

Abstract
A robust, click‐chemistry‐inspired procedure for radiolabeling of cyclic ureas was developed. This protocol, suitable for all carbon isotopes (11C, 13C, 14C), is based on the direct functionalization of carbon dioxide: the universal building block for carbon radiolabeling. The strategy is operationally simple and reproducible in different radiochemistry centers, exhibits remarkably wide substrate scope with short reaction times, and demonstrates superior reactivity as compared to previously reported systems. With this procedure, a variety of pharmaceuticals and an unprotected peptide were labeled with high radiochemical efficiency.
Keywords: carbon dioxide, carbon-11, carbon-14, heterocycles, isotopic labeling
Radioisotope labeling has a remarkable impact on our society and particularly on public health: from the collection of precious preclinical absorption, distribution, metabolism, excretion (ADME), and toxicological data required for drug development and registration with long‐lived β− isotopes,1 to the early diagnosis of disease with non‐invasive positron emission tomography (PET) imaging with short‐lived β+ radiotracers.2 Late‐stage labeling is the most effective strategy to introduce the radioisotope into the desired organic molecule: insertion at the last step of the synthesis has a beneficial impact on the overall efficiency of the process. Recent developments in late‐stage tritium (3H)3, 4 and fluorine‐18 (18F)5, 6 labeling clearly showcased the benefits of such an approach.
Carbon is ubiquitously present in nature and is the overriding choice for isotopic radiolabeling of pharmaceuticals and agrochemicals. Two radioisotopes with diametrically opposite physical properties are commonly used: carbon‐14 (β− emitter, half‐life 5730 years) and carbon‐11 (β+ emitter, half‐life 20.4 min). Carbon‐14 is a favored isotope in drug development and is often preferred to tritium because of its higher metabolic stability. This natural radioisotope is generated as Ba[14C]CO3, which is further converted into [14C]CO2, the universal precursor of all 14C‐labeled compounds. Traditionally, [14C]CO2 functionalization requires multistep approaches and results in the production of long‐lasting radioactive waste.7 Carbon‐11 would be the most general radioisotope for PET tracers but its narrow half‐life makes 11C‐labeled radiopharmaceuticals extremely challenging to prepare.8 One major 11C primary precursor produced by cyclotrons is [11C]CO2,9 an almost chemically inert molecule that is not trivial to introduce directly into complex molecules, such as pharmaceuticals. Owing to such restrictions, the most frequently used method for the introduction of 11C into organic molecules is methylation.10 [11C]CO2 is transformed into a methylating agent (typically [11C]CH3I or [11C]CH3OTf) by a series of reactions, which are time‐ and material‐consuming. Alternatives to methylation are known but seldom applied to pharmaceutically relevant molecules and biomolecules.11, 12, 13 The development of methodologies capable of converting CO2 directly into the desired scaffolds, in a single operation, at a late‐stage of the synthesis would be highly beneficial for radiolabeling with carbon isotopes.
Urea is a fundamental functional group in organic chemistry and commonly found in pharmaceuticals and dyes (Scheme 1 a).14 Despite its chemical and metabolic stability, no general and efficient labeling protocol has been reported so far. Cyclic ureas have been traditionally labeled using [11C and 14C]‐phosgene,15 a highly toxic radioactive reagent, which can be synthesized only in a rather limited number of laboratories on a routine basis. More recently, a number of methods using carbon monoxide [11C and 14C]CO and metal catalysts or selenium at high pressures have been described (Scheme 1 b).11, 16 Alternatives involving the direct use of [11C and 14C]CO2 in the presence of 2‐tert‐butylimino‐2‐diethylamino‐1,3‐dimethylperhydro‐1,3,2‐diazaphosphorine (BEMP) and POCl3 display limited functional‐group tolerance.17
Scheme 1.
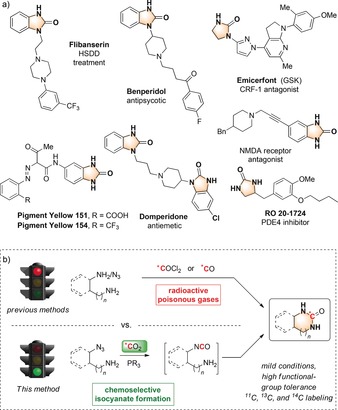
a) Relevant examples of cyclic ureas; b) synthetic strategies to access radiolabeled ureas. Bn=benzyl.
Herein, we describe a one‐pot operationally simple labeling procedure for the synthesis of cyclic ureas on the basis of a sequential Staudinger/aza‐Wittig (SAW) approach directly from [11C and 14C]CO2. This example of late‐stage labeling proved to be broad in scope, highly tolerant towards functional groups, suitable for all isotopes of carbon, and effective for the labeling of drugs and an unprotected peptide.
At the outset, we aimed to develop a general approach to radiolabel cyclic ureas, and we reasoned that click chemistry might be a source of inspiration. Azides play a central role in click chemistry, and the Staudinger ligation shows high substrate compatibility and is effective even in complex biological media.18 The o‐azidoaniline 1 a was identified as an ideal building block. When 1 a is treated with CO2 in the presence of a phosphine, the resulting iminophosphorane undergoes an aza‐Wittig reaction to generate an intermediate isocyanate, and subsequent intramolecular nucleophile addition delivers the cyclized urea product. Since in radiochemistry CO2 is the limiting reagent, optimization of the reaction was performed using a Tritec manifold to precisely deliver stoichiometric amounts of 13C‐labeled gas (see the Supporting Information for details).
From preliminary screening experiments, dimethylphenylphosphine (PMe2Ph) was identified as a superior reducing agent (Table 1; see Supporting Information). As compared to less nucleophilic and more sterically hindered Ph3P and MePh2P (Table 1, entry 1–4), PMe2Ph was highly effective in delivering the desired benzoimidazolone [13C]2 a in only 5 min at room temperature (Table 1, entry 6; see the Supporting Information for more details). Solvent screening revealed that MeCN and N,N‐dimethylformamide (DMF) were both suitable for the transformation. Under the optimized reaction conditions in the presence of radiolabeled [14C]CO2, 1 a was converted into the labeled urea [14C]2 a in a remarkable radiochemical yield (RCY) of 95 %. The transformation required only a stoichiometric amount of [14C]CO2 and a single purification step, thus minimizing the radioactive waste generated in a rare example of environmentally sustainable radiolabeling.
Table 1.
Optimization of the Staudinger/aza‐Wittig reaction with stoichiometric CO2.[a]

| Entry | *CO2 | Phosphine | T [° C] | t | Yield [%] |
|---|---|---|---|---|---|
| 1 | [13C]CO2 | PPh3 | 65 | 2 h | 90 |
| 2 | [13C]CO2 | PPh3 | 25 | 2 h | 80 |
| 3 | [13C]CO2 | PPh2Me | 25 | 2 h | 95 |
| 4 | [13C]CO2 | PPh2Me | 25 | 1 h | 84 |
| 5 | [13C]CO2 | PPhMe2 | 25 | 1 h | 95 |
| 6 | [13C]CO2 | PPhMe2 | 25 | 5 min | 95 |
| 7 | [14C]CO2 | PPhMe2 | 25 | 5 min | 95[b] |
| 8 | [11C]CO2 | PPhMe2 | 25 | 5 min | 79[c] |
[a] Carbon‐13 and carbon‐14 experiments were performed in the presence of stoichiometric amounts of [13C and 14C]CO2; for carbon‐11 labeling, precursor 1 a and the phosphine were typically used in 100‐fold excess relative to [11C]CO2 (see the Supporting Information for details). [b] Radiochemical yield. [c] Radiochemical conversion.
Encouraged by the exceptionally short time required to reach full conversion, we next looked at the application of the transformation to 11C labeling. In stark contrast to 14C, [11C]CO2 is generated with a cyclotron in nanomolar amounts, thus forcing complete modification of the stoichiometry of the reaction and the use of 1 a and phosphine in large excess as compared to [11C]CO2. We were pleased to observe that this protocol was straightforward to implement, and [11C]2 a was obtained with 79 % radiochemical conversion (RCC), without the need for CO2‐trapping agents.19 The radiosynthesis was carried out in automated modules and proved to be compliant with good manufacturing practice (GMP).
We next investigated the scope of the reaction by testing a variety of substituted aromatic o‐azidoanilines, synthesized according to reported procedures (Scheme 2).20 A whole range of benzoimidazolones 2 a–n were labeled in good to excellent yields both with carbon‐13 and carbon‐11. Not surprisingly, electron‐rich anilines proved to be competent substrates for the transformation (products 2 e,f). The presence of halogens (products 2 b–d) and substituents ortho to the reactive azide (products 2 i–k) did not affect the transformation, whereas substrates with electron‐withdrawing substituents were converted into the products in moderate yields (products 2 g,h). When secondary anilines were used, the desired products 2 l–n were obtained in 52–78 % yield for carbon‐13 and 55–62 % RCC for carbon‐11. The use of simple, reproducible, and easy to implement protocols is highly desirable in radiochemistry and particularly for positon emitters, for which isotope manipulation is restricted by the use of automated facilities. The current technology was successfully implemented in two PET centers, with different automated systems, thus clearly highlighting the robustness of this protocol (see the Supporting Information for details).
Scheme 2.
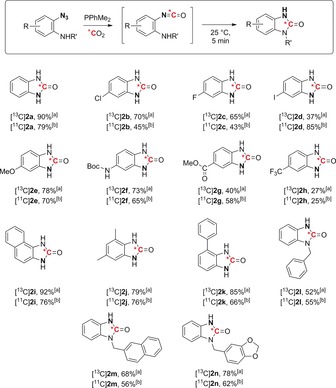
Late‐stage labeling of benzoimidazolones with carbon isotopes. The position of the azide on the o‐azidoaniline precursor is highlighted in bold. [a] Yield of the isolated product. [b] Radiochemical conversion. Boc=tert‐butoxycarbonyl.
A variety of aliphatic ureas were also successfully labeled using this procedure (Scheme 3). Six‐membered derivatives 3 a–d were isolated in good to excellent yields. Interestingly, the presence of a guanine ring did not affect the efficiency of the transformation (product 3 e). The five‐membered derivative 3 f was obtained in 51 and 99 % yield with carbon‐13 and carbon‐11, respectively. Notably, tricyclic urea 3 g was obtained from the corresponding benzimidazole precursor.
Scheme 3.
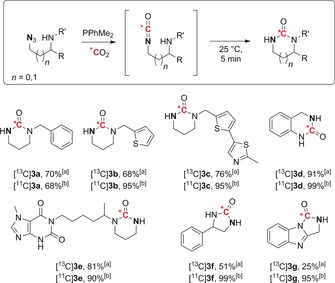
Late‐stage labeling of aliphatic ureas with carbon isotopes. The position of the azide on the o‐azidoaniline precursor is highlighted in bold. [a] Yield of the isolated product. [b] Radiochemical conversion.
The promising functional‐group orthogonality of this approach together with the successful application to both carbon‐13 and carbon‐11 prompted the evaluation of the isotopic labeling of pharmaceutically relevant ureas (Scheme 4). Oxatomide, an orally active antihistaminic, was labeled in 82 % yield with carbon‐13 and 66 % radiochemical yield (RCY) with carbon‐14,21 while [11C]4 a was obtained ready‐to‐inject in 45 % RCY (molar activity: 75 GBq⋅μmol−1) within 30 min from the end of bombardment (EOB). Domperidone, a commercially available antiemetic drug, was successfully labeled in 57 % RCY from [14C]CO2 and 43 % RCY from [11C]CO2. The 5‐HT7 antagonist 4 c, whose fluorinated analogue was previously labeled with the short‐lived PET isotope fluorine‐18,22 was obtained in 89 and 39 % RCY, respectively, using carbon‐14 and carbon‐11 isotopes. Flibanserin, a medication approved for the treatment of premenopausal women with hypoactive sexual desire disorder (HSDD), was readily labeled in 95 and 48 % RCY with carbon‐14 and carbon‐11, respectively. Finally, CGP12177A, a tracer for β‐adrenergic receptor was isolated in 35 % RCY with a molar activity of 32 GBq⋅μmol−1. In comparison with the previously reported radiosynthesis of 11C‐CGP12177A using [11C]phosgene,23 the SAW approach afforded higher yields and a shorter process without the use of hazardous chemicals. In all cases, the ready‐to‐inject labeled drugs were isolated in high chemical and radiochemical purities. Furthermore, the azide precursors were readily synthetized in two linear steps from commercially available anilines.
Scheme 4.
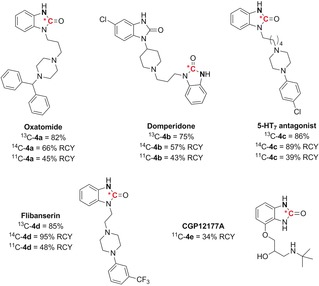
Late‐stage carbon isotope labeling of pharmaceutically relevant ureas. RCY: radiochemical yield. The position of the azide on the o‐azidoaniline precursor is highlighted in bold.
The use of radiolabeled peptides and proteins as imaging tools for drug development and clinical diagnostics has recently attracted considerable attention.24 In 2017, two major contributions from Buchwald, Hooker, and co‐workers24b using H[11C]CN as a labeled building block and from Antoni, Skrydstrup, and co‐workers24c utilizing [11C]CO were reported. Both methods use efficient metal catalysts for the insertion of the desired tag on a series of peptides. Considering the high efficiency and orthogonality of the SAW sequence, we applied it to a designed peptide sequence bearing the desired azido–amine group and most of the classical reactive moieties displayed by amino acids (alcohol, carboxylic acid, amine, and indole groups). A very clean reaction took place under the optimized reaction conditions to afford radiolabeled 5 in 43 % yield from [14C]CO2 and 23 % radiochemical conversion with [11C]CO2 (Scheme 5).
Scheme 5.
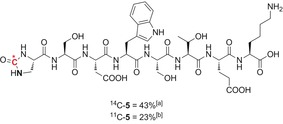
Late‐stage labeling of peptide 5. The position of the azide on the o‐azidoaniline precursor is highlighted in bold. [a] Yield of the isolated product. [b] Radiochemical conversion.
In conclusion, we have shown that late‐stage carbon labeling through a Staudinger/aza‐Wittig reaction can provide access to functionalized molecules that are particularly challenging to synthesize by conventional procedures. The advantages of the current method are its applicability to all isotopes of carbon (11C, 13C, 14C), the simple and easy‐to‐reproduce protocol, and the broad scope of the transformation, as showcased by the labeling of drug candidates and the insertion of carbon tags into an unprotected peptide.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This project received funding from the Horizon 2020 research and innovation program of the European Union under Marie Sklodowska‐Curie grant agreement No. 675071 and the CEA (program DRF Impulsion). We thank David‐Alexandre Buisson and Elodie Marcon for excellent analytical support and Dr. Catriona Wimberley for kind proofreading.
A. Del Vecchio, F. Caillé, A. Chevalier, O. Loreau, K. Horkka, C. Halldin, M. Schou, N. Camus, P. Kessler, B. Kuhnast, F. Taran, D. Audisio, Angew. Chem. Int. Ed. 2018, 57, 9744.
Dedicated to Dr. Louis Pichat on the occasion of his 92nd birthday
References
- 1.
- 1a. Isin E. M., Elmore C. S., Nilsson G. N., Thompson R. A., Weidolf L., Chem. Res. Toxicol. 2012, 25, 532–542; [DOI] [PubMed] [Google Scholar]
- 1b. Penner N., Xu L., Prakash C., Chem. Res. Toxicol. 2012, 25, 513–531; [DOI] [PubMed] [Google Scholar]
- 1c. Atzrodt J., Derdau V., Kerr W. J., Reid M., Angew. Chem. Int. Ed. 2018, 57, 1758–1784; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 1774–1802. [Google Scholar]
- 2.
- 2a. Miller P. W., Long N. J., Vilar R., Gee A. D., Angew. Chem. Int. Ed. 2008, 47, 8998–9033; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 9136–9172; [Google Scholar]
- 2b. Ametamey S. M., Honer M., Schubiger P. A., Chem. Rev. 2008, 108, 1501–1516. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Pony Yu R., Hesk D., Rivera N., Pelczer I., Chirik P. J., Nature 2016, 529, 195–199; [DOI] [PubMed] [Google Scholar]
- 3b. Loh Y. Y., Nagao K., Hoover A. J., Hesk D., Rivera N. R., Colletti S. L., Davies I. W., MacMillan D. W. C., Science 2017, 10.1126/science.aap9674; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3c. Pieters G., Taglang C., Bonnefille E., Gutmann T., Puente C., Berthet J. C., Dugave C., Chaudret B., Rousseau B., Angew. Chem. Int. Ed. 2014, 53, 230–234; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 234–238. [Google Scholar]
- 4. Atzrodt J., Derdau V., Kerr W. J., Reid M., Angew. Chem. Int. Ed. 2018, 57, 3022–3047; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 3074–3101. [Google Scholar]
- 5.
- 5a. Brooks A. F., Topczewski J. J., Ichiishi N., Sanford M. S., Scott P. J. H., Chem. Sci. 2014, 5, 4545–4553; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Campbell M. G., Ritter T., Org. Process Res. Dev. 2014, 18, 474–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Taylor N. J., Emer E., Preshlock S., Schedler M., Tredwell M., Verhoog S., Mercier J., Genicot C., Gouverneur V., J. Am. Chem. Soc. 2017, 139, 8267–8276; [DOI] [PubMed] [Google Scholar]
- 6b. Verhoog S., Kee C. W., Wang Y., Khotavivattana T., Wilson T. C., Kersemans V., Smart S., Tredwell M., Davis B. G., Gouverneur V., J. Am. Chem. Soc. 2018, 140, 1572–1575; [DOI] [PubMed] [Google Scholar]
- 6c. Lee E., Kamlet A. S., Powers D. C., Neumann C. N., Boursalian G. B., Furuya T., Choi D. C., Hooker J. M., Ritter T., Science 2011, 334, 639–642; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6d. McCammant M. S., Thompson S., Brooks A. F., Krska S. W., Scott P. J. H., Sanford M. S., Org. Lett. 2017, 19, 3939–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Voges R., Heys J. R., Moenius T., Preparation of Compounds Labeled with Tritium and Carbon-14, Wiley, Hoboken, 2009, p. 393. [Google Scholar]
- 8. Scott P. J. H., Angew. Chem. Int. Ed. 2009, 48, 6001–6004; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 6115–6118. [Google Scholar]
- 9. Rotstein B. H., Liang S. H., Holland J. P., Collier T. L., Hooker J. M., Wilson A. A., Vasdev N., Chem. Commun. 2013, 49, 5621–5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tu Z., Mach R. H., Curr. Top. Med. Chem. 2010, 10, 1060–1095. [DOI] [PubMed] [Google Scholar]
- 11. Rotstein B. H., Liang S. H., Placzek M. S., Hooker J. M., Gee A. D., Dollé F., Wilson A. A., Vasdev N., Chem. Soc. Rev. 2016, 45, 4708–4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For recent labeling methods using [11C]CN−, see:
- 12a. Makaravage K. J., Shao X., Brooks A. F., Yang L., Sanford M. S., Scott P. J. H., Org. Lett. 2018, 20, 1530–1533; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Ma L., Placzek M. S., Hooker J. M., Vasdev N., Liang S. H., Chem. Commun. 2017, 53, 6597–6600; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12c. Lee H. G., Milner P. J., Placzek M. S., Buchwald S. L., Hooker J. M., J. Am. Chem. Soc. 2015, 137, 648–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.For labeling methods using [11C]CO2, see:
- 13a. Hooker J. M., Reibel A. T., Hill S. M., Schueller M. J., Fowler J. S., Angew. Chem. Int. Ed. 2009, 48, 3482–3485; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 3534–3537; [Google Scholar]
- 13b. Riss P. J., Lu S., Telu S., Aigbirhio F. I., Pike V. W., Angew. Chem. Int. Ed. 2012, 51, 2698–2702; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 2752–2756; [Google Scholar]
- 13c. Mossine A. V., Brooks A. F., Jackson I. M., Quesada C. A., Sherman P., Cole E. L., Donnelly D. J., Scott P. J. H., Shao X., Bioconjugate Chem. 2016, 27, 1382–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.
- 14a. Bansal Y., Silakari O., Bioorg. Med. Chem. 2012, 20, 6208–6236; [DOI] [PubMed] [Google Scholar]
- 14b. Wright J. L., Gregory T. F., Kesten S. R., Boxer P. A., Serpa K. A., Meltzer L. T., Wise L. D., Espitia S. A., Konkoy C. S., Whittemore E. R., Woodward R. M., J. Med. Chem. 2000, 43, 3408–3419; [DOI] [PubMed] [Google Scholar]
- 14c. Weisner J., Gontla R., Westhuizen L. v. d., Oeck S., Ketzer J., Janning P., Richters A., Mühlenberg T., Fang Z., Taher A., Jendrossek V., Pelly S. C., Bauer S., Otterlo W. A. L. v., Rauh D., Angew. Chem. Int. Ed. 2015, 54, 10313–10316; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10452–10456. [Google Scholar]
- 15.
- 15a. Awata N., Satomi O., J. Labelled Compd. Radiopharm. 1987, 24, 331–338; [Google Scholar]
- 15b. Labas R., Sobrio F., Bramoullé Y., Hérard A. S., Guillermier M., Hantraye P., Dollé F., Barré L., J. Labelled Compd. Radiopharm. 2010, 53, 63–67. [Google Scholar]
- 16. Kealey S., Husbands S. M., Bennacef I., Gee A. D., Passchier J., J. Labelled Compd. Radiopharm. 2014, 57, 202–208. [DOI] [PubMed] [Google Scholar]
- 17.No examples of cyclic ureas were reported with this method: Wilson A. A., Garcia A., Houle S., Sadovski O., Vasdev N., Chem. Eur. J. 2011, 17, 259–264. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Sletten E. M., Bertozzi C. R., Acc. Chem. Res. 2011, 44, 666–676; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. van Berkel S. S., van Eldijk M. B., van Hest J. C. M., Angew. Chem. Int. Ed. 2011, 50, 8806–8827; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 8968–8989. [Google Scholar]
- 19. van Tilburg E. W., Windhorst A. D., van der Mey M., Herscheid J. D. M., J. Labelled Compd. Radiopharm. 2006, 49, 321–330. This report describes the synthesis of [11C]phenylisocyanate by trapping [11C]CO2 in a solution of phenyltriphenylphosphinimine. The method was applied to four linear substrates with moderate yields and may be constrained by the lack of structurally diverse triphenylphosphinimines. [Google Scholar]
- 20.
- 20a. Tang C., Jiao N., J. Am. Chem. Soc. 2012, 134, 18924–18927; [DOI] [PubMed] [Google Scholar]
- 20b. Fan Y., Wan W., Ma G., Gao W., Jiang H., Zhu S., Hao J., Chem. Commun. 2014, 50, 5733–5736; [DOI] [PubMed] [Google Scholar]
- 20c. Fang H., Dou Y., Ge J., Chhabra M., Sun H., Zhang P., Zheng Y., Zhu Q., J. Org. Chem. 2017, 82, 11212–11217. [DOI] [PubMed] [Google Scholar]
- 21.Oxatomide was previously labeled at the same position using [14C]urea under much more drastic conditions (190 °C, 3 h): Meuldermanst W., Hendrickx J., Knaeps F., Lauwerss W., Heykants J., Grindel J. M., Xenobiotica 1984, 14, 445–462. [DOI] [PubMed] [Google Scholar]
- 22. Deau E., Robin E., Voinea R., Percina N., Satała G., Finaru A.-L., Chartier A., Tamagnan G., Alagille D., Bojarski A. J., Morisset-Lopez S., Suzenet F., Guillaumet G., J. Med. Chem. 2015, 58, 8066–8096. [DOI] [PubMed] [Google Scholar]
- 23. Nishijima K., Kuge Y., Seki K., Ohkura K., Morita K., Nakada K., Tamaki N., Nucl. Med. Commun. 2004, 25, 845–849. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Cornilleau T., Audrain H., Guillemet A., Hermange P., Fouquet E., Org. Lett. 2015, 17, 354–357; [DOI] [PubMed] [Google Scholar]
- 24b. Zhao W., Lee H. G., Buchwald S. L., Hooker J. M., J. Am. Chem. Soc. 2017, 139, 7152–7155; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24c. Andersen T. L., Nordeman P., Christoffersen H. F., Audrain H., Antoni G., Skrydstrup T., Angew. Chem. Int. Ed. 2017, 56, 4549–4553; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4620–4624. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary