

Abstract
Oxidation chemistry using enzymes is approaching maturity and practical applicability in organic synthesis. Oxidoreductases (enzymes catalysing redox reactions) enable chemists to perform highly selective and efficient transformations ranging from simple alcohol oxidations to stereoselective halogenations of non‐activated C−H bonds. For many of these reactions, no “classical” chemical counterpart is known. Hence oxidoreductases open up shorter synthesis routes based on a more direct access to the target products. The generally very mild reaction conditions may also reduce the environmental impact of biocatalytic reactions compared to classical counterparts. In this Review, we critically summarise the most important recent developments in the field of biocatalytic oxidation chemistry and identify the most pressing bottlenecks as well as promising solutions.
Keywords: Baeyer–Villiger oxidation, biocatalysis, halogenation, oxidation, oxyfunctionalisation
1. Introduction: The Scope of Biocatalytic Oxidation Catalysis
Nature's arsenal of catalysts (enzymes) not only enables life as we know it but also can be gainfully used in organic synthesis. Nowadays, industrial and academic chemists are well aware of the benefits of hydrolases as stereoselective catalysts operating under mild reaction conditions. Oxidoreductases are now at the edge of becoming truly useful catalysts for selective oxidation and reduction reactions. Especially oxidative enzymes provide the synthetic chemist with unforeseen means for selective oxidation and oxyfunctionalisation reactions.
In this Review, we critically evaluate the current state of the art and point out promising new developments, but also name current limitations and areas where improvements are needed.
2. The Catalysts
Mechanistically, biocatalytic oxidation reactions fall into two categories: dehydrogenation (Scheme 1) and oxyfunctionalisation reactions (Scheme 2). In the dehydrogenative case, an acceptor molecule (mostly a nicotinamide or flavin cofactor; see the Supporting Information, Figure S1) abstracts a hydride from a heteroatom‐substituted carbon atom. Laccase‐ and peroxidase‐catalysed hydrogen‐atom abstraction reactions, mostly on phenols, are also known, resulting in phenoxy radicals as primary products.
Scheme 1.
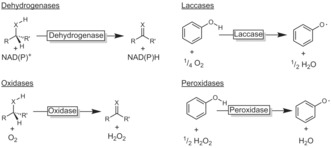
Biocatalytic oxidation by dehydrogenation. Dehydrogenases and oxidases mediate a hydride abstraction from alcohols and amines (X=O, NH); laccases and peroxidases mediate H atom abstraction reactions from phenolic starting materials.
Scheme 2.
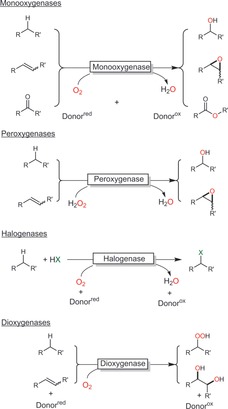
Biocatalytic oxidation by oxyfunctionalisation.
Oxyfunctionalisation pathways rely on the reductive activation of molecular oxygen (in the case of mono‐ and dioxygenases) or on already reduced hydrogen peroxide (in the case of peroxygenases) and subsequent electrophilic insertion into the starting material.
2.1. Dehydrogenation Catalysts
Alcohol dehydrogenases (ADHs, E.C. 1.1.1) and imine reductases (IREDs, E.C. 1.4.1) are classical dehydrogenation catalysts, mediating the reversible transfer of a hydride from an alcohol or amine carbon atom to the nicotinamide cofactor (NAD(P)+; Scheme 1 and Figure S2). Compared to their use in reductive processes (generating chiral centres), oxidative applications (destroying a chiral centre) are far less common.
Flavin‐dependent oxidases (E.C. 1.1.3; Scheme 1 and Figure S3) utilise an enzyme‐bound flavin moiety (generally flavin adenine dinucleotide, FAD) as the primary hydride acceptor.1 Regeneration of the oxidised cofactor occurs by direct aerobic reoxidation, yielding H2O2 as a byproduct (Figure S3). At first sight, H2O2 as a stoichiometric byproduct may appear as a challenge for enzyme robustness. However, in most cases, simple addition of catalase is sufficient to circumvent such issues. More importantly, the poor O2 solubility in aqueous media (ca. 0.25 mm) makes efficient O2 intake essential to avoid limitations.2
Copper‐dependent oxidases (E.C. 1.1.3, Figure S3) are also well‐known. Galactose oxidase (GaOx) is certainly one of the most frequently used copper‐dependent oxidases.3 Especially its engineered variants show significant potential for kinetic resolution reactions.4
Laccases (E.C. 1.10.3.2) are copper‐dependent oxidases, reducing molecular oxygen to water (Figure S4). The substrate scope of laccases is rather restricted to phenols and similarly activated compounds. Furthermore, phenoxy radicals are formed as the primary products, which then further react chemically to oligomers or quinoid products. Therefore, preparative applications of laccases are mainly focussed on so‐called laccase mediator systems (LMSs) wherein the laccase oxidatively regenerates a low‐molecular‐weight redox mediator acting as the actual oxidant (Figure S4).
2.2. Oxyfunctionalisation Catalysts
Oxygenases are, from a synthetic point of view, the most exciting enzymes. They catalyse the selective insertion of oxygen into C−H, C−C, and C=C bonds, thereby generating new functional groups instead of “just” converting existing ones. They utilise activated electrophilic oxygen species such as organic peroxides or high‐valent metal oxo complexes (Scheme 3).5
Scheme 3.
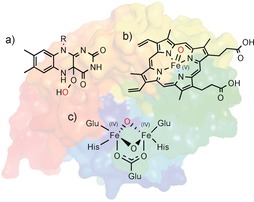
The most common active species used by oxygenases: a) 4a‐peroxoflavins, b) oxyferryl heme species, and c) compound Q in non‐heme iron monooxygenases.5 The electrophilic oxygen atoms are shown in red.
In the case of mono‐ and dioxygenases, these activated species are formed by reduction of O2. The reducing equivalents are derived from reduced nicotinamide cofactors (NAD(P)H). As mentioned above, the high cost of the latter has driven the development of a range of in situ regeneration systems (Table S2).
Flavin‐dependent monooxygenases catalyse a broad range of epoxidation, hydroxylation, and halogenation reactions of activated arenes, heteroatom oxygenations, and Baeyer–Villiger oxidations.6 They depend on NAD(P)H to reduce the enzyme‐bound flavin cofactor, which, after reaction with O2, forms the oxygenating 4a‐peroxoflavin (Scheme 3 a and Figure S5). Unproductive oxidation of NAD(P)H, yielding H2O2 and wasting reducing equivalents, can occur but is generally not as pronounced as with P450 monooxygenases.
Heme‐dependent monooxygenases are more powerful oxyfunctionalisation catalysts as they also act on non‐activated C−H bonds.7 The formation of the active species (compound I, Scheme 3 b and Figure S6) is more complex than with flavin‐dependent monooxygenases. FeIII, in contrast to flavins, cannot accept a hydride from NAD(P)H directly but requires a relay enzyme in between to transform the hydride donation event (by NAD(P)H) into two successive single‐electron‐transfer processes (Scheme 4).
Scheme 4.
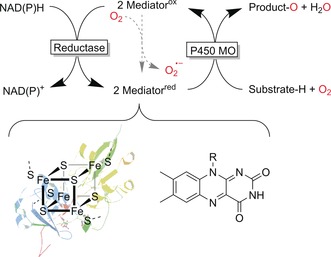
Generalised regeneration scheme for P450 monooxygenases. The reducing equivalents are derived from NAD(P)H and delivered sequentially via single electron mediators (e.g., ferredoxin or flavins) to the monooxygenase subunit.
P450 monooxygenases are prone to the “oxygen dilemma”.8 The mediators are single‐electron mediators, which react with molecular oxygen in a diffusion‐controlled process, hence uncoupling the electron transport chain. As a consequence, not only reactive oxygen species are formed but also valuable reducing equivalents are wasted. This is possibly one of the reasons for the comparably low turnover numbers for P450 monooxygenases, limiting their general preparative usefulness.
Peroxygenases are structurally related to the P450 monooxygenases as they also contain a heme moiety and rely on compound I as the oxygenating species.9 In contrast to P450 monooxygenases, however, peroxygenases form compound I from H2O2 (in the so‐called peroxide shunt pathway; Figure S6), and in principle thereby enable much simpler reaction schemes.9, 10 H2O2, however, is also a strong inactivator of heme enzymes (Figure S6), necessitating adjusted provision with H2O2 to enable the peroxygenase reaction while minimising the oxidative inactivation. A range of in situ H2O2 generation systems have been developed to address this challenge (Table S3).
Aside from heme‐dependent oxygenases, a range of non‐heme‐iron‐dependent mono‐ and dioxygenases also exist.5 While the actual oxyfunctionalisation mechanism differs somewhat from those of the heme‐dependent enzymes (Figure S7), the electron supply mechanisms (together with the issues outlined above) are similar. Copper‐dependent lytic polysaccharide monooxygenases (LPMOs) are receiving tremendous research interest as their contribution to a more efficient use of biomass is envisaged.11
3. General Considerations and Concepts
3.1. Cofactor/Enzyme costs
For decades, the high cost of the nicotinamide cofactors has been seen as the major challenge towards economical oxidoreductase reactions. As a consequence, a broad range of in situ regeneration systems have been developed that enable the use of NAD(P) in catalytic amounts (Table S1). As a rule of thumb, the turnover number for NAD(P) should exceed 1000.
More recently, enzyme costs are also coming to the forefront. As pointed out by Woodley and co‐workers,12 biocatalyst prices are subject to the economy of scale, and enzymes produced on very large scale only cost around several hundred euros per kilogram. To estimate the cost contribution of an enzyme to the final product, its turnover number is a good indication. Table 1 gives an overview of the approximate turnover numbers (TNs) minimally needed for an enzyme to meet the requirements of a given product category. It should be emphasised here that these numbers are meant as a rule of thumb.
Table 1.
Enzyme performance requirements.12
| Industry segment | Product price range [€ kgprod. −1] |
Allowable cost contribution of the catalyst [€ kgprod. −1] |
Minimal productivity [kgProd. kgcat. −1][a] |
Minimal productivity [TN][b] |
|---|---|---|---|---|
| pharma | >100 | 10 | 20 | 4000 |
| fine chemicals | >15 | 1.5 | 133 | 26 666 |
| speciality chemicals | 5 | 0.25 | 800 | 16 0000 |
| bulk chemicals | 1 | 0.05 | 4000 | 80 0000 |
[a] Assuming an average catalyst cost of 200 € kg−1 (for crude enzyme preparations). [b] Assuming an enzyme molecular weight of 40 kDa and a product molecular weight of 200 g mol−1. TN=mol(product)×mol(enzyme)−1.
3.2. Whole‐Cell Catalysis versus Isolated Enzymes
The choice of the enzyme formulation, either as whole, metabolically active cells or as isolated enzymes, is a fundamental decision to be taken at an early stage of the synthesis planning. Both approaches have specific (dis)advantages. Using whole cells, for example, makes external cofactors and their regeneration obsolete. On the other hand, whole‐cell biotransformations require specialised equipment, which is not readily available in chemical laboratories. Moreover, undesired side reactions are frequently observed owing to metabolisation of the reagents by endogenous host enzymes. With isolated enzymes, higher productivities can often be reached, albeit at the expense of lower biocatalyst robustness under the process conditions.
Overall, a general recommendation for either biocatalyst formulation cannot be given. The ideal system depends on specific factors related to the existing infrastructure and the requirements of the reaction.
3.3. Substrate Scope
Depending on their natural function, enzymes can be highly specific for one substrate, exhibiting poor or no activity on anything else but the natural substrate. This is especially true for enzymes originating from the primary metabolism. Other enzymes, however, exhibit a much more relaxed substrate scope, converting a broad range of starting materials. Also, the natural selectivity of a given enzyme not necessarily meets the requirements of the envisaged transformation. Furthermore, enzyme activity or stability may not be sufficient.
Protein engineering is the method of choice to address these issues. The past two decades have seen an astonishing development of protein engineering methods.13 Some examples are discussed throughout this Review.
3.4. Substrate Loading
The majority of the reagents of interest are rather hydrophobic, with poor solubilities in the aqueous reaction media required for many oxidoreductase reactions. For those cases where non‐aqueous biocatalysis is not possible, the two‐liquid phase system (2LPS) is an appropriate compromise, enabling overall high reagent loadings (Scheme 5).
Scheme 5.
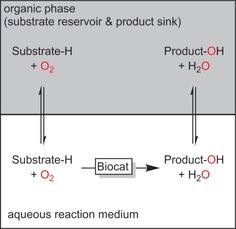
The two‐liquid‐phase system (2LPS) comprising an aqueous reaction medium with biocatalysts and a hydrophobic organic phase serving as the substrate reservoir and product sink.
Furthermore, 2LPS can help shift unfavourable reaction equilibria, remove reactive species (thus preventing undesired side reactions), and reduce toxic effects of the reagents on the catalysts. 2LPS will be discussed throughout this Review.
3.5. Cascade Reactions
Combining oxidative reactions with further (bio)catalytic transformations represents a sustained trend in academic research.14 The potential of such cascade reactions is related to their efficiency in terms of reactor use (several transformations occurring simultaneously instead of sequentially) and environmental impact (downstream processing and product purification are usually rather material‐intensive). Some interesting cascades will be discussed later.
4. Oxidation of C−H Bonds
Selective transformations of C−H bonds into C−X bonds are a challenging task for organic synthesis. The inertness of C−H bonds even in allylic or benzylic positions necessitates rather potent oxidation agents for which discrimination between similar C−H bonds is difficult. Particularly, the presence of C−H bonds that are more activated than that of the position of interest reduces the selectivity of the desired oxyfunctionalisation reaction. Hence, the outcome of many “chemical” oxidation reactions is often dominated by the properties of the starting material rather than by the oxidation catalyst. In this respect, enzymatic C−H bond activations excel because the oxidation agent is embedded within the well‐defined structure of an enzyme active site. Hence, positioning of the starting material relative to the reactive agent can override the chemical reactivity of the starting material.
A broad range of biocatalytic C−H activation reactions are known today, and a representative selection will be discussed in the following Section.
4.1. Oxidation of C−H to C−OH
4.1.1. Hydroxylation of C(sp3)−H bonds
(Non‐)heme monooxygenases catalyse the regio‐ and stereospecific hydroxylation of non‐activated C−H bonds. Especially, P450 monooxygenases have received significant attention, resulting in vast knowledge with regard to their catalytic mechanisms as well as various wild‐type and mutant enzymes with tailored properties.7, 15 Scheme 6 gives a representative, yet incomplete, overview of the versatility and often superb selectivity of monooxygenases in hydroxylation reactions. In essence, any given starting material can be hydroxylated with P450 monooxygenases.
Scheme 6.
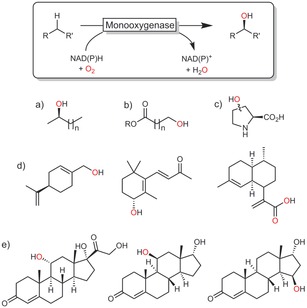
Selection of monooxygenase‐catalysed hydroxylation reactions: a) ω‐1 hydroxylation,16 b) ω hydroxylation,17 c) stereospecific 3‐ or 4‐hydroxylation of, for example, proline,18 d) allylic hydroxylation,19 e) steroid hydroxylation.20
Not every wild‐type enzyme exhibits the desired regio‐ and/or enantioselectivity for a given starting material. Recent advances in protein engineering, however, enable tailoring the selectivity of a given P450 monooxygenase. In this respect, the P450 monooxygenase from Bacillus megaterium (P450BM3) is one of the most intensively studied enzymes.21 Scheme 7 shows some selected examples that impressively demonstrate the power of protein engineering to adjust the selectivity of P450BM3.
Scheme 7.
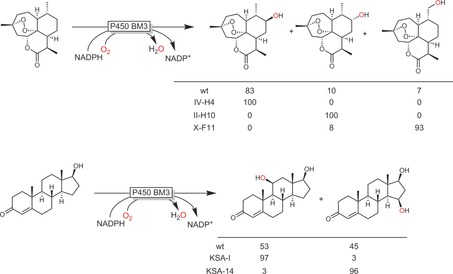
Selected examples of P450BM3 engineering to change the selectivity.19c, 20
In addition to protein engineering, reaction and substrate engineering also efficiently modulate the reactivity and selectivity of P450 monooxygenases. Decoy molecules, for example, are experiencing some interest in facilitating the conversion of small substrates.22 Watanabe, Reetz, and others have reported that perfluorinated fatty acids can accelerate P450BM3‐catalysed oxyfunctionalisation reactions of small aromatics,23 short‐chain alkanes,24 and even methane.25 The rationale behind these results is that the inert decoy compounds partially fill up the spacious active site of the enzyme and thereby facilitate the orientation of the small substrate to the heme active site.
Another non‐protein‐engineering‐based approach to improve the activity or selectivity of an enzyme towards non‐natural substrates is based on the temporary introduction of bulky protecting/directing groups. Early contributions by Griengl and co‐workers demonstrated that the hydroxylation of cyclopentanone does not occur using the P450 monooxygenase from Beauveria bassiana whereas the corresponding spirooxazolidine was converted rather efficiently.26 Similarly, Bell and co‐workers achieved the selective 4‐hydroxylation of ester‐ or ether‐protected cyclohexanols.27 Very impressive changes in the regioselectivity of a P450 monooxygenase‐catalysed hydroxylation of macrolides have been achieved with this approach (Scheme 8).28
Scheme 8.
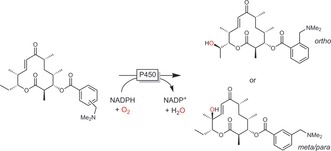
Controlling the regioselectivity of a macrolide hydroxylation with ester directing groups.28
Aside from the selectivity issues, P450 monooxygenase reactions also suffer from the generally low turnover numbers of the oxyfunctionalisation catalysts. Typically, TNs of no more than 50 000 are reported.29 This also explains why preparative‐scale30 or even industrial‐scale applications of P450 monooxygenase reactions today are still predominantly realised in the pharmaceutical industry. Two exceptions developed by BASF are summarised in Scheme 9.
Scheme 9.
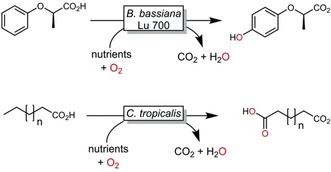
Examples of industrial P450 monooxygenase catalysed hydroxylation reactions.31
A possible solution to this may be the use of peroxygenases (E.C. 1.11.2.1). Already in the 1960s, but especially in the 1990s, the peroxygenase from Caldariomyces fumago (CfuUPO) received attention as an alternative to P450 monooxygenases.32 More recently, Hofrichter and co‐workers reported on a significantly more active peroxygenase from Agrocybe aegerita (AaeUPO),9, 33 which was soon followed by further peroxygenases from other sources, such as Marasmius rotula (MroUPO)34 or Coprinopsis cinerea (CciUPO).35 These enzymes exhibit interesting differences in their selectivity: Alkane hydroxylation by AaeUPO occurs preferentially in ω‐1 and ω‐2 position whereas MroUPO is highly selective for the terminal ω position. The degree of overoxidation also differs significantly. For example, selective monohydroxylation of cyclohexane to cyclohexanol was observed with AaeUPO whereas MroUPO exhibited higher overoxidation activity and consequently generated larger amounts of cyclohexanone.36
Gutierrez and co‐workers compared the dramatic selectivity difference of two peroxygenases (AaeUPO and CciUPO) in the hydroxylation of cholecalciferol (vitamin D).35b Whereas AaeUPO was rather unselective, giving at least three different hydroxylated isomers, CciUPO gave only one product (25‐OH‐cholecalciferol). These observations were rationalized by differences between the molecular architectures and the resulting differences in the mobilities of the substrates in the active sites.
We expect that our systematic knowledge and understanding of structural determinants for peroxygenase activity and selectivity will increase in the near future. Already today, TNs of more than 400 000 have been achieved. Together with efficient in situ H2O2 generation systems,37 this will render peroxygenases truly preparatively useful hydroxylation catalysts.
The cofactor independence of peroxygenases brings about another advantage over P450 monooxygenases. Peroxygenases can be applied under non‐aqueous, or even neat, reaction conditions. In a proof‐of‐concept study, enantiomerically pure products were obtained in up to 120 mm from such a neat reaction system using immobilised AaeUPO.38
4.1.2. Hydroxylation of Aromatic C(sp2)−H Bonds
4.1.2.1. Hydroxylation of Non‐Activated Arenes
Hydroxylations of non‐phenolic aromatic compounds are clearly the domain of (non‐)heme iron monooxygenases. Only these enzymes exhibit the oxidation power required for the conversion of non‐activated C−H bonds. The generally accepted mechanism for this reaction comprises the formation of an arene oxide, which spontaneously rearranges into the phenol product (NIH shift). This oxidative power, however, also frequently causes selectivity issues, especially when alkyl‐substituted arenes are employed as starting materials (ring hydroxylation vs. benzylic hydroxylation). Recent advances in (semi‐)rational enzyme engineering, however, demonstrate that this issue can be solved.39
With peroxygenases,40 further conversion of the phenol products into phenoxy radicals (and further radical reactions) represents an undesired side reaction, which can be circumvented either by co‐administration of antioxidants or, more elegantly, by protein engineering.41
Dioxygenases (DOs, E.C. 1.13.11) are very powerful catalysts for the selective cis dihydroxylation of aromatic compounds. Several hundreds of DO products have been reported.42 Mostly toluene dioxygenases (TDOs), naphthalene dioxygenases (NDOs), benzoate dioxygenases (BDOs), and biphenyl dioxygenases (BPDOs) from various organisms are used (Scheme 10). Owing to their complex electron‐transport chains (Scheme 4), the majority of DO reactions build on recombinant, whole‐cell catalysts. Dioxygenases are widely used in natural product synthesis.43
Scheme 10.
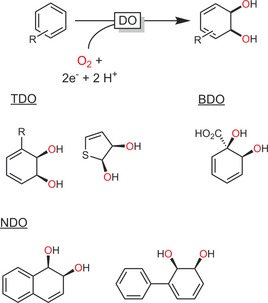
Dioxygenase (DO)‐catalysed cis dihydroxylation of aromatic compounds and a representative selection of DO‐derived products. TDO: toluene dioxygenase;44 BDO: benzoate dioxygenase;45 NDO: naphthalene dioxygenase.46
4.1.2.2. Hydroxylation of Activated Arenes
Flavin‐dependent monooxygenases (FMOs, E.C. 1.14.13.x) are mostly used for the selective ortho or para hydroxylation of phenols.47 Well‐known representatives of the FMO class are 4‐hydroxybenzoate 3‐hydroxylase (PHBA),48 hydroxyl biphenyl‐3‐monooxygenase (HbpA),49 and 3‐hydroxybenzoic acid‐6‐hydroxylase (3HB6H),50 which all catalyse the selective ortho or para hydroxylation of various phenols.
Thus far, FMO‐catalysed hydroxylations of phenols have been reported to occur only in combination with rearomatisation, hence giving aromatic products. Very recently, Narayan and co‐workers, however, reported various FMOs catalysing the oxidative dearomatisation of substituted phenols, giving access to a whole world of new synthons for organic synthesis (Scheme 11).51 Very exciting new reactions may be expected in the future.
Scheme 11.
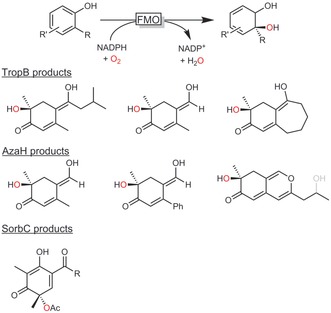
ortho‐Quinol products obtained by oxidative dearomatisation.51 TropB: FMO from Talaromyces stipitatus; AzaH: FMO from Aspegillus niger; SorbC: FMO from Penicillium chrysogenum.
Aside from hydroxylation, the halogenation of activated aromatics is also an emerging field of research. Haloperoxidases, for example, initiate the halogenation of activated aromatic compounds. The catalytic mechanism involves oxidative activation of halides to hypohalites, which then, in a non‐enzymatic follow‐up reaction, form the halogenated products (Scheme 12). Aside from phenols, also pyrroles52 and indoles53 have been converted in this fashion. As the halogenation does not occur within the enzyme active site, the selectivity of the halogenation reaction is dictated by the chemical reactivity of the starting material rather than by the enzyme, challenging its preparative scope. Nevertheless, the very high catalytic activities (TN>2 000 000 and TFaverage>25 s−1) that have been achieved are very convincing.37d, 52, 54
Scheme 12.
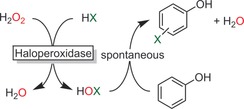
Chemoenzymatic halogenation of phenols by haloperoxidase‐catalysed hypohalite formation.
Selective halogenations of aniline derivates are catalysed by a range of flavin‐dependent halogenases.55 Most interestingly, halogenases with complementary regioselectivities have been reported. For example, tryptophan halogenases with 5‐,56 6‐,57 or 7‐selectivity57b are known.
Micklefield and co‐workers recently demonstrated that both the substrate scope and the regioselectivity of flavin‐dependent halogenases can be controlled by protein engineering (Scheme 13).58
Scheme 13.
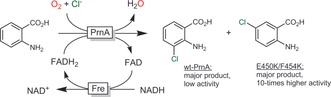
Halogenation of “non‐natural” anthranilate substrates using the tryptophan halogenase from Pseudomonas fluorescens (PrnA) together with an FADH2‐recycling system (Fre).58
Seebach and co‐workers have focused on the preparative application of halogenases by designing elegant multi‐enzyme cascades to convert indoles into halogenated tryptophan derivatives59 on gram scale.60 Halogenases have also been used in more complex chemoenzymatic reactions, particularly by using the halogenated products as starting materials for Suzuki‐type cross‐coupling reactions.61
Nevertheless, despite the undoubted synthetic potential of this enzyme class, the very low catalytic activity of flavin‐dependent halogenases (k cat values that are rather in the range of h−1 than s−1) still challenges their broad applicability.
Finally, aromatic nitration is worth mentioning. Arnold and co‐workers have reported mutants of the P450 monooxygenase from Streptomyces scabies (TxtE) exhibiting different regioselectivities in the nitration of tryptophan (Scheme 14).62
Scheme 14.
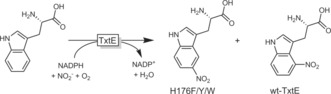
Enzymatic nitration of tryptophan with TxtE. The regioselectivity of the wild‐type enzyme was altered by protein engineering.62a
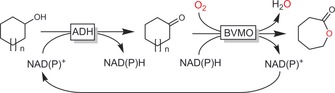
4.2. Oxidation of C−H to C=O
Double oxidation of (cyclo)alkanes to the corresponding aldehydes or ketones is possible through the combination of NAD(P)H‐dependent monooxygenases catalysing the C−H hydroxylation with NAD(P)+‐dependent dehydrogenases to transform the intermediate alcohol into the carbonyl product (Scheme 15). Such redox‐neutral reaction schemes are attractive especially for the production of low‐value‐added compounds such as cyclohexanone, where any additional costs for cosubstrates would make any possible production system unattractive from scratch.
Scheme 15.
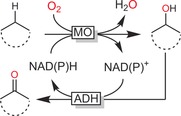
Double oxidation of alkanes to ketones. The combination of NAD(P)H‐dependent monooxygenases with NAD(P)+‐dependent alcohol dehydrogenases enables overall redox‐neutral reactions.
This strategy has been applied for the conversion of (cyclo)alkanes,63 of α‐isophorone into ketoisophorone,19b and of valencene into nootkatone.64
Reetz and co‐workers have extended this system to obtain all possible stereoisomers of 1,2‐cyclohexanediol from cyclohexane (Scheme 16).65
Scheme 16.
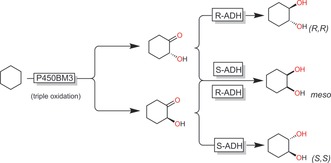
Cascade P450BM3‐catalysed, three‐step oxyfunctionalisation of cyclohexane with stereospecific ADH‐catalysed reduction of the resulting hydroxyketones to yield all possible cyclohexane‐1,2‐diol isomers.65
The selective oxidation of CH3 groups to the corresponding aldehydes can be a challenge using whole‐cell biocatalysts. This is due to the presence of endogenous aldehyde dehydrogenases catalysing the undesired overoxidation. Aside from microbial strain optimisation, the 2LPS approach is a simple solution to this issue as the hydrophobic aldehyde is extracted into the hydrophobic organic phase and thereby removed from the oxidation catalyst.66
4.3. Desaturation of CH−CH to C=C Bonds
Desaturations of carbonyl compounds to the corresponding α,β‐unsaturated enones are principally possible by exploiting the reversibility of flavin‐dependent Old Yellow Enzyme‐like ene reductases.67
Even more interesting than the aforementioned desaturations of “activated” C−C bonds is the desaturation of non‐activated C−C bonds as, for example, found in saturated fatty acids. In principle, Nature already provides suitable catalysts for this reaction in the form of the so‐called fatty acid desaturases (FADs). These non‐heme diiron enzymes are structurally and mechanistically related to some non‐heme diiron monooxygenases (Figure S8). Most FADs do not act on the free fatty acid but rather on the corresponding thioester with a so‐called acyl carrier protein. Therefore, in vitro applications of FADs do not appear practical nowadays. A few reports deal with the in vivo overexpression of some desaturases in whole cells, generating a few mg L−1 of, for example, α‐linoleic acid.68
Overall, oxidative desaturation is an interesting but not yet very far developed reaction.
4.4. Amination (C−H to C−NH2)
Indirect C−H amination reactions can be achieved by cascades of monooxygenase‐catalysed hydroxylation followed by ADH‐catalysed oxidation to the aldehyde/ketone and reductive amination (Scheme 17).69
Scheme 17.
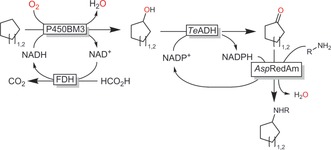
Enzyme cascade for the transformation of cycloalkanes into secondary amines.69
More elegantly, Arnold and co‐workers recently developed a method for the direct amination of C−H bonds by designing P450 variants (particularly with a distal Ser instead of the natural Cys ligand; named P411) capable of forming nitrenoid intermediates that act as nitrene transfer agents (Scheme 18).70 An intramolecular variant of this reaction is also catalysed by other heme‐dependent enzymes.71, 72
Scheme 18.
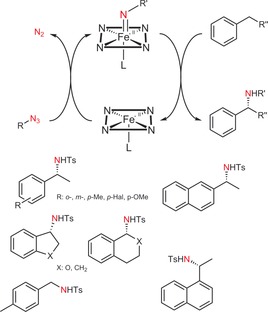
Enantiospecific nitrene transfer to alkyl‐substituted arenes catalysed by engineered P450 monooxygenases. The P411 enzyme has been generated from a P450 monooxygenase by ligand substitution. Note that the reduced (FeII) oxidation state is the catalytically active one. Below: Preliminary product scope; all chiral products are essentially enantiopure.70
Although the turnover numbers reported for the P450 mutants tend to be low (ca. 1000), exciting future developments and synthetic applications are expected.
4.5. Halogenation (C−H to C−X)
Selective halogenation, especially of non‐activated C−H bonds, represents a major challenge for organic synthesis. In contrast, Nature has developed an arsenal of halogenating enzymes (halogenases) that combine oxidative power with selectivity.55a, 73 In addition to the halogenases/haloperoxidases discussed above, also a range of α‐ketoglutarate‐dependent non‐heme iron halogenases capable of halogenating non‐activated C−H bonds are known. Their catalytic mechanism resembles that of α‐KG‐dependent monooxygenases, with the exception that the rebound of the C‐centred radical does not occur with an iron‐bound OH group but rather with an iron‐bound halogen atom (Figure S7). α‐KG‐dependent halogenases are a relatively young class of enzymes first reported in 2005 by Walsh and co‐workers.74 Since then, a range of selective halogenations (mostly chlorinations) have been reported (Scheme 19).73
Scheme 19.
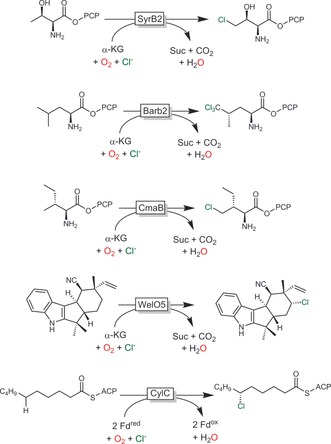
Selection of selective halogenation reactions mediated by halogenases. PCP: peptidyl carrier protein; ACP: acyl carrier protein; SyrB2: halogenase from Pseudomonas syringae;74 Barb2: halogenase from Lyngbya majuscule (involved in barbamide biosynthesis);75 CmaB: halogenase from Pseudomonas syringae (involved in coronatine biosynthesis);76 WelO5: halogenase from Hapalosiphon welwitschii (involved in welwitindolinone biosynthesis);77 CylC: non‐α‐KG‐dependent halogenase from Cylindrospermum licheniforme.78
Preparative applications of this enzyme class are challenged by their very high to exclusive substrate specificity (including the need for tethering of the starting material to a peptidyl carrier protein). In this respect, the recent discovery of WelO5 from Hapalosiphon welwitschii (Scheme 19),77 catalysing halogenation on a freestanding (i.e., non‐carrier‐protein‐bound) substrate, may lead to further developments. Their mechanistic and structural similarity to α‐KG‐dependent monooxygenases also offers the possibility of engineering of the latter into halogenases.79 This may well lead to a very significant broadening of the scope of biocatalytic halogenation chemistry.
Overall, biocatalytic halogenation of non‐activated C−H bonds is a very young, but dynamically developing field. Current limitations, such as the narrow substrate scope and the rather poor turnover numbers of the biocatalysts reported thus far, are expected to be solved in the coming years.
5. Oxidation of C=C Bonds
5.1. Epoxidation Reactions
Epoxidations of alkenes have been investigated with various enzyme systems, ranging from peroxygenases and monooxygenases to lipase systems (Scheme 20).
Scheme 20.
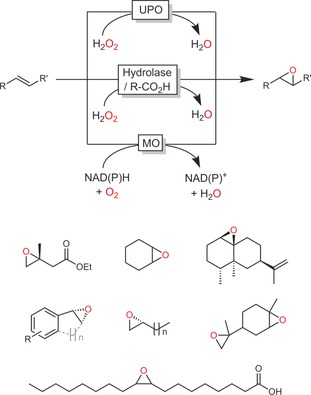
Most common biocatalytic epoxidation methods employing monooxygenases,80 peroxygenases,81 or lipases in the “perhydrolase” approach and some representative products.
Schmid and co‐workers, for example, pioneered preparative‐scale epoxidation reactions using the SMO from Pseudomonas sp.80c, 82
Recombinant whole‐cell systems in 2LPSs are efficient in producing enantiomerically pure styrene epoxides.80c Growing cells provide sufficient reductive power to promote the monooxygenase reaction while the organic phases not only increase the overall substrate loading but also alleviate toxic effects of the reagents on the biocatalyst. Thus more than 600 mm of the enantiomerically pure product can be obtained in excellent yields, pointing towards economically compatible processes.82a
The substrate scope of the SMO from Pseudomonas sp. VLB120 is limited to styrene derivates. This gap was closed by a new SMO from Rhodococcus sp.80b, 83 enlarging the product scope to include aliphatic terminal alkenes.
P450 monooxygenases80a, 84 and peroxygenases81, 85 also catalyse the epoxidation of C=C double bonds. However, allylic hydroxylation is a frequently observed side reaction. Again, protein engineering is a very powerful tool to overcome this limitation as, for example, demonstrated by Urlacher and co‐workers for the P450BM3‐catalysed epoxidation of limonene (Scheme 21).86
Scheme 21.
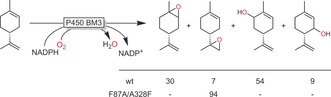
Engineered P450 BM3 (F87A/A328F) for highly (regio)selective epoxidation (as compared to the wild‐type (wt) enzyme).86
Increasing attention is being paid to hydrolase‐mediated chemoenzymatic epoxidation reactions. In the presence of H2O2, many hydrolases catalyse the conversion of carboxylic acid (esters) into the corresponding peracids (perhydrolase activity).87 The latter can then undergo a chemical (non‐stereospecific) epoxidation reaction (Prilezhaev reaction; Scheme 22).
Scheme 22.
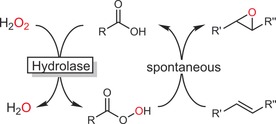
Chemoenzymatic Prilezhaev reaction using hydrolase‐(re)generated peracids.
The high robustness of many hydrolases and the cofactor independence of this reaction make it an interesting alternative to the above‐mentioned epoxidation approaches (provided that racemic products are acceptable),88 for example in the epoxidation of vegetable oils.89
Epoxides are excellent starting materials for further chemical transformations. Li and co‐workers have demonstrated this in their recent elegant work on the development of multi‐enzyme cascades to transform alkenes into (chiral) diols,90 amino alcohols,90 (amino) acids,90a and more (Scheme 23).91
Scheme 23.
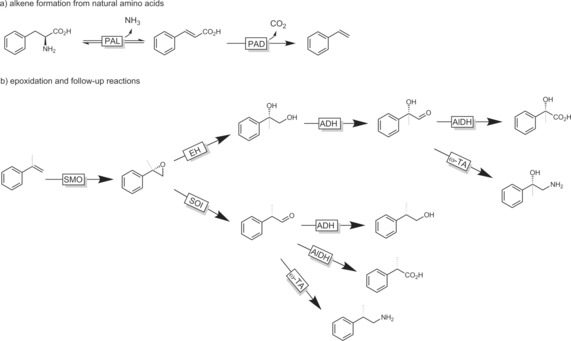
Combinatorial “explosion” of products attainable from alkenes by the smart combination of different biocatalysts. PAL: phenylalanine ammonia lyase; PAD: phenylacetic acid decarboxylase; SMO: styrene monooxygenase; EH: epoxide hydrolase; ADH: alcohol dehydrogenase; AldDH: aldehyde dehydrogenase; ω‐TA: transaminase; SOI: styrene oxide isomerase.91 For reasons of clarity, cosubstrates and regeneration systems have been omitted.
5.2. Aziridination Reactions
The Arnold group has recently developed some very exciting new abiotic reactions based on P450 mutants.92
For example, P450 mutants capable of tosyl azide driven aziridination reactions proceeding via iron nitrenoids have been reported (Scheme 24).93 A broad range of styrene derivates were converted into essentially enantiomerically pure aziridines.
Scheme 24.
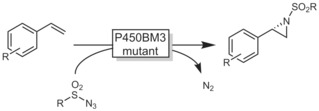
Enzymatic aziridination with P450BM3 mutants.93
It is not very surprising that this exciting new reactivity has already found applications, even on preparative scale.94
5.3. Hydroxyhalogenation Reactions
Hydroxyhalogenation of alkenes (and alkynes) is possible using haloperoxidases.73 These enzymes catalyse the H2O2‐driven oxidation of halides to hypohalites, which then spontaneously react with C=C double bonds to form halohydrins (Scheme 25).95
Scheme 25.
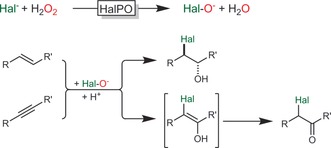
Haloperoxidase‐catalysed hypohalite formation followed by spontaneous, non‐enzymatic reactions with C=C and C≡C bonds.
Especially the vanadium‐dependent haloperoxidases excel over their heme counterparts owing their robustness against H2O2. The V‐dependent choroperoxidase from Curvularia inaequalis (CiVCPO) was used for the hydroxyhalogenation of various alkenes.96 On preparative scale, the enzyme performed more than 2 000 000 turnovers at an average turnover frequency of more than 20 s−1. This high activity, however, comes at the expense of low selectivity as the actual halohydrin formation occurs spontaneously outside the enzyme active site. Some exceptions prove the rule.97
5.4. Ozonolysis of Alkenes to Aldehydes
Oxidative cleavage of C=C double bonds has been well documented in various biosynthetic and degenerative pathways as in the cleavage of carotene to retinal.98 Preparative applications, however, are scarce.
In 2006, Kroutil and co‐workers reported that a MnIII‐dependent enzyme from Trametes hirsuta (AlkCE) is able to oxidatively cleave C=C double bonds, yielding the corresponding aldehydes or ketones (Scheme 26).99
Scheme 26.
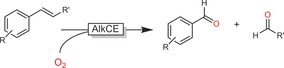
Aerobic cleavage of styrenes with AlkCE from Trametes hirsuta.99
5.5. Wacker Oxidation
Arnold and co‐workers engineered a P450 monooxygenase to perform anti‐Markovnikov Wacker oxidations of alkenes instead of the natural epoxidation reaction (Scheme 27).100
Scheme 27.
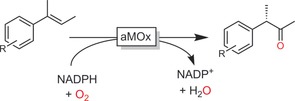
Use of an engineered P450 monooxygenase (aMOx) to catalyse the anti‐Markovnikov Wacker–Tsuji oxidation of alkenes.100
Despite the early stage of development, this approach not only represents an interesting alternative to chemical (Markovnikov‐type) Wacker–Tsuij oxidations101 but also sets the stage for further (enzymatic) transformations of the reactive aldehydes and ketones.
6. Oxidation of Alcohols
The importance of alcohol oxidation in preparative organic chemistry also explains the significance of this reaction in biocatalysis.102 The following Section concentrates on some recent concepts and selected examples for the oxidation of primary alcohols to aldehydes and acids. In addition, oxidative kinetic resolutions will be discussed briefly. Recent examples of oxidation/reduction cascades to transform alcohols into (chiral) amines and for the stereoinversion/deracemisation of alcohols have been discussed elsewhere.103
6.1. Oxidation of Primary Alcohols to Aldehydes
The majority of biocatalysts used for the oxidation of alcohols to aldehydes follow a dehydrogenative reaction mechanism. As a consequence, with isolated dehydrogenases or oxidases, overoxidation into the carboxylic acid is generally not observed as the aldehyde H atom cannot be abstracted as a hydride. However, using whole cells, overoxidation is frequently observed. When overoxidation to the carboxylic acid is not desired, the 2LPS approach has proven very efficient in removing hydrophobic aldehydes from the reaction medium.66b, 104
Often, the high reactivity of aldehydes is seen as a challenge for the stability of the biocatalysts used. In recent years, this has been more and more seen as an opportunity to generate more complex products. In situ generated aldehydes, for example, can be coupled to lyase‐catalysed C−C bond formation, generating chiral acyloins.105 Combinations with an aldolase106 or an organocatalytic aldol reaction107 are also possible (Scheme 28). Even combinations with a metal‐catalysed coupling reaction are feasible.108 The last reaction, however, had to be performed in a sequential fashion owing to the incompatibility of the enzymes used and the indium catalyst.
Scheme 28.
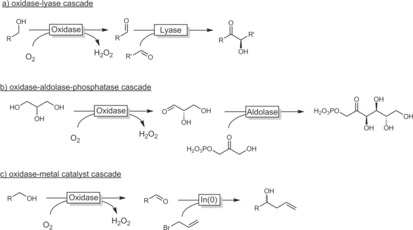
Selected cascade reactions involving an oxidase‐catalysed alcohol oxidation step coupled to a further catalytic transformation of the aldehyde generated.
Finally, the oxidative dealkylation of methyl ethers is worth mentioning. In particular P450 monooxygenases have been used for the specific hydroxylation of the methoxy C atom, yielding an instable hemiacetal, which spontaneously decomposes into formaldehyde and the deprotected alcohol. Very recently, Bornscheuer and co‐workers reported on a very interesting application of this reaction for the selective deprotection of carbohydrate ethers.177
6.2. Oxidation of Primary Alcohols to Acids
The aldehyde H atom cannot simply be abstracted as a hydride. Therefore, the enzymes commonly used for alcohol oxidation (operating through a hydride abstraction mechanism) are not directly applicable for the full oxidation of primary alcohols to carboxylic acids. Nevertheless, this reaction can be achieved by using a combination of alcohol and aldehyde dehydrogenases.
Especially with microbial catalysts (containing endogenous aldehyde dehydrogenases), full oxidations of alcohols to carboxylic acids are reported frequently. For example, the microbial oxidation of glycerol can lead to (R)‐glyceric acid using Acetobacter tropicalis or to dihydroxyacetone if Gluconobacter oxydans is used.102b
A similar reaction was recently reported by Pyo and co‐workers, who used Gluconobacter oxydans for the oxidation of 2‐methylpropane‐1,2‐diol to 3‐hydroxy‐2‐methylpropionic acid, which was followed by a thermal, TiO2‐catalysed dehydration of the intermediate hydroxy acid to methyl methacrylate (Scheme 29).110
Scheme 29.

Chemoenzymatic cascade for the production of methacrylic acid from 2‐methyl‐1,3‐propanediol.110
Using isolated enzymes generally requires the combination of alcohol dehydrogenases/oxidases and aldehyde dehydrogenases. As early as the 1980s Wong and co‐workers reported on the productive combination of alcohol and aldehyde dehydrogenases for the oxidation of primary alcohols to carboxylic acids.111 Later on, Ohta and co‐workers further developed this approach.112
More recently, Turner, Carnell, and co‐workers developed a chemoenzymatic toolbox to convert alcohols into acids or amides (Scheme 30).113 The authors impressively demonstrated that aryl aldehydes (generated in situ from the corresponding benzyl alcohols) can be further oxidised to either the acid using xanthine dehydrogenase or amides using peroxides in the presence of an amine.
Scheme 30.
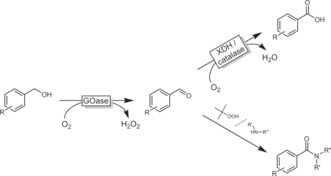
(Chemo)enzymatic cascades to transform (benzyl) alcohols into carboxylic acids or amides, depending on the reaction conditions. GOase: evolved galactose oxidase; XDH: xanthine dehydrogenase.113
Aldehydes react reversibly with nucleophiles to form adducts (e.g., gem‐diols, hemiacetals) that contain abstractable H atoms and consequently can be oxidised by dehydrogenases and oxidases. This strategy is used by the above‐mentioned aldehyde dehydrogenases (forming thio adducts during the catalytic cycle) and can be mimicked by using alcohol dehydrogenases. One example is the ADH‐catalysed oxidation of 1,n‐diols to lactones (Scheme 31). The initially formed aldehyde reversibly forms a hemiacetal (lactol), which is then oxidised once more to the corresponding lactone.
Scheme 31.
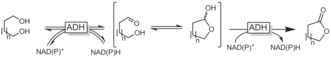
Oxidative lactonisation of 1,n‐diols using ADHs exploiting spontaneous lactol formation.
This approach has been studied extensively with the ADH from horse liver (HLADH) and other ADHs for the (enantioselective) oxidative lactonisation of various diols.114
Oxidative lactonisation reactions of diols have also been reported with the laccase–TEMPO system.115 As the alcohol oxidation step is non‐enzymatic, any chiral discrimination can be excluded.
Lactams, in principle, should be accessible through the strategy outlined in Scheme 31. The intermediate adduct, however, tends to undergo dehydration to yield imines, and is therefore not accessible for a second dehydrogenation step. This issue can be overcome by a bienzymatic strategy combining two oxidases (Scheme 32).106
Scheme 32.
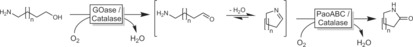
Bienzymatic synthesis of lactams from amino alcohols.106
In line with the current interest in furan dicarboxylic acid (FDCA) as a substitute for non‐renewable polymer building blocks such as phthalates, a range of biocatalytic transformations of glucose‐derived hydroxymethyl furfural (HMF) into FDCA have been developed (Scheme 33). Aside from whole‐cell approaches,116 oxidases117 and combinations of oxidases/peroxygenases118 have been employed.
Scheme 33.
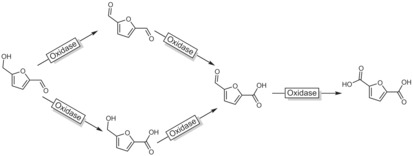
Generalised scheme for the oxidation of HMF to FDCA. For reasons of clarity, the actual catalysts (peroxygenase or oxidase) as well as the oxidants (H2O2 or O2) have been omitted.
6.3. Oxidation of Secondary Alcohols to Ketones
The oxidation of chiral secondary alcohols goes hand in hand with the destruction of a chiral centre and is therefore far less popular than the reverse reaction (i.e., enantiospecific reduction or reductive amination) of ketones. Nevertheless, enantioselective oxidation of only one enantiomer (i.e. kinetic resolution) keeps receiving considerable attention for the synthesis of enantiomerically pure alcohols.119 The challenge of a maximal theoretical yield of only 50 % remains and is being addressed by an increasing number of dynamic kinetic resolution methods.103
meso Compounds obviously represent an exception to the aforementioned yield limitation in the oxidation of secondary alcohols. For example, 1,2‐cycloalkanediols can be oxidised to the enantiomerically pure α‐hydroxy ketones (Scheme 34).120
Scheme 34.
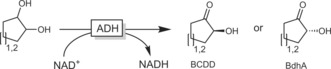
ADH‐catalysed oxidation of 1,2‐diols to the corresponding (R)‐ or (S)‐α‐hydroxy ketones depending on the ADH used. BdhA: butane diol DH from B. subtilis;119 BCDD: cis‐diol DH from P. putida.120a
6.4. Oxidation of Phenols
In addition to the above‐described hydroxylation of phenols, the radical polymerisation of phenols is also worth being mentioned.
Predominantly laccases and peroxidases have been reported for H‐atom abstraction reactions of phenols to generate phenoxy radicals, which undergo secondary, non‐enzymatic follow‐up reactions, mostly polymerisations (Scheme 35).121
Scheme 35.
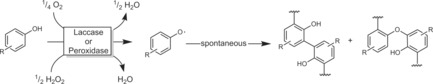
Phenol radical polymerisation initiated by laccases or peroxidases.
The radical mechanism also implies that usual advantages of enzyme catalysis such as selectivity may not be expected from this approach. In fact, aside from the biocatalyst concentration (correlating with the rate of the radical initiation reaction), factors such as solvent and oxygen concentration have a greater influence on the polymer properties than the nature of the biocatalyst used.121
7. Oxidation of Amines
Transaminases and imine reductases catalyse the reversible interconversion of amines and imines. Very similar to the situation with ADHs, the reductive direction is much more popular as here a chiral centre is being generated from a prochiral starting material whereas the oxidative direction only allows for a kinetic‐resolution‐type reaction to obtain optically pure amines. Therefore, examples of transaminase‐122 and IRED‐catalysed123 amine oxidations are scare.
Amino acid oxidases have received some attention as catalysts for the kinetic resolution of (un)natural amino acids. Both d‐ and l‐AAOx systems are available, giving access to both enantiomers of a broad range of α‐amino acids (as, for example, obtained from Strecker synthesis).124
In the pharmaceutical industry, d‐AAOx enzymes are used for the oxidative deamination of cephalosporin C, yielding the β‐lactam antibiotic precursor 7‐aminocephalosporanic acid.125
(Mono)amine oxidases exhibit a more relaxed substrate scope compared to AAOx systems. The monoamine oxidase from Aspegillus niger (MAO‐N) is by far the most widely studied and best‐understood amine oxidase. About 15 years ago, Turner and co‐workers turned their attention to MAO‐N,124a, 126 whose substrate scope and enantioselectivity were improved dramatically by several rounds of directed evolution.127
The poor yields of the MAO‐N‐catalysed kinetic resolution were also efficiently improved by designing (chemo)enzymatic deracemisation protocols.128
More recent developments focus around embedding MAO‐N into more complex cascade reactions. On the one hand, the MAO‐N substrate can be generated in situ in enzymatic129 and chemical130 steps. On the other hand, the reactive imines formed by MAO‐N can also undergo chemical follow‐up reactions to generate more complex products. For example, MAO‐N‐catalysed desymmetrisation of some pyrrolidines leads to enantiomerically pure Δ1‐pyrrolines. Nucleophilic attack by cyanide131 or imidazole132 predominantly yields the trans adducts (Scheme 36 a). The MAO‐N‐derived Δ1‐pyrrolines have also been embedded into multicomponent reactions such as the Ugi reaction (Scheme 36 a).133
Scheme 36.
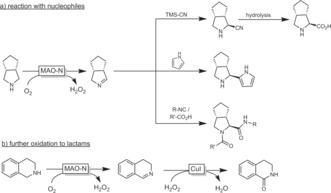
Embedding MAO‐N‐generated imines into more complex multistep chemoenzymatic syntheses.
Very recently, the further chemical oxidation of MAO‐N‐derived imines into lactams was demonstrated (Scheme 36 b).134 Similarly, Chen and co‐workers reported a whole‐cell oxidation of some amines to lactams.135
An intramolecular version of the aforementioned electrophilic attack at in situ generated imines and iminium ions is catalysed by the so‐called berberine bridging enzyme (BBE).136
A very interesting new development in the oxidation of amines was reported by Asano and co‐workers.137 Using a mutant of the d‐amino acid oxidase from porcine kidney in the presence of cyanides, they demonstrated the conversion of amines into α‐aminonitriles, serving as starting materials for further reactions (Scheme 37).
Scheme 37.
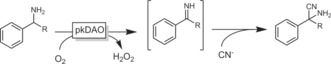
Conversion of amines into α‐aminonitriles using a combination of d‐amino acid oxidase (mutant, pkDAO) and cyanide.137
The oxidative conversion of amino acids into nitriles using haloperoxidases is also worth mentioning here (Scheme 38).138 If further developed, this might become a novel route to bio‐based nitriles.
Scheme 38.
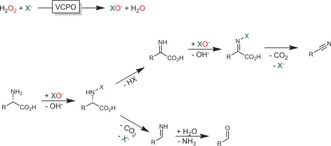
Haloperoxidase‐initiated oxidative decarboxylation of amino acids leading to either the C1‐shortened nitrile or the aldehyde via the corresponding imine. VCPO: vanadium‐dependent haloperoxidase.138
8. Oxidation of Aldehydes and Ketones
8.1. Oxidation of Aldehydes to Acids
As mentioned above, aldehydes are generally unreactive in dehydrogenase‐catalysed oxidations. If, however, the gem‐diol form is present in significant amounts, ADH‐catalysed oxidation becomes feasible. For example, racemic 2‐aryl propionals can be oxidised by ADHs in a dynamic kinetic resolution to enantiomerically pure 2‐aryl propionic acids (Scheme 39).139 Under slightly alkaline conditions, not only the aldehyde is hydrated but also enolisation takes place, paving the way for in situ racemisation.
Scheme 39.
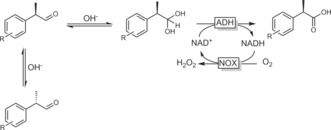
Dynamic kinetic resolution of profen aldehydes to the acids using ADHs.139
In a similar approach, Hall, Faber, and co‐workers reported an ADH‐catalysed Cannizzaro‐type disproportionation of aldehydes.140
8.2. Baeyer–Villiger Oxidation
Two enzyme classes dominate the field of enzymatic Baeyer–Villiger oxidations: the so‐called Baeyer–Villiger monooxygenases (BVMOs) and hydrolases (lipases, esterases) in the perhydrolase approach (Scheme 40).
Scheme 40.
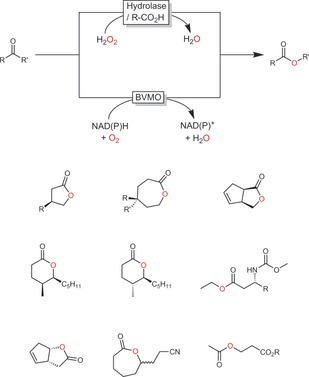
Biocatalytic Baeyer–Villiger oxidations using flavin‐dependent Baeyer–Villiger monooxygenases (BVMO) or the perhydrolase approach. Below some selected preparative examples are shown.
8.2.1. Baeyer–Villiger Monooxygenases (BVMOs)
BVMOs are flavin‐dependent monooxygenases (Figure S5).141 The deprotonated 4a‐peroxoflavin acts as a nucleophile in attacking the carbonyl group, which yields a Criegee intermediate. The migration tendency of the two alkyl substituents, however, not only depends on their substitution pattern but can also be influenced by the enzyme active site. Hence, BVMO‐mediated Baeyer–Villiger oxidations give access to products inaccessible by chemical pathways. Furthermore, even if a given enzyme does not reach the desired selectivity, it can be engineered.142
Amongst the numerous examples demonstrating how BVMO engineering can be applied to obtain both the “normal” and the “abnormal” lactone products, two examples reported by the Reetz and Bornscheuer groups should be mentioned here. Using CHMO as the catalyst, Reetz and co‐workers achieved the discrimination of E/Z‐configured, trisubstituted alkenes containing keto groups (Scheme 41).142a 4‐Methylene cyclohexanone was selectively converted into the E (wt‐CHMO) or Z isomer (mutant‐CHMO). By redesigning the active site of CHMO, Bornscheuer and co‐workers succeeded in changing the selectivity of this enzyme for the Baeyer–Villiger oxidation of (+)‐trans‐dihydrocarvone.143 With only three mutations obtained from rational design, the authors could completely invert the regioselectivity from the “abnormal” lactone to the “normal” lactone (Scheme 41).
Scheme 41.
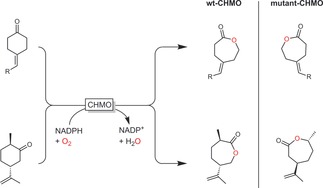
Examples of BVMO engineering to change the stereoselectivity.142a, 143
In the past, the majority of BVMO applications have centred around their stereoselectivity, amongst them desymmetrisation reactions of para‐substituted cyclobutanones144 or cyclohexanones145 and kinetic resolution reactions.146
Mihovilovic and co‐workers have explored the combination of classical chemical catalysis with BVMOs for aroma synthesis.146a, 147 For example, in a flow‐chemistry approach, they combined the rhodium‐catalysed cis hydrogenation of disubstituted cyclopentenones with a BVMO‐catalysed kinetic resolution either directly or following an isomerization step (cis–trans over a cation exchange resin; Scheme 42). Hence, some value‐added aroma lactones such as Aerangis lactones were obtained in good productivities.
Scheme 42.
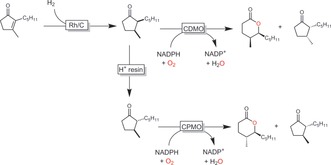
Chemoenzymatic synthesis of Aerangis lactones with chemoenzymatic cascades in a flow process.147
BVMOs are also increasingly investigated for the synthesis of bulk chemicals such as lactones. The low added value of the products necessitates most economical reaction schemes by design, challenging the use of cofactor‐dependent BVMOs. Therefore, a current trend is to design sequential, redox‐neutral reaction sequences starting from cycloalkanols (Scheme 43). The groups of Gröger, Bornscheuer, and Liese have developed cascades of this sort starting from cycloalkanols,63b, 148 but also from non‐functionalised alkanes.63c
Scheme 43.
Redox‐neutral cascade transforming cycloalkanols into lactones by combining an ADH‐catalysed oxidation of the starting alcohol and BVMO‐catalysed lactonisation.
Whereas product concentrations in the range of several hundred millimolar are possible, these systems are challenged by the rather low turnover numbers of the enzymes and cofactors. To reduce the enzyme production costs, Fraaije and co‐workers proposed ADH–BVMO fusion proteins, reaching very promising product titres (>200 mm) and respectable turnover numbers for the fusion protein and nicotinamide cofactor of 20 000 and 1000, respectively.149
Another issue is substrate and/or product inhibition. The first limitation can relatively easily be overcome by a fed‐batch strategy.150 To circumvent inhibitory effects of the product (lactone), either in situ extraction into a hydrophobic organic phase may be used or in situ oligomerisation using a lipase.151 Very decent product titres of up to 20 g L−1 were thus reported. Chiral oligomers can also be obtained through this approach.151b Recent developments move into the design of whole cells comprising the entire enzymatic machinery to generate lactones from non‐alkanes.152
A very creative synthesis of lactones from waste products was reported by Rudroff and co‐workers.153 Starting from limonene (a waste product from orange peels), carvolactone was produced (Scheme 44). Although this conversion necessitated a mixed‐culture fermentation (comprising P. putida catalysing the allylic hydroxylation of limonene and E. coli catalysing the remaining steps to carvolactone) and the best results were obtained in a sequential (time‐consuming) fermentation procedure, it represents a very promising concept for the production of value‐added products from agricultural waste products.
Scheme 44.
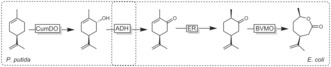
Mixed‐culture approach to convert limonene (waste) into chiral carvolactone. The first hydroxylation step is catalysed by recombinant P. putida overexpressing a dioxygenase; this is followed by a sequence of alcohol oxidation (ADH), ene reduction (ER), and BV oxidation (BVMO) mediated by recombinant E. coli. For reasons of simplicity, the cofactors and cosubstrates have been omitted.153
The redox‐neutral cascades discussed above are sequential (i.e., linear). Kara and co‐workers have come up with a convergent alternative wherein both the BVMO as well as the ADH reaction yield the same product (ϵ‐caprolactone; Scheme 45).114e, 154
Scheme 45.
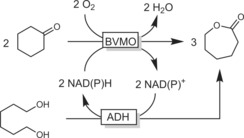
Convergent cascade transforming cyclohexanone and 1,6‐hexanediol into ϵ‐caprolactone.154
While lactones are interesting building blocks, lactams are even more interesting. Therefore, Kroutil and co‐workers aimed at an extension of the above‐mentioned self‐sufficient cascades by catalytic ring opening of the lactone followed by their aza‐Mitsunobu sequence (Scheme 46).155 A respectable overall 24 % yield based on the starting material was achieved.
Scheme 46.
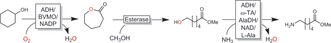
Conversion of cyclohexanol into ω‐aminohexanoic acid (ester). The reaction sequence comprises two redox‐self‐sufficient cascades. First, the ADH/BVMO combination generates ϵ‐caprolactone; the second cascade comprises esterase‐catalysed methanolysis of the lactone followed by ADH‐catalysed oxidation of the terminal OH group and reductive amination catalysed by an ω‐TA. Alanine dehydrogenase (AlaDH) regenerates both the ADH and the ω‐TA (alanine/pyruvate system).155
Starting from fatty acid esters, Schmid, Bühler, and co‐workers reported a similar whole‐cell cascade that yielded ω‐amino acid esters.156
Finally, a very interesting combination of various enzymes by Park and co‐workers is worth mentioning. In an artificial biosynthetic pathway, the authors combined recombinant hydratases with alcohol dehydrogenases and BVMOs to transform oleic acid into either ω‐hydroxy acids (C9) or α,ω‐dicarboxylic acids (C10; Scheme 47).157
Scheme 47.
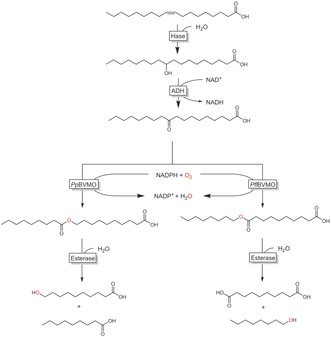
Artificial biotransformation pathway converting oleic acid into ω‐hydroxy acids or α,ω‐dicarboxylic acids. Hase: hydratase; ADH: alcohol dehydrogenase; BVMO: Baeyer–Villiger monooxygenase from P. putida or P. fluorescens).157a
In both pathways, oleic acid (originating from lipase‐hydrolysed olive oil) was hydrated and oxidised to the 10‐keto acid. Using one of two regiocomplementary Baeyer–Villiger monooxygenases, either the normal or the abnormal ester was formed to selectively generate the desired products in rather decent yields.
8.2.2. Perhydrolase Approaches to BV Oxidation
In recent years, chemoenzymatic Baeyer–Villiger oxidation reactions via the so‐called perhydrolysis pathway have also received increasing attention. In this system, a hydrolase (mostly lipases) catalyses the formation of a percarboxylic acid from carboxylic acid (esters) and hydrogen peroxide. The latter performs the chemical Baeyer–Villiger oxidation (Scheme 48).158
Scheme 48.
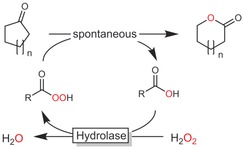
Chemoenzymatic Baeyer–Villiger oxidation using peracids in situ generated by hydrolases.
The most obvious advantage of the chemoenzymatic reaction scheme is the cofactor independence of hydrolases together with their frequently high activity and stability under non‐aqueous reaction conditions, enabling high substrate loadings. Water, however, cannot be fully omitted from the reaction simply because it represents a stoichiometric byproduct (Scheme 48).
One issue frequently observed with this approach is the hydrolytic activity of the hydrolases, which leads to insufficient perhydrolysis rates in aqueous media and to undesired hydrolysis of the lactone products. This may be addressed either by protein engineering159 or by solvent engineering.159c, 160 The scope of this reaction is very broad, and many (cyclo)alkanones can be converted in high yields.88b, 161
Another issue of the chemoenzymatic Baeyer–Villiger reaction is that the actual oxidation reaction is not enzyme‐related. Hence, typical benefits from enzymatic reactions such as enantioselectivity do not apply. This, however, can to some extent be addressed by using chiral carboxylic acids.162
9. Oxidation of Carboxylic Acids
Thus far, oxidations of carboxylic acids play a minor role in biocatalytic oxidation chemistry. Oxidative decarboxylation of long‐chain fatty acids, however, is receiving increased attention. For example, OleT from Pseudomonas 163 and other organisms164 catalyses the formation of terminal alkenes from carboxylic acids. As a P450 peroxygenase, it can be regenerated either by the classical P450 regeneration cycle,165 or directly with H2O2 (Scheme 49).166
Scheme 49.
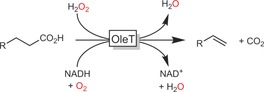
Oxidative decarboxylation of carboxylic acids using the P450 peroxygenase OleT.
Dicarboxylic acids are converted into α,ω‐dienes.167 The substrate scope can also be engineered.168
10. Heteroatom Oxidation
Most heteroatom oxidations reported in the literature centre around thioethers, particularly on their oxyfunctionalisation. (Enantioselective) oxyfunctionalisations of allylic chalconides (S or Se) can also be used for the chemoenzymatic synthesis of chiral allylic alcohols. Arnold and co‐workers reported on an aza variant of this reaction using evolved P450 enzymes.169 Similarly, myoglobins can catalyse a biocatalytic Doyle–Kirmse reaction.170
Codexis engineered a BVMO to generate pharmaceuticals by enantiospecific thioether sulfoxidation (Scheme 50).171
Scheme 50.
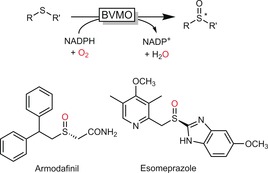
Enantiospecific sulfoxidation of thioethers to generate enantiomerically pure active pharmaceutical ingredients (APIs).
Aside from the sulfur oxidation reactions described above, the oxidation of organoboron172 and organoselenium173 compounds and the oxygenation of amines and pyridines174 should be mentioned.
11. Final Remarks
Biocatalysis has a lot to offer to the organic chemist. In particular oxyfunctionalisations of non‐activated C−H bonds can be performed in a highly selective manner, thereby dramatically simplifying synthesis schemes.175
Many of the limitations seen by organic chemists have been solved, or at least some promising solutions are in sight. The sometimes narrow substrate scopes of enzymes for example are no longer a major limitation thanks to the ever more efficient protein engineering protocols.13, 176 The impressive examples presented here demonstrate the power of protein engineering to obtain tailored biocatalysts.
Enzyme costs are often mentioned as a major drawback of biocatalysis. This, however, is not true per se as the enzyme performance (e.g., in terms of TNs) has to be taken into consideration (Table 1). In some of the examples presented here, TNs of several millions have been achieved, raising hope for industrial applicability.
Another issue requiring more attention is the generally low substrate loading used in biocatalytic experiments, which is due to the hydrophobic nature of many reagents. Millimolar product concentrations are neither economical nor ecologically favourable!176 Using water‐soluble cosolvents to increase the aqueous solubility of the reagents is highly questionable in view of further downstream processing. 2LPS systems or even reactions under non‐aqueous conditions are, in our opinion, most promising to demonstrate the practical feasibility of a given reaction.
Finally, the ongoing trend towards cascade reactions is very promising to us. Cascade reactions are of interest as they bear the promise of more efficient and more elegant synthesis schemes.14b, 14c Especially chemoenzymatic cascades may synergistically combine the best of both worlds. Overall, biocatalytic redox chemistry remains an exciting and challenging research area for the future.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Dr. Frank Hollmann is Associate Professor in the biocatalysis group of the Department for Biotechnology at the Delft University of Technology. The biocatalysis group brings together scientists from the areas of biocatalysis, enzymology, organic chemistry, and molecular imaging. Their research towards a fundamental understanding of enzymes and the application thereof in organic chemistry forms the basis for the application of enzymes in industrial processes, with green chemistry as one of their leitmotifs.

Acknowledgements
We gratefully acknowledge funding by the European Research Commission (ERC consolidator grant, No. 648026), the European Union (H2020‐BBI‐PPP‐2015‐2‐1‐720297), and the Netherlands Organisation for Scientific Research (VICI grant, No. 724.014.003).
J. Dong, E. Fernández-Fueyo, F. Hollmann, C. E. Paul, M. Pesic, S. Schmidt, Y. Wang, S. Younes, W. Zhang, Angew. Chem. Int. Ed. 2018, 57, 9238.
Dedicated to Professor Manfred T. Reetz on the occasion of his 75th birthday
References
- 1.
- 1a. Dijkman W., Gonzalo G., Mattevi A., Fraaije M., Appl. Microbiol. Biotechnol. 2013, 97, 5177–5188; [DOI] [PubMed] [Google Scholar]
- 1b. Pickl M., Fuchs M., Glueck S. M., Faber K., Appl. Microbiol. Biotechnol. 2015, 99, 6617–6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pedersen A. T., Birmingham W. R., Rehn G., Charnock S. J., Turner N. J., Woodley J. M., Org. Process Res. Dev. 2015, 19, 1580–1589. [Google Scholar]
- 3. Siebum A., van Wijk A., Schoevaart R., Kieboom T., J. Mol. Catal. B 2006, 41, 141–145. [Google Scholar]
- 4. Escalettes F., Turner N. J., ChemBioChem 2008, 9, 857–860. [DOI] [PubMed] [Google Scholar]
- 5. Blank L. M., Ebert B. E., Bühler K., Bühler B., Antioxid. Redox Signaling 2010, 13, 349–394. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Gygli G., van Berkel W. J. H., Curr. Biotechnol. 2015, 4, 100–110; [Google Scholar]
- 6b. Huijbers M. M. E., Montersino S., Westphal A. H., Tischler D., van Berkel W. J. H., Arch. Biochem. Biophys. 2014, 544, 2–17. [DOI] [PubMed] [Google Scholar]
- 7. Bernhardt R., Urlacher V. B., Appl. Microbiol. Biotechnol. 2014, 98, 6185–6203. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Holtmann D., Hollmann F., ChemBioChem 2016, 17, 1391–1398; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Morlock L. K., Böttcher D., Bornscheuer U. T., Appl. Microbiol. Biotechnol. 2018, 102, 985–994. [DOI] [PubMed] [Google Scholar]
- 9. Hofrichter M., Ullrich R., Curr. Opin. Chem. Biol. 2014, 19, 116–125. [DOI] [PubMed] [Google Scholar]
- 10. Wang Y., Lan D., Durrani R., Hollmann F., Curr. Opin. Chem. Biol. 2017, 37, 1–9. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Hemsworth G. R., Davies G. J., Walton P. H., Curr. Opin. Struct. Biol. 2013, 23, 660–668; [DOI] [PubMed] [Google Scholar]
- 11b. Tan T.-C., Kracher D., Gandini R., Sygmund C., Kittl R., Haltrich D., Hallberg B. M., Ludwig R., Divne C., Nat. Commun. 2015, 6, 7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tufvesson P., Lima-Ramos J., Nordblad M., Woodley J. M., Org. Process Res. Dev. 2011, 15, 266–274. [Google Scholar]
- 13. Bornscheuer U. T., Huisman G. W., Kazlauskas R. J., Lutz S., Moore J. C., Robins K., Nature 2012, 485, 185–194. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Schmidt S., Castiglione K., Kourist R., Chem. Eur. J. 2018, 24, 1755–1768; [DOI] [PubMed] [Google Scholar]
- 14b. Rudroff F., Mihovilovic M. D., Gröger H., Snajdrova R., Iding H., Bornscheuer U. T., Nat. Catal. 2018, 1, 12–22; [Google Scholar]
- 14c. Schrittwieser J. H., Velikogne S., Hall M., Kroutil W., Chem. Rev. 2017, 118, 270–348. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Urlacher V. B., Girhard M., Trends Biotechnol. 2012, 30, 26–36; [DOI] [PubMed] [Google Scholar]
- 15b. Fasan R., ACS Catal. 2012, 2, 647–666; [Google Scholar]
- 15c. Jung S. T., Lauchli R., Arnold F. H., Curr. Opin. Biotechnol. 2011, 22, 809–817; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15d. Schmid R. D., Urlacher V. B., Modern Biooxidation: Enzymes, Reactions and Applications, Wiley-VCH, Weinheim, 2007. [Google Scholar]
- 16. Tieves F., Erenburg I. N., Mahmoud O., Urlacher V. B., Biotechnol. Bioeng. 2016, 113, 1845–1852. [DOI] [PubMed] [Google Scholar]
- 17. Hoschek A., Bühler B., Schmid A., Angew. Chem. Int. Ed. 2017, 56, 15146–15149; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15343–15346. [Google Scholar]
- 18. Klein C., Hüttel W., Adv. Synth. Catal. 2011, 353, 1375–1383. [Google Scholar]
- 19.
- 19a. Willrodt C., Halan B., Karthaus L., Rehdorf J., Julsing M. K., Bühler K., Schmid A., Biotechnol. Bioeng. 2017, 114, 281–290; [DOI] [PubMed] [Google Scholar]
- 19b. Tavanti M., Parmeggiani F., Castellanos J. R. G., Mattevi A., Turner N. J., ChemCatChem 2017, 9, 3338–3348; [Google Scholar]
- 19c. Zhang K. D., Shafer B. M., Demars M. D., Stern H. A., Fasan R., J. Am. Chem. Soc. 2012, 134, 18695–18704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kille S., Zilly F. E., Acevedo J. P., Reetz M. T., Nat. Chem. 2011, 3, 738–743. [DOI] [PubMed] [Google Scholar]
- 21. Roiban G.-D., Reetz M. T., Chem. Commun. 2015, 51, 2208–2224. [DOI] [PubMed] [Google Scholar]
- 22. Shoji O., Yanagisawa S., Stanfield J. K., Suzuki K., Cong Z., Sugimoto H., Shiro Y., Watanabe Y., Angew. Chem. Int. Ed. 2017, 56, 10324–10329; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 10460–10465. [Google Scholar]
- 23.
- 23a. Munday S. D., Shoji O., Watanabe Y., Wong L. L., Bell S. G., Chem. Commun. 2016, 52, 1036–1039; [DOI] [PubMed] [Google Scholar]
- 23b. Munday S. D., Dezvarei S., Bell S. G., ChemCatChem 2016, 8, 2789–2796; [Google Scholar]
- 23c. Suzuki K., Stanfield J. K., Shoji O., Yanagisawa S., Sugimoto H., Shiro Y., Watanabe Y., Catal. Sci. Technol. 2017, 7, 3332–3338. [Google Scholar]
- 24. Kawakami N., Shoji O., Watanabe Y., Angew. Chem. Int. Ed. 2011, 50, 5315–5318; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 5427–5430. [Google Scholar]
- 25. Zilly F. E., Acevedo J. P., Augustyniak W., Deege A., Häusig U. W., Reetz M. T., Angew. Chem. Int. Ed. 2011, 50, 2720–2724; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 2772–2776. [Google Scholar]
- 26. de Raadt A., Griengl H., Curr. Opin. Biotechnol. 2002, 13, 537–542. [DOI] [PubMed] [Google Scholar]
- 27. Bell S. G., Spence J. T. J., Liu S. L., George J. H., Wong L. L., Org. Biomol. Chem. 2014, 12, 2479–2488. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a. Negretti S., Narayan A. R. H., Chiou K. C., Kells P. M., Stachowski J. L., Hansen D. A., Podust L. M., Montgomery J., Sherman D. H., J. Am. Chem. Soc. 2014, 136, 4901–4904; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28b. Hall E. A., Sarkar M. R., Lee J. H. Z., Munday S. D., Bell S. G., ACS Catal. 2016, 6, 6306–6317. [Google Scholar]
- 29. Ströhle F. W., Kranen E., Schrader J., Maas R., Holtmann D., Biotechnol. Bioeng. 2016, 113, 1225–1233. [DOI] [PubMed] [Google Scholar]
- 30. Kaluzna I., Schmitges T., Straatman H., van Tegelen D., Muller M., Schurmann M., Mink D., Org. Process Res. Dev. 2016, 20, 814–819. [Google Scholar]
- 31.
- 31a. Dingler C., Ladner W., Krei G. A., Cooper B., Hauer B., Pestic. Sci. 1996, 46, 33–35; [Google Scholar]
- 31b. Schorken U., Kempers P., Eur. J. Lipid Sci. Technol. 2009, 111, 627–645. [Google Scholar]
- 32. van Rantwijk F., Sheldon R. A., Curr. Opin. Biotechnol. 2000, 11, 554–564. [DOI] [PubMed] [Google Scholar]
- 33. Ullrich R., Nüske J., Scheibner K., Spantzel J., Hofrichter M., Appl. Environ. Microbiol. 2004, 70, 4575–4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.
- 34a. Olmedo A., Aranda C., del Rio J. C., Kiebist J., Scheibner K., Martinez A. T., Gutierrez A., Angew. Chem. Int. Ed. 2016, 55, 12248–12251; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 12436–12439; [Google Scholar]
- 34b. Gröbe G., Ullrich R., Pecyna M. J., Kapturska D., Friedrich S., Hofrichter M., Scheibner K., AMB Express 2011, 1, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.
- 35a. Babot E. D., del Río J. C., Kalum L., Martínez A. T., Gutiérrez A., Biotechnol. Bioeng. 2013, 110, 2323–2332; [DOI] [PubMed] [Google Scholar]
- 35b. Lucas F., Babot E. D., Canellas M., del Rio J. C., Kalum L., Ullrich R., Hofrichter M., Guallar V., Martinez A. T., Gutierrez A., Catal. Sci. Technol. 2016, 6, 288–295. [Google Scholar]
- 36. Peter S., Karich A., Ullrich R., Grobe G., Scheibner K., Hofrichter M., J. Mol. Catal. B 2014, 103, 47–51. [Google Scholar]
- 37.
- 37a. Zhang W., Fernández-Fueyo E., Ni Y., van Schie M., Gacs J., Renirie R., Wever R., Mutti F. G., Rother D., Alcalde M., Hollmann F., Nat. Catal. 2018, 1, 55–62; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37b. Zhang W., Burek B. O., Fernández-Fueyo E., Alcalde M., Bloh J. Z., Hollmann F., Angew. Chem. Int. Ed. 2017, 56, 15451–15455; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15654–15658; [Google Scholar]
- 37c. Ni Y., Fernández-Fueyo E., Baraibar A. G., Ullrich R., Hofrichter M., Yanase H., Alcalde M., van Berkel W. J. H., Hollmann F., Angew. Chem. Int. Ed. 2016, 55, 798–801; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 809–812; [Google Scholar]
- 37d. Getrey L., Krieg T., Hollmann F., Schrader J., Holtmann D., Green Chem. 2014, 16, 1104–1108. [Google Scholar]
- 38. Fernández-Fueyo E., Ni Y., Gomez Baraibar A., Alcalde M., van Langen L. M., Hollmann F., J. Mol. Catal. B 2016, 134, 347–352. [Google Scholar]
- 39.
- 39a. Rühlmann A., Antovic D., Müller T. J. J., Urlacher V. B., Adv. Synth. Catal. 2017, 359, 984–994; [Google Scholar]
- 39b. Dennig A., Weingartner A. M., Kardashliev T., Müller C. A., Tassano E., Schürmann M., Ruff A. J., Schwaneberg U., Chem. Eur. J. 2017, 23, 17981–17991. [DOI] [PubMed] [Google Scholar]
- 40.
- 40a. Kinne M., Ullrich R., Hammel K. E., Scheibner K., Hofrichter M., Tetrahedron Lett. 2008, 49, 5950–5953; [Google Scholar]
- 40b. Barková K., Kinne M., Ullrich R., Hennig L., Fuchs A., Hofrichter M., Tetrahedron 2011, 67, 4874–4878. [Google Scholar]
- 41. Molina-Espeja P., Canellas M., Plou F. J., Hofrichter M., Lucas F., Guallar V., Alcalde M., ChemBioChem 2016, 17, 341–349. [DOI] [PubMed] [Google Scholar]
- 42.
- 42a. Hudlicky T., Gonzalez D., Gibson D. T., Aldrichimica Acta 1999, 32, 35–62; [Google Scholar]
- 42b. Gibson D. T., Parales R. E., Curr. Opin. Biotechnol. 2000, 11, 236–243; [DOI] [PubMed] [Google Scholar]
- 42c. Boyd D. R., Sharma N. D., Allen C. C. R., Curr. Opin. Biotechnol. 2001, 12, 564–573; [DOI] [PubMed] [Google Scholar]
- 42d. Boyd D. R., Bugg T. D. H., Org. Biomol. Chem. 2006, 4, 181–192. [DOI] [PubMed] [Google Scholar]
- 43.
- 43a. Boyd D. R., Sharma N. D., McIntyre P. B. A., Stevenson P. J., McRoberts W. C., Gohil A., Hoering P., Allen C. C. R., Adv. Synth. Catal. 2017, 359, 4002–4014; [Google Scholar]
- 43b. Zia M. F., Vasko A. G., Riedl Z., Hametner C., Hajos G., Mereiter K., Mihovilovic M. D., Tetrahedron 2016, 72, 7348–7355; [Google Scholar]
- 43c. White L. V., Banwell M. G., J. Org. Chem. 2016, 81, 1617–1626; [DOI] [PubMed] [Google Scholar]
- 43d. Boyd D. R., Sharma N. D., Malone J. F., McIntyre P. B. A., McRoberts C., Floyd S., Allen C. C. R., Gohil A., Coles S. J., Horton P. N., Stevenson P. J., J. Org. Chem. 2015, 80, 3429–3439; [DOI] [PubMed] [Google Scholar]
- 43e. Varghese V., Hudlicky T., Angew. Chem. Int. Ed. 2014, 53, 4355–4358; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4444–4447. [Google Scholar]
- 44.
- 44a. Boyd D. R., Sheldrake G. N., Nat. Prod. Rep. 1998, 15, 309–324; [Google Scholar]
- 44b. Boyd D. R., Sharma N. D., Brannigan I. N., Haughey S. A., Malone J. F., Clarke D. A., Dalton H., Chem. Commun. 1996, 2361–2362. [Google Scholar]
- 45.
- 45a. Jenkins G. N., Ribbons D. W., Widdowson D. A., Slawin A. M. Z., Williams D. J., J. Chem. Soc. Perkin Trans. 1 1995, 2647–2655; [Google Scholar]
- 45b. Ghavre M., Froese J., Murphy B., Simionescu R., Hudlicky T., Org. Lett. 2017, 19, 1156–1159; [DOI] [PubMed] [Google Scholar]
- 45c. Froese J., Overbeeke C., Hudlicky T., Chem. Eur. J. 2016, 22, 6180–6184. [DOI] [PubMed] [Google Scholar]
- 46. Hudlicky T., Endoma M. A. A., Butora G., Tetrahedron: Asymmetry 1996, 7, 61–68. [Google Scholar]
- 47. Montersino S., Tischler D., Gassner G. T., van Berkel W. J. H., Adv. Synth. Catal. 2011, 353, 2301–2319. [Google Scholar]
- 48. Moonen M. J. H., Fraaije M. W., Rietjens I. M. C. M., Laane C., van Berkel W. J. H., Adv. Synth. Catal. 2002, 344, 1023–1035. [Google Scholar]
- 49. Meyer A., Schmid A., Held M., Westphal A. H., Rothlisberger M., Kohler H. P. E., van Berkel W. J. H., Witholt B., J. Biol. Chem. 2002, 277, 5575–5582. [DOI] [PubMed] [Google Scholar]
- 50. Montersino S., Orru R., Barendregt A., Westphal A. H., van Duijn E., Mattevi A., van Berkel W. J. H., J. Biol. Chem. 2013, 288, 26235–26245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Baker Dockrey S. A., Lukowski A. L., Becker M. R., Narayan A. R. H., Nat. Chem. 2017, 10.1038/nchem.2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wischang D., Radlow M., Hartung J., Dalton Trans. 2013, 42, 11926–11940. [DOI] [PubMed] [Google Scholar]
- 53. Médici R., Garaycoechea J. I., Dettorre L. A., Iribarren A. M., Lewkowicz E. S., Biotechnol. Lett. 2011, 33, 1999–2003. [DOI] [PubMed] [Google Scholar]
- 54.
- 54a. Fernández-Fueyo E., van Wingerden M., Renirie R., Wever R., Ni Y., Holtmann D., Hollmann F., ChemCatChem 2015, 7, 4035–4038; [Google Scholar]
- 54b. Frank A., Seel C. J., Groll M., Gulder T., ChemBioChem 2016, 17, 2028–2032. [DOI] [PubMed] [Google Scholar]
- 55.
- 55a. Weichold V., Milbredt D., van Pée K.-H., Angew. Chem. Int. Ed. 2016, 55, 6374–6389; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 6482–6498; [Google Scholar]
- 55b. van Pée K. H., Milbredt D., Patallo E. P., Weichold V., Gajewi M. in Methods Enzymol., Vol. 575 (Ed.: E. O. C. Sarah), Academic Press, San Diego, 2016, pp. 65–92; [DOI] [PubMed] [Google Scholar]
- 55c. Schnepel C., Sewald N., Chem. Eur. J. 2017, 23, 12064–12086. [DOI] [PubMed] [Google Scholar]
- 56. Zehner S., Kotzsch A., Bister B., Sussmuth R. D., Mendez C., Salas J. A., van Pee K. H., Chem. Biol. 2005, 12, 445–452. [DOI] [PubMed] [Google Scholar]
- 57.
- 57a. Zeng J., Zhan J., Biotechnol. Lett. 2011, 33, 1607–1613; [DOI] [PubMed] [Google Scholar]
- 57b. Heemstra J. R., Walsh C. T., J. Am. Chem. Soc. 2008, 130, 14024–14025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shepherd S. A., Karthikeyan C., Latham J., Struck A.-W., Thompson M. L., Menon B. R. K., Styles M. Q., Levy C., Leys D., Micklefield J., Chem. Sci. 2015, 6, 3454–3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Frese M., Guzowska P. H., Voß H., Sewald N., ChemCatChem 2014, 6, 1270–1276. [Google Scholar]
- 60. Frese M., Sewald N., Angew. Chem. Int. Ed. 2015, 54, 298–301; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 302–305. [Google Scholar]
- 61.
- 61a. Latham J., Henry J. M., Sharif H. H., Menon B. R. K., Shepherd S. A., Greaney M. F., Micklefield J., Nat. Commun. 2016, 7, 11873; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61b. Runguphan W., O'Connor S. E., Org. Lett. 2013, 15, 2850–2853; [DOI] [PubMed] [Google Scholar]
- 61c. Durak L. J., Payne J. T., Lewis J. C., ACS Catal. 2016, 6, 1451–1454; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61d. Corr M. J., Sharma S. V., Pubill-Ulldemolins C., Bown R. T., Poirot P., Smith D. R. M., Cartmell C., Abou Fayad A., Goss R. J. M., Chem. Sci. 2017, 8, 2039–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.
- 62a. Dodani S. C., Kiss G., Cahn J. K. B., Su Y., Pande V. S., Arnold F. H., Nat. Chem. 2016, 8, 419–425; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62b. Tomita H., Katsuyama Y., Minami H., Ohnishi Y., J. Biol. Chem. 2017, 292, 15859–15869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.
- 63a. Müller C. A., Dennig A., Welters T., Winkler T., Ruff A. J., Hummel W., Gröger H., Schwaneberg U., J. Biotechnol. 2014, 191, 196–204; [DOI] [PubMed] [Google Scholar]
- 63b. Staudt S., Burda E., Giese C., Müller C. A., Marienhagen J., Schwaneberg U., Hummel W., Drauz K., Gröger H., Angew. Chem. Int. Ed. 2013, 52, 2359–2363; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 2415–2419; [Google Scholar]
- 63c. Müller C. A., Akkapurathu B., Winkler T., Staudt S., Hummel W., Gröger H., Schwaneberg U., Adv. Synth. Catal. 2013, 355, 1787–1798; [Google Scholar]
- 63d. Pennec A., Hollmann F., Smit M. S., Opperman D. J., ChemCatChem 2015, 7, 236–239; [Google Scholar]
- 63e. Pennec A., Jacobs C. L., Opperman D. J., Smit M. S., Adv. Synth. Catal. 2015, 357, 118–130. [Google Scholar]
- 64. Schulz S., Girhard M., Gassmeyer S. K., Jager V. D., Schwarze D., Vogel A., Urlacher V. B., ChemCatChem 2015, 7, 601–604. [Google Scholar]
- 65. Li A., Ilie A., Sun Z., Lonsdale R., Xu J.-H., Reetz M. T., Angew. Chem. Int. Ed. 2016, 55, 12026–12029; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 12205–12208. [Google Scholar]
- 66.
- 66a. Bühler B., Bollhalder I., Hauer B., Witholt B., Schmid A., Biotechnol. Bioeng. 2003, 82, 833–842; [DOI] [PubMed] [Google Scholar]
- 66b. Bühler B., Bollhalder I., Hauer B., Witholt B., Schmid A., Biotechnol. Bioeng. 2003, 81, 683–694. [DOI] [PubMed] [Google Scholar]
- 67. Schittmayer M., Glieder A., Uhl M. K., Winkler A., Zach S., Schrittwieser J. H., Kroutil W., Macheroux P., Gruber K., Kambourakis S., Rozzell J. D., Winkler M., Adv. Synth. Catal. 2011, 353, 268–274. [Google Scholar]
- 68. Chen G., Qu S., Wang Q., Bian F., Peng Z., Zhang Y., Ge H., Yu J., Xuan N., Bi Y., He Q., Biotechnol. Biofuels 2014, 7, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tavanti M., Mangas-Sanchez J., Montgomery S. L., Thompson M. P., Turner N. J., Org. Biomol. Chem. 2017, 15, 9790–9793. [DOI] [PubMed] [Google Scholar]
- 70. Prier C. K., Zhang R. K., Buller A. R., Brinkmann-Chen S., Arnold F. H., Nat. Chem. 2017, 9, 629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. McIntosh J. A., Coelho P. S., Farwell C. C., Wang Z. J., Lewis J. C., Brown T. R., Arnold F. H., Angew. Chem. Int. Ed. 2013, 52, 9309–9312; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 9479–9482. [Google Scholar]
- 72.
- 72a. Singh R., Kolev J. N., Sutera P. A., Fasan R., ACS Catal. 2015, 5, 1685–1691; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72b. Singh R., Bordeaux M., Fasan R., ACS Catal. 2014, 4, 546–552; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72c. Hyster T. K., Farwell C. C., Buller A. R., McIntosh J. A., Arnold F. H., J. Am. Chem. Soc. 2014, 136, 15505–15508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Latham J., Brandenburger E., Shepherd S. A., Menon B. R. K., Micklefield J., Chem. Rev. 2018, 118, 232–269. [DOI] [PubMed] [Google Scholar]
- 74.
- 74a. Vaillancourt F. H., Yin J., Walsh C. T., Proc. Natl. Acad. Sci. USA 2005, 102, 10111–10116; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74b. Vaillancourt F. H., Yeh E., Vosburg D. A., O'Connor S. E., Walsh C. T., Nature 2005, 436, 1191–1194. [DOI] [PubMed] [Google Scholar]
- 75. Galonić D. P., Vaillancourt F. H., Walsh C. T., J. Am. Chem. Soc. 2006, 128, 3900–3901. [DOI] [PubMed] [Google Scholar]
- 76. Budde I. P., Rohde B. H., Bender C. L., Ullrich M. S., J. Bacteriol. 1998, 180, 1360–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.
- 77a. Hillwig M. L., Liu X. Y., Nat. Chem. Biol. 2014, 10, 921–923; [DOI] [PubMed] [Google Scholar]
- 77b. Hillwig M. L., Fuhrman H. A., Ittiamornkul K., Sevco T. J., Kwak D. H., Liu X. Y., ChemBioChem 2014, 15, 665–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Nakamura H., Schultz E. E., Balskus E. P., Nat. Chem. Biol. 2017, 13, 916–921. [DOI] [PubMed] [Google Scholar]
- 79. Mitchell A. J., Dunham N. P., Bergman J. A., Wang B., Zhu Q., Chang W. C., Liu X. Y., Boal A. K., Biochemistry 2017, 56, 441–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.
- 80a. Zhang C., Liu P.-X., Huang L.-Y., Wei S.-P., Wang L., Yang S.-Y., Yu X.-Q., Pu L., Wang Q., Chem. Eur. J. 2016, 22, 10969–10975; [DOI] [PubMed] [Google Scholar]
- 80b. Toda H., Imae R., Itoh N., Adv. Synth. Catal. 2014, 356, 3443–3450; [Google Scholar]
- 80c. Schmid A., Hofstetter K., Feiten H.-J., Hollmann F., Witholt B., Adv. Synth. Catal. 2001, 343, 732–737. [Google Scholar]
- 81. Peter S., Kinne M., Ullrich R., Kayser G., Hofrichter M., Enzyme Microb. Technol. 2013, 52, 370–376. [DOI] [PubMed] [Google Scholar]
- 82.
- 82a. Kuhn D., Kholiq M. A., Heinzle E., Bühler B., Schmid A., Green Chem. 2010, 12, 815–827; [Google Scholar]
- 82b. Hofstetter K., Lutz J., Lang I., Witholt B., Schmid A., Angew. Chem. Int. Ed. 2004, 43, 2163–2166; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 2215–2218; [Google Scholar]
- 82c. Schmutzler K., Kupitz K., Schmid A., Buehler K., Microb. Biotechnol. 2017, 10, 735–744; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82d. Gross R., Buehler K., Schmid A., Biotechnol. Bioeng. 2013, 110, 424–436. [DOI] [PubMed] [Google Scholar]
- 83.
- 83a. Toda H., Imae R., Itoh N., Tetrahedron: Asymmetry 2012, 23, 1542–1549; [Google Scholar]
- 83b. Toda H., Imae R., Komio T., Itoh N., Appl. Microbiol. Biotechnol. 2012, 96, 407–418; [DOI] [PubMed] [Google Scholar]
- 83c. Toda H., Ohuchi T., Imae R., Itoh N., Appl. Environ. Microbiol. 2015, 81, 1919–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.
- 84a. Li A. T., Wu S. K., Adams J. P., Snajdrova R., Li Z., Chem. Commun. 2014, 50, 8771–8774; [DOI] [PubMed] [Google Scholar]
- 84b. Polic V., Cheong K. J., Hammerer F., Auclair K., Adv. Synth. Catal. 2017, 359, 3983–3989. [Google Scholar]
- 85.
- 85a. Kluge M., Ullrich R., Scheibner K., Hofrichter M., Green Chem. 2012, 14, 440–446; [Google Scholar]
- 85b. Lindborg J., Tanskanen A., Kanerva L. T., Biocatal. Biotransform. 2009, 27, 204–210; [Google Scholar]
- 85c. Liu Y., Wang Y., Jiang Y., Hu M., Li S., Zhai Q., Biotechnol. Prog. 2015, 31, 724–729. [DOI] [PubMed] [Google Scholar]
- 86. Seifert A., Vomund S., Grohmann K., Kriening S., Urlacher V. B., Laschat S., Pleiss J., ChemBioChem 2009, 10, 853–861. [DOI] [PubMed] [Google Scholar]
- 87. Björkling F., Godtfredsen S. E., Kirk O., J. Chem. Soc. Chem. Commun. 1990, 1301–1303. [Google Scholar]
- 88.
- 88a. Ranganathan S., Tebbe J., Wiemann L., Sieber V., Process Biochem. 2016, 51, 1479–1485; [Google Scholar]
- 88b. Meyer-Wassewitz J., Hohmann D., Ansorge-Schumacher M. B., Kraume M., Drews A., Biochem. Eng. J. 2017, 126, 68–77; [Google Scholar]
- 88c. Méndez-Sánchez D., Rios-Lombardia N., Gotor V., Gotor-Fernández V., Tetrahedron 2014, 70, 1144–1148. [Google Scholar]
- 89.
- 89a. Kern S., Himmelspach A., Grammann K., Thum O., Liese A., Org. Process Res. Dev. 2016, 20, 1930–1936; [Google Scholar]
- 89b. Milchert E., Malarczyk K., Klos M., Molecules 2015, 20, 21481–21493; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89c. Zhou P., Wang X., Zeng C., Wang W., Yang B., Hollmann F., Wang Y., ChemCatChem 2017, 9, 934–936; [Google Scholar]
- 89d. Aouf C., Durand E., Lecomte J., Figueroa-Espinoza M. C., Dubreucq E., Fulcrand H., Villeneuve P., Green Chem. 2014, 16, 1740–1754; [Google Scholar]
- 89e. Tang Q., Popowicz G. M., Wang X., Liu J., Pavlidis I. V., Wang Y., ChemistrySelect 2016, 1, 836–839. [Google Scholar]
- 90.
- 90a. Wu S., Zhou Y., Wang T., Too H.-P., Wang D. I. C., Li Z., Nat. Commun. 2016, 7, 11917; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90b. Wu S. K., Liu J., Li Z., ACS Catal. 2017, 7, 5225–5233. [Google Scholar]
- 91. Wu S. K., Liu J., Li Z., Synlett 2016, 27, 2644–2658. [Google Scholar]
- 92.
- 92a. Brandenberg O. F., Fasan R., Arnold F. H., Curr. Opin. Biotechnol. 2017, 47, 102–111; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92b. Hammer S. C., Knight A. M., Arnold F. H., Curr. Opin. Green Sustain. Chem. 2017, 7, 23–30; [Google Scholar]
- 92c. Renata H., Wang Z. J., Arnold F. H., Angew. Chem. Int. Ed. 2015, 54, 3351–3367; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 3408–3426. [Google Scholar]
- 93. Farwell C. C., Zhang R. K., McIntosh J. A., Hyster T. K., Arnold F. H., ACS Cent. Sci. 2015, 1, 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.
- 94a. Gober J. G., Ghodge S. V., Bogart J. W., Wever W. J., Watkins R. R., Brustad E. M., Bowers A. A., ACS Chem. Biol. 2017, 12, 1726–1731; [DOI] [PubMed] [Google Scholar]
- 94b. Wang Z. J., Renata H., Peck N. E., Farwell C. C., Coelho P. S., Arnold F. H., Angew. Chem. Int. Ed. 2014, 53, 6810–6813; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 6928–6931; [Google Scholar]
- 94c. Tinoco A., Steck V., Tyagi V., Fasan R., J. Am. Chem. Soc. 2017, 139, 5293–5296; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94d. Bajaj P., Sreenilayam G., Tyagi V., Fasan R., Angew. Chem. Int. Ed. 2016, 55, 16110–16114; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 16344–16348; [Google Scholar]
- 94e. Bordeaux M., Tyagi V., Fasan R., Angew. Chem. Int. Ed. 2015, 54, 1744–1748; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1764–1768. [Google Scholar]
- 95. Leblanc C., Vilter H., Fournier J. B., Delage L., Potin P., Rebuffet E., Michel G., Solari P. L., Feiters M. C., Czjzek M., Coord. Chem. Rev. 2015, 301–302, 134–146. [Google Scholar]
- 96. Dong J. J., Fernandez-Fueyo E., Li J., Guo Z., Renirie R., Wever R., Hollmann F., Chem. Commun. 2017, 53, 6207–6210. [DOI] [PubMed] [Google Scholar]
- 97.
- 97a. Kaysser L., Bernhardt P., Nam S.-J., Loesgen S., Ruby J. G., Skewes-Cox P., Jensen P. R., Fenical W., Moore B. S., J. Am. Chem. Soc. 2012, 134, 11988–11991; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97b. Diethelm S., Teufel R., Kaysser L., Moore B. S., Angew. Chem. Int. Ed. 2014, 53, 11023–11026; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 11203–11206; [Google Scholar]
- 97c. Teufel R., Kaysser L., Villaume M. T., Diethelm S., Carbullido M. K., Baran P. S., Moore B. S., Angew. Chem. Int. Ed. 2014, 53, 11019–11022; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 11199–11202. [Google Scholar]
- 98. Kim Y. S., Park C. S., Oh D. K., Biotechnol. Lett. 2010, 32, 957–961. [DOI] [PubMed] [Google Scholar]
- 99.
- 99a. Mang H., Gross J., Lara M., Goessler C., Schoemaker H. E., Guebitz G., Kroutil W., Angew. Chem. Int. Ed. 2006, 45, 5201–5203; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 5325–5328; [Google Scholar]
- 99b. Paul C. E., Rajagopalan A., Lavandera I., Gotor-Fernandez V., Kroutil W., Gotor V., Chem. Commun. 2012, 48, 3303–3305; [DOI] [PubMed] [Google Scholar]
- 99c. Rajagopalan A., Schober M., Emmerstorfer A., Hammerer L., Migglautsch A., Seisser B., Glueck S. M., Niehaus F., Eck J., Pichler H., Gruber K., Kroutil W., ChemBioChem 2013, 14, 2427–2430; [DOI] [PubMed] [Google Scholar]
- 99d. Rajagopalan A., Lara M., Kroutil W., Adv. Synth. Catal. 2013, 355, 3321–3335. [Google Scholar]
- 100. Hammer S. C., Kubik G., Watkins E., Huang S., Minges H., Arnold F. H., Science 2017, 358, 215–218. [DOI] [PubMed] [Google Scholar]
- 101. Sato H., Hummel W., Gröger H., Angew. Chem. Int. Ed. 2015, 54, 4488–4492; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 4570–4574. [Google Scholar]
- 102.
- 102a. Liu J., Wu S., Li Z., Curr. Opin. Chem. Biol. 2018, 43, 77–86; [DOI] [PubMed] [Google Scholar]
- 102b. Romano D., Villa R., Molinari F., ChemCatChem 2012, 4, 739–749. [Google Scholar]
- 103. Turner N. J., Curr. Opin. Chem. Biol. 2010, 14, 115–121. [DOI] [PubMed] [Google Scholar]
- 104.
- 104a. Gandolfi R., Ferrara N., Molinari F., Tetrahedron Lett. 2001, 42, 513–514; [Google Scholar]
- 104b. Bühler B., Schmid A., J. Biotechnol. 2004, 113, 183–210. [DOI] [PubMed] [Google Scholar]
- 105.
- 105a. Pérez-Sánchez M., Müller C. R., Domínguez de María P., ChemCatChem 2013, 5, 2512–2516; [Google Scholar]
- 105b. Schmidt S., Pedroso de Almeida T., Rother D., Hollmann F., Green Chem. 2017, 19, 1226–1229. [Google Scholar]
- 106. Herter S., McKenna S. M., Frazer A. R., Leimkuhler S., Carnell A. J., Turner N. J., ChemCatChem 2015, 7, 2313–2317. [Google Scholar]
- 107. Hafenstine G. R., Ma K., Harris A. W., Yehezkeli O., Park E., Domaille D. W., Cha J. N., Goodwin A. P., ACS Catal. 2017, 7, 568–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Fuchs M., Schober M., Pfeffer J., Kroutil W., Birner-Gruenberger R., Faber K., Adv. Synth. Catal. 2011, 353, 2354–2358. [Google Scholar]
- 109. Reisky L., Büchsenschütz H. C., Engel J., Song T., Schweder T., Hehemann J.-H., Bornscheuer U. T., Nat. Chem. Biol. 2018, 14, 342–344. [DOI] [PubMed] [Google Scholar]
- 110. Pyo S.-H., Dishisha T., Dayankac S., Gerelsaikhan J., Lundmark S., Rehnberg N., Hatti-Kaul R., Green Chem. 2012, 14, 1942–1948. [Google Scholar]
- 111. Wong C. H., Matos J. R., J. Org. Chem. 1985, 50, 1992–1994. [Google Scholar]
- 112.
- 112a. Hirano J.-I., Miyamoto K., Ohta H., Appl. Microbiol. Biotechnol. 2007, 76, 357–363; [DOI] [PubMed] [Google Scholar]
- 112b. Hirano J.-I., Miyamoto K., Ohta H., Tetrahedron Lett. 2008, 49, 1217–1219. [Google Scholar]
- 113. Bechi B., Herter S., McKenna S., Riley C., Leimkuhler S., Turner N. J., Carnell A. J., Green Chem. 2014, 16, 4524–4529. [Google Scholar]
- 114.
- 114a. Díaz-Rodríguez A., Iglesias-Fernández J., Rovira C., Gotor-Fernández V., ChemCatChem 2014, 6, 977–980; [Google Scholar]
- 114b. Könst P., Kara S., Kochius S., Holtmann D., Arends I. W. C. E., Ludwig R., Hollmann F., ChemCatChem 2013, 5, 3027–3032; [Google Scholar]
- 114c. Kara S., Spickermann D., Schrittwieser J. H., Weckbecker A., Leggewie C., Arends I. W. C. E., Hollmann F., ACS Catal. 2013, 3, 2436–2439; [Google Scholar]
- 114d. Kara S., Spickermann D., Schrittwieser J. H., Leggewie C., Van Berkel W. J. H., Arends I. W. C. E., Hollmann F., Green Chem. 2013, 15, 330–335; [Google Scholar]
- 114e. Huang L., Romero E., Ressmann A. K., Rudroff F., Hollmann F., Fraaije M. W., Kara S., Adv. Synth. Catal. 2017, 359, 2142–2148; [Google Scholar]
- 114f. Romano D., Contente M., Granato T., Remelli W., Zambelli P., Molinari F., Monatsh. Chem. 2013, 144, 735–737; [Google Scholar]
- 114g. Boratyński F., Dancewicz K., Paprocka M., Gabryś B., Wawrzeńcz C., PLoS One 2016, 11, e0146160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.
- 115a. Díaz-Rodríguez A., Martínez-Montero L., Lavandera I., Gotor V., Gotor-Fernández V., Adv. Synth. Catal. 2014, 356, 2321–2329; [Google Scholar]
- 115b. Díaz-Rodríguez A., Lavandera I., Kanbak-Aksu S., Sheldon R. A., Gotor V., Gotor-Fernández V., Adv. Synth. Catal. 2012, 18, 3405–3408. [Google Scholar]
- 116. Zhang X. Y., Zong M. H., Li N., Green Chem. 2017, 19, 4544–4551. [Google Scholar]
- 117.
- 117a. Dijkman W. P., Groothuis D. E., Fraaije M. W., Angew. Chem. Int. Ed. 2014, 53, 6515–6518; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 6633–6636; [Google Scholar]
- 117b. McKenna S. M., Leimkuhler S., Herter S., Turner N. J., Carnell A. J., Green Chem. 2015, 17, 3271–3275. [Google Scholar]
- 118. Carro J., Ferreira P., Rodriguez L., Prieto A., Serrano A., Balcells B., Arda A., Jimenez-Barbero J., Gutierrez A., Ullrich R., Hofrichter M., Martinez A. T., FEBS J. 2015, 282, 3218–3229. [DOI] [PubMed] [Google Scholar]
- 119. Hollmann F., Arends I. W. C. E., Buehler K., Schallmey A., Bühler B., Green Chem. 2011, 13, 226–265. [Google Scholar]
- 120.
- 120a. Boyd D. R., Sharma N. D., Berberian M. V., Cleij M., Hardacre C., Ljubez V., McConville G., Stevenson P. J., Kulakov L. A., Allen C. C. R., Adv. Synth. Catal. 2015, 357, 1881–1894; [Google Scholar]
- 120b. Zhang J., Wu S., Wu J., Li Z., ACS Catal. 2015, 5, 51–58. [Google Scholar]
- 121. Hollmann F., Arends I. W. C. E., Polymer 2012, 4, 759–793. [Google Scholar]
- 122. Contente M. L., Dall′Oglio F., Tamborini L., Molinari F., Paradisi F., ChemCatChem 2017, 9, 3843–3848. [Google Scholar]
- 123.
- 123a. Höhne M., Kuhl S., Karen R., Bornscheuer U. T., ChemBioChem 2008, 9, 363–365; [DOI] [PubMed] [Google Scholar]
- 123b. Aleku G. A., Mangas-Sanchez J., Citoler J., France S. P., Montgomery S. L., Heath R. S., Thompson M. P., Turner N. J., ChemCatChem 2018, 10, 515–519. [Google Scholar]
- 124.
- 124a. Turner N. J., Chem. Rev. 2011, 111, 4073–4087; [DOI] [PubMed] [Google Scholar]
- 124b. Gasparini G., Archer I., Jones E., Ashe R., Org. Process Res. Dev. 2012, 16, 1013–1016; [Google Scholar]
- 124c. Pollegioni L., Molla G., Trends Biotechnol. 2011, 29, 276–283. [DOI] [PubMed] [Google Scholar]
- 125. Henderson R. K., Jiminez-Gonzalez C., Preston C., Constable D. J. C., Woodley J. M., Ind. Biotechnol. 2008, 4, 180–192. [Google Scholar]
- 126. Alexeeva M., Enright A., Dawson M. J., Mahmoudian M., Turner N. J., Angew. Chem. Int. Ed. 2002, 41, 3177–3180; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 3309–3312. [Google Scholar]
- 127.
- 127a. Ghislieri D., Green A. P., Pontini M., Willies S. C., Rowles I., Frank A., Grogan G., Turner N. J., J. Am. Chem. Soc. 2013, 135, 10863–10869; [DOI] [PubMed] [Google Scholar]
- 127b. Carr R., Alexeeva M., Enright A., Eve T. S. C., Dawson M. J., Turner N. J., Angew. Chem. Int. Ed. 2003, 42, 4807–4810; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 4955–4958; [Google Scholar]
- 127c. Li G. Y., Ren J., Yao P. Y., Duan Y. T., Zhang H. L., Wu Q. Q., Feng J. H., Lau P. C. K., Zhu D. M., ACS Catal. 2014, 4, 903–908. [Google Scholar]
- 128. Heath R. S., Pontini M., Hussain S., Turner N. J., ChemCatChem 2016, 8, 117–120. [Google Scholar]
- 129. O'Reilly E., Iglesias C., Ghislieri D., Hopwood J., Galman J. L., Lloyd R. C., Turner N. J., Angew. Chem. Int. Ed. 2014, 53, 2447–2450; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 2479–2482. [Google Scholar]
- 130. Scalacci N., Black G. W., Mattedi G., Brown N. L., Turner N. J., Castagnolo D., ACS Catal. 2016, 6, 1295–1300. [Google Scholar]
- 131.
- 131a. Köhler V., Bailey K. R., Znabet A., Raftery J., Helliwell M., Turner N. J., Angew. Chem. Int. Ed. 2010, 49, 2182–2184; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 2228–2230; [Google Scholar]
- 131b. Li T., Liang J., Ambrogelly A., Brennan T., Gloor G., Huisman G., Lalonde J., Lekhal A., Mijts B., Muley S., Newman L., Tobin M., Wong G., Zaks A., Zhang X., J. Am. Chem. Soc. 2012, 134, 6467–6472. [DOI] [PubMed] [Google Scholar]
- 132. de Graaff C., Oppelaar B., Peruch O., Vande Velde C. M. L., Bechi B., Turner N. J., Ruijter E., Orru R. V. A., Adv. Synth. Catal. 2016, 358, 1555–1560. [Google Scholar]
- 133. Znabet A., Ruijter E., de Kanter F. J. J., Köhler V., Helliwell M., Turner N. J., Orru R. V. A., Angew. Chem. Int. Ed. 2010, 49, 5289–5292; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 5417–5420. [Google Scholar]
- 134. Zajkoska P., Cardenas-Fernandez M., Lye G. J., Rosenberg M., Turner N. J., Rebros M., J. Chem. Technol. Biotechnol. 2017, 92, 1558–1565. [Google Scholar]
- 135. Zheng D. J., Zhou X. J., Cui B. D., Han W. Y., Wan N. W., Chen Y. Z., ChemCatChem 2017, 9, 937–940. [Google Scholar]
- 136.
- 136a. Gandomkar S., Fischereder E. M., Schrittwieser J. H., Wallner S., Habibi Z., Macheroux P., Kroutil W., Angew. Chem. Int. Ed. 2015, 54, 15051–15054; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 15265–15268; [Google Scholar]
- 136b. Schrittwieser J. H., Groenendaal B., Resch V., Ghislieri D., Wallner S., Fischereder E. M., Fuchs E., Grischek B., Sattler J. H., Macheroux P., Turner N. J., Kroutil W., Angew. Chem. Int. Ed. 2014, 53, 3731–3734; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3805–3809; [Google Scholar]
- 136c. Schrittwieser J. H., Resch V., Sattler J. H., Lienhart W.-D., Durchschein K., Winkler A., Gruber K., Macheroux P., Kroutil W., Angew. Chem. Int. Ed. 2011, 50, 1068–1071; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 1100–1103. [Google Scholar]
- 137. Kawahara N., Yasukawa K., Asano Y., Green Chem. 2017, 19, 418–424. [Google Scholar]
- 138.
- 138a. But A., Le Nôtre J., Scott E. L., Wever R., Sanders J. P. M., ChemSusChem 2012, 5, 1199–1202; [DOI] [PubMed] [Google Scholar]
- 138b. But A., van Noord A., Poletto F., Sanders J. P. M., Franssen M. C. R., Scott E. L., Mol. Catal. 2017, 443, 92–100. [Google Scholar]
- 139. Könst P., Merkens H., Kara S., Kochius S., Vogel A., Zuhse R., Holtmann D., Arends I. W. C. E., Hollmann F., Angew. Chem. Int. Ed. 2012, 51, 9914–9917; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 10052–10055. [Google Scholar]
- 140. Wuensch C., Lechner H., Glueck S. M., Zangger K., Hall M., Faber K., ChemCatChem 2013, 5, 1744–1748. [Google Scholar]
- 141. Bučko M., Gemeiner P., Schenkmayerova A., Krajčovič T., Rudroff F., Mihovilovič M. D., Appl. Microbiol. Biotechnol. 2016, 100, 6585–6599. [DOI] [PubMed] [Google Scholar]
- 142.
- 142a. Zhang Z.-G., Roiban G.-D., Acevedo J. P., Polyak I., Reetz M. T., Adv. Synth. Catal. 2013, 355, 99–106; [Google Scholar]
- 142b. Reetz M. T., Wu S., J. Am. Chem. Soc. 2009, 131, 15424–15432; [DOI] [PubMed] [Google Scholar]
- 142c. Balke K., Baumgen M., Bornscheuer U. T., ChemBioChem 2017, 18, 1627–1638; [DOI] [PubMed] [Google Scholar]
- 142d. Wang J. B., Li G. Y., Reetz M. T., Chem. Commun. 2017, 53, 3916–3928. [DOI] [PubMed] [Google Scholar]
- 143. Balke K., Schmidt S., Genz M., Bornscheuer U. T., ACS Chem. Biol. 2016, 11, 38–43. [DOI] [PubMed] [Google Scholar]
- 144.
- 144a. Rudroff F., Fink M. J., Pydi R., Bornscheuer U. T., Mihovilovic M. D., Monatsh. Chem. 2017, 148, 157–165; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144b. Rodríguez-Mata M., Lavandera I., Gotor-Fernández V., Gotor V., García-Cerrada S., Mendiola J., de Frutos O., Collado I., Tetrahedron 2016, 72, 7268–7275. [Google Scholar]
- 145. Rudroff F., Bianchi D. A., Moran-Ramallal R., Iqbal N., Dreier D., Mihovilovic M. D., Tetrahedron 2016, 72, 7212–7221. [Google Scholar]
- 146.
- 146a. Fink M. J., Rudroff F., Mihovilovic M. D., Bioorg. Med. Chem. 2011, 21, 6135–6138; [DOI] [PubMed] [Google Scholar]
- 146b. Rehdorf J., Mihovilovic M. D., Bornscheuer U. T., Angew. Chem. Int. Ed. 2010, 49, 4506–4508; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 4609–4611. [Google Scholar]
- 147. Fink M. J., Schön M., Rudroff F., Schnürch M., Mihovilovic M. D., ChemCatChem 2013, 5, 724–727. [Google Scholar]
- 148.
- 148a. Reimer A., Wedde S., Staudt S., Schmidt S., Höffer D., Hummel W., Kragl U., Bornscheuer U. T., Gröger H., J. Heterocycl. Chem. 2017, 54, 391–396; [Google Scholar]
- 148b. Mallin H., Wulf H., Bornscheuer U. T., Enzyme Microb. Technol. 2013, 53, 283–287. [DOI] [PubMed] [Google Scholar]
- 149. Aalbers F. S., Fraaije M. W., Appl. Microbiol. Biotechnol. 2017, 101, 7557–7565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Scherkus C., Schmidt S., Bornscheuer U. T., Gröger H., Kara S., Liese A., ChemCatChem 2016, 8, 3446–3452. [Google Scholar]
- 151.
- 151a. Schmidt S., Scherkus C., Muschiol J., Menyes U., Winkler T., Hummel W., Gröger H., Liese A., Herz H. G., Bornscheuer U. T., Angew. Chem. Int. Ed. 2015, 54, 2784–2787; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 2825–2828; [Google Scholar]
- 151b. Schmidt S., Buchsenschutz H. C., Scherkus C., Liese A., Gröger H., Bornscheuer U. T., ChemCatChem 2015, 7, 3951–3955; [Google Scholar]
- 151c. Scherkus C., Schmidt S., Bornscheuer U. T., Gröger H., Kara S., Liese A., Biotechnol. Bioeng. 2017, 114, 1215–1221. [DOI] [PubMed] [Google Scholar]
- 152. Karande R., Salamanca D., Schmid A., Buehler K., Biotechnol. Bioeng. 2018, 115, 312–320. [DOI] [PubMed] [Google Scholar]
- 153. Oberleitner N., Ressmann A. K., Bica K., Gartner P., Fraaije M. W., Bornscheuer U. T., Rudroff F., Mihovilovic M. D., Green Chem. 2017, 19, 367–371. [Google Scholar]
- 154.
- 154a. Bornadel A., Hatti-Kaul R., Hollmann F., Kara S., ChemCatChem 2015, 7, 2442–2445; [Google Scholar]
- 154b. Bornadel A., Hatti-Kaul R., Hollmann F., Kara S., Tetrahedron 2016, 72, 7222–7228. [Google Scholar]
- 155. Sattler J. H., Fuchs M., Mutti F. G., Grischek B., Engel P., Pfeffer J., Woodley J. M., Kroutil W., Angew. Chem. Int. Ed. 2014, 53, 14153–14157; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 14377–14381. [Google Scholar]
- 156. Schrewe M., Ladkau N., Bühler B., Schmid A., Adv. Synth. Catal. 2013, 355, 1693–1697. [Google Scholar]
- 157.
- 157a. Song J.-W., Jeon E.-Y., Song D.-H., Jang H.-Y., Bornscheuer U. T., Oh D.-K., Park J.-B., Angew. Chem. Int. Ed. 2013, 52, 2534–2537; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 2594–2597; [Google Scholar]
- 157b. Koppireddi S., Seo J.-H., Jeon E.-Y., Chowdhury P. S., Jang H.-Y., Park J.-B., Kwon Y.-U., Adv. Synth. Catal. 2016, 358, 3084–3092; [Google Scholar]
- 157c. Kim S. U., Kim K. R., Kim J. W., Kim S., Kwon Y. U., Oh D. K., Park J. B., J. Agric. Food Chem. 2015, 63, 2773–2781; [DOI] [PubMed] [Google Scholar]
- 157d. Oh H. J., Kim S. U., Song J. W., Lee J. H., Kang W. R., Jo Y. S., Kim K. R., Bornscheuer U. T., Oh D. K., Park J. B., Adv. Synth. Catal. 2015, 357, 408–416. [Google Scholar]
- 158. Carboni-Oerlemans C., Domínguez de María P., Tuin B., Bargeman G., van der Meer A., van Gemert R., J. Biotechnol. 2006, 126, 140–151. [DOI] [PubMed] [Google Scholar]
- 159.
- 159a. Bernhardt P., Halt K., Kazlauskas R. J., Angew. Chem. Int. Ed. 2005, 44, 2742–2746; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 2802–2806; [Google Scholar]
- 159b. Yin D. L., Bernhardt P., Morley K. L., Jiang Y., Cheeseman J. D., Purpero V., Schrag J. D., Kazlauskas R. J., Biochemistry 2010, 49, 1931–1942; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159c. Wang X.-P., Zhou P.-F., Li Z.-G., Yang B., Hollmann F., Wang Y.-H., Sci. Rep. 2017, 7, 44599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Zhou P. F., Lan D. M., Popowicz G. M., Wang X. P., Yang B., Wang Y. H., Appl. Microbiol. Biotechnol. 2017, 101, 5689–5697. [DOI] [PubMed] [Google Scholar]
- 161.
- 161a. Markiton M., Boncel S., Janas D., Chrobok A., ACS Sustainable Chem. Eng. 2017, 5, 1685–1691; [Google Scholar]
- 161b. Drożdż A., Hanefeld U., Szymańska K., Jarzębski A., Chrobok A., Catal. Commun. 2016, 81, 37–40; [Google Scholar]
- 161c. Meyer J., Horst A. E. W., Steinhagen M., Holtmann D., Ansorge-Schumacher M. B., Kraume M., Drews A., Eng. Life Sci. 2017, 17, 759–767; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161d. Meyer J., Holtmann D., Ansorge-Schumacher M. B., Kraume M., Drews A., Bioche. Eng. J. 2017, 118, 34–40; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161e. Zhong J. R., Xu F., Wang J. F., Li Y. Y., Lin X. F., Wu Q., RSC Adv. 2014, 4, 8533–8540. [Google Scholar]
- 162. Drozdz A., Chrobok A., Chem. Commun. 2016, 52, 1230–1233. [DOI] [PubMed] [Google Scholar]
- 163. Rui Z., Li X., Zhu X., Liu J., Domigan B., Barr I., Cate J. H. D., Zhang W., Proc. Natl. Acad. Sci. USA 2014, 111, 18237–18242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.
- 164a. Xu H. F., Ning L. L., Yang W. X., Fang B., Wang C., Wang Y., Xu J., Collin S., Laeuffer F., Fourage L., Li S. Y., Biotechnol. Biofuels 2017, 10, 15; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164b. Kang M. K., Nielsen J., J. Ind. Microbiol. Biotechnol. 2017, 44, 613–622; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164c. Herman N. A., Zhang W. J., Curr. Opin. Chem. Biol. 2016, 35, 22–28; [DOI] [PubMed] [Google Scholar]
- 164d. Zhu Z. W., Zhou Y. J. J., Kang M. K., Krivoruchko A., Buijs N. A., Nielsen J., Metab. Eng. 2017, 44, 81–88. [DOI] [PubMed] [Google Scholar]
- 165. Dennig A., Kuhn M., Tassoti S., Thiessenhusen A., Gilch S., Bülter T., Haas T., Hall M., Faber K., Angew. Chem. Int. Ed. 2015, 54, 8819–8822; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 8943–8946. [Google Scholar]
- 166. Zachos I., Gassmeyer S., Bauer D., Sieber V., Hollmann F., Kourist R., Chem. Commun. 2015, 51, 1918–1921. [DOI] [PubMed] [Google Scholar]
- 167. Dennig A., Kurakin S., Kuhn M., Dordic A., Hall M., Faber K., Eur. J. Org. Chem. 2016, 3473–3477. [Google Scholar]
- 168. Wang J., Lonsdale R., Reetz M. T., Chem. Commun. 2016, 52, 8131–8133. [DOI] [PubMed] [Google Scholar]
- 169.
- 169a. Prier C. K., Hyster T. K., Farwell C. C., Huang A., Arnold F. H., Angew. Chem. Int. Ed. 2016, 55, 4711–4715; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 4789–4793; [Google Scholar]
- 169b. Farwell C. C., McIntosh J. A., Hyster T. K., Wang Z. J., Arnold F. H., J. Am. Chem. Soc. 2014, 136, 8766–8771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Tyagi V., Sreenilayam G., Bajaj P., Tinoco A., Fasan R., Angew. Chem. Int. Ed. 2016, 55, 13562–13566; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 13760–13764. [Google Scholar]
- 171. Bong Y. K., Clay D., Collier S., Mijts B., Vogel M., Zhang X., Zhu J., Nazor J., Smith D., Song S., WO2011071982, 2011.
- 172.
- 172a. Brondani P. B., de Gonzalo G., Fraaije M. W., Andrade L. H., Adv. Synth. Catal. 2011, 353, 2169–2173; [Google Scholar]
- 172b. Brondani P. B., Dudek H., Reis J. S., Fraaije M. W., Andrade L. H., Tetrahedron: Asymmetry 2012, 23, 703–708. [Google Scholar]
- 173.
- 173a. Andrade L. H., Pedrozo E. C., Leite H. G., Brondani P. B., J. Mol. Catal. B 2011, 73, 63–66; [Google Scholar]
- 173b. Brondani P. B., Guilmoto N. M. A. F., Dudek H. M., Fraaije M. W., Andrade L. H., Tetrahedron 2012, 68, 10431–10436. [Google Scholar]
- 174.
- 174a. Méndez-Sánchez D., Lavandera I., Gotor V., Gotor-Fernández V., Org. Biomol. Chem. 2017, 15, 3196–3201; [DOI] [PubMed] [Google Scholar]
- 174b. Ullrich R., Dolge C., Kluge M., Hofrichter M., FEBS Lett. 2008, 582, 4100–4106. [DOI] [PubMed] [Google Scholar]
- 175. Turner N. J., O'Reilly E., Nat. Chem. Biol. 2013, 9, 285–288. [DOI] [PubMed] [Google Scholar]
- 176.
- 176a. Hönig M., Sondermann P., Turner N. J., Carreira E. M., Angew. Chem. Int. Ed. 2017, 56, 8942–8973; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 9068–9100; [Google Scholar]
- 176b. de Souza R., Miranda L. S. M., Bornscheuer U. T., Chem. Eur. J. 2017, 23, 12040–12063. [DOI] [PubMed] [Google Scholar]
- 177. Ni Y., Holtmann D., Hollmann F., ChemCatChem 2014, 6, 930–943. [Google Scholar]