

Abstract
Objective
To develop an objective, readily measurable pharmacodynamic biomarker of glucocorticoid (GC) activity.
Methods
Genes modulated by prednisolone were identified from in vitro studies using peripheral blood mononuclear cells from normal healthy volunteers. Using the criteria of a >2‐fold change relative to vehicle controls and an adjusted P value cutoff of less than 0.05, 64 up‐regulated and 18 down‐regulated genes were identified. A composite score of the up‐regulated genes was generated using a single‐sample gene set enrichment analysis algorithm.
Results
GC gene signature expression was significantly elevated in peripheral blood leukocytes from normal healthy volunteers following oral administration of prednisolone. Expression of the signature increased in a dose‐dependent manner, peaked at 4 hours postadministration, and returned to baseline levels by 48 hours after dosing. Lower expression was detected in normal healthy volunteers who received a partial GC receptor agonist, which is consistent with the reduced transactivation potential of this compound. In cohorts of patients with systemic lupus erythematosus and patients with rheumatoid arthritis, expression of the GC signature was negatively correlated with the percentages of peripheral blood lymphocytes and positively correlated with peripheral blood neutrophil counts, which is consistent with the known biology of the GC receptor. Expression of the signature largely agreed with reported GC use in these populations, although there was significant interpatient variability within the dose cohorts.
Conclusion
The GC gene signature identified in this study represents a pharmacodynamic marker of GC exposure.
Glucocorticoids (GCs) are effective antiinflammatory drugs that are used extensively to treat many human diseases, including rheumatoid arthritis (RA), inflammatory bowel disease, psoriasis, asthma, and systemic lupus erythematosus (SLE) 1. However, the utility of these drugs is limited by their toxicities, which include diabetes, osteoporosis, muscle wasting, fat redistribution, and suppression of the hypothalamic–pituitary–adrenal gland (HPA) axis 2. The risk for harmful side effects increases with higher doses and more prolonged use 3, 4. Despite the potential for adverse effects, GCs remain a key standard‐of‐care treatment.
GCs mediate their biologic effects via interactions with a nuclear hormone receptor, GC receptor (GR). GR is a ligand‐activated transcription factor that induces transcription by binding as a homodimer to GC‐responsive elements 5. Many GR‐activated genes have antiinflammatory activity 6, 7, 8. However, transactivated genes are also associated with side effects 9. GR has also been shown to inhibit the activity of several proinflammatory transcription factors, including NF‐κB, activator protein 1 (AP‐1), interferon regulatory factor 3, CREB, NF‐AT, STAT, T‐Bet, and GATA‐3, independently of DNA binding, in a process referred to as transrepression 10. In an attempt to broaden the therapeutic window, several synthetic GCs with reduced transactivation but intact transrepression activity have been developed 11.
In addition to the risk of damaging effects, long‐term GC use is also associated with tissue‐specific resistance 12. Several resistance mechanisms have been described, including down‐regulation of GR expression as well as up‐regulation of a dominant‐negative isoform of the receptor 13. Polymorphisms of the GR that modulate sensitivity to agonists have also been described 14. Given the heterogeneity of clinical responses to GCs, it would be extremely valuable to have a companion biomarker of GC biologic activity.
In the current study, we developed a gene signature based on genes modulated by treatment of peripheral blood mononuclear cells (PBMCs) from normal healthy volunteer (NHV) donors with prednisolone. We confirmed the sensitivity of this signature by analyzing postdose whole blood gene expression in healthy participants given either prednisolone or a partial GR agonist. Expression of the signature was higher in healthy subjects who received prednisolone than in those who received the partial agonist, which is consistent with the transactivation potential of the compounds. Expression of the signature in whole blood from patients with SLE and patients with RA correlated with known GC‐mediated pharmacodynamic effects, including higher levels of peripheral blood neutrophils and lower levels of peripheral blood lymphocytes. Expression of the signature also aligned with the reported use and dose of prednisolone in these cohorts. These data suggest that the GC gene signature may provide a sensitive biomarker to monitor pharmacodynamic responses to GCs.
Patients and methods
Study approval. These studies were performed in accordance with the Declaration of Helsinki and approved by the institutional review boards of Brigham and Women's Hospital and Northwell Health. Participants provided written informed consent prior to sample collection.
Identification of GC‐regulated genes. PBMCs were isolated from the blood of 10 independent donors, using Ficoll density‐gradient centrifugation. Cells were cultured at 5 million lymphocytes/well in a 96‐well flat‐bottomed block plate (Qiagen) in 500 μl assay media (RPMI 1640 with GlutaMAX containing 10% charcoal‐stripped fetal bovine serum; Gibco). Cells were cultured for 6 hours with either dimethyl sulfoxide (DMSO) vehicle or 1 μM prednisolone. After 6 hours, cells were pelleted and resuspended in 1 ml of nucleic acid purification lysis solution (Applied Biosystems) diluted 1:2 with calcium‐free and magnesium‐free phosphate buffered saline (Invitrogen). Cells were incubated in lysis buffer for 10 minutes at room temperature followed by storage at −80°C. RNA was isolated using a Qiagen RNeasy Isolation Kit according to the instructions of the manufacturer.
For profiling of whole blood, anticoagulant citrate dextrose solution A–containing whole blood from 4 NHVs was cultured with either DMSO vehicle, 200 nM prednisolone, 1 μM prednisolone, 5 μM prednisolone, 5 μM BMS‐791826, or 10 μM BMS‐776532 for 5 hours, followed by transfer to a PAXgene tube. Total RNA was isolated, treated with DNase I, and cleaned using a Qiagen RNeasy MinElute Cleanup Kit. RNA concentrations were determined using NanoDrop (Thermo Fisher), and RNA quality was evaluated using an Experion electrophoresis system (Bio‐Rad). All target‐labeling reagents were purchased from Affymetrix.
Double‐stranded complementary DNAs (cDNAs) were synthesized from 1 μg of total RNA by reverse transcription with an oligo(dT) primer containing the T7 RNA polymerase promoter and converted to double‐strand using a cDNA Synthesis System (Invitrogen). Biotin‐labeled complementary RNA (cRNA) was generated from the cDNA and was used to probe a Human Genome HT_HG‐U133A plate (Affymetrix), consisting of 96 single HG‐U133A arrays in a 96‐well plate. All cDNA and cRNA target preparation steps were processed on a Caliper GeneChip Array Station (Affymetrix). Array hybridization, washing, and scanning were performed according to the recommendations of the manufacturer. Data are available in the NCBI Gene Expression Omnibus database (accession nos. GSE110098, GSE110156, GSE110157).
Gene signature development and scoring. CEL files from the Affymetrix Array Station were processed and normalized using the Robust Multi‐array Average algorithm 15 and the “affy” package in R version 3.2.1 16 and Bioconductor 17 with custom CDF files from BrainArray (version 18.0.0) 18. Differential gene expression analysis was performed to compare gene expression levels in prednisolone‐treated versus control samples, using a moderated t‐test 19 in Array Studio (OmicSoft). P values were adjusted using the multiple testing correction method, which is also called the false discovery rate (FDR) 20. Genes that were up‐regulated or down‐regulated by at least 2‐fold with an adjusted P value of less than 0.05 across experiments were reported as the GC gene signatures.
To score an individual sample according to the enrichment level of GC gene signatures, we adapted the single‐sample gene set enrichment analysis (ssGSEA) algorithm 21 to generate a composite score, which was implemented using the Gene Set Variation Analysis package in R (version 3.4.0) 22. This algorithm ranks genes in the transcriptome within each sample and scores genes of interest according to the ranks. The higher the ranks of individual GC genes are, the higher the ssGSEA GC signature score is. We modified the algorithm so that enrichment scores fell between –1 and 1, representing the lowest to the highest possible rankings of genes in the transcriptome.
Mammalian 2‐hybrid analysis. Sequences encoding either full‐length human peroxisome proliferator–activated receptor γ coactivator 1α (PGC‐1α) or full‐length human transcription intermediary factor 2 (TIF‐2) were cloned in frame with the Gal‐4 DNA–binding domain in the vector pM (Clontech). Full‐length human GR was cloned in frame with the VP16 activation domain in the vector pVP16 (Clontech). Human SK‐N‐MC neuroblastoma cells (American Type Culture Collection) were cotransfected with these plasmids and a Gal‐4–dependent luciferase reporter (pGF‐luc; Promega). Transfectants were stimulated with either 200 nM dexamethasone or different concentrations of prednisolone, BMS‐791826, or BMS‐776532. Luciferase activity was measured 48 hours posttransfection.
Chromatin immunoprecipitation. For chromatin immunoprecipitations, A549 cells were cultured for 1 hour with either DMSO, 1 μM prednisolone, 1 μM BMS‐791826, or 2 μM BMS‐776532 in RPMI with 10% charcoal‐stripped fetal calf serum. Cells were fixed with formaldehyde and sent to Active Motif for analysis of the GR and TIF‐2 recruitment to specific promoter sequences, using quantitative polymerase chain reaction (qPCR).
SLE and RA cross‐sectional cohorts. Peripheral blood samples were obtained in 2014 and 2015 from 82 patients with SLE during routine visits at Northwell Health. These patients were receiving standard‐of‐care treatment for general SLE or lupus nephritis that included hydroxychloroquine, mycophenolate mofetil, GCs, and/or belimumab. The characteristics of the patients were as follows. The mean ± SD age was 45 ± 14 years, 85% were female, the mean ± SD SLE Disease Activity Index 2000 23 score was 3.7 ± 3.2, 43% had a history of lupus nephritis, and the mean ± SD disease duration was 15 ± 13 years.
In 2014 and 2015, blood samples were also obtained from 84 patients with RA during routine visits at either Brigham and Women's Hospital or Northwell Health. These patients were receiving standard‐of‐care treatment for RA that included methotrexate, hydroxychloroquine, tofacitinib, abatacept, anti–tumor necrosis (anti‐TNF) biologics, tocilizumab, GCs, and/or nonsteroidal antiinflammatory drugs. The characteristics of the patients were as follows. The mean ± SD age was 57 ± 14 years, 77% were female, the mean ± SD American College of Rheumatology/European League Against Rheumatism 2010 classification 24 score was 7.8 ± 1.6, and the mean ± SD disease duration was 17 ± 10 years. Blood was collected in heparin and in PAXgene tubes at each visit. Blood was shipped overnight to Bristol‐Myers Squibb and, upon arrival, was processed for fluorescence‐activated cell sorting analysis. Blood from age‐ and sex‐matched NHVs was collected in PAXgene tubes (Bristol‐Myers Squibb). RNA was isolated from PAXgene tubes and used to probe Affymetrix HG‐U219 gene arrays, using the protocols described above. Data are available in the NCBI Gene Expression Omnibus database (accession no. GSE110169).
IM101‐042 Abatacept SLE clinical cohort (ClinicalTrials.gov identifier: NCT00119678). Baseline PAXgene collections and complete blood cell counts were obtained for 144 adults with SLE meeting the criteria of the British Isles Lupus Assessment Group 25 with a score of A or B. The population at baseline consisted of 53% of patients with polyarthritis, 35% with discoid lupus, and 12% with serositis. Overall, 87% of patients were receiving prednisone, 50% were receiving hydroxychloroquine, and 41% were receiving immunosuppressive agents (methotrexate, azathioprine, or mycophenolate mofetil). Expression data are available in the NCBI Gene Expression Omnibus database (accession no. GSE110174).
IM124‐001 cohort (ClinicalTrials.gov identifier: NCT03196557). Male NHVs were randomly assigned (6 participants per group) to receive daily doses of 5, 10, or 30 mg prednisolone for 7 days. Two participants received placebo. Blood was collected in PAXgene tubes before dosing and 2, 4, 8, 48, 144, and 216 hours postadministration. Expression data are available in the NCBI Gene Expression Omnibus database (accession no. GSE110160).
IM125‐001 cohort (ClinicalTrials.gov identifier: NCT03198013). Male NHVs were randomly assigned to receive either a placebo of polyethylene glycol 400 (PEG 400) solution (4 participants), a single daily oral dose of BMS‐791826 (150 mg or 300 mg) as a PEG 400 solution (6 participants/dose), or a single daily dose of 10 mg prednisolone (4 participants) for 3 consecutive days. PAXgene tubes were collected before dosing and 4 hours postdose on day 1. Expression data are available in the NCBI Gene Expression Omnibus database (accession no. GSE110161).
Heparinized whole blood was stained with premixed cocktails of antibodies, followed by lysis and fixation. Antibodies used for the SLE panel included phycoerythrin–Cy7–conjugated CD4 (clone OKT4; BioLegend), allophycocyanin‐H7–conjugated CD8 (clone SK1; BD Biosciences), and Brilliant Violet 421 (BV421)–conjugated CD19 (clone HIB19; BioLegend). The antibodies used for the RA panel included BV421‐conjugated CD19, Alexa Fluor 700–conjugated CD3 (clone OKT3; BioLegend), PerCP–Cy5.5–conjugated CD4 (clone RPA‐T4; eBioscience), and BV785‐conjugated CD8 (clone RPA‐T8; BioLegend).
Statistical analysis. All statistical analyses of GC gene signature scores were performed in statistical programming language R (version 3.4.0) 16 with Bioconductor packages 17. GC gene signature scores in different treatment groups were compared using linear regression (lm function in the Stats Package). When samples from matching donors were included, a linear mixed‐effect model (lme function in the nlme package) was used, with donors as a random factor. Spearman's correlation and P values comparing GC gene signature scores with CD4+ T cells, CD8+ T cells, B cells, and neutrophil counts were calculated using the cor.test function in the Stats Package. Analysis of chromatin immunoprecipitation data was performed using GraphPad Prism version 7.
Results
Identification of GC‐regulated genes. In order to monitor GC‐dependent responses in peripheral blood, we focused on genes modulated by prednisolone in human PBMCs. PBMCs from 10 independent NHV donors were treated with either DMSO control or 1 μM prednisolone for 6 hours. Using a cutoff value of >2‐fold change and an FDR‐corrected P value of less than 0.05, 64 up‐regulated genes (Figure 1A) and 18 down‐regulated genes (Figure 1B) were identified. Many of these genes were known GC‐regulated genes 26. However, half of the up‐regulated genes had not been previously linked to GC regulation. Several of the up‐regulated genes have previously been associated with antiinflammatory activity, including DUSP1 7, TSC22D3 8, IRAK3 27, and CD163 28, while several of the down‐regulated genes encoded chemokines, chemokine receptors, and other proinflammatory mediators. Network analysis of the regulated genes indicated enrichment for immune response pathways (data not shown).
Figure 1.
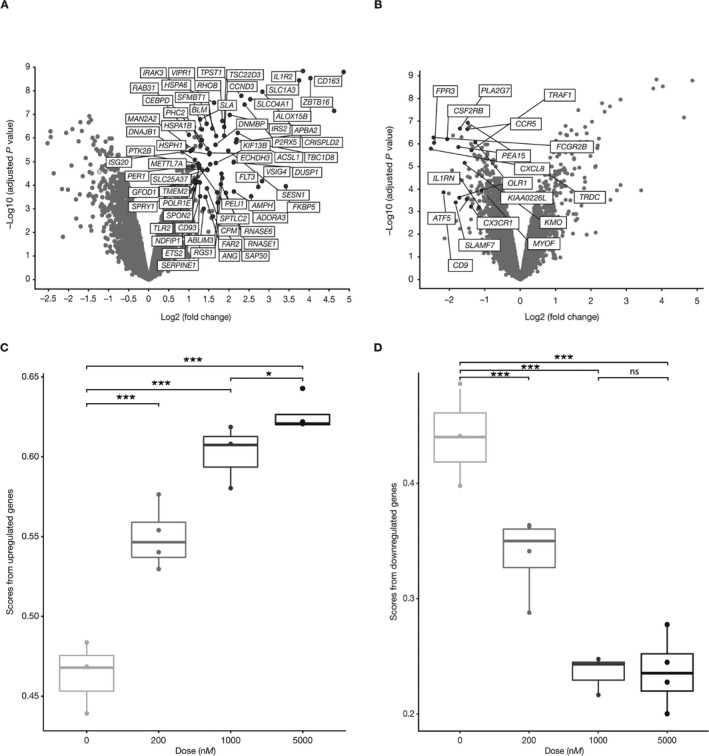
Identification of glucocorticoid (GC)–regulated genes. Peripheral blood mononuclear cells from normal healthy volunteers were cultured in vitro for 6 hours with either 1 μM prednisolone or DMSO vehicle alone. RNA was analyzed for gene expression using Affymetrix profiling. Analyses of genes modulated by prednisolone compared with vehicle are shown. Axes represent the false discovery rate (FDR)–adjusted log10‐transformed P value versus fold change. A and B, Genes up‐regulated (A) and down‐regulated (B) >2‐fold by prednisolone versus vehicle with an FDR‐adjusted P value of ≤0.05. C and D, Single‐sample gene set enrichment analysis scores for genes up‐regulated (C) and down‐regulated (D) in whole blood samples stimulated with increasing concentrations of prednisolone in vitro. Data in C and D are presented as box plots, where the boxes represent the 25th to 75th percentiles, the lines within the boxes represent the median, and the lines outside the boxes extend to the minimum or maximum values after excluding outliers. * = P = 0.027; *** = P < 0.001. NS = not significant.
We used the ssGSEA algorithm to generate a composite score for enrichment of these genes in the transcriptomes of individual samples 21. This algorithm ranks genes in the transcriptome within each sample and scores genes of interest according to the ranks; the higher the ranks of individual GC genes are, the higher the composite ssGSEA GC signature score is. Whole blood was stimulated with different concentrations of prednisolone in vitro for 5 hours, and the expression levels of up‐regulated and down‐regulated genes were calculated. The ssGSEA score for the up‐regulated genes increased dose dependently (Figure 1C). Similarly, expression of the down‐regulated genes decreased in a dose‐dependent manner (Figure 1D). The up‐regulated gene module appeared to have a larger dynamic range based on the ability of this gene module to distinguish differences between the higher doses of prednisolone in the experiments using whole blood samples (Figure 1C). The down‐regulated gene module was not significantly different between these prednisolone concentrations. Therefore, we focused on the up‐regulated gene module for all other analyses.
In order to provide further mechanistic evidence that this gene module accurately reflected GR activity, we analyzed the activity of partial GR agonists. Our group previously described the in vitro and in vivo activities of 2 selective GR modulators, BMS‐776532 and BMS‐791826 29. Both compounds potently bound to GR and potently repressed AP‐1– and NF‐κB–dependent reporters but demonstrated significantly weaker induction of a GR‐dependent reporter as compared with prednisolone. BMS‐791826 was more potent in transrepression and transactivation assays as compared with BMS‐776532.
We used a mammalian 2‐hybrid system as well as a chromatin immunoprecipitation assay to characterize the transactivation potential of these compounds. We focused on 2 co‐regulators associated with GR activity, TIF‐2 30 and PGC‐1α 31. Compared with prednisolone, BMS‐791826 and BMS‐776532 recruited significantly less PGC‐1α and TIF‐2 to the GR, peaking at 30–75% of the level recruited by prednisolone (Figures 2A and B). Compared with BMS‐776532, BMS‐791826 recruited more TIF‐2 (50% versus 30%) but similar amounts of PGC‐1α. In a chromatin immunoprecipitation assay, both compounds recruited significantly lower amounts of GR (Figure 2C) as well as TIF‐2 (Figure 2D) to the promoters of 3 target genes as compared with prednisolone, confirming the reduced transactivation potential of these compounds. Whole blood from 2 independent NHV donors was stimulated in vitro with these compounds and prednisolone for 5 hours, followed by RNA isolation and gene expression profiling. The GC gene signature scores for these samples aligned well with the transactivation potential of the compounds: prednisolone greater than BMS‐791826, which is greater than BMS‐776532 (Figure 2E).
Figure 2.
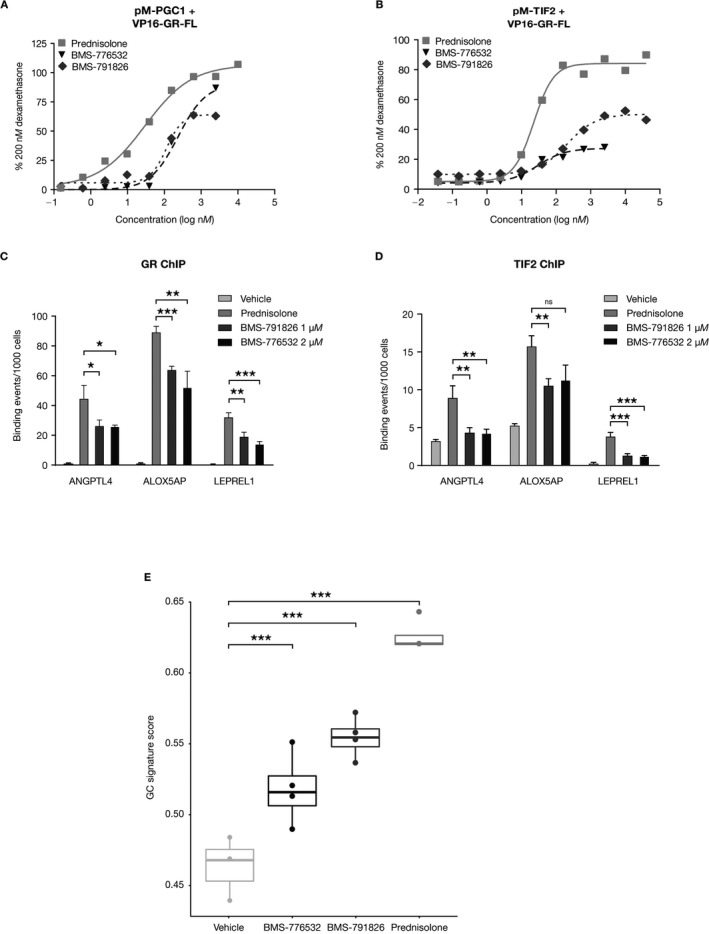
Validation of the glucocorticoid (GC) gene signature using partial GC receptor (GR) agonists. A and B, Mammalian 2‐hybrid analysis of peroxisome proliferator–activated receptor γ coactivator 1 (PGC1) (A) and transcription intermediary factor 2 (TIF‐2) (B) recruitment by prednisolone, BMS‐776532, and BMS‐791826. Values are the means of triplicate wells, normalized to the activity induced by 200 nM dexamethasone. Results are from a representative experiment of 2 independent experiments performed. C and D, Analysis of GR (C) and TIF‐2 (D) recruitment to the promoters of ANGPTL4,ALOX5AP, and LEPREL1 by 1 μM prednisolone, 1 μM BMS‐791826, and 2 μM BMS‐776532, as analyzed by chromatin immunoprecipitation (ChIP) assay followed by quantitative polymerase chain reaction analysis. Values are the mean ± SD of triplicate reactions. Binding values are normalized to input values. E, GC gene signature scores for whole blood samples cultured in vitro with either DMSO vehicle, 5 μM prednisolone, 5 μM BMS‐776532, or 10 μM BMS‐791826. Data are presented as box plots, where the boxes represent the 25th to 75th percentiles, the lines within the boxes represent the median, and the lines outside the boxes extend to the minimum or maximum values after excluding outliers. * = P < 0.05; ** = P < 0.01; *** = P < 0.001 versus prednisolone, by Student's t‐test. FL = full‐length; NS = not significant.
In vivo assessment of the GC gene signature. Because the GC signature accurately captured GR agonist activity in vitro, we examined the behavior of the signature in vivo following the administration of specific compounds. NHVs received placebo, 10 mg prednisolone, or 150 or 300 mg BMS‐791826. Blood was drawn before dosing and 4 hours postdose, and RNA was analyzed by Affymetrix gene expression profiling. The GC signature scores for participants who received prednisolone were significantly elevated at the 4‐hour time point relative to predose levels and those in the placebo group (Figure 3A). The signature scores for participants who were treated with BMS‐791826 were higher than predose levels and higher than those for participants given placebo but lower than those for participants in the prednisolone group.
Figure 3.
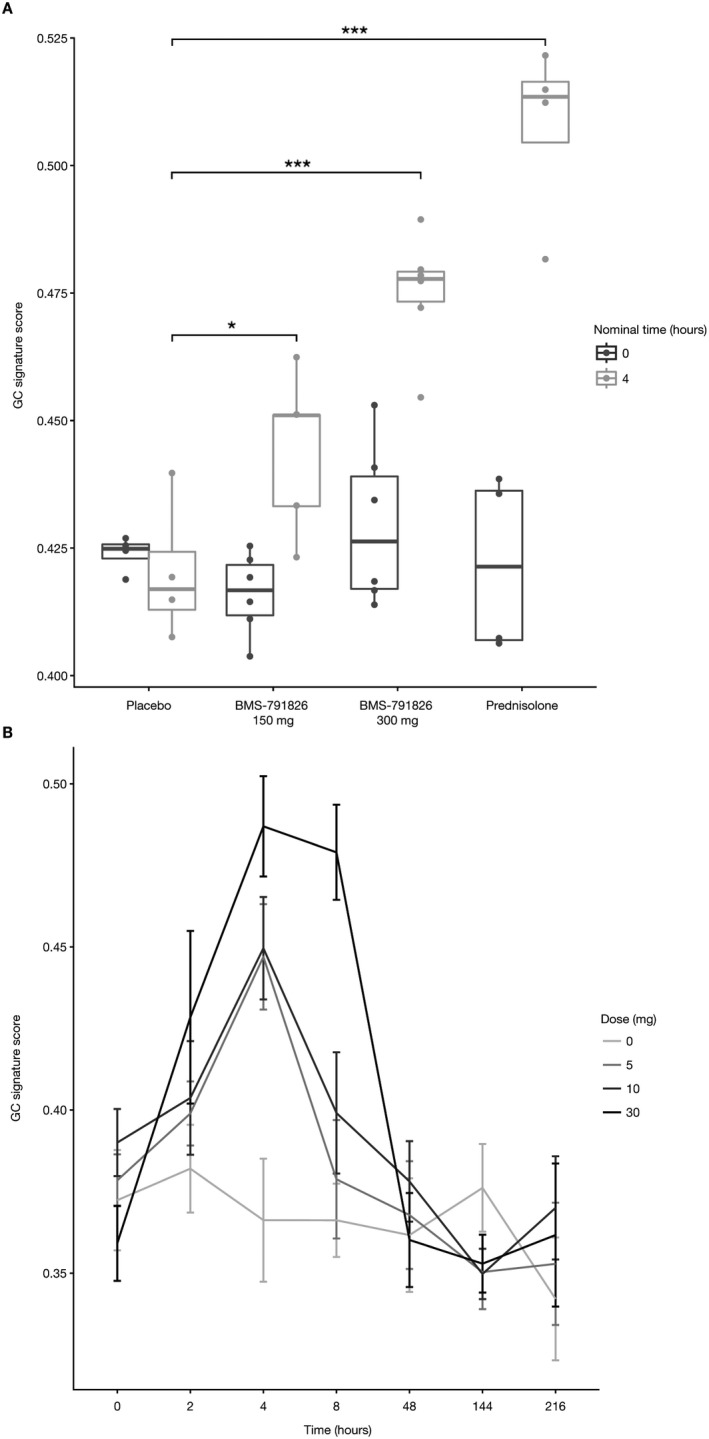
In vivo validation of the GC gene signature. A, Normal healthy volunteers (NHVs) were administered an oral dose of 150 mg or 300 mg BMS‐791826, 10 mg prednisolone, or placebo. Blood samples were collected before administration and 4 hours postdose. Whole blood expression profiles were analyzed for the GC gene signature. Data are presented as box plots, where the boxes represent the 25th to 75th percentiles, the lines within the boxes represent the median, and the lines outside the boxes extend to the minimum or maximum values after excluding outliers. B, NHVs were administered 5 mg, 10 mg, or 30 mg prednisolone or placebo (i.e., 0 mg). Blood was drawn before administration and at different time points postdose (2, 4, 8, 48, 144, and 216 hours). Whole blood expression profiles were analyzed for the GC gene signature. Bars show the mean ± SEM. * = P = 0.027; *** = P < 0.001. See Figure 1 for definitions.
To address the kinetics of the GC gene signature response, we analyzed the whole blood RNA profiles of NHVs who were administered different doses of prednisolone. The GC gene signature score increased dose dependently and peaked at 4 hours postdose (Figure 3B). For all but the highest dose of prednisolone, GC gene signature scores had returned to baseline levels by 8 hours postdose. The signature score was at baseline levels in all groups by 48 hours postdose. We conclude that the GC signature score is a sensitive measure of in vivo responses to GC administration.
Relationship between the GC gene signature and reported GC use. To determine whether the GC signature could differentiate patients based on treatment status, we analyzed expression of the signature in cross‐sectional cohorts of patients with SLE or RA. Relative to either normal healthy controls or patients treated with other standard‐of‐care medications, patients with SLE or RA who were prescribed GCs had elevated signature scores (Figure 4A). We did not observe significant differences in the pattern of gene expression for the signature genes across these cohorts. Although the GC signatures were elevated, there was significant interpatient variability in the signature scores. We also analyzed baseline samples from a phase II study of abatacept in SLE 32 for expression of the GC gene signature (Figure 4B). The GC gene signature scores generally aligned well with the reported prednisone dose when categorized into high (>30 mg), medium (10–30 mg), and low (<10 mg) doses. However, there was again significant interpatient variability in GC gene signature scores in all groups. This could reflect steroid resistance in some patients or deviations from the stated GC doses.
Figure 4.
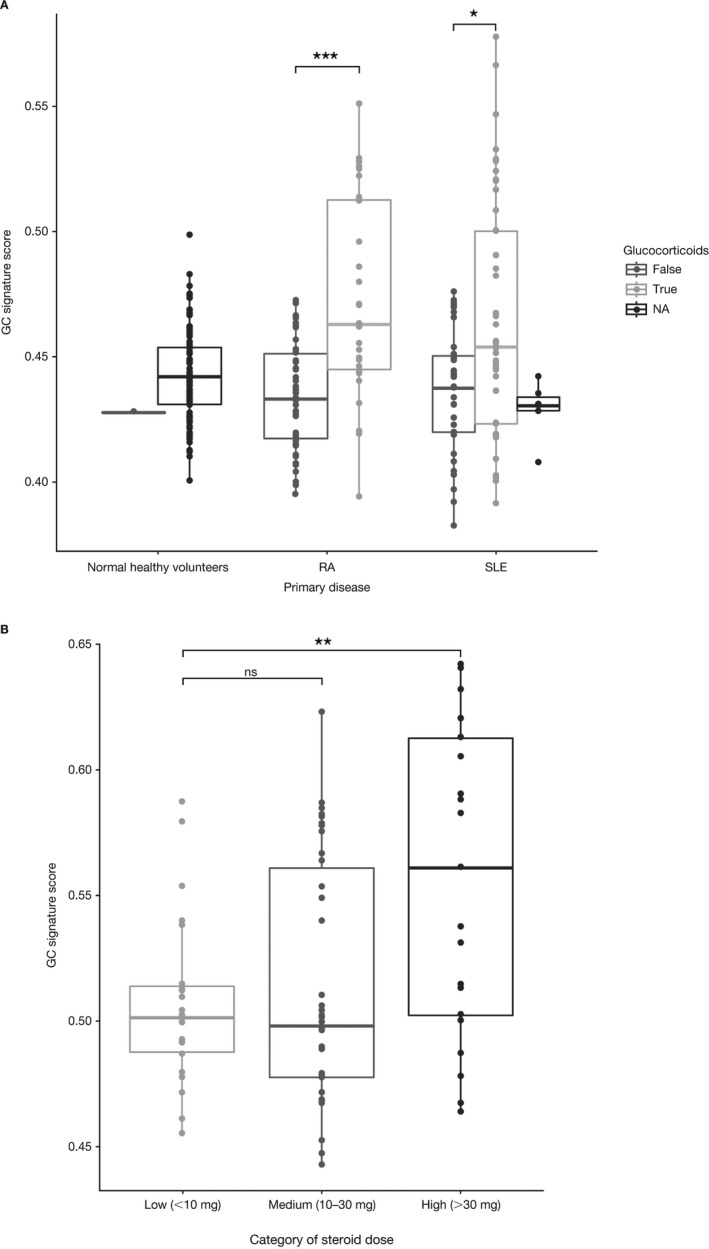
Relationship between the GC gene signature and GC use in the rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE) cohorts. A, GC gene signature expression in cross‐sectional cohorts of patients with RA or SLE. Whole blood was collected from normal healthy volunteers, patients with RA, and patients with SLE. RNA was isolated and used to probe Affymetrix HG‐219 arrays. GC gene signature scores are categorized as patients currently receiving GCs (true) versus patients receiving other standard‐of‐care treatments (false). Patients without treatment information are indicated as not available (NA). B, GC gene signature scores for baseline samples from the IM101‐042 abatacept SLE phase II trial, according to GC dose (low, medium, or high). Data are presented as box plots, where the boxes represent the 25th to 75th percentiles, the lines within the boxes represent the median, and the lines outside the boxes extend to the minimum or maximum values after excluding outliers. * = P = 0.01; ** = P = 0.001; *** = P < 0.001. See Figure 1 for other definitions.
Correlation of the GC gene signature with other pharmacodynamic end points. GCs are known to cause redistribution of leukocyte subsets through demargination of neutrophils from the bone marrow or sequestration of lymphocyte populations in lymphoid organs 33, 34. To determine whether the GC signature correlated with these pharmacodynamic end points, we analyzed peripheral blood samples obtained from SLE and RA patients for CD4+ T cells, CD8+ T cells, and CD19+ B cells. Expression of the GC signature was negatively correlated with the percentages of these subsets in the peripheral blood of SLE patients (Figure 5A) and RA patients (Figure 5B). In the abatacept SLE study, the GC signature scores were positively correlated with neutrophil counts (Figure 5C). Therefore, expression of the GC gene signature correlates with the known biology of GCs in both patients with SLE and patients with RA.
Figure 5.
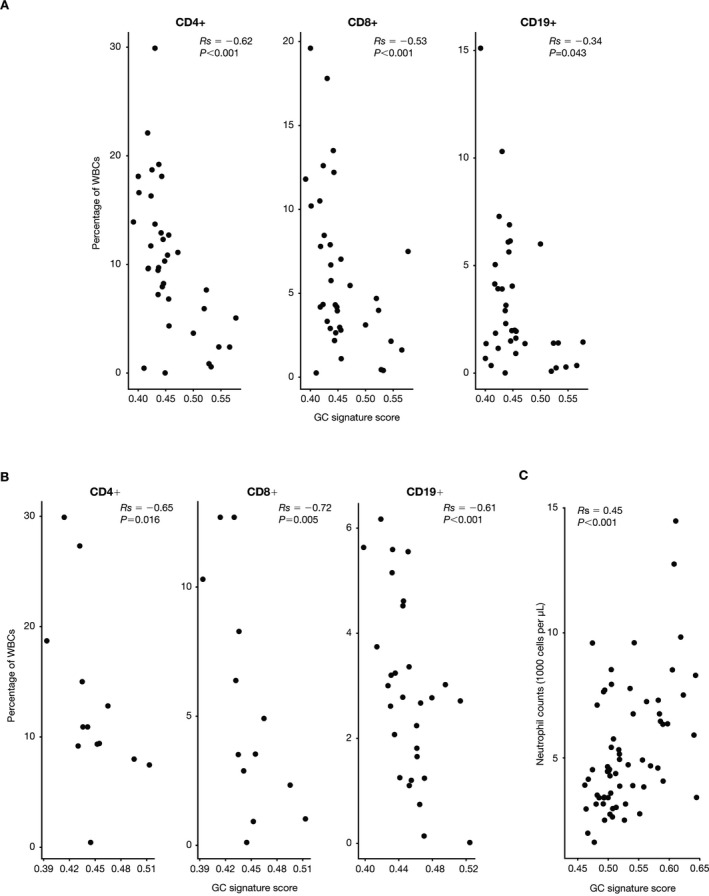
Glucocorticoid (GC) gene signature correlations with T and B cell subsets. A and B, Percentages of peripheral blood CD4+ T cells, CD8+ T cells, and CD19+ B cells from patients with systemic lupus erythematosus (SLE) (A) and patients with rheumatoid arthritis (B), plotted relative to the GC gene signature score for each patient. C, Peripheral blood neutrophil counts from the IM101‐042 abatacept SLE study baseline samples, plotted relative to the GC gene signature score for each patient. Correlations were analyzed using Spearman's rank correlation coefficients. Each data point represents an individual patient. WBCs = white blood cells.
Refinement of the GC gene signature. Having identified a gene signature that reflected the pharmacodynamic effects of GCs, we sought to further refine the signature in order to facilitate its implementation in the clinic. We refined the list of 64 up‐regulated genes to those genes that were induced by >1.5‐fold with an FDR‐adjusted P value of less than 0.05, comparing patients who received prednisolone with those who received placebo in the IM125‐001 trial. We further filtered for detectable expression in the abatacept IM101‐042 SLE trial. Of the initial 64 genes, 18 met these criteria (see Supplementary Table 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40476/abstract). The top 8 genes from the list (FKBP5, ECHDC3, IL1R2, ZBTB16, IRS2, IRAK3, ACSL1, DUSP1) were then used to calculate ssGSEA scores. Analysis of the IM125‐001 study of the partial GR agonist with this abbreviated signature fully captured the behavior of the 64‐gene signature (Figure 6A). Similar to the signature generated with the 64 up‐regulated genes, the 8‐gene signature accurately reflected the transactivation potential of the partial agonist and prednisolone following in vivo administration of these compounds. The 8‐gene signature also was positively correlated with peripheral blood neutrophil counts from the abatacept IM101‐042 SLE trial, with a similar P value to that for the correlation generated with the 64‐gene list (Figure 6B). We conclude that a qPCR assay for these 8 genes would be a sensitive biomarker of GC pharmacodynamic activity that can be implemented with a simple whole blood collection.
Figure 6.
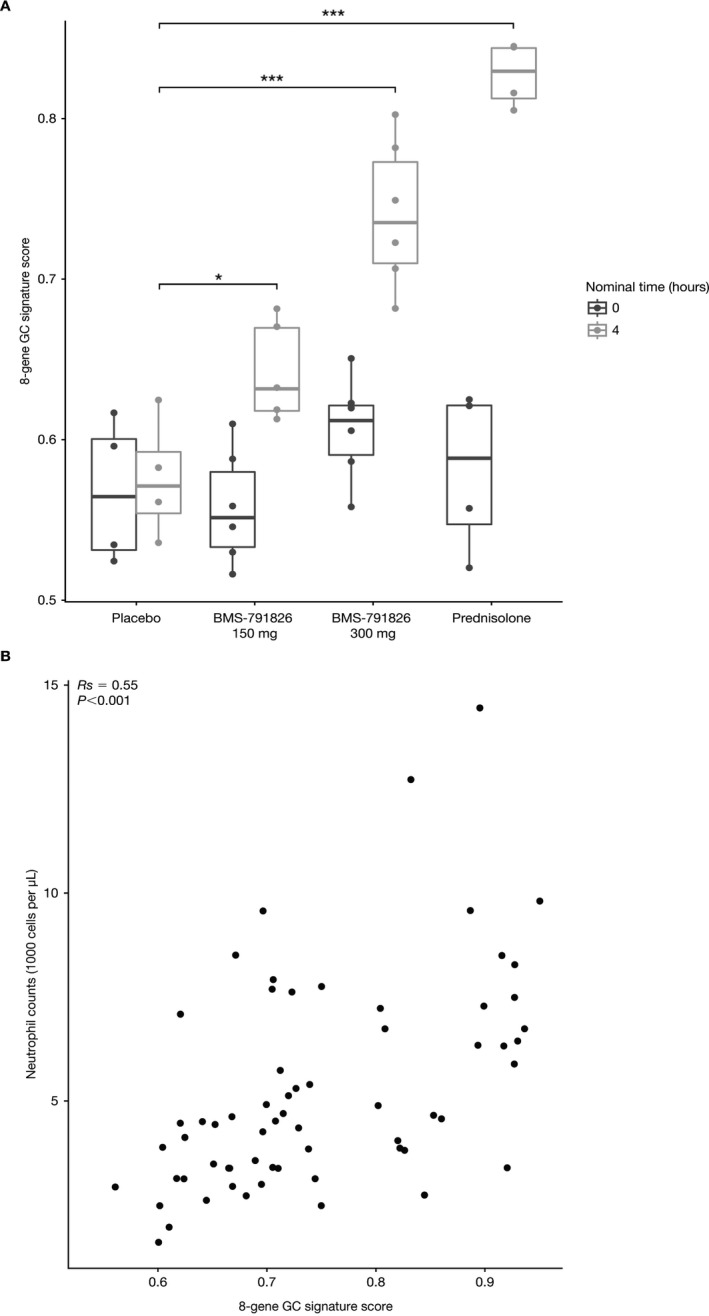
Validation of the 8‐gene glucocorticoid (GC) signature. A, GC gene signature scores using an abbreviated list of 8 genes for participants from the IM125‐001 study who received placebo, 150 or 300 mg BMS‐791826, or 10 mg prednisolone. Data are presented as box plots, where the boxes represent the 25th to 75th percentiles, the lines within the boxes represent the median, and the lines outside the boxes extend to the minimum or maximum values after excluding outliers. B, GC gene signature scores using the 8‐gene list versus peripheral blood neutrophil counts for participants from the IM101‐042 abatacept systemic lupus erythematosus study. Correlations were calculated using Spearman's rank correlation coefficients. Each data point represents an individual patient. * = P = 0.015; *** = P < 0.001.
Discussion
GCs remain a mainstay of treatment for many autoimmune and inflammatory diseases, due to their potent antiinflammatory activity. Long‐term treatment is, however, associated with an increased risk of toxic effects. Given this risk and the significant interpatient variability in the clinical response to GCs, there is a need for a sensitive, objective pharmacodynamic biomarker that will facilitate proper dose selection. In this report, we describe the development of a gene signature that can be applied to whole blood RNA analysis.
We developed the gene signature based on in vitro expression‐profiling experiments using PBMCs derived from NHVs. We focused on genes induced by prednisolone treatment rather than down‐regulated genes, due to a larger dynamic range across donors. We used the ssGSEA algorithm to generate a composite score that can be applied to individual samples or patients. This algorithm appeared to sensitively detect GC‐dependent transcriptional responses, based on several observations. The GC signature score accurately reflected the transactivation potential of synthetic partial GR agonists from both in vitro whole blood profiling studies and in vivo using samples obtained following oral administration of full and partial GR agonists. The signature scores also captured the dose response to prednisolone both in vitro and in vivo. When applied to samples from cross‐sectional cohorts of patients with SLE and patients with RA, GC signature scores were higher in patients receiving GCs compared with those receiving other non‐GC standard‐of‐care medications. In baseline samples from the IM101‐042 abatacept SLE study, GC signature scores progressively increased as steroid doses increased.
Other biomarkers of GR agonism have been described. In a randomized, placebo‐controlled trial of prednisone in NHVs, cortisol concentrations in plasma declined rapidly following administration of prednisone, with a maximal reduction by 8–12 hours postdose 34. Peripheral blood leukocyte populations also rapidly reacted to prednisone administration. Peripheral blood neutrophil counts increased significantly by 12 hours postdose and returned to baseline levels by 24 hours. Lymphocyte counts decreased significantly as early as 2 hours postdose, peaked at 4 hours, and recovered to baseline levels by 12 hours. Similar shifts in peripheral blood leukocyte populations have been observed following intravenous administration of hydrocortisone 33. Consistent with these effects, we observed negative correlations between the GC signature score and the percentages of peripheral blood CD4+ T cells, CD8+ T cells, and CD19+ B cells in cross‐sectional cohorts of both RA patients and SLE patients. GC signature scores were also positively correlated with peripheral blood neutrophil counts in baseline samples from the abatacept SLE study (IM101‐042).
Although changes in circulating cell populations represent potential pharmacodynamic responses to GCs, these changes may be difficult to assess in patients with autoimmune disease, because many of these patients have lymphopenia 35. Furthermore, many of the proinflammatory cytokines associated with autoimmune diseases, including interleukin‐1, interleukin‐6, and TNF, have been shown to impact the HPA axis 36, thereby confounding the use of serum cortisol levels as a pharmacodynamic response biomarker. We propose that the GC gene signature provides an objective measure of the downstream effects of the GR that can be applied to all patient populations.
In addition to reflecting GR agonism, the signature may also provide insight into efficacy. Our in vitro gene profiling studies were not biased to identify transrepressed genes, because we did not include a proinflammatory stimulus in the experiment. However, it is clear that transactivated genes also contribute to the efficacy of GCs. Three genes in our final 8‐gene signature have been implicated in mediating the antiinflammatory effects of GCs (DUSP1 [7], IRAK3 37, and IL1R2 38). To confirm the ability of the signature to capture GC efficacy, the signature analysis could be included in the context of a clinical trial that has a GC comparator arm.
The GC gene signature we have developed has utility not only as part of clinical practice but also in helping to determine the potential confounding effects of steroids in clinical trials. In baseline samples from the abatacept SLE study, GC gene signature scores generally correlated with the reported steroid dosage. However, significant interpatient variability within each dose group was observed. One limitation of our study is that GC use is physician‐reported and may not accurately reflect patient use. This heterogeneity could also be attributable to steroid resistance. A significant percentage of patients with autoimmune disease exhibit steroid resistance 12. Alternatively, the heterogeneity could reflect nonadherence to the study protocol. Given the strong antiinflammatory effects of GCs, trials often include a requirement to taper or even discontinue GC treatment.
The GC gene signature provides an objective method with which to assess compliance to study protocols. We have also observed that commonly used co‐medications such as hydroxychloroquine do not appear to interfere with expression of the signature (data not shown). Furthermore, we have refined the signature to a list of 8 genes. Calculation of the 8‐gene signature score could easily be conducted using qPCR or other platforms using whole blood collections. In summary, we believe that the gene signature we have developed has broad utility for monitoring responses to GCs in the many indications for which they are prescribed.
Author Contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Nadler had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Hu, Carman, Bandyopadhyay, Nadler.
Acquisition of data
Holloway, Kansal, Fan, Goldstine, Lee, Somerville, Latek, Townsend, Johnsen, Connolly, Shadick, Weinblatt, Furie.
Analysis and interpretation of data
Hu, Carman, Bandyopadhyay, Nadler.
Role of the Study Sponsor
The study design, data collection, and analysis were approved by Bristol‐Myers Squibb. Bristol‐Myers Squibb approved submission and publication of the manuscript. Publication of this article was not contingent upon approval by Bristol‐Myers Squibb.
Supporting information
Acknowledgments
We would like to thank all of the patients who participated in and provided samples for these studies. We also thank Ferva Abidi, Lauren Dewey, and Miranda Girard for coordinating patient recruitment.
ClinicalTrials.gov identifiers: NCT00119678; NCT03198013; NCT03196557.
Supported by Bristol‐Myers Squibb.
Drs. Hu and Carman contributed equally to this work.
Drs. Hu, Carman, Latek, Townsend, Johnsen, Connolly, and Nadler, and Ms Holloway, Ms Kansal, Ms Fan, Ms Goldstine, and Ms Lee own stock or stock options in Bristol‐Myers Squibb. Dr. Shadick has received consulting fees from Bristol‐Myers Squibb (less than $10,000) and grant support from Amgen, Mallinckrodt, the BRASS registry (funded by Bristol‐Myers Squibb), UCB, Sanofi, Crescendo Biosciences, and dexTerity. Dr. Weinblatt has received consulting fees (less than $10,000) and grant support from Bristol‐Myers Squibb. Dr. Furie has received consulting fees and/or honoraria from Bristol‐Myers Squibb (less than $10,000).
References
- 1. Buttgereit F. A fresh look at glucocorticoids: how to use an old ally more effectively. Bull NYU Hosp Jt Dis 2012;70 Suppl 1:S26–9. [PubMed] [Google Scholar]
- 2. Desmet SJ, De Bosscher K. Glucocorticoid receptors: finding the middle ground. J Clin Invest 2017;127:1136–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bijlsma JW, Buttgereit F. Adverse events of glucocorticoids during treatment of rheumatoid arthritis: lessons from cohort and registry studies. Rheumatology (Oxford) 2016;55 Suppl 2:ii3–5. [DOI] [PubMed] [Google Scholar]
- 4. Ruiz‐Arruza I, Ugarte A, Cabezas‐Rodriguez I, Medina JA, Moran MA, Ruiz‐Irastorza G. Glucocorticoids and irreversible damage in patients with systemic lupus erythematosus. Rheumatology (Oxford) 2014;53:1470–6. [DOI] [PubMed] [Google Scholar]
- 5. Weikum ER, Knuesel MT, Ortlund EA, Yamamoto KR. Glucocorticoid receptor control of transcription: precision and plasticity via allostery. Nat Rev Mol Cell Biol 2017;18:159–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Colotta F, Re F, Muzio M, Bertini R, Polentarutti N, Sironi M, et al. Interleukin‐1 type II receptor: a decoy target for IL‐1 that is regulated by IL‐4. Science 1993;261:472–5. [DOI] [PubMed] [Google Scholar]
- 7. Abraham SM, Lawrence T, Kleiman A, Warden P, Medghalchi M, Tuckermann J, et al. Antiinflammatory effects of dexamethasone are partly dependent on induction of dual specificity phosphatase 1. J Exp Med 2006;203:1883–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beaulieu E, Morand EF. Role of GILZ in immune regulation, glucocorticoid actions and rheumatoid arthritis. Nat Rev Rheum 2011;7:340–8. [DOI] [PubMed] [Google Scholar]
- 9. Cain DW, Cidlowski JA. Immune regulation by glucocorticoids. Nat Rev Immunol 2017;17:233–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Greulich F, Hemmer MC, Rollins DA, Rogatsky I, Uhlenhaut NH. There goes the neighborhood: assembly of transcriptional complexes during the regulation of metabolism and inflammation by the glucocorticoid receptor. Steroids 2016;114:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Strehl C, van der Goes MC, Bijlsma JW, Jacobs JW, Buttgereit F. Glucocorticoid‐targeted therapies for the treatment of rheumatoid arthritis. Exp Opin Invest Drugs 2017;26:187–95. [DOI] [PubMed] [Google Scholar]
- 12. Rodriguez JM, Monsalves‐Alvarez M, Henriquez S, Llanos MN, Troncoso R. Glucocorticoid resistance in chronic diseases. Steroids 2016;115:182–92. [DOI] [PubMed] [Google Scholar]
- 13. Dendoncker K, Libert C. Glucocorticoid resistance as a major driver in sepsis pathology. Cytokine Growth Factor Rev 2017;35:85–96. [DOI] [PubMed] [Google Scholar]
- 14. Straub RH, Cutolo M. Glucocorticoids and chronic inflammation. Rheumatology (Oxford) 2016;55 Suppl 2:ii6–14. [DOI] [PubMed] [Google Scholar]
- 15. Gautier l, Cope L, Bolstad BM, Irizarry RA. Affy—analysis of Affymetrix gene chip data at the probe level. Bioinformatics 2004;20:307–15. [DOI] [PubMed] [Google Scholar]
- 16. Team RDC. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2010. [Google Scholar]
- 17. Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 2004;5:R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dai M, Wang P, Boyd AD, Kostov G, Athey B, Jones EG, et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res 2005;33:e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. Limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B 1995;57:289–300. [Google Scholar]
- 21. Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS‐driven cancers require TBK1. Nature 2009;462:108–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA‐seq data. BMC Bioinformatics 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gladman DD, Ibanez D, Urowitz MB. Systemic Lupus Erythematosus Disease Activity Index 2000. J Rheumatol 2002;29:288–91. [PubMed] [Google Scholar]
- 24. Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO III, et al. 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 2010;62:2569–81. [DOI] [PubMed] [Google Scholar]
- 25. Symmons DP, Coppock JS, Bacon PA, Bresnihan B, Isenberg DA, Maddison P, et al, and Members of the British Isles Lupus Assessment Group (BILAG) . Development and assessment of a computerized index of clinical disease activity in systemic lupus erythematosus. Q J Med 1988;69:927–37. [PubMed] [Google Scholar]
- 26. Chinenov Y, Coppo M, Gupte R, Sacta MA, Rogatsky I. Glucocorticoid receptor coordinates transcription factor‐dominated regulatory network in macrophages. BMC Genomics 2014;15:656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Miyata M, Lee JY, Susuki‐Miyata S, Wang WY, Xu H, Kai H, et al. Glucocorticoids suppress inflammation via the upregulation of negative regulator IRAK‐M. Nat Commun 2015;6:6062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schaer DJ, Boretti FS, Hongegger A, Poehler D, Linnscheid P, Staege H, et al. Molecular cloning and characterization of the mouse CD163 homologue, a highly glucocorticoid‐inducible member of the scavenger receptor cysteine‐rich family. Immunogenetics 2001;53:170–7. [DOI] [PubMed] [Google Scholar]
- 29. Weinstein DS, Gong H, Doweyko AM, Cunningham M, Habte S, Wang JH, et al. Azaxanthene based selective glucocorticoid receptor modulators: design, synthesis, and pharmacological evaluation of (S)‐4‐(5‐(1‐((1,3,4‐thiadiazol‐2‐yl)amino)‐2‐methyl‐1‐oxopropan‐2‐yl)‐5H‐chromeno[2,3‐b]pyridin‐2‐yl)‐2‐fluoro‐N, N‐dimethylbenzamide (BMS‐776532) and its methylene homologue (BMS‐791826). J Med Chem 2011;54:7318–33. [DOI] [PubMed] [Google Scholar]
- 30. Khan SH, Awasthi S, Guo C, Goswami D, Ling J, Griffin PR, et al. Binding of the N‐terminal region of coactivator TIF2 to the intrinsically disordered AF1 domain of the glucocorticoid receptor is accompanied by conformational reorganizations. J Biol Chem 2012;287:44546–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Knutti D, Kaul A, Kralli A. A tissue‐specific coactivator of steroid receptors, identified in a functional genetic screen. Mol Cell Biol 2000;20:2411–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Merrill JT, Burgos‐Vargas R, Westhovens R, Chalmers A, D'Cruz D, Wallace DJ, et al. The efficacy and safety of abatacept in patients with non–life‐threatening manifestations of systemic lupus erythematosus: results of a twelve‐month, multicenter, exploratory, phase IIb, randomized, double‐blind, placebo‐controlled trial. Arthritis Rheum 2010;62:3077–87. [DOI] [PubMed] [Google Scholar]
- 33. Olnes MJ, Kotliarov Y, Biancotto A, Cheung F, Chen J, Shi R, et al. Effects of systemically administered hydrocortisone on the human immunome. Sci Rep 2016;6:23002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fleishaker DL, Mukherjee A, Whaley FS, Daniel S, Zeiher BG. Safety and pharmacodynamic dose response of short‐term prednisone in healthy adult subjects: a dose ranging, randomized, placebo‐controlled, crossover study. BMC Musculoskelet Disord 2016;17:293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Merayo‐Chalico J, Rajme‐López S, Barrera‐Vargas A, Alcocer‐Varela J, Díaz‐Zamudio M, Gómez‐Martin D. Lymphopenia and autoimmunity: a double‐edged sword. Hum Immunol 2016;77:921–9. [DOI] [PubMed] [Google Scholar]
- 36. Spies CM, Straub RH, Cutolo M, Buggereit F. Circadian rhythms in rheumatology: a glucocorticoid perspective. Arthritis Res Ther 2014;16 Suppl 2:S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Miyata M, Lee J, Susuki‐Miyata S, Wang WY, Xu H, Kai H, et al. Glucocorticoids suppress inflammation via the upregulation of negative regulator IRAK‐M. Nat Commun 2015;6:6062–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vambutas A, DeVoti J, Goldofsky E, Gordon M, Lesser M, Bonagura V. Alternate splicing of interleukin‐1 receptor type II (IL1R2) in vitro correlates with clinical glucocorticoid responsiveness in patients with AIED. PLoS One 2009;4:e5293. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials