductions or oxidations observed for nitro or amino derivatives, respectively. The substituents also impacted on the optical properties, with absorption maxima varying from λ=528 to 640 nm and fluorescence being shifted from the yellow to the red range, up to λ=656 nm.
Keywords: dyes/pigments, electrochemistry, fluorescence, substituent effects, synthesis design
Introduction
Cationic heterocyclic triangulenes are well‐known chromophores studied in a variety of fields from biology,1 supramolecular chemistry,2 electrogenerated chemiluminescence,3 photosensitization,4 to materials science.5 These derivatives (Figure 1), which differ by the number of bridging oxygen and nitrogen atoms, are commonly named TOTA, ADOTA, DAOTA, and TATA in view of their trioxa, azadioxa, diazaoxa, and triaza nature, respectively.6 They present 1) exceptionally high chemical stabilities for carbenium ions (pK R + up to 23.7),7 and 2) intense fluorescence in the visible range characterized by particularly long lifetimes.8 Syntheses are straightforward from simple triarylcarbenium precursors (one to three steps in total). In terms of fluorescence, DAOTA moieties are noticeable because they present the most redshifted emission (λ=590 nm), and the highest brightness (ϵΦ≈7000 m −1 cm−1) of the series. These features are particularly relevant for applications in fluorescence microscopy in the transparency window of biological media.9
Figure 1.
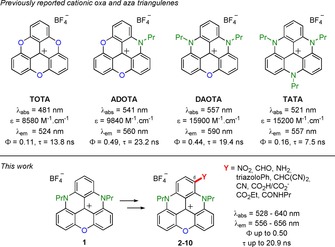
Top: Cationic triangulenes with corresponding optical properties in acetonitrile. Bottom: Scope of the study.
So far, molecular engineering of DAOTA structures has focused mainly on the atoms or residues attached to the nitrogen atoms. Specific side chains can be introduced to provide efficient fluorophores for bioconjugation to bovine serum album (BSA),10 fluorescence lifetime imaging (FLIM) of G‐quadruplex,1h, 11 or pH‐controlled selective imaging of late endosomes.1i The C‐functionalized triangulenes have only been scarcely reported and such derivatives can be made from prefunctionalized TOTA and ADOTA precursors.12 The N‐functionalized triaryl‐TATA derivatives react under oxidative chlorination conditions with moderate levels of chemo‐ and regioselectivity.5c Similar behavior is observed with TOTA moieties.13 Recently, the iridium‐catalyzed C−H borylation of TATA moieties has been achieved to give access, after cross‐coupling chemistry, to C‐functionalized triaryl derivatives. This sequence of reactions is, to the best of our knowledge, the only example of regioselective late‐stage functionalization of a triangulene skeleton.14
Herein, inspired by studies on the postfunctionalization of related cationic [6]‐ and [4]helicenes,15 we report on the regioselective nitration and formylation of DAOTA 1 that has opened up unprecedented access to various triangulenes functionalized at position 6 (compounds 2–10). The C‐substituents influence the solid‐state conformations and packing of the structures. The added functional groups also impact strongly on the physicochemical properties of the DAOTA core in solution. This late‐stage functionalization strategy thus allows, for the first time, in the DAOTA series, fine‐tuning of both electrochemical and optical properties of this important class of dyes and luminophores.
Results and Discussion
Synthesis
Initial attempts at functionalizing 1 were performed with halogenating reagents, such as N‐chloro‐ or N‐bromosuccinimide. However, as observed for TOTA,13 TATA,5c and diaza[4]helicene analogues,15b mixtures of mono‐ and polyhalogenated species are obtained. With substrate 1, separation of the various products was impossible. To overcome this hurdle, it was decided to avoid polysubstitution reactions as a whole. The introduction of strong electron‐withdrawing groups was considered, such that, after the first substitution, these substituents would deactivate the triangulene core and stop further reactivity. Upon nitration of 1 under biphasic conditions (CH2Cl2/HNO3 (13:2)) the orange‐colored mononitro derivative 2 was obtained in excellent yield of 98 % (Scheme 1). Experimentally, due to the low solubility of 1 in dichloromethane, dilution (0.05 m) and prolonged reaction times are required.16 Following the same strategy, compound 1 was treated under Vilsmeier–Haack conditions with large excesses of POCl3 and DMF at 90 °C for 14 h to afford formylated derivative 3. Recrystallization from a mixture of dichloromethane/toluene was necessary to isolate the product as a pink solid in yields of up to 65 %.17
Scheme 1.
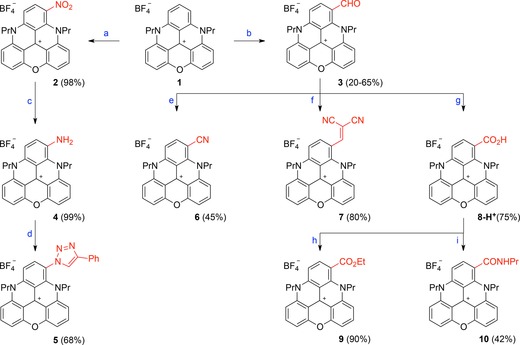
Synthesis of functionalized DAOTAs. Reagents and conditions: a) HNO3 (60 % aqueous solution), CH2Cl2, 25 °C, 12 h. b) POCl3 (24 equiv), DMF (12 equiv), 90 °C, 14 h. c) H2, Pd/C (20 mol %), CH2Cl2/MeOH, 25 °C, 30 min. d) tBuONO (1.5 equiv), trimethylsilyl azide (TMSN3; 2 equiv), CH3CN, 0 to 25 °C, 1 h, then PhC≡CH, CuSO4 ⋅5 H2O (0.1 equiv), ascorbic acid (0.2 equiv), NaHCO3 (0.2 equiv), CH3CN/H2O, 25 °C, 30 min. e) NaN3 (1.5 equiv), trifluoromethanesulfonic acid (TfOH; 3 equiv), CH3CN, 25 °C, 5 min. f) NCCH2CN (3 equiv), Ph3P (20 mol %), CH3CN, 130 °C (microwave (MW)), 25 °C, 1 h. g) NaH2PO4 (1 equiv), NaClO2 (2 equiv), H2O2, CH3CN, 60 °C, 1 h, then HBF4 (1 m aqueous solution). h) SOCl2 (6 equiv), CH2Cl2, 25 °C, 10 min, then EtOH (6 equiv), 25 °C, 10 min. i) SOCl2 (6 equiv), CH2Cl2, 25 °C, 10 min, then PrNH2 (18 equiv), 0 °C then 25 °C, 15 min.
With nitro and aldehyde derivatives 2 and 3, respectively, in hand, we performed further derivatizations. For instance, compound 2 was converted efficiently into (light green) amino derivative 4 by hydrogenation over Pd/C (99 % yield). To prepare 5, a two‐step procedure was necessary. First, amino 4 was transformed into an azido derivative by treatment with tert‐butyl nitrite and azidotrimethylsilane in acetonitrile for 1 h. This compound was unstable and quickly engaged in a copper‐catalyzed azide–alkyne cycloaddition. Treatment with phenylacetylene, CuSO4, NaHSO4, and ascorbic acid afforded 5 in 68 % overall yield.
Formylated 3 was derivatized into cyano 6 under Schmidt conditions, with NaN3 and TfOH in acetonitrile (45 % yield).18, 19 A Knoevenagel condensation was achieved by treatment of 3 with malonitrile (3 equiv) under PPh3 catalysis (20 mol %) in acetonitrile (130 °C, MW irradiation, 1 h) to afford 7 in 80 % yield. Compound 8‐H+ was obtained under Pinnick–Kraus conditions, by treating 3 with NaH2PO4 and NaClO2, in the presence of H2O2 as a scavenger. After acidic workup, the product was isolated as the carboxylic acid in 75 % yield. Finally, compound 8‐H+ was converted into ester 9 and amide 10 by using a stepwise protocol. In both cases, the acid was converted into the corresponding acyl chloride derivative by treatment of 8‐H+ with SOCl2 in dichloromethane. Upon reactions with ethanol (excess) or n‐propylamine, products 9 and 10 were obtained in 90 and 42 % yields, respectively.
Overall, in very few steps, a rather large chemical diversity of cationic DAOTA derivatives could be afforded by this late‐stage functionalization strategy. The observed regioselectivity, in favor of diaza‐ rather than azaoxa‐substituted phenyl rings, is in agreement with that previously observed on related helicenes.15a,15b, 20
Solid‐state analysis
Crystals of DAOTA 1, nitro 2, aldehyde 3, and amino 4 derivatives were obtained by slow diffusion of hexane into solutions of the corresponding compounds in dichloromethane. The substituents influenced the stacking of the triangulene cores and the conformations of the N‐alkyl side chains in the solid state. The X‐ray structure of 1 is presented in Figure S1 in the Supporting Information and relevant values for all compounds are summarized in Table 1; complementary data can be found in the Supporting Information.
Table 1.
Selected solid‐state parameters for 1–4.
| |||
|---|---|---|---|
| Molecule[a] | Y | Distance[a] [Å] | RMSD[b] [Å] |
| 1 | H | 3.5 | 0.024 |
| 2 | NO2 | 3.4 | 0.123 |
| 3 | CHO | 3.4 | 0.041 |
| 4 | NH2 | 3.7 | 0.131 |
[a] Interplanar distance of stacking. [b] Root‐mean‐square deviation (RMSD) of the atomic positions from the calculated least‐squares plane.
As already reported for TATA and ADOTA, DAOTA 1 is a quasi‐planar system,7b with an average RMSD value of 0.024 Å. Both side chains are disposed in the syn conformation, and extensive π–π overlap means that DAOTA stacks in dimers that are slightly shifted, with close interplanar distances of 3.5 Å, in a head to tail disposition (Figure 2 a). The dimers stack in layers running along the a axis, with a distance of 4.7 Å between the dimers. Hydrogen bonds are observed between the BF4 − counterions and side chains (Figure 2 b; tables of the hydrogen bonds are reported in the Supporting Information). A similar situation is observed for aldehyde derivative 3, which also stacks in dimers in a head to tail disposition with an equal interplanar distance of 3.4 Å (Figure 2 c). In this case, the propyl side chains are disposed in an anti conformation, layers form along the a axis, and hydrogen‐bonding interactions are observed with the BF4 − counterions (Figure 2 d). The introduction of the formyl group provokes a slight increase of the RMSD value (0.041 Å).
Figure 2.
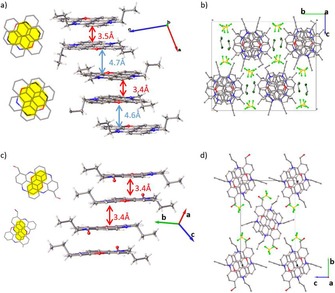
a) Extensive π–π stacking of 1, and b) view along the a axis, showing the column of triangulene molecules formed. c) The π–π stacking of 3 and d) view along the a axis, showing the triangulene molecules. Overlap between the π systems of close neighboring molecules is shown in yellow.
Somewhat surprisingly, and in contrast to the diaza [4]helicene analogue of 3,15b the predominant conformation of the CHO group is s‐trans, rather than s‐cis. In the solid state, the formyl group and adjacent C−H of the same molecule interact, but with a C−H⋅⋅⋅O angle of 102°. More linear geometries are observed for intermolecular hydrogen bonds (Figure 3). In solution, both conformations occur, as shown by 1H NOESY NMR spectroscopy experiments (CD3CN; Figure 4). Through‐space correlation between the H of the formyl group, H9, and H10 indicates the occurrence of the s‐trans geometry, whereas close proximity with H7 and H8 demonstrates the s‐cis conformation.
Figure 3.
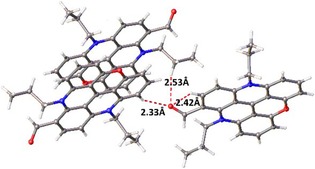
Inter‐ and intramolecular hydrogen bonds of aldehyde derivative 3.
Figure 4.
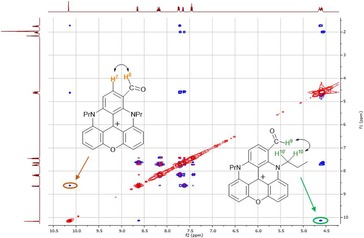
1H NOESY map of 3 (CD3CN, 400 MHz, 298 K) showing the minor (left, s‐cis) and major (right, s‐trans) conformers.
Nitro derivative 2 presents a greater RSMD value of 0.123 Å. The π–π interactions are still present in this derivative; however, layered arrangements are observed rather than columns, as characterized by a head to tail disposition with an interplane distance of 3.4 Å (Figure 5 a). These layers are held together by hydrogen‐bonding interactions with the BF4 − counterions (Figure 5). Amino derivative 4 also presents a greater RSMD value of 0.131 Å. The dimer is held in head to tail disposition by π–π interactions, forming columns, as observed for 1 and 3. The distance is slightly increased to 3.7 Å (Figure 5 c). Again, hydrogen bonds are observed with the BF4 − counterions (Figure 5 d).
Figure 5.
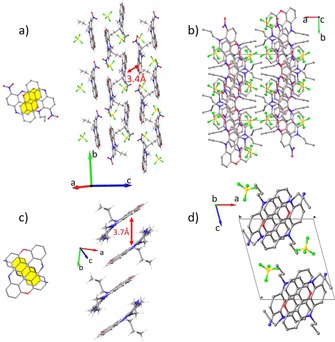
a) The π–π stacking of nitro 2 and b) view along the c axis of the columns. c) The π–π stacking of amino 4 and d) view along the c axis of the columns. Overlap between the π systems of close neighboring molecules is shown in yellow. Disorder was omitted for clarity.
Overall, in the four structures, π–π interactions generate dimers in a head to tail disposition. They induce the formation of columns in DAOTA 1, aldehyde 3, and amino 4, or layers in the case of nitro derivative 2. The BF4 − counterions act as hydrogen‐bond donors, generating interactions between and within the columns (layers for dye 2).
Electrochemical properties
The electrochemical behavior of 1–4 was analyzed by means of cyclic voltammetry (CV). The CV curves presented in Figure 6 were recorded in degassed solutions containing 10−3 m of each of the DAOTAs in acetonitrile and 0.1 m tetrabutylammonium hexafluorophosphate, [TBA][PF6], as the supporting electrolyte (electrochemical data are gathered in Table 2). It is noteworthy that repeated cleaning and polishing of the electrode surface is mandatory to record accurate and reproducible cyclic voltammograms because these compounds have a strong tendency to adsorb at the surface of the working electrode.
Figure 6.
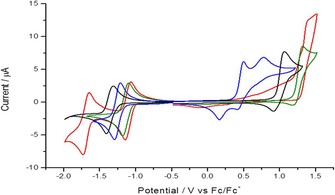
Cyclic voltammograms measured in solutions containing 10−3 m of 1 (black), 2 (red), 3 (green), and 4 (blue) in acetonitrile ([TBA][PF6]; 10−1 m). Data are recorded at the Pt working electrode (ν=0.1 V s−1).
Table 2.
Oxidation and reduction half‐wave potential values [mV] measured by CV for 1–4 (10−3 m) in acetonitrile ([TBA][PF6]; 10−1 m) at a Pt electrode (Ø=1.5 mm, ν=0.1 V s−1), E, versus the ferrocene/ferrocenium redox couple (Fc/Fc+). Redn and Oxn represent the n successive reduction and oxidation processes observed in the tested electrochemical window.
| Dye | Y | Reduction | Oxidation | |||||
|---|---|---|---|---|---|---|---|---|
| Red1 | ΔE(Red1)[a] | Red2 | Ox1 | ΔE(Ox1)[a] | Ox2 | |||
| 1 | H | −1359 | 0 | – | 992[b] | 0 | – | |
| 2 | NO2 | −1109 | −250 | −1690 | 1383[c] | 391 | – | |
| 3 | CHO | −1140 | −219 | – | 1258[b] | 266 | – | |
| 4 | NH2 | −1250 | −109 | – | 333[b] | −659 | 600[c] | |
[a] Potential difference relative to the first reduction or oxidation waves of reference compound 1. [b] Not a fully reversible process. [c] Irreversible process.
Unfunctionalized DAOTA 1, used as a reference, exhibits relatively simple redox behavior. This compound is typically reduced at E red 1/2=−1.36 V versus Fc/Fc+, with a peak to peak separation of 89 mV (ΔE peaks). This provides evidence of a quasi‐reversible monoelectronic transfer that generates a neutral species from the cation. Oxidation shows a slow, but still reversible behavior at the tested scan rate, with E ox 1/2=0.99 V versus Fc/Fc+. The difference in intensity between both peaks indicates a deviation from an ideal fully reversible system, which is attributed to a lower stability of the oxidized form. Evaluation of such an oxidative decomposition of the triangulene core was not investigated further, but already observed with several analogues.3a, 11
With nitro derivative 2, a reversible one‐electron reduction is also observed first, albeit proving to be an easier reduction process (E red 1/2=−1.11 V vs. Fc/Fc+ and ΔE peaks=88 mV). A second reduction wave appeared at more cathodic potential (E red 1/2=−1.69 V vs. Fc/Fc+ and ΔE peaks=88 mV). This second wave can be assigned to the monoelectronic reduction of the nitro group of 2, as already observed for nitro derivatives of [4]helicenes.15b The reduction of nitro compounds to form a stable radical species has been widely studied in nonaqueous media.21 On the other hand, a fully irreversible oxidation process is observed, which occurs at a large anodic overpotential (E ox 1/2=1.38 V vs. Fc/Fc+). With aldehyde derivative 3, the reductive behavior is apparently unchanged, in comparison to 1, revealing a single one‐electron reversible reduction, which occurs at a more cathodic potential (E red 1/2=−1.14 V vs. Fc/Fc+ and ΔE peaks=87 mV). A partially irreversible oxidation process is observed, with a very positive E ox 1/2 value (1.26 V vs. Fc/Fc+). Finally, the electrochemical characterization of amino derivative 4 reveals a slightly easier reversible reduction process than that of 1 (E red 1/2=−1.25 V vs. Fc/Fc+ and ΔE peaks=82 mV), and two partially irreversible oxidations at very mild E ox 1/2 values (0.33 and 0.60 V). Such an easier reduction is somehow unexpected in comparison with 1 and the corresponding amino derivative 4.
The introduction of functional groups onto the cationic triangulene core does affect the reversibility of the oxidation pathways because the various products of oxidation do not exhibit the same stability. By comparing the CV data gathered in Figure 6, nitro derivative 2 is the only one that exhibits a fully irreversible oxidation at a scan rate of 0.1 V s−1. On the other hand, the oxidation processes of electron‐deficient nitro 2 and aldehyde 3 are above that of DAOTA 1, with cathodic shifts of 0.39 and 0.27 V, respectively. Amino derivative 4 shows more complex oxidation behavior, with two anodic waves at less positive potentials. This may be attributed to the oxidation of the amino group,22 as already observed for amino derivatives of [4]helicenes.15b In terms of the reduction process, most of the compounds feature a single, fully reversible, electron transfer, as seen for aldehyde 3 and amino 4, and as observed for reference compound DAOTA 1. Nevertheless, nitro derivative 2 displays two reversible monoelectronic reductions; the second of which is assigned to the NO2 group.
Photophysical properties
Electronic absorption and fluorescence properties of the functionalized DAOTA derivatives were recorded in acetonitrile (ca. 10−5 m) at 20 °C. Selected spectra are presented in Figure 7 and a summary of the data is provided in Table 3. The effect of the nine substituents follows a similar trend to that previously reported for functionalized diaza [4]helicenes.15b Electron‐withdrawing functional groups induce a moderate hypsochromic effect of the absorption maximum, up to λ=528 nm for derivative 2, corresponding to a 990 cm−1 blueshift relative to that of unsubstituted 1. The intensity of the lower energy transition is weakly influenced, with ϵ between 10 000 and 16 100 m −1 cm−1. On the contrary, electron‐donating substituents give rise to a noticeable low‐energy shift of the band with maxima centered at λ=591 and 640 nm (1030 and 2330 cm−1 redshifts relative to 1) for carboxylate and amino derivatives 8 and 4, respectively. The broadening of the transition presumably indicates the establishment of intramolecular charge transfer from the substituent towards the cationic skeleton.15c
Figure 7.
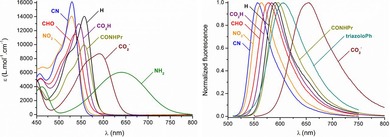
Selected electronic absorption (left) and normalized fluorescence (right) spectra in acetonitrile.
Table 3.
Photophysical properties of 1–10 in acetonitrile.
| Dye | Y | λ abs [nm] | ϵ [m −1 cm−1] | λ em [nm] | Stokes shift [cm−1] | Φ [a] | τ [ns] | k R (106 s−1)[b] | k NR (106 s−1)[b] |
|---|---|---|---|---|---|---|---|---|---|
| 1 | H | 557 | 14 000 | 591 | 1030 | 0.42 | 19.6 | 21.4 | 29.6 |
| 2 | NO2 | 528 | 14 100 | 564 | 1170 | 0.09 | 3.7 | 24.3 | 246 |
| 3 | CHO | 535 | 11 500 | 578 | 1390 | 0.26 | 11.7 | 22.2 | 63.3 |
| 4 | NH2 | 640 | 6000 | – | – | – | – | – | – |
| 5 | triazolo‐Ph | 543 | 11 400 | 607 | 1940 | 0.29 | 18.3 | 15.9 | 38.8 |
| 6 | CN | 529 | 16 100 | 556 | 920 | 0.26 | 16.2 | 16.1 | 45.7 |
| 7 | CHC(CN)2 | 546 | 11 400 | 590 | 1370 | 0.22 | 13.0 | 16.9 | 60.0 |
| 8‐H+ [c] | CO2H | 545 | 13 000 | 584 | 1230 | 0.46 | 18.4 | 25.0 | 29.4 |
| 8 [d] | CO2 ‐ | 591 | 8700 | 656 | 1680 | 0.04[e] | 2.7[f] | 14.8 | 356 |
| 9 | CO2Et | 547 | 13 200 | 584 | 1160 | 0.49 | 19.2 | 25.5 | 26.6 |
| 10 | CONHPr | 557 | 10 000 | 596 | 1180 | 0.50 | 20.9 | 23.9 | 23.3 |
[a] Reference: rhodamine B (Φ=0.49 in ethanol). [b] With k R=φ/τ and k NR=(1−φ)/τ. [c] 0.1 m trifluoroacetic acid (TFA) in CH3CN. [d] 0.1 m Et3N in CH3CN. [e] Reference: oxazine 725 (Φ=0.11 in ethanol). [f] Biexponential fit: 2.7 (63), 5.5 ns (37 %).
In term of fluorescence, the compounds carrying electron‐withdrawing moieties present fluorescence profiles quasi‐similar to that of 1, centered between λ=556 (6) and 596 nm (10), with Stokes shifts in the range of 900–1400 cm−1. The fluorescence quantum yields and lifetimes are sensibly equivalent to that of 1, with, for instance, Φ≈0.5 for 9–10, but are noticeably lowered for compounds 3, 6, and 7 (Φ≈0.3). Surprisingly, nitro derivative 2 is characterized by a dramatic loss of fluorescence intensity and lifetime due to increased nonradiative de‐excitation pathways. On the other hand, the dyes featuring triazolo‐Ph (5) and carboxylate (8) presents redshifted emission bands centered at λ=607 and 656 nm, respectively, with higher Stokes shifts (ca. 1700–1900 cm1). Their diminished intensity and lifetimes, together with the absence of signal for amino derivative 4, is consistent with previous observations on cationic diaza [4]‐ and [6]helicenes introducing strong donor groups.15b,15d Finally, it is noteworthy to underline that deprotonation of carboxylic derivative 8‐H+ to zwitterionic form 8 leads to 46 and 72 nm (1430 and 1880 cm−1) redshifts of absorption and fluorescence maxima, respectively. Such a system may find particular interest as a pH‐sensitive probe for biological applications.1i
Conclusion
A simple and efficient derivatization strategy has been developed to afford C‐functionalized cationic DAOTA derivatives (9 examples). The electronic effects caused by the added substituents are visible by CV, which has revealed a tuning of the redox potentials with, in a general manner, easier reductions of the substituted dyes. The tuning was also effective for the absorption and fluorescence properties, with a cutoff emission reaching the near‐infrared region for carboxylate derivative 8. By analogy with related cationic diaza [4]helicene derivatives, the direct introduction of functional groups onto the DAOTA skeleton paves the way to future chemical engineering of these dyes for bioconjugation or vectorization in biological media.23
Experimental Section
Reagents
Compound 1 was prepared according to a reported procedure.7b Column chromatography purifications were performed by using Siliaflash P60 silica gel (40–63 μm, 60 Å).
Analytical methods and instruments
Melting points were measured in open capillary tubes with a Buchi B‐550 melting points apparatus and are uncorrected. Absorption spectra were recorded on a JASCO V‐650 spectrophotometer at 20 °C in analytical‐grade solvents (ca. 10−5 m). IR spectra were recorded on a PerkinElmer 1650 FTIR spectrometer by using a diamond attenuated total reflectance (ATR) Golden Gate sampling attachment. NMR spectra were recorded on Brucker Advance II+ AMX‐500 and AMX‐400 spectrometers at room temperature. NMR chemical shifts are given in ppm (δ) relative to Me4Si with solvent resonances as internal standards (CD2Cl2: δ=5.32 ppm for 1H and δ=53.8 ppm for 13C; CD3CN: δ=1.94 ppm for 1H, δ=118.3 ppm for 13C, δ=−164.7 ppm for 19F, considering an external calibration with C6F6 as a reference).24 Electrospray mass spectra were obtained on a Finnigan SSQ 7000 spectrometer QSTAR pulsar i (AB/ MDS Sciex), ESI (TIS)/nanoESI/APCI‐QqTof by the Department of Mass Spectroscopy of the University of Geneva.
Crystallography
All data were collected on an Agilent supernova dual source diffractometer equipped with an Atlas detector, by using CuKα radiation. Data reduction was carried out in the CrysAlis Pro software.25 Structure solution was made by using direct methods (sir2004),26 dual‐space methods (SHELXT), or charge‐flipping (OLEX2). Refinements were carried out in SHELXL27 within the OLEX228 software. https://www.ccdc.cam.ac.uk/services/structures?id=doi:10.1002/chem.201801486‐1824707, 1443634, 1824709, and 1824706 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from http://www.ccdc.cam.ac.uk/.
Electrochemistry
Voltammetric experiments were performed with a PGSTAT30 Autolab potentiostat connected to a conventional three‐electrode cell, consisting of a silver wire pseudoreference electrode, a platinum wire auxiliary electrode, and a 1.5 mm diameter platinum disk working electrode. Prior to measurements, Pt disks were polished with alumina slurry of different sizes, rinsed thoroughly with Milli‐Q water between each polishing step, and sonicated in water and ethanol successively, followed by a final rinse with acetonitrile, and dried with a stream of N2. All solutions were degassed for 10 min before measurements.
Fluorescence spectroscopy
Steady‐state fluorescence spectra were measured by using a Varian Cary 50 Eclipse spectrofluorimeter. All fluorescence spectra were corrected for the wavelength‐dependent sensitivity of the detection. Fluorescence quantum yields, Φ, were measured in diluted solutions (at least five different concentrations for each sample) with an optical density lower than 0.1 by using the following equation: Φ x/Φ r=[A r(λ)/A x(λ)](n x 2/n r 2)(D x/D r), in which A is the absorbance at the excitation wavelength (λ), n is the refractive index, and D the integrated intensity; r and x indicate reference and sample, respectively. The fluorescence quantum yields were measured relative to rhodamine B in ethanol (Φ=0.49) for all compounds, except for 8, which was measured relative to oxazine 725 in ethanol (Φ=0.11). Excitation of reference and sample compounds was performed at the same wavelength (λ=520 nm, except for 8 and oxazine 725, for which excitation was performed at λ=590 nm). Short luminescence decay was monitored by using a Horiba‐Jobin–Yvon Fluorolog‐3 iHR320 fluorimeter equipped with the TC‐SPC Horiba apparatus and by using Ludox in distilled water to determine the instrumental response function used for deconvolution. Excitation was performed by using a λ=495 nm nano‐light‐emitting diode (NanoLED; peak wavelength: λ=490 nm; pulse duration: <250 ps), and deconvolution was performed by using the DAS6 fluorescence‐decay analysis software.
Compound 2
An aqueous solution of HNO3 (2 mL, 60 %, 19 mmol) was added to a solution of 1 (300 mg, 0.7 mmol) in CH2Cl2 (13 mL, 0.05 m). After 12 h, the reaction mixture was quenched by the addition of an aqueous solution of NaOH (1 m), then it was extracted with CH2Cl2 (3×20 mL) and washed with 1 m HBF4. The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The desired product was obtained as an orange solid after dissolution in the minimum amount of CH2Cl2 and precipitation with Et2O (320 mg, 98 %). R f=0.2 (SiO2, CH2Cl2/MeOH, 98:2); m.p. 187 °C (decomp); UV/Vis (CH3CN): λ max (ϵ max)=528 nm (14 100 L mol−1 cm−1); IR (CH2Cl2): =3080, 2974, 2880, 1612, 1588, 1514, 1459, 1313, 1273, 1166, 1042, 822, 753 cm−1; 1H NMR (400 MHz, CD2Cl2): δ=8.79 (d, J=9.6 Hz, 1 H; CH), 8.30 (t, J=8.5 Hz, 1 H; CH), 8.20 (t, J=8.5 Hz, 1 H; CH), 7.72 (d, J=8.8 Hz, 1 H; CH), 7.64–7.47 (m, 4 H; 4×CH), 4.74–4.58 (m, 2 H; CH2), 4.25 (t, J=7.0 Hz, 2 H; CH2), 2.16–2.00 (m, 2 H; CH2), 1.82–1.68 (m, 2 H; CH2), 1.26 (t, J=7.4 Hz, 3 H; CH3), 0.64 ppm (t, J=7.3 Hz, 3 H; CH3); 13C NMR (101 MHz, CD2Cl2): δ=153.0 (C), 152.8 (C), 143.8 (C), 140.0 (CH), 140.0 (C), 139.9 (C), 139.2 (C), 138.9 (CH), 138.1 (C), 136.8 (CH), 131.8 (C), 112.6 (CH), 112.4 (C), 110.8 (CH), 110.8 (CH), 110.1 (CH), 109.4 (C), 108.9 (C), 106.8 (CH), 57.9 (CH2), 50.8 (CH2), 21.1 (CH2), 19.7 (CH2), 10.7 (CH3), 10.3 ppm (CH3); 19F NMR (282 MHz, CD3CN): δ=−149.6 (20 %), −149.7 ppm (80 %); HRMS (ESI+): m/z calcd for C25H22N3O3 + [M]+: 412.1656; found: 412.1661.
Compound 3
POCl3 (2.5 mL, 26.42 mmol) was added to a flask containing a solution of salt 1 (500 mg, 1.1 mmol) in DMF (1.0 mL, 13.21 mmol) at 90 °C. After being stirred for 14 h at this temperature, H2O (10 mL) was added to the reaction mixture at 0 °C. The obtained solution was stirred for 30 min at 25 °C. Then, the reaction mixture was washed with a 5 % aqueous solution of LiCl and extracted with CH2Cl2 (3×20 mL). The organic layer was washed with 1 m HBF4, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude material was purified by flash chromatography on silica gel (CH2Cl2/MeOH 98:2) and recrystallization by slow diffusion in a CH2Cl2/toluene system. The product was obtained as a purple solid (20–65 %).17 R f=0.3 (SiO2, CH2Cl2/MeOH, 98:2); m.p. 217 °C (decomp); UV/Vis (CH3CN): λ max (ϵ max)=535 nm (11 500 L mol−1 cm−1); IR (CH2Cl2): =3001, 1741, 1615, 1456, 1369, 1276, 1263, 1217, 837, 755 cm−1; 1H NMR (400 MHz, CD2Cl2): δ=10.16 (s, 1 H; CH), 8.67 (d, J=9.1 Hz, 1 H; CH), 8.26–8.15 (m, 2 H; 2×CH), 7.70–7.60 (m, 2 H; 2×CH), 7.57 (d, J=9.1 Hz, 1 H; CH), 7.55–7.45 (m, 2 H; 2×CH), 4.71–4.55 (m, 4 H; CH2), 2.17–1.98 (m, 2 H; CH2), 1.85–1.75 (m, 2 H; CH2), 1.26 (t, J=7.4 Hz, 3 H; CH3), 0.67 ppm (t, J=7.3 Hz, 3 H; CH3); 13C NMR (101 MHz, CD2Cl2): δ=187.2 (CHO), 153.8 (C), 153.6 (C), 145.2 (C), 144.9 (CH), 144.5 (C), 141.2 (C), 140.9 (C), 140.3 (C), 140.0 (CH), 139.3 (CH), 118.8 (C), 113.6 (CH), 113.4 (C), 111.0 (CH), 110.8 (CH), 110.7 (CH), 110.4 (C), 109.8 (C), 107.9 (CH), 59.9 (CH2), 51.0 (CH2), 22.1 (CH2), 20.1 (CH2), 11.1 (CH3), 10.5 ppm (CH3); 19F NMR (282 MHz, CD3CN): δ=−150.1 (20 %), −150.2 ppm (80 %); HRMS (ESI+): m/z calcd for C23H22N2O2 + [M]+: 395.1754; found: 395.1753.
Compound 4
Pd/C (12.8 mg, 10 wt %, 20 mol %) was added to solution of 2 (300 mg, 0.6 mmol) in CH2Cl2/MeOH (10:10 mL) under a N2 atmosphere. Then H2 was bubbled through the reaction mixture for 5 min and the solution was stirred for an additional 30 min at 25 °C under a H2 atmosphere. Then the reaction mixture was purged with N2 and filtered through Celite, which was then washed with CH2Cl2. The product was concentrated under reduced pressure. The desire product was obtained as a light green solid (280 mg, 99 %). R f=0.2 (SiO2, CH2Cl2/MeOH, 98:2); m.p. 257 °C (decomp); UV/Vis (CH3CN): λ max (ϵ max)=640 nm (6000 L mol−1 cm−1); IR (CH2Cl2): =3007, 2990, 1611, 1462, 1276, 1262, 1163, 1068, 755 cm−1; 1H NMR (500 MHz, CD3CN): δ=7.96 (t, J=8.2 Hz, 1 H; CH), 7.90 (t, J=8.4 Hz, 1 H; CH), 7.79 (d, J=9.0 Hz, 1 H; CH), 7.49–7.37 (m, 3 H; 3×CH), 7.13 (d, J=8.0 Hz, 1 H; CH), 7.08 (d, J=8.0 Hz, 1 H; CH), 4.76–4.71 (m, 2 H; CH2), 4.46 (s, 2 H; NH2), 4.40–4.31 (m, 2 H; CH2), 1.93–1.84 (m, 2 H; CH2), 1.65–1.55 (m, 2 H; CH2), 1.16 (t, J=7.3 Hz, 3 H; CH3), 0.67 ppm (t, J=7.3 Hz, 3 H; CH3); 13C NMR (126 MHz, CD3CN): δ=153.4 (C), 153.3 (C), 144.7 (C), 140.9 (C), 140.3 (CH), 138.9 (CH), 138.8 (CH), 135.0 (C), 132.5 (C), 132.0 (CH), 129.4 (C), 115.4 (C), 112.0 (CH), 109.7 (CH), 109.1 (C), 109.0 (CH), 108.8 (CH), 108.6 (C), 108.2 (CH), 51.0 (CH2), 50.1 (CH2), 22.7 (CH2), 20.1 (CH2), 11.1 (CH3), 11.0 ppm (CH3); 19F NMR (282 MHz, CD3CN): δ=−150.1 (20 %), −150.2 ppm (80 %); HRMS (ESI+): m/z calcd for C25H24N3O+ [M]+: 382.1914; found: 382.1918.
Compound 5
tBuONO (8 μL, 0.08 mmol) and TMSN3 (13 μL, 0.10 mmol) were added dropwise to a solution of 4 (24 mg, 0.05 mmol) in CH3CN (60 μL) at 0 °C and in the absence of light. The reaction mixture was stirred for 5 min at 25 °C. Then the addition of Et2O led to the precipitation of azido intermediate, which was separated from the mother liquor by centrifugation. The solid was dissolved in MeOH (40 μL) and phenylacetylene (5.5 μL, 0.05 mmol) was added. A solution of CuSO4 ⋅5 H2O (1.0 mg, 8 mol %), ascorbic acid (1.9 mg, 21 mol %), and NaHCO3 (0.9 mg, 21 mol %) in water (40 μL) was added dropwise. The reaction mixture was stirred for 30 min. The crude product was dissolved in a minimum amount of CH2Cl2 and then the addition of Et2O led to the precipitation of the product, which was separated from the mother liquor by centrifugation. Then it was purified by flash chromatography on silica gel (CH2Cl2/MeOH 98:2) and recrystallized by slow diffusion in a biphasic system of CH2Cl2 and hexane. The desire product was obtained as a light pink solid (20.3 mg, 68 % yield from 4). R f=0.2 (SiO2, CH2Cl2/MeOH, 98:2); m.p. 205 °C (decomp); UV/Vis (CH3CN): λ max (ϵ max)=543 nm (11 400 L mol−1 cm−1); IR (CH2Cl2): =2988, 2922, 2852, 1616, 1465, 1276, 1262, 1059, 755 cm−1; 1H NMR (500 MHz, CD3CN): δ=8.62 (s, 1 H; CH), 8.24–8.13 (m, 2 H; 2×CH), 8.09 (t, J=8.4 Hz, 1 H; CH), 8.03–7.98 (m, 2 H; 2×CH), 7.72 (d, J=8.7 Hz, 1 H; CH), 7.67 (d, J=9.1 Hz, 1 H; CH), 7.59–7.50 (m, 3 H; 3×CH), 7.48–7.42 (m, 2 H; 2×CH), 7.41 (d, J=8.1 Hz, 1 H; CH), 4.64–4.52 (m, 2 H; CH2), 3.79–3.65 (m, 2 H; CH2), 2.07–1.97 (m, 2 H; CH2), 1.60–1.47 (m, 3 H; CH2), 1.20 (t, J=7.3 Hz, 3 H; CH3), 0.52 ppm (t, J=7.3 Hz, 3 H; CH3); 13C NMR (126 MHz, CD3CN): δ=153.7 (C), 153.6 (C), 149.3 (C), 143.5 (C), 142.3 (C), 141.8 (C), 141.1 (CH), 140.5 (C), 140.1 (CH), 139.7 (CH), 138.4 (C), 131.1 (C), 130.2 (CH), 129.7 (CH), 126.7 (CH), 125.3 (CH), 117.7 (C), 114.4 (C), 112.1 (CH), 110.6 (CH), 110.4 (CH), 110.3 (CH), 109.7 (C), 109.6 (C), 108.4 (CH), 53.2 (CH2), 51.1 (CH2), 21.7 (CH2), 20.1 (CH2), 11.1 (CH3), 10.7 ppm (CH3); 19F NMR (282 MHz, CD3CN): δ=−150.1 (20 %), −150.2 ppm (80 %); HRMS (ESI+): m/z calcd for C33H28N5O+ [M]+: 510.2288; found: 510.2306.
Compound 6
TfOH (55 μL, 0.62 mmol) was added to a flask containing a solution of salt 3 (100 mg, 0.21 mmol) and NaN3 (20.2 mg, 0.31 mmol) in CH3CN (414 μL, 0.5 m). The obtained mixture was stirred for 5 min at 25 °C. Then the mixture was extracted with CH2Cl2 (3×10 mL) and the organic layer was washed with 1 m HBF4, dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude material was dissolved in the minimum amount of CH2Cl2 and precipitated with Et2O. After flash chromatography on silica gel (CH2Cl2/MeOH 98:2), the product was obtained as a purple solid (44.7 mg, 45 % yield). R f=0.2 (SiO2, CH2Cl2/MeOH, 98:2); m.p. 183 °C (decomp); UV/Vis (CH3CN): λ max (ϵ max)=529 nm (16 100 L mol−1 cm−1); IR (CH2Cl2): =2930, 2210, 1739, 1615, 1530, 1460, 1327, 1266, 1154, 1030, 821, 753 cm−1; 1H NMR (500 MHz, CD3CN): δ=8.42 (d, J=9.2 Hz, 1 H; CH), 8.14–8.21 (m, 2 H; 2×CH), 7.71–7.66 (m, 2 H; 2×CH), 7.57 (d, J=9.2 Hz, 1 H; CH), 7.46–7.43 (m, 2 H; 2×CH), 5.3–4.6 (m, 2 H; CH2), 4.58–4.47 (m, 2 H; CH2), 2.17–2.11 (m, 2 H; CH2), 2.00–1.95 (m, 2 H; CH2), 1.25–1.15 ppm (m, 6 H; 2×CH3); 13C NMR (126 MHz, CD3CN): δ=153.6 (C), 153.4 (C), 146.8 (CH), 145.3 (C), 143.5 (C), 141.2 (C), 140.8 (C), 140.2 (CH), 140.2 (CH), 140.1 (C), 119.9 (C), 118.3 (C), 112.8 (CH), 111.2 (CH), 111.1 (CH), 111.0 (CH), 111.0 (CH), 109.7 (C), 109.5 (C), 108.5 (CH), 53.1 (CH2), 51.2 (CH2), 22.2 (CH2), 20.0 (CH2), 11.0 (CH3), 9.7 ppm (CH3); 19F NMR (282 MHz, CD3CN): δ=−150.3 (20 %), −150.3 ppm (80 %); HRMS (ESI+): m/z calcd for C26H22N3O+ [M]+: 392.1757; found: 392.1759.
Compound 7
Ph3P (11 mg, 20 mol %) was added to a flask containing a solution of salt 3 (100 mg, 0.21 mmol) and malononitrile (41.1 mg, 0.62 mmol) in CH3CN (70 μL, 3 m). The obtained mixture was heated under MW irradiation at 130 °C. The crude material was purified by selective precipitation (Et2O addition to a CH2Cl2 solution). The desired product was obtained as a pink solid (77 mg, 80 % yield). R f=0.3 (SiO2, CH2Cl2/MeOH, 98:2); m.p. 133 °C (decomp); UV/Vis (CH3CN): λ max (ϵ max)=546 nm (11 400 L mol−1 cm−1); IR (CH2Cl2): =3364, 2930, 2230, 1612, 1577, 1460, 1344, 1276, 1173, 1113, 930, 894, 781, 752 cm−1; 1H NMR (500 MHz, CD3CN): δ=8.57–8.45 (m, 2 H; 2×CH), 8.22–8.12 (m, 2 H; 2×CH), 7.76–7.70 (m, 2 H; 2×CH), 7.67 (d, J=9.2 Hz, 1 H; CH), 7.46–7.40 (m, 2 H; 2×CH), 4.67–7.53 (m, 4 H; 2×CH2), 2.03–1.96 (m, 2 H; CH2), 1.86–1.77 (m, 2 H; CH2), 1.19 (t, J=7.3 Hz, 3 H; CH3), 0.71 ppm (t, J=7.3 Hz, 3 H; CH3); 13C NMR (126 MHz, CD3CN): δ=159.6 (CH), 153.8 (C), 153.6 (C), 145.1 (C), 144.7 (C), 141.5 (CH), 141.4 (C), 140.9 (C), 140.2 (C), 140.2 (CH), 139.6 (CH), 115.0 (C), 113.9 (C), 113.5 (CH), 113.4 (C), 112.7 (C), 111.1 (CH), 111.0 (CH), 110.8 (CH), 110.1 (C), 110.0 (C), 108.6 (CH), 83.7 (C), 58.6 (CH2), 51.2 (CH2), 22.9 (CH2), 20.3 (CH2), 11.1 (CH3), 10.5 ppm (CH3); 19F NMR (282 MHz, CD3CN): δ=−150.2 (20 %), −150.4 ppm (80 %); HRMS (ESI+): m/z calcd for C29H23N4O+ [M]+: 443.1866; found: 443.1860.
Compound 8‐H+
Compound 3 (200 mg, 0.37 mmol.) was dissolved in CH3CN (8 mL, 0.05 m). NaH2PO4 (44 mg, 0.37 mmol), H2O2 (83 μL, 0.74 mmol), and NaClO4 (90 mg, 0.74 mmol) were added separately to this solution. The reaction was stirred for 1 h at 60 °C, then concentrated under reduced pressure. The residue was purified by column chromatography on silica gel with CH2Cl2/MeOH (90/10) as the eluent. The obtained solid was dissolved in CH2Cl2, washed with an aqueous solution of HBF4 (1 m), dried over Na2SO4, filtered, and concentrated under reduced pressure, to afford the pure product as a pink solid (154 mg, 75 %). R f=0.2 (SiO2, CH2Cl2/MeOH, 98:2); m.p. 146 °C (decomp); UV/Vis (CH3CN): λ max (ϵ max)=545 nm (13 000 L mol−1 cm−1); IR (neat): =2927, 1708, 1616, 1460, 1347, 1276, 1261, 1223, 1169, 1112, 840, 755 cm−1; 1H NMR (400 MHz, CD3CN): δ=8.51 (d, J=9.1 Hz, 1 H; CH), 8.04–7.96 (m, 2 H; 2×CH), 7.57‐ 7.51 (m, 2 H; 2×CH), 7.46 (d, J=9.2 Hz, 1 H; CH), 7.21–7.17 (m, 2 H; 2×CH), 4.47–4.40 (m, 4 H; 2×CH2), 1.98–1.87 (m, 2 H; CH2), 1.73–1.60 (m, 2 H; CH2), 1.18 (t, J=7.4 Hz, 3 H; CH3), 0.56 ppm (t, J=7.3 Hz, 3 H; CH3); 13C NMR (101 MHz, CD3CN): δ=167.7 (C), 153.4 (C), 153.2 (C), 143.7 (C), 143.3 (C), 142.4 (CH), 141.1 (C), 140.4 (C), 140.1 (C), 139.9 (CH), 139.3 (CH), 113.7 (C), 113.3 (C), 113.2 (CH), 110.6 (CH), 110.2 (CH), 110.1 (CH), 109.5 (C), 109.1 (C), 107.3 (CH), 58.1 (CH2), 50.8 (CH2), 21.5 (CH2), 20.1 (CH2), 11.1 (CH3), 10.6 ppm (CH3); 19F NMR (282 MHz, CD3CN): δ=−150.2 (20 %), −150.4 ppm (80 %); HRMS (ESI+): m/z calcd for C26H23N2O4 [M]+: 411.1703; found: 411.1675.
Compound 9
Compound 8‐H+ (25 mg, 0.04 mmol) was dissolved in anhydrous CH2Cl2 (1 mL, 0.05 m). SOCl2 (27 μL, 0.22 mmol) was added to this solution, and the reaction was stirred for 10 min at 25 °C. Then distilled ethanol (13 μL, 0.22 mmol) was added and, after 10 min of stirring at 25 °C, the mixture was concentrated under reduced pressure. The residue was diluted with CH2Cl2 and washed with an aqueous diluted solution of HBF4 (1 m), dried over Na2SO4, filtered, and finally evaporated. Purification by column chromatography on silica gel with CH2Cl2/MeOH (95/5) as the eluent afforded the pure product as a pink solid (23 mg, 90 %). R f=0.3 (SiO2, CH2Cl2/MeOH, 98:2); m.p. 186 °C; UV/Vis (CH3CN): λ max (ϵ max)=547 nm (13 200 L mol−1 cm−1); IR (neat): =3625, 2979, 2880, 1710, 1613, 1527, 1461, 1345, 1262, 1222, 1171, 1105, 1046, 820, 756 cm−1; 1H NMR (500 MHz, CD3CN): δ=8.49 (d, J=9.1 Hz, 1 H; CH), 8.13–8.06 (m, 2 H; 2×CH), 7.65–7.60 (m, 2 H; 2×CH), 7.52 (d, J=9.1 Hz, 1 H; CH), 7.36–7.31 (m, 2 H; 2×CH), 4.54–4.47 (m, 4 H; 2×CH2), 4.44–4.37 (m, 2 H; CH2), 2.01–1.95 (m, 2 H; CH2), 1.74–1.65 (m, 2 H; CH2), 1.45 (t, J=7.1 Hz, 3 H; CH3), 1.18 (t, J=7.4 Hz, 3 H; CH3), 0.58 ppm (t, J=7.3 Hz, 3 H; CH3); 13C NMR (126 MHz, CD3CN): δ=167.5 (C), 153.7 (C), 153.6 (C), 143.8 (C), 143.2 (C), 142.2 (CH), 141.3 (C), 140.9 (C), 140.4 (C), 139.9 (CH), 139.3 (CH), 114.1 (C), 113.6 (C), 113.1 (CH), 110.6 (CH), 110.2 (CH), 110.2 (CH), 109.8 (C), 109.4 (C), 109.3 (C), 107.4 (CH), 63.3 (CH2), 57.9 (CH2), 50.8 (CH2), 21.4 (CH2), 20.1 (CH2), 14.5 (CH3), 11.1 (CH3), 10.6 ppm (CH3); 19F NMR (282 MHz, CD3CN): δ=−150.1 (20 %), −150.2 ppm (80 %); HRMS (ESI+): m/z calcd for C24H27N2O3 + [M]+: 439.2016; found: 439.2012.
Compound 10
Compound 8‐H+ (25 mg, 0.04 mmol) was dissolved in anhydrous CH2Cl2 (1 mL, 0.05 m). SOCl2 (27 μL, 0.22 mmol) was added to this solution, and the reaction was stirred for 10 min at 25 °C. Then distilled propylamine (40 μL, 0.7 mmol) was added at 0 °C and, after 15 min of stirring at 25 °C, the mixture was quenched with water. The organic layer was extracted and washed with a 1 m aqueous diluted solution of HBF4 (1 m), dried over Na2SO4, filtered, and concentrated under reduced pressure. Purification by column chromatography on silica gel with CH2Cl2/MeOH (95/5) as the eluent afforded the pure product as a pink solid (11 mg, 42 %). R f=0.4 (SiO2, CH2Cl2/MeOH, 98:2); m.p. 146 °C; UV/Vis (CH3CN): λ max (ϵ max)=557 nm (10 000 L mol−1 cm−1); IR (neat): =3409, 2969, 2980, 1615, 1526, 1461, 1344, 1275, 1171, 1119, 1077, 967, 839, 755 cm−1; 1H NMR (CD2Cl2, 500 MHz): δ=8.17 (d, J=8.8 Hz, 1 H; CH), 8.13–8.05 (m, 2 H; 2×CH), 7.65–7.55 (m, 2 H; 2×CH), 7.49 (d, J=9.1, 1 H; CH), 7.38–7.30 (m, 3 H; 3×CH), 4.55–4.45 (m, 4 H; 2×CH2), 3.44–3.38 (m, 2 H; CH2), 12.0–1.90 (m, 2 H; CH2), 1.86–1.74 (m, 2 H; CH2), 1.74–1.65 (m, 2 H; CH2), 1.18 (t, J=7.3 Hz, 3 H; CH3), 1.03 (t, J=7.4 Hz, 3 H; CH3), 0.78 ppm (t, J=7.3 Hz, 3 H; CH3); 13C NMR (126 MHz, CD3CN): δ=169.1 (C), 153.7 (C), 153.6 (C), 142.7 (C), 141.7 (C), 141.4 (C), 141.2 (CH), 140.6 (C), 139.7 (CH), 139.3 (CH), 119.3 (C), 113.8 (C), 112.1 (CH), 110.3 (CH), 109.9 (CH), 109.7 (CH), 109.6 (C), 109.2 (C), 107.0 (CH), 54.4 (CH2), 50.6 (CH2), 42.7 (CH2), 23.2 (CH2), 21.4 (CH2), 19.9 (CH2), 12.0 (CH3), 11.1 (CH3), 10.8 ppm (CH3); 19F NMR (CD3CN, 282 MHz): δ=−150.0 (20 %), −150.1 (80 %); HRMS (ESI+): m/z calcd for C29H30N3O2 + [M]+: 452.2333; found: 452.2340.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the University of Geneva and Swiss National Science Foundation for funding. We would also like to acknowledge the Mass Spectrometry platform at the University of Geneva for mass spectroscopy analysis. We also thank the Bordeaux INP, the University of Bordeaux, and the Centre National de la Recherche Scientifique for their financial support. S.P. thanks the “Laboratoire de Chimie” at the ENS Lyon for access to NanoLED and TS‐SPC equipment.
I. H. Delgado, S. Pascal, C. Besnard, S. Voci, L. Bouffier, N. Sojic, J. Lacour, Chem. Eur. J. 2018, 24, 10186.
References
- 1.
- 1a. Dileesh S., Gopidas K. R., Chem. Phys. Lett. 2000, 330, 397–402; [Google Scholar]
- 1b. Pothukuchy A., Ellapan S., Gopidas K. R., Salazar M., Bioorg. Med. Chem. Lett. 2003, 13, 1491–1494; [DOI] [PubMed] [Google Scholar]
- 1c. Reynisson J., Schuster G. B., Howerton S. B., Williams L. D., Barnett R. N., Cleveland C. L., Landman U., Harrit N., Chaires J. B., J. Am. Chem. Soc. 2003, 125, 2072–2083; [DOI] [PubMed] [Google Scholar]
- 1d. Dileesh S., Gopidas K. R., J. Photochem. Photobiol. A 2004, 162, 115–120; [Google Scholar]
- 1e. Pothukuchy A., Mazzitelli C. L., Rodriguez M. L., Tuesuwan B., Salazar M., Brodbelt J. S., Kerwin S. M., Biochemistry 2005, 44, 2163–2172; [DOI] [PubMed] [Google Scholar]
- 1f. Maliwal B. P., Fudala R., Raut S., Kokate R., Sørensen T. J., Laursen B. W., Gryczynski Z., Gryczynski I., PLoS One 2013, 8, e63043; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1g. Sørensen T. J., Thyrhaug E., Szabelski M., Luchowski R., Gryczynski I., Gryczynski Z., Laursen B. W., Methods Appl. Fluoresc. 2013, 1, 025001; [DOI] [PubMed] [Google Scholar]
- 1h. Shivalingam A., Izquierdo M. A., Le Marois A., Vysniauskas A., Suhling K., Kuimova M. K., Vilar R., Nat. Commun. 2015, 6, 8178; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1i. Wallabregue A., Moreau D., Sherin P., Lorente P. M., Jarolímová Z., Bakker E., Vauthey E., Gruenberg J., Lacour J., J. Am. Chem. Soc. 2016, 138, 1752–1755. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Sørensen T. J., Laursen B. W., Luchowski R., Shtoyko T., Akopova I., Gryczynski Z., Gryczynski I., Chem. Phys. Lett. 2009, 476, 46–50; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Haketa Y., Sasaki S., Ohta N., Masunaga H., Ogawa H., Mizuno N., Araoka F., Takezoe H., Maeda H., Angew. Chem. Int. Ed. 2010, 49, 10079–10083; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 10277–10281; [Google Scholar]
- 2c. Folmar M., Shtoyko T., Fudala R., Akopova I., Gryczynski Z., Raut S., Gryczynski I., Chem. Phys. Lett. 2012, 531, 126–131; [Google Scholar]
- 2d. Hamacek J., Besnard C., Mehanna N., Lacour J., Dalton Trans. 2012, 41, 6777–6782; [DOI] [PubMed] [Google Scholar]
- 2e. Dong B., Maeda H., Chem. Commun. 2013, 49, 4085–4099; [DOI] [PubMed] [Google Scholar]
- 2f. Haketa Y., Tamura Y., Yasuda N., Maeda H., Org. Biomol. Chem. 2016, 14, 8035–8038; [DOI] [PubMed] [Google Scholar]
- 2g. Qiao B., Hirsch B. E., Lee S., Pink M., Chen C.-H., Laursen B. W., Flood A. H., J. Am. Chem. Soc. 2017, 139, 6226–6233. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Adam C., Wallabregue A., Li H., Gouin J., Vanel R., Grass S., Bosson J., Bouffier L., Lacour J., Sojic N., Chem. Eur. J. 2015, 21, 19243–19249; [DOI] [PubMed] [Google Scholar]
- 3b. Li H., Voci S., Wallabregue A., Adam C., Labrador G. M., Duwald R., Delgado I. H., Pascal S., Bosson J., Lacour J., Bouffier L., Sojic N., ChemElectroChem 2017, 4, 1750–1756. [Google Scholar]
- 4. Gueret R., Poulard L., Oshinowo M., Chauvin J., Dahmane M., Dupeyre G., Lainé P. P., Fortage J., Collomb M.-N., ACS Catal. 2018, 8, 3792–3802. [Google Scholar]
- 5.
- 5a. Baisch B., Raffa D., Jung U., Magnussen O. M., Nicolas C., Lacour J., Kubitschke J., Herges R., J. Am. Chem. Soc. 2009, 131, 442–443; [DOI] [PubMed] [Google Scholar]
- 5b. Raut S. L., Rich R., Shtoyko T., Bora I., Laursen B. W., Sorensen T. J., Borejdo J., Gryczynski Z., Gryczynski I., Nanoscale 2015, 7, 17729–17734; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c. Hu X.-M., Chen Q., Sui Z.-Y., Zhao Z.-Q., Bovet N., Laursen B. W., Han B.-H., RSC Adv. 2015, 5, 90135–90143. [Google Scholar]
- 6.Recently, carbon-bridged triangulenium dyes were reported, see: Rosenberg M., Rostgaard K. R., Liao Z., Madsen A. O., Martinez K. L., Vosch T., Laursen B. W., Chem. Sci. 2018, 9, 3122–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Laursen B. W., Krebs F. C., Angew. Chem. Int. Ed. 2000, 39, 3432–3434; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 3574–3576; [Google Scholar]
- 7b. Laursen B. W., Krebs F. C., Chem. Eur. J. 2001, 7, 1773–1783; [DOI] [PubMed] [Google Scholar]
- 7c. Bosson J., Gouin J., Lacour J., Chem. Soc. Rev. 2014, 43, 2824–2840; [DOI] [PubMed] [Google Scholar]
- 7d. Hitzenberger J. F., Drewello T., Uhl A., Schatz J., J. Mass Spectrom. 2017, 52, 174–181. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Thyrhaug E., Sørensen T. J., Gryczynski I., Gryczynski Z., Laursen B. W., J. Phys. Chem. A 2013, 117, 2160–2168; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Bogh S. A., Simmermacher M., Westberg M., Bregnhøj M., Rosenberg M., De Vico L., Veiga M., Laursen B. W., Ogilby P. R., Sauer S. P. A., Sørensen T. J., ACS Omega 2017, 2, 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Bricks J. L., Kachkovskii A. D., Slominskii Y. L., Gerasov A. O., Popov S. V., Dyes Pigm. 2015, 121, 238–255; [Google Scholar]
- 9b. Ni Y., Wu J., Org. Biomol. Chem. 2014, 12, 3774–3791; [DOI] [PubMed] [Google Scholar]
- 9c. Guo Z., Park S., Yoon J., Shin I., Chem. Soc. Rev. 2014, 43, 16–29; [DOI] [PubMed] [Google Scholar]
- 9d. Yuan L., Lin W., Zheng K., He L., Huang W., Chem. Soc. Rev. 2013, 42, 622–661. [DOI] [PubMed] [Google Scholar]
- 10. Bora I., Bogh S. A., Rosenberg M., Santella M., Sorensen T. J., Laursen B. W., Org. Biomol. Chem. 2016, 14, 1091–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shivalingam A., Vyšniauskas A., Albrecht T., White A. J. P., Kuimova M. K., Vilar R., Chem. Eur. J. 2016, 22, 4129–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Laursen B. W., Krebs F. C., Nielsen M. F., Bechgaard K., Christensen J. B., Harrit N., J. Am. Chem. Soc. 1998, 120, 12255–12263; [Google Scholar]
- 12b. Laursen B. W., Sørensen T. J., J. Org. Chem. 2009, 74, 3183–3185; [DOI] [PubMed] [Google Scholar]
- 12c. Sørensen T. J., Laursen B. W., J. Org. Chem. 2010, 75, 6182–6190; [DOI] [PubMed] [Google Scholar]
- 12d. Westerlund F., Hildebrandt C. B., Sørensen T. J., Laursen B. W., Chem. Eur. J. 2010, 16, 2992–2996. [DOI] [PubMed] [Google Scholar]
- 13. Lofthagen M., VernonClark R., Baldridge K. K., Siegel J. S., J. Org. Chem. 1992, 57, 61–69. [Google Scholar]
- 14.
- 14a. Hirose T., Sasatsuki K., Noguchi H., Yokoyama S., Matsuda K., Chem. Lett. 2016, 45, 1090–1092; [Google Scholar]
- 14b. Noguchi H., Hirose T., Yokoyama S., Matsuda K., CrystEngComm 2016, 18, 7377–7383. [Google Scholar]
- 15.
- 15a. Torricelli F., Bosson J., Besnard C., Chekini M., Bürgi T., Lacour J., Angew. Chem. Int. Ed. 2013, 52, 1796–1800; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 1840–1844; [Google Scholar]
- 15b. Delgado I. H., Pascal S., Wallabregue A., Duwald R., Besnard C., Guénée L., Nancoz C., Vauthey E., Tovar R. C., Lunkley J. L., Muller G., Lacour J., Chem. Sci. 2016, 7, 4685–4693; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15c. Pascal S., Besnard C., Zinna F., Di Bari L., Le Guennic B., Jacquemin D., Lacour J., Org. Biomol. Chem. 2016, 14, 4590–4594; [DOI] [PubMed] [Google Scholar]
- 15d. Bosson J., Labrador G. M., Pascal S., Miannay F.-A., Yushchenko O., Li H., Bouffier L., Sojic N., Tovar R. C., Muller G., Jacquemin D., Laurent A. D., Le Guennic B., Vauthey E., Lacour J., Chem. Eur. J. 2016, 22, 18394–18403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.In the [6]helicene series, bis- rather than mono-nitration is routinely achieved; see ref. [15a].
- 17.A second regioisomer of formylation of the triangulene is observed. Substitution occurs on azaoxa- rather than diaza-substituted phenyl rings. This minor derivative can only be removed by recrystallizing desired product 3 in a mixture of toluene/dichloromethane. This recrystallization is somewhat capricious; hence, the wide variation in yields.
- 18. Rokade B. V., Prabhu K. R., J. Org. Chem. 2012, 77, 5364–5370. [DOI] [PubMed] [Google Scholar]
- 19.A formanilide byproduct (Y=NHCHO) can be observed as a minor component. It presents a characteristic green color.
- 20.On cationic triangulenes and related helicenes, the position of the substituents is important for subsequent control of the physicochemical properties; refs. [6, 15d], see also: Duwald R., Pascal S., Bosson J., Grass S., Besnard C., Bürgi T., Lacour J., Chem. Eur. J. 2017, 23, 13596–13601. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Kraiya C., Singh P., Evans D. H., J. Electroanal. Chem. 2004, 563, 203–212; [Google Scholar]
- 21b. Chauhan B. G., Fawcett W. R., Lasia A., J. Phys. Chem. 1977, 81, 1476–1481. [Google Scholar]
- 22. Sasaki K., Newby W. J., J. Electroanal. Chem. Interfacial Electrochem. 1969, 20, 137–165. [Google Scholar]
- 23.
- 23a. Babič A., Pascal S., Duwald R., Moreau D., Lacour J., Allémann E., Adv. Funct. Mater. 2017, 27, 1701839; [Google Scholar]
- 23b. Bauer C., Duwald R., Labrador G. M., Pascal S., Lorente P. M., Bosson J., Lacour J., Rochaix J.-D., Org. Biomol. Chem. 2018, 16, 919–923. [DOI] [PubMed] [Google Scholar]
- 24.A value of +1.7 ppm was added to all measured 19F signals in consideration of the external calibration.
- 25.CrysalisPro Software system, Agilent Technologies UK Ltd., Oxford, UK.
- 26. Burla M. C., Caliandro R., Camalli M., Carrozzini B., Cascarano G. L., De Caro L., Giacovazzo C., Polidori G., Spagna R., J. Appl. Crystallogr. 2005, 38, 381–388. [Google Scholar]
- 27.
- 27a. Sheldrick G., Acta Crystallogr. Sect. C 2015, 71, 3–8; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27b. Sheldrick G., Acta Crystallogr. Sect. A 2008, 64, 112–122. [DOI] [PubMed] [Google Scholar]
- 28. Dolomanov O. V., Bourhis L. J., Gildea R. J., Howard J. A. K., Puschmann H., J. Appl. Crystallogr. 2009, 42, 339–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary