

Abstract
The identification of unknown molecules has been one of the cornerstone applications of mass spectrometry for decades. This tutorial reviews the basics of the interpretation of electrospray ionization‐based MS and MS/MS spectra in order to identify small‐molecule analytes (typically below 2000 Da). Most of what is discussed in this tutorial also applies to other atmospheric pressure ionization methods like atmospheric pressure chemical/photoionization. We focus primarily on the fundamental steps of MS‐based structural elucidation of individual unknown compounds, rather than describing strategies for large‐scale identification in complex samples. We critically discuss topics like the detection of protonated and deprotonated ions ([M + H]+ and [M − H]−) as well as other adduct ions, the determination of the molecular formula, and provide some basic rules on the interpretation of product ion spectra. Our tutorial focuses primarily on the fundamental steps of MS‐based structural elucidation of individual unknown compounds (eg, contaminants in chemical production, pharmacological alteration of drugs), rather than describing strategies for large‐scale identification in complex samples. This tutorial also discusses strategies to obtain useful orthogonal information (UV/Vis, H/D exchange, chemical derivatization, etc) and offers an overview of the different informatics tools and approaches that can be used for structural elucidation of small molecules. It is primarily intended for beginning mass spectrometrists and researchers from other mass spectrometry sub‐disciplines that want to get acquainted with structural elucidation are interested in some practical tips and tricks.
Keywords: atmospheric pressure ionization, electrospray ionization, identification, MS/MS, small molecules, structural elucidation
Abbreviations
- AIF
all ion fragmentation
- APCI
atmospheric pressure chemical ionization
- API
atmospheric pressure ionization
- APPI
atmospheric pressure photoionization
- CCS
collisional cross‐section
- CE
capillary electrophoresis
- CI
chemical ionization
- CID
collision induced dissociation
- DBE
double bond equivalents
- DESI
desorption electrospray ionization
- DFT
density functional theory
- EI
electron ionization
- ESI
electrospray ionization
- FT‐ICR
Fourier transform ion cyclotron resonance
- GC
gas chromatography
- H/D exchange
hydrogen/deuterium exchange
- IM
ion mobility
- IR
infrared
- LC
liquid chromatography
- m/z
mass‐to‐charge
- MDF
mass‐defect filtering
- MS
mass spectrometry
- MS/MS
tandem mass spectrometry
- MW
molecular weight
- NMR
nuclear magnetic resonance
- PDA
photodiode array
- ppm
parts‐per‐million
- QTOF
quadrupole time‐of‐flight
- RDB
rings and/or double bonds
- SWATH
sequential windowed acquisition of all theoretical fragment ion mass spectra
- UHPLC
ultra‐high pressure liquid chromatography
- UV
ultraviolet
- Vis
visible
- XIC
extracted ion chromatogram
1. INTRODUCTION
Structural identification and quantification of unknown chemical entities is a constant question in analytical science. Four classes of analytes can be distinguished, based on the answer to two yes/no questions: (1) has the unknown compound ever been identified before (ie, is the chemical entity present in a database/knowledgebase)? and (2) can the compound be expected by the analyst based on a priori knowledge on the sample and the conditions? These four different classes are presented in Table 1. The first class is that of the “known” analytes; that is, compounds that are expected in a certain sample and whose structure is known (class 1). The analytical goal for these compounds is confirmation of the presence of the molecule studied and/or quantification of the analyte in a given sample. Although the strategies to confirm presence of these compounds by MS share similarities with structure elucidation, we keep this class outside of the scope of this tutorial. The “unknown” compounds can be classified into three more classes: “Known not expected” (class 2); that is, compounds that are described for which mass spectral information is available in databases. Typical examples are common contaminants in solvents, small molecules that leach from plastic consumables, and the like. The class “unknown but expected substances” (class 3) contains compounds in the studied sample that can be rationalized by metabolism and/or chemical side reactions of a known compound present in the sample. The last class 4 of unknowns is that of the “unknowns and unexpected compounds,” where there is no a priori knowledge on the structure and origin. Identification of the molecular structure of compounds in each class requires different techniques and approaches.
Table 1.
Stratification of unknown compounds into different classes, based on a priori available information known to the scientist and/or its presence in knowledge bases
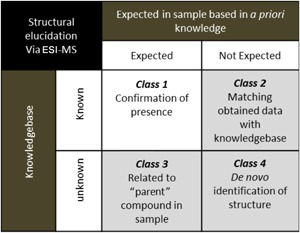
Table 1 illustrates that the availability of meta‐information (or a priori information) is often crucial in the identification process. For example, in the analysis of substances that migrate from plastic baby bottles,1 it is important to acknowledge the fact that we deal with polymers. The analyst does not know the individual analytes. However, the identification of these Class 2 compounds is enhanced by searching for compounds with known applications in polymer industry (ie, the knowledgebase: including catalysts, monomers and small oligomers, optical brighteners, plasticizers, anti‐oxidantia, etc). In a similar way, knowledge of the process in which the unknown analytes are produced and the chemical reactivity of the molecules involved is of great value for the identification of unknown compounds; for example, in the analysis of unknown hydrolysis products of a known chemotherapy drug melphalan (ie, the “parent” compound in the sample, resulting in Class 3 unknowns).2
Questions for structural elucidation of small molecules arise in research laboratories where new molecules are synthesized on a small scale. The analyte can contain intended molecules, but also unexpected structures and unknown side products that are produced and must be studied to understand the chemistry of the reaction at hand. On a much larger scale, the same questions must be answered in the quality control of bulk chemicals that must meet certain technical specifications. The latter becomes even more stringent in, for example, the pharmaceutical industry when product and processes must meet the requirements of regulatory authorities. We focus on the fundamentals of MS‐based structural elucidation of individual unknown small molecules, strategies for high‐throughput identification in metabolomics,3 peptidomics,4 proteomics,5, 6 glycomics,7 and lipidomics8, 9 are discussed elsewhere.
“Together with ultraviolet/visible (UV/Vis), infrared (IR), and nuclear magnetic resonance (NMR) spectroscopy, mass spectrometry (MS) is a key technology in the identification of unknown compounds” could be the opening sentence of an undergraduate course on the use of analytical techniques to identify unknown small organic molecules. This statement is largely valid, but the practical implication implies often a lot more than what can be studied in even advanced courses. On the one hand, this complexity is due to the enormous diversity of analytes and molecules, but on the other hand mass spectrometry is no longer a single technique. The analytical chemist has a wide range of mass spectrometric instruments available that differ in ionization technique, type of mass analyzer, and the way in which the analyzers are combined into tandem mass spectrometry (MS/MS) instruments. Sometimes, the MS instruments are even combined with other analytical technologies (ion mobility (IM), IR spectroscopy, etc). In a traditional approach, a particular ionization technique is used in combination with a single mass analyzer. To cover all available approaches in a single course for analytical chemists‐to‐be is impossible, and so it is for a single review like ours. Instead, we will focus on the applications of electrospray ionization (ESI)—high resolution mass spectrometry for small molecule structural elucidation. In this tutorial, we focus on high resolution (and mass accuracy) mass spectrometry because this is pivotal to obtain the elemental composition of an unknown (see section 2.2). Also, it is important to note that, in a strict sense, structural elucidation implies full structure identification. This full‐blown structure identification is usually not feasible via MS‐based techniques alone, because it is practically impossible to assign, for example, stereochemistry or to discriminate between regioisomers or rotamers, at least not without extensive background information or in conjunction with other techniques such as (chiral) chromatography, NMR, etc. Therefore, we will focus on the structural information that can be extracted from MS experiments.
Until the late eighties of last century and the development of “soft” ionization techniques like ESI10 and matrix‐assisted laser desorption ionization (MALDI),11 MS relied on electron ionization (EI) and chemical ionization (CI) as ionization techniques and gas chromatography (GC) as separation technique. These techniques are still very valuable, but have limitations with regard to the range of analytes that can be handled. Nowadays, sample introduction into the mass spectrometer is often realized via the liquid phase often accompanied with a liquid chromatography (LC) system. This liquid‐phase introduction to the MS is a choice driven by the properties of the analytes (eg, polar analytes, often found for drug compounds) but is also driven by the ease of use and availability of the technology. The LC systems up front of the MS can be used to introduce pure analytes, but often are necessary to separate a mixture of analytes prior to MS analysis. It is important to note that certain classes of compounds (eg, volatile compounds or compounds that tend to hydrolyze under aqueous conditions) are not compatible with LC‐MS. The most commonly used LC‐MS interface and ionization technique is ESI. Nevertheless, other atmospheric‐pressure ionization techniques like atmospheric pressure chemical ionization (APCI) and atmospheric pressure photoionization (APPI) can be coupled to LC as well. The choice for a certain ionization technique is mainly dependent on the polarity in solution and proton affinity in the gas phase of the analytes that can be expected in the sample.12 Apart from the analyte properties, the specific design of the ionization source will also influence the suitability of an actual ionization source for the analysis of a specific compound. For structural characterization, it is important to realize that these atmospheric‐pressure ionization methods all result in protonated or deprotonated molecules and other adducts; in contrast, EI generates radical cations (molecular ions). The nature of the ions generated in ESI (protonated/deprotonated, Na+‐, K+‐adducts, etc) depends on the characteristics of the analyte and on the experimental conditions used, and can either be problematic or put to an advantage in the analytical question at hand. Whereas EI most often fragments the molecular ion into product ions, the protonated ion from ESI mostly has a limited excess energy input that results in MS spectra that contain few or no product ions. Therefore, little or no structural information is available in ESI mass spectra; however, this feature is advantageous to identify the molecular mass of the analytes.
Obviously, the selected mass analyzer has an important effect on mass spectral information. The accuracy and resolving power of the instrument determine whether only the nominal mass can be measured or whether it also allows determination of an elemental composition. In the last decade, a revolution has occurred in the resolving power and accuracy of high‐end mass spectrometers in routine measurements. Novel time‐of‐flight mass analyzers have a resolving power of several tens of thousands and a low ppm mass accuracy. These features limit the number of possible elemental formula for a measured m/z value. The newest generation of Orbitrap mass analyzers achieves a resolving power of approximately 500 000, which can to resolve the fine structure of isotopes for most small molecules. Recent FT‐ICR instruments can achieve an even higher resolving power (>2 000 000). This isotopic fine structure further helps to determine the correct elemental formula.13, 14
A second very important factor is the use of fragmentation techniques in mass spectrometry. Because ESI mostly yield intact protonated/deprotonated molecules (or other adducts of the intact molecule) with very little excess internal energy, there is none or very little fragmentation that results in none or very few product ions available that can reveal structural information. Therefore, techniques such as in‐source fragmentation, tandem mass spectrometry (MS/MS), and MSn are applied to induce the formation of product ions that can be used to study the structure of the analyte of interest. Various ion activation modes exist in tandem mass spectrometry. Nevertheless, collision‐induced dissociation (CID), including higher energy collisional dissociation (HCD, in orbitrap instruments) is by far the most widespread technique for the analysis of small molecules.15 Therefore, this ion activation/fragmentation technique will be used in the examples discussed in this review. Electron‐based ion activation methods like electron capture/transfer dissociation are more commonly used for larger molecules such as peptides but can be also useful in certain small molecule applications, as was recently reviewed by Qi and Volmer.16
Several techniques can be applied to gain additional and orthogonal information that might lead to identification of the correct structure. The techniques discussed further in this tutorial are limited to those that are routinely coupled to mass spectrometry in the quest for the correct molecular structure.
Apart from the technical aspects to acquire MS and tandem MS data, the interpretation of this information is the actual process of characterization/identification of the unknown molecules. Therefore, informatics and software‐aided identification becomes increasingly important and ever more performant in the speed of analysis and in the quality of the interpretation of the data. Also of importance for correct and efficient interpretation of the MS data can be other data available on the analytes studied. Integration of these data with a combination of analytical techniques (eg, LC‐MS) or with careful alignment of all sources of information (IR and/or UV/Vis spectra, retention times, solubility, reaction conditions, etc) will greatly facilitate the identification of the unknown analytes.
2. THE MASS SPECTRUM
2.1. Strategies to find the ions of interest
Mass spectrometry can to detect an almost infinitely wide range of molecules. This broad applicability, in combination with its intrinsically high sensitivity and high throughput, results in very rich and complex data sets from which the analyte(s) of interest must be extracted.
In cases where a compound, detected as a chromatographic peak with another LC‐coupled detector (eg, UV/Vis) must be identified, one can focus on the specific retention time of interest and take into account the potential retention time shift due the transfer time between the detectors (online) or different dead volume of the systems (offline). An average spectrum at the peak top with or without background subtraction before and/or after the peak is expected to provide a spectrum that contains the ion of interest accompanied by co‐eluting (background) ions. The retention time of the peak top and peak shape of the extracted ion chromatograms (XIC's) that correspond to the ions present in the average spectrum will reveal whether the signals are compound‐ or background‐related (Figures 1A and 1B). Background ions can come from the sample matrix, but also from impurities or contamination (eg, bacterial growth) in solvents, leachables of recipients (eg, phthalates), degradation of LC columns, cross‐contamination or carryover of previous samples, etc. Often, background ions show an aberrant chromatographic behavior. Either they show no chromatographic profile at all (ie, their signal is present at all retention times in similar intensities, see Figure 1C), or peaks are broader than other chromatographic peaks, peaks are asymmetric, overloaded (high intensity) or show a non‐consistent, varying retention time. Multiple lists can be found that address common background ions. Keller et al made a comprehensive compilation that can also be downloaded in a table format.17 Background “ions” can also originate from electronic noise and artifacts in transformation of the data.18, 19 The amount and nature of this noise depends on the type of MS system used. They can usually be recognized as spikes (one data point, that resembles a triangle in the XIC; Figure 1C), but can also be omnipresent, usually at the same m/z and similar intensity. For identification, it is important to rely only on ions that are detected several times in the time frame of a chromatographic peak, and that exceed the intrinsic background levels of the MS system.
Figure 1.
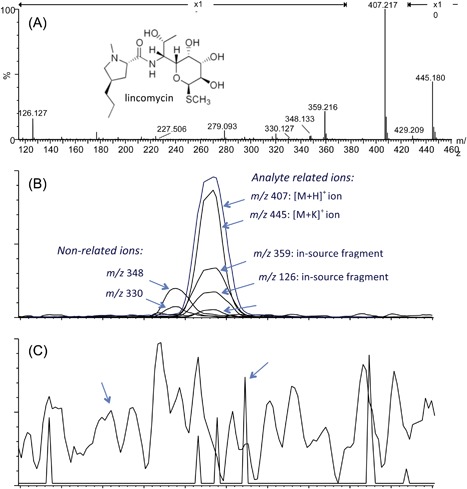
LC/MS analysis of lincomycin in plasma. (A) MS spectrum obtained by averaging the spectra over the chromatographic peak of lincomycin. The extracted ion chromatograms (XICs) of the individual ions in the spectrum show (B) which ions are aligned in retention time and peak shape and, thus, are analyte related (adduct ions: m/z 429 and 445 or in source fragments: m/z 126 and 359) and which ions are derived from partially co‐eluting compounds (m/z 348 and 330) or (C) background ions (m/z 279) or noise (m/z 227). In (B) all XICs were magnified 20‐fold relative to the [M + H]+ signal
The signal of the molecule of interest is often distributed over multiple entities in the mass spectrum. Besides the distribution over the different isotopes, adducts, in‐source fragments, and dimers are often present (Figures 1A and 2A and 2B). The most commonly encountered adducts are Na+, K+, and NH4 + in the positive‐ion mode and Cl−, and CH3COO− in the negative‐ion mode. Many lists of potential adducts can be found in the literature17, 20 or on the web. Larger molecules (usually >MW 1000 Da) often bear multiple charges, but multiple charging can also be observed in small molecules with multiple functional groups that are ionic or very prone to ionization. Because mass‐to‐charge (m/z) ratios are measured in MS, multiply charged ions will be detected at lower m/z; For instance, a doubly charged ion will be present at an m/z that corresponds to [M + 2H]2+/2, which can easily be recognized by a 0.5 amu (1 amu divided by the charge) mass difference between the monoisotopic and the first isotope peak.
Figure 2.
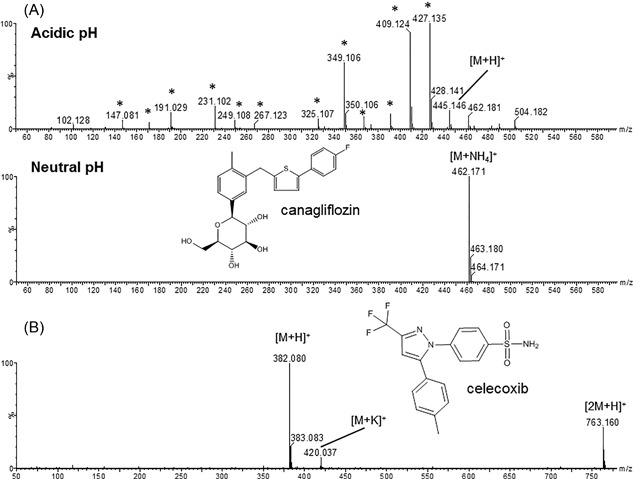
MS spectrum of canagliflozin obtained under acidic (0.1% v/v TFA in water) and neutral pH (10 mM ammonium acetate in water, pH 7) (A). In acidic conditions, many in‐source fragments are formed (*), In the ammonium acetate buffer the ammonium adduct [M + NH4]+ is the main ion. (B) Shows the MS spectrum of celecoxib under neutral conditions (10 mM ammonium acetate in water) next to the protonated molecule [M + H]+, a kalium adduct [M + K]+ and an in source dimer [2M + H]+ can be observed
It should of course also be taken into account that sometimes the ions of the molecule of interest are absent from the spectrum because the compound is not ionized under the conditions used, its signal is below the limit of detection because its concentration is too low or it is suppressed by the ionization of competing (co‐eluting) molecules.21 Since the compound cannot be detected under these conditions, comparison with an orthogonal detection method is important. For example, the presence of a UV signal can show the presence of an unknown that is not detected in the mass spectrum. A change of the ionization mode (positive or negative), source type, solvent conditions (pH, make‐up flow with more organic solvent, etc), sample preparation, derivatization, or electrochemical oxidation can be considered in order to solve this issue.
A univariate peak‐by‐peak comparison between individual samples is often applied in cases where degradation or metabolism of a specific compound is studied. Compound‐related ions are extracted from the analyte‐derived sample by comparison with a control sample (eg, animal dosed with only the formulation) that (ideally) contains the same matrix.22 The analyte‐control comparison is also more effective for complex samples (eg, a biological matrix) than mere background subtraction approaches, which will lead to many false positives due to variability between analyses with regard to retention times and MS sensitivity. The advantage of such an untargeted approach over a targeted search is that unexpected metabolites will not be missed. These might be missed in a targeted approach, when one only looks for expected and known metabolites.
Other untargeted approaches like isotope filtration and mass‐defect filtering (MDF) can be applied to reduce the background or to selectively filter out the ions of interest. Isotope filtration is an easy and efficient technique available in most standard instrument software packages, and is useful when the isotope distribution of the “parent” compound deviates from most background ions due to the presence of atoms with a distinct and characteristic isotope distribution such as B, Cl, Br, S, metal ions, isotopic labels, multiply‐charged ions, etc.23, 24, 25, 26 MDF is another technique often used in drug metabolite profiling.27, 28 The term “mass defect” refers to the difference between the exact mass of an element or molecule and its integer value. Because the decimal portion of the exact mass of metabolites often only changes in a limited way relative to the “parent” compound, filtering on a mass defect similar to that of the parent compound can be useful to deplete the data from a large part of the background ions. It should be noted that large metabolic cleavages or conjugations can have a significant impact on the mass defect of metabolites. Therefore, in cases where these cleavages are to be expected, a separate MDF for every cleavage product or application of a chemically intelligent MDF that takes these cleavages into account is preferred.29 Likewise, a similar strategy can be developed for process impurities and degradation products. For an in depth discussion on the use of the mass defect we refer the reader to the review by Sleno.30
Searching for product ions (shared fragment ions) or neutral losses that are common with a known (“parent”) compound can also be a strategy to find ions from related compounds in a complex matrix. Although these searches can be performed in many different ways on different instruments, high‐resolution MS with alternate low‐ and high‐energy CID data results in the richest dataset. The high‐energy CID data are obtained via continuous recording of all product ions in the whole mass range31 or by multiplexing broader isolation windows.32 These fragmentation methods are further elaborated in section 3.1. Extracted ion chromatograms of product ions of the “parent” compound reveal potential compound‐related peaks from which the precursor ion can be found at the same retention time in the low‐energy trace. Similarly, ions with an exact mass difference that corresponds to a neutral loss observed in the “parent” compound fragmentation can be extracted with different methods. Because most of the latter tools discussed are somewhat biased by the “parent” compound structure or thresholds and settings used, a combination of different tools that apply broader ranges or lower thresholds can often be more powerful.33
2.2. Elemental composition assignment
Once the ions of interest of the unknown analytes are detected, the ideal first step in the identification of the molecules is to perform some mass spectral interpretation to find out whether a protonated or deprotonated ion is selected for identification or rather an adduct with other ions (eg, Na+, K+, acetate). Then, the elemental composition of the selected ions is to be assessed based on accurate mass measurements on a high‐resolution MS system. In a strict sense, this assignment of elemental composition also is a computational problem (elaborated in section 5), but because all MS vendors include elemental composition calculators in their standard software packages, we will discuss them here. It is worth to note that, next to those offered by the MS vendors, academic labs have developed high quality elemental composition calculators, such as SIRIUS and BRAIN.34, 35 An elemental composition calculator is used to provide a list of all potential elemental compositions within the expected range of the measured accurate mass (ie, the mass accuracy) of the instrument and the minimum and maximum number of potential elements set by the user. It is important to note that, for high resolving power mass spectra, the m/z value must be compared to the exact (monoisotopic) mass of a candidate structure instead of the average mass. The selection of potential elements largely depends on the predefined knowledge of the mass spectrometrist of the sample and molecule to be identified. The smaller the a priori knowledge, the broader the selection criteria will be. Some elements such as chlorine, bromine, sulfur, and boron have characteristic isotope ratios. This implies that the presence of these elements in the unknown analyte can be deduced from the isotope ratios obtained from the MS spectrum. Also, the mass defect of the ion can provide some indication on the elemental composition; for example, the presence of iodine, a halogen, but unlike chlorine and bromine without characteristic isotope pattern, can sometimes be suspected when the mass defect of the unknown is lower than anticipated for its molecular weight. It should also be taken into account that ions can also result from in‐source fragmentation of a larger molecule or that compounds are modified (eg, reduced or oxidized) under the conditions used for ESI. It is even possible that the precursor ion is not a dominant peak in the spectrum due to this effect. The extent of adduct formation and in‐source fragmentation can also be dependent on the ionization conditions used (eg, mobile phase and cone voltage; see Figure 2A). Because Na+ and K+ adducts yield far less product ions than their protonated analogs due to loss of the cation, the co‐occurrence of these adducts in a mass spectrum (Figures 2A and 2B) is valuable information to recognize that the ion of interest is not a result of in‐source fragmentation, based on the mass difference among the H+, Na+, and K+ adducts. In ESI, proton‐bound dimers with formula [2M + H]+ (see Figure 2B), and their corresponding Na+ and K+ adducts often appear in the spectrum of highly concentrated analytes. It can be useful to modify the collision energy or cone voltage in order to distinguish proton‐bound dimers from, or convert them to, a protonated monomer [M + H]+.
It is obvious that a higher mass accuracy of the MS system will produce a shorter list of potential elemental compositions. This fact is illustrated by an example in Table 2, where for the m/z of protonated celecoxib (C17H15O2N3F3S, theoretical m/z 382.08316), 37 potential hits were obtained within a 5 ppm error window, 16 within 2 ppm, and 5 within a 1 ppm window for the same elemental search settings. Even with a good mass accuracy of <1 ppm, the accurate mass alone is often not sufficient to determine the elemental composition of an unknown.36 The lower the mass the ions of interest are, the more likely a single or a very limited number of potential elemental compositions can be assigned. As a consequence, the number of potential elemental formulas can also be reduced by examining product ions because they are lower in molecular weight. The combination of elemental compositions for different product ions can be combined to obtain the most likely elemental composition for the intact precursor ion.37, 38, 39, 40 This feature was exploited by Böcker and Rasche when they first introduced the concept spectral fragmentation trees.41
Table 2.
Potential elemental compositions for the exact monoisotopic mass of (protonated) celecoxib within a 5, 2, and 1 ppm error window and the following elemental search parameters were used: C (0‐30), H (0‐50), O (0‐10), N (0‐5), F (0‐3), S (0‐3)
| Number | 5 ppm | Number | 2 ppm | Number | 1 ppm |
|---|---|---|---|---|---|
| 1 | C17 H15 O2 N3 F3 S | 1 | C17 H15 O2 N3 F3 S | 1 | C17 H15 O2 N3 F3 S |
| 2 | C10 H17 O10 N2 F3 | 2 | C10 H17 O10 N2 F3 | 2 | C10 H17 O10 N2 F3 |
| 3 | C22 H16 O2 F2 S | 3 | C22 H16 O2 F2 S | 3 | C22 H16 O2 F2 S |
| 4 | C19 H13 O5 N3 F | 4 | C19 H13 O5 N3 F | 4 | C19 H13 O5 N3 F |
| 5 | C16 H18 O7 N2 S | 5 | C16 H18 O7 N2 S | 5 | C16 H18 O7 N2 S |
| 6 | C24 H14 O5 | 6 | C24 H14 O5 | ||
| 7 | C12 H21 O2 N5 F S3 | 7 | C12 H21 O2 N5 F S3 | ||
| 8 | C11 H17 O7 N5 F S | 8 | C11 H17 O7 N5 F S | ||
| 9 | C14 H20 O4 N2 F2 S2 | 9 | C14 H20 O4 N2 F2 S2 | ||
| 10 | C10 H24 O10 N S2 | 10 | C10 H24 O10 N S2 | ||
| 11 | C17 H22 O2 N2 S3 | 11 | C17 H22 O2 N2 S3 | ||
| 12 | C25 H11 N F3 | 12 | C25 H11 N F3 | ||
| 13 | C9 H19 O4 N5 F3 S2 | 13 | C9 H19 O4 N5 F3 S2 | ||
| 14 | C11 H21 O5 N2 F3 S2 | 14 | C11 H21 O5 N2 F3 S2 | ||
| 15 | C8 H18 O8 N5 F2 S | 15 | C8 H18 O8 N5 F2 S | ||
| 16 | C15 H20 O N5 S3 | 16 | C15 H20 O N5 S3 | ||
| 17 | C8 H22 O9 N4 S2 | ||||
| 18 | C11 H25 O6 N F S3 | ||||
| 19 | C13 H19 O8 N2 F S | ||||
| 20 | C22 H12 O4 N3 | ||||
| 21 | C25 H15 O F S | ||||
| 22 | C20 H17 N3 F S2 | ||||
| 23 | C20 H14 O N3 F2 S | ||||
| 24 | C25 H18 S2 | ||||
| 25 | C13 H16 O9 N2 F2 | ||||
| 26 | C19 H17 O3 F3 S | ||||
| 27 | C16 H14 O6 N3 F2 | ||||
| 28 | C8 H15 O9 N5 F3 | ||||
| 29 | C21 H15 O6 F | ||||
| 30 | C9 H22 O3 N5 F2 S3 | ||||
| 31 | C14 H16 O6 N5 S | ||||
| 32 | C17 H19 O3 N2 F S2 | ||||
| 33 | C25 H10 O N4 | ||||
| 34 | C14 H23 O3 N2 F S3 | ||||
| 35 | C11 H20 O6 N5 S2 | ||||
| 36 | C12 H18 O3 N5 F2 S2 | ||||
| 37 | C15 H21 N2 F3 S3 |
Next to mass accuracy, the isotope pattern is also important to obtain the correct elemental formula. In most elemental composition calculators, a function is provided that scores the remaining possible molecular formulas for a given mass accuracy by matching their simulated isotope patterns with the observed one. This isotope pattern originates from the fact that molecules incorporate stable elemental ions according to their natural abundances of the isotopes in the environment where the molecules are made; that is, for terrestrial molecules the abundances of the isotopes is as they are found on earth. For example, a molecule that incorporates one 13C isotope at a position where an otherwise identical molecule has a 12C atom, has a mass of 1.003355 Dalton (Da) higher due to the additional neutron in 13C. Thus, a molecule can have different isotope variants with different masses that depend on the number of different isotopes of the elements that compose the molecule.42 The probability of occurrence of these isotope variants can be calculated given the atomic composition and the known elemental isotope abundances, as described by IUPAC.43 The result of this calculation is known as the isotope distribution. Prior information on potential elements and/or their distribution (minimum and/or maximum values) can be used for elemental composition determination because the masses, intensities, and peak envelopes calculated for a hypothetical molecule can be compared and scored against the observed isotope pattern.44 A more‐exhaustive overview on isotope distribution calculations is provided in the review articles by Valkenborg et al45 and Rockwood and Palmblad.46 However, a few aspects are worth discussing here.
First, the instrument resolving power will determine the type of isotope distribution that should be computed. High resolution instruments (10 000‐50 000 resolving power) can resolve an isotopic envelope (ie, the ensemble of all naturally occurring isotopes), but do not provide information on the isotopic fine structure (ie, the mass spectral signature that arises from naturally occurring isotopes, including those low abundant such as 15N, and 18O, of the analyzed molecule). Closely related isotopes lump together in so called aggregated isotopes. Spectra acquired on ultra‐high resolution instruments (>100 000 resolving power) are more informative and thus better suited for compositional determination.
This effect of resolution is illustrated in Figure 3, where the measured and calculated isotope distributions for protonated celecoxib (C17H15O2N3F3S, theoretical mass 382.0832 Da) are illustrated at 10 000, 50 000, and 500 000 resolving power (FWHM). At 10 000 resolving power (Figure 3A), there is no separation within the different isotope peaks and only their relative ratios are indicative for the underlying elemental composition. At 50 000 resolving power (Figure 3B), a slight separation can be noticed within the M + 2 isotope between m/z 384.0792 (mainly 34S) and 384.0888 (mainly 13C). This partial separation within the M + 2 peak with this mass difference and abundance ratio points to the presence of one sulfur in the molecule and limits the potential elemental compositions to 10 at 5 ppm, 5 at 2 ppm, and 3 at 1 ppm mass measurement window (see Table 1). At 500 000 resolving power (Figure 3C), the fine structure in all isotope peaks is so detailed (the different isotopes are baseline resolved) that in most cases only one possible elemental composition remains. With increasing mass, however, the number of isotope peaks and their overlap will gradually increase as well as the number of potential elemental compositions.
Figure 3.
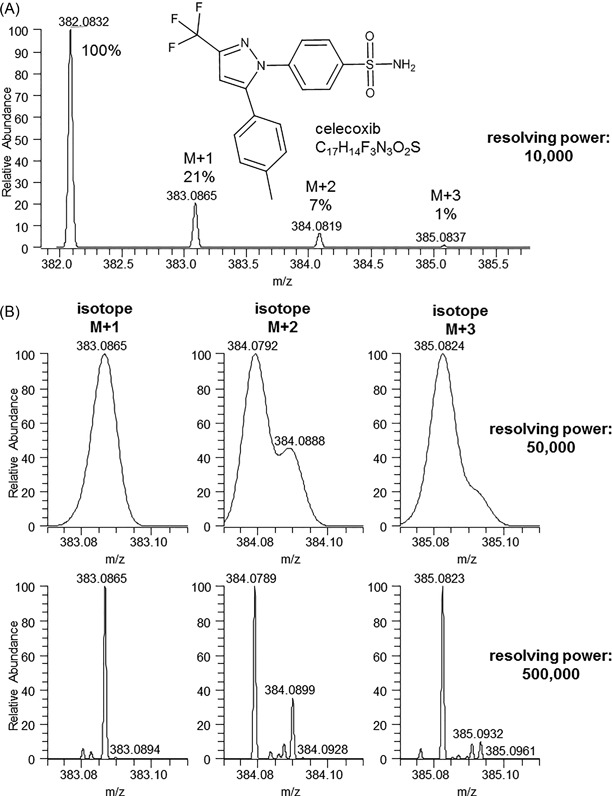
Theoretical isotope distribution for celecoxib at (A) 10 000 resolving power and zoom of the +2, +3, and +4 isotope peak at (B) 50 000 and (C) 500 000 resolving power (FWHM), calculated with Thermo XCalibur 3.0
Second, a user that relies on software to compare isotope distributions should be aware of the spectral abundance accuracy of the instrument (ie, variations in the relative intensity of the isotopes) because several types of error can compromise the observed abundances within the isotope pattern. In 2009, Böcker et al reported that the isotope ratio intensity accuracies obtained on a TOF instrument were higher compared to those measured with a fourier‐transform ion cyclotron resonance (FT‐ICR) instrument.34 More recently, similar isotope intensity accuracies were found when comparing an Orbitrap instrument to a QTOF instrument.47 It is to be noted that, in general, the accuracy of the isotope ratio intensities are heavily dependent on the intensity of the studied ions. This variable isotope ratio intensity accuracy will add uncertainty to the scoring scheme and might corrupt the molecular identification.
Next to the accurate mass and isotope distribution, other parameters can be taken into account to determine the elemental composition of an unknown. The “degree of unsaturation,” usually represented as rings and/or double bonds (RDB) or double‐bond equivalents (DBE), of an impurity, degradation product or metabolite is closely related to that of the “parent” analyte unless a significant part of the structure is lost.48
Atmospheric pressure ionization processes generate even‐electron ions (ie, ions with an even number of electrons as opposed to the radical ions that contain an uneven number of electrons that are generated via EI). This even‐electron rule can be used as a filtering criterion when potential elemental compositions are considered for a measured accurate mass. By applying the nitrogen rule, these even‐electron structures also imply that an ion with an even m/z will have an odd number of nitrogen atoms and vice versa.49, 50 This practical rule can be used to further narrow down the number of potential elemental formulas within a specific mass accuracy. More elaborated guidelines that help in the selection of the correct elemental composition were published by Kind and Fiehn in 2007.
3. THE PRODUCT‐ION SPECTRUM
3.1. Generation of MS/MS and MSn spectra
Even when the correct elemental composition can be assigned to an unknown analyte via the MS spectrum, a myriad of different chemical structures might still be in the running for the compounds identity. Therefore, tandem (MS/MS) or multistage (MSn) mass spectrometry combined with fragmentation of the unknown analyte is a pivotal next step in structural elucidation.
Different mass spectrometric fragmentation techniques exist and might lead to ion activation of alternative fragmentation mechanisms for the same unknown analyte. For structural elucidation of small molecules, beam‐type CID in a collision cell (in triple‐quadrupole (QQQ) or quadrupole‐time‐of‐flight (Q‐TOF) instruments) and resonant‐excitation type CID (in linear or three‐dimensional ion traps and ICR cells) are most frequently used. It is important to note that there can be significant differences between MS/MS spectra obtained via beam‐type CID and resonant excitation CID. However, the approach towards structural identification remains the same. A detailed discussion of all fragmentation techniques is outside of the scope of this tutorial, but was the subject of an extensive review.51 For large molecules, electron‐based ion activation methods are often applied as well.52
In CID, it is critical to carefully select the precursor‐ion mass (usually the protonated (+) or deprotonated (−) molecule) and the “laboratory collision energy” that determines the extent of fragmentation. The isolation window (ie, the m/z region that is selected for fragmentation) can be very narrow in a way that only the monoisotopic peak is fragmented; for example, to obtain high selectivity in complex matrices. In these complex matrices, like biological samples (drug metabolite identification, metabolomics, food, plant material, etc) many compounds enter the mass spectrometer simultaneously and might end up in the product ion spectrum together if the isolation window is too wide. Alternatively, a wider range can be selected to include the entire isotopic envelope, in order to maintain the specific isotope patterns in the fragmentation spectra (eg, for chlorine‐ or bromine‐containing compounds). Another important consequence of the selected isolation width is product ion abundance; the application of very narrow isolation windows, desirable for a high selectivity, will result in lower intensity product ions, which might hamper subsequent structural elucidation due to poor signal‐to‐noise ratios. Therefore, it is important to strike the right balance between selectivity and product ion abundance, especially when one is confronted with complex (biological) matrices.53
Resonant excitation CID (“tandem in time”) MS/MS spectra are produced after isolation of a precursor ion in a trap. This precursor ion is excited by application of an AC potential with a frequency dependent on the m/z of the precursor ion, and allowed to collide with an inert gas present in the trap to induce fragmentation. Fragment ions with a different m/z will no longer be in resonance with the applied excitation frequency and will not fragment further. Specific fragment ions can, in their turn, be isolated, excited, and fragmented to generate an MS3 spectrum. This process can be further iterated, and is generally referred to as multistage fragmentation or MSn.
Beam‐type CID (“tandem in Space”) is accomplished by accelerating the analyte precursor ions, after selection of a precursor ion m/z in the first analyzer, into a cell where they collide with an inert gas to induce fragmentation. Any ion in the cell, including the generated product ions, will be accelerated and activated by subsequent collisions. A wide range of collision energies can be applied, and higher collision energies or longer collision cells will result in multiple rounds of fragmentation inside the collision cell (ie, multiple‐collision conditions CID) that can form second generation product ions within a single MS/MS spectrum. The amount of collision energy applied depends on the kinetic energy of the ions that pass the collision cell and the type of collision gas applied (usually N2, He, Ar, or Xe). Therefore, spectra obtained from different MS systems or at different conditions on the same system can show large differences in the kind and relative abundance of fragments. This feature complicates spectral library searches, as discussed in section 5 of the tutorial.
MS/MS experiments generally imply prior knowledge of the unknown analyte ion (and its corresponding m/z) and therefore require multiple analyses: a first analysis to obtain molecular masses (and m/z values) for the unknowns and a second experiment to obtain product‐ion data. In the past this resulted in at least two experiments; a first injection and LC separation, with acquisition of MS data that was processed manually and from which ions of interest were selected manually. Data analysis was followed by a separate injection and acquisition that contained one or more MS/MS experiments.
Nowadays, data‐dependent MS/MS acquisition (DDA) modes acquire MS/MS spectra based on information obtained from the previous MS spectrum in the same analysis. This can only be done by using sufficiently fast software and MS hardware. Modern tandem MS capable systems all include these features. For a detailed discussion on the advantages and drawbacks of DDA modes, the reader is referred to the recent review by Kind et al.3
A combination of ultra‐high resolution chromatography (UHPLC) with computational tools allows one to cluster ions based on their chromatographic profile. By switching the CID conditions between a low‐ and high‐energy regime, alternate spectra can be generated that contain intact precursor ions (low energy) and their product ions (high energy). With time alignment, product ions can be correlated with their intact precursor. This data‐independent acquisition (DIA) mode provides full MS (unfragmented) data as well as fragments of all ions in one analysis. When this collision‐energy switching is performed over a wide m/z range in one scan event, it is called MSe, MSall, or All Ion Fragmentation (AIF).54, 55, 56 When this collision‐energy switching is performed in multiple, smaller, m/z windows to increase selectivity, this is termed “Sequential Windowed Acquisition of All Theoretical Fragment Ion Mass Spectra” (SWATH).32, 57
As explained earlier in this section, resonant‐excitation CID in ion traps can be iterated to produce MSn spectra. This feature can be exploited in structural elucidation via the generation of “mass spectral trees.”58 When product ions are subjected to a subsequent round of CID, a hierarchy of product ions can be constructed, which eventually leads to prototypic product ions of substructures that can be searched via algorithms.59
In beam‐type CID, selective higher order MSn cannot be easily applied. However, work‐around strategies were developed and applied. For example, an increased cone (or nozzle/skimmer) voltage will accelerate ions that will subsequently collide with neutrals in the ESI source to produce in‐source fragmentations, prior to mass selection. These product ions can then be selected (eg, with a quadrupole) and further fragmented in a collision cell, to produce so‐called “pseudo‐MS3” spectra, which is a strategy that has been around since the early nineties.60, 61 The geometry of some Quadrupole Time‐of‐Flight (QTOF) mass spectrometers equipped with a traveling wave ion‐mobility cell surrounded by two collision cells even allows “pseudo‐MS4” spectra to be generated by application of ion mobility between two rounds of CID and a subsequent match of the mobility drift times.62 The uses of ion mobility in combination with MS are further elaborated in section 4.
3.2. Interpretation of product‐ion spectra
The ease or difficulty of the interpretation of an MS/MS spectrum that leads to a structure proposal depends on a number of parameters. First of all, structural elucidation is dependent on the available background information on the unknown analyte. De novo identification of a structure from a product ion‐spectrum without any a priori knowledge can be very challenging. However, comparison of the fragmentation spectrum from the unknown analyte with that of a related known structure (eg, a drug metabolite or degradation product compared with the drug itself) can drastically help the interpretation. It is important to note that the rules for assignment of the elemental composition, described in section 2.2, also apply for tandem MS spectra. The elemental composition of product ions will greatly facilitate structural elucidation (eg, presence of a chlorine isotope pattern in a product ion).
The CID fragmentation of [M + H]+, [M − H]−, or other adduct ions that are generated by ESI or APCI differs from the fragmentation induced by EI, which generates ions. The fragmentation of ions has been the subject of intensive study and was described in a comprehensive monograph by McLafferty and Turecek.63 Due to the absence of radical formation, fragmentation mechanisms of ESI (or APCI)‐generated ions are significantly different from EI.
An overview of small‐molecule fragmentation mechanisms of [M + H]+ ions has been the subject of specific studies and reviews,64, 65, 66 and falls outside of the scope of this tutorial. For an overview of the scarce information on fragmentation mechanisms of deprotonated [M − H]− molecules, the reader is referred to the review of Wilfried Niessen67 and references therein. Nevertheless, some basic rules that apply to fragmentation of ions generated in ESI and other API sources are described below.
Fragmentation of an ion generated via ESI results in the formation of a charged species, which is observed as the product ion, and a neutral species, which is lost for MS detection. As such, the location of the charge in the molecule upon ionization largely determines the product ions that are formed, a process called “charge‐directed fragmentation.” Therefore, it is important to have a good understanding of the sites and functional groups in a candidate structure that can be (de)protonated. In ESI, the ease with which a functional group within a molecule is ionized is dependent on its basicity/acidity. Up until now, it is not clear whether the basicity in solution or in the gas phase is the most determining factor.68, 69 It must be noted that there also exist gas‐phase decompositions in ESI that occur physically remote from the charge site of the ion. This “charge‐remote fragmentation” phenomenon occurs less frequently for (de)protonated ions but is extensively described in a review by Cheng and Gross.70
The nature of the product ions observed and their relative abundance mainly depends on the number and strength of the bonds that must be broken, and the stability (lowest energy structures) of the product ions and neutral losses. Therefore, aliphatic bonds are usually more easily cleaved than aromatic bonds or bonds that are part of a conjugated system, whereas carbon—carbon bonds are generally more stable than carbon—heteroatom bonds. As stated in the first rule, the neutral part of the molecule and its related chemical information are lost in the MS/MS experiment. Nevertheless, the neutral losses, observed as (exact) mass differences between product ions and their precursor ion can also give crucial information on the ”parent” structure. A comprehensive table of neutral losses was reported by Levsen et al.64
Fragmentation of protonated or deprotonated molecules generates even‐electron ions in the majority of cases (parity rule). Therefore, the same logic as described in section 2.2 for precursor ions can be used to understand product‐ion spectra: for example, the nitrogen rule states that a fragment with even m/z will have an odd number of nitrogen atoms or vice versa.49, 71 As an exception, odd‐electron ions can be observed for specific functional groups and classes of molecules, such as the loss of halogen or nitro‐radicals from (poly)aromatic compounds, where the resulting radical fragment ion can be stabilized.64
The cleavage of a carbon‐heteroatom bond (such as N, O, and S) with charge migration to the α‐carbon represents an important fragmentation mechanism of ESI‐generated ions, also known as inductive cleavage. Other major fragments are formed by fragmentation of the same bond with charge retention on the heteroatom by proton rearrangement. In some cases, both fragments can be observed simultaneously, and the sum of the nominal m/z of both fragments equals the nominal value of ([M + H]+ + 1) or ([M − H]− − 1).72 This fragmentation pattern is commonly observed for carbon‐heteroatom bond‐containing functional groups like ethers, esters, thioesters, amines, amides, glycosides, etc.
Cleavage of a C—C bond adjacent to a heteroatom frequently occurs as well. Here, charge retention at the heteroatom is likely to occur with formation of an iminium or an oxonium ion as a result.
Bond cleavages in low energy CID often result in the addition or subtraction of protons, a process known as “hydrogen rearrangement.” Tsugawa et al formulated nine rules for the hydrogen rearrangement of hydrogens to the most common elements C, N, O, P, and S in positive and negative ion modes.73 These hydrogen rearrangement rules inherently capture most of the other rules mentioned above.
Next to these basic rules, a more‐detailed list of ESI fragmentation rules is given in the work of Weissberg and Dagan.66 It is worth noting that more complex fragmentation reactions or rearrangements also frequently occur in ESI MS/MS spectra. Therefore, the interpretation of small‐molecule CID fragmentation spectra remains a skill that is acquired through a lot of practice. In this regard, the reviews by Wilfried Niessen and coworkers, which describes the positive‐ion mode fragmentation of therapeutic drugs,72 drugs of abuse,74 and pesticides,75 are excellent starting points to get acquainted with small molecule fragmentation spectra. They also nicely demonstrate that fragmentation behavior can be very specific for a given class of molecules.
As an example to illustrate some of the fragmentation rules described in this section, a beam‐type CID MS/MS spectrum of the antipsychotic drug haloperidol is depicted in Figure 4A. Here, a major fragment ion is detected at m/z 165 that corresponds to the cleavage of a carbon—heteroatom bond with charge migration. The complementary fragment would be detected at m/z 212 (that obeys the [M + H]+ +1 rule), but shows a neutral loss of water (−18.01 Da) to form the minor ion observed at m/z 194 as a second generation product‐ion characteristic to beam‐type CID instruments. This ion shows the characteristic isotopic pattern of a chlorine‐containing molecule. Also, the precursor ion directly loses a water molecule to form the ion at m/z 358. Another intense fragment is at m/z 123 results from formation of the fluorobenzyl oxonium ion after cleavage of the C‐C bond adjacent to the ketone.
Figure 4.
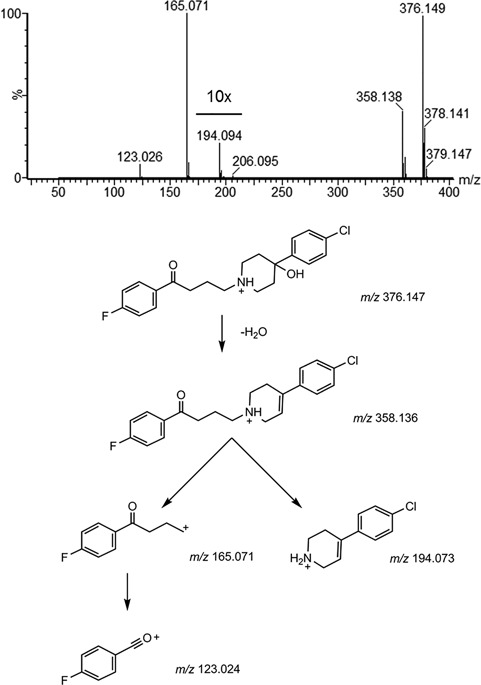
(A) Product ion spectrum and (B) proposed fragmentation scheme of haloperidol
The product‐ion spectrum of a derivatized azide anion from sodium azide is shown in Figure 5A as an example of more‐seldom observed fragmentation behavior. First, azide was derivatized with the polyaromatic sulfonamide dansyl chloride to enhance chromatographic retention and ESI efficiency.76 The initial loss of N2 to form the ion at m/z 249 probably occurs via a five‐membered ring rearrangement of the sulfonamide nitrogen. Afterwards, two competing fragmentation reactions occur: expulsion of SO2 to result in the product ion at m/z 185, similar to what was described by Wang et al,77 and loss of a methyl radical (−CH3•, 15 Da) to produce the odd‐electron product ion at m/z 234. When both reactions occur, the odd electron product ion at m/z 170 is formed. This example shows the formation of odd‐electron product ions (m/z 234, 170) and rearrangements (m/z 248) that are not covered by the basic rules described above.
Figure 5.
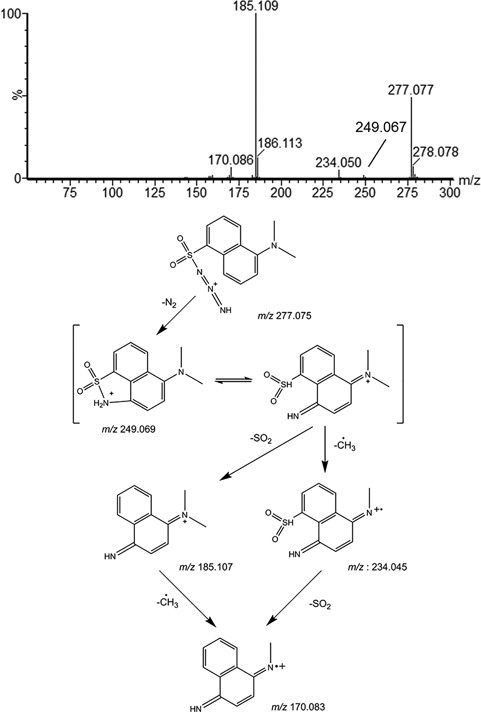
(A) Product ion spectrum and (B) proposed fragmentation scheme of azide derivatized with dansyl chloride. Note that the location of the charge in the fragments is localized for better understanding of the fragmentation mechanism. In reality, the charge will be delocalized over the aromatic system
After acquisition of the MS/MS data, careful analysis of the product‐ion spectra can eventually lead to the construction of a fragmentation pathway for the proposed compound. This pathway provides an explanation of the major fragment ions. It takes into account all the data that were gathered, including elemental compositions, nitrogen rule, and degree of unsaturation (RDB/DBE) of the precursor ion and its fragments, plausible fragmentation sites, and all data gathered by any orthogonal techniques (eg, these described in section 4). An example of proposed fragmentation pathways for the observed MS/MS spectrum of haloperidol (Figure 4A) and dansylated azide (Figure 5A) are depicted in Figures 4B and 5B, respectively. A similar strategy can be followed for the generation of mass spectral trees after acquisition of MSn data.
4. ORTHOGONAL INFORMATION
When MS techniques alone do not suffice for the structural elucidation of an unknown analyte, additional, orthogonal information can be crucial to determine the correct structure. In this tutorial, we will focus on those techniques that can be readily combined with mass spectrometry. Extra information can be based on the coupling with other separation methods (ion mobility) or detectors (UV/Vis). Also, chemical interventions that aid structure identification (H/D exchange, chemical derivatization) will be discussed.
4.1. UV/Vis detection
Photodiode array detectors (PDA) or other UV/Vis spectrophotometers are amongst the most widely available detectors for LC. These detectors are non‐destructive, and can easily be coupled with MS in a serial (ie, the LC column is connected to the UV/Vis detector, which is further coupled to the MS) or parallel fashion (ie, the liquid flow is split after the LC column, and partially goes to the MS and UV/Vis detectors). Because the absorption of light in the UV and/or visible region of the electromagnetic spectrum depends on the conjugated π‐electron system present in an organic compound, the UV spectrum renders orthogonal information that can be of added value in addition to the information obtained with MS. It is often correlated with the degree of unsaturation (RDB/DBE) that can be calculated from the elemental composition as obtained from the accurate mass data.
UV spectra are especially valuable for identification of class III unknown analytes (Table 1), in which differences in the UV spectra between the “parent” compound and its unknown derivatives (eg, metabolites, impurities, and degradation products) could reveal pivotal information on the site of modification. Moreover, unknown analytes might not ionize under the applied ionization conditions; but, if these compounds contain a conjugated system, they can at least be detected and traced by UV/Vis detection.
4.2. Ion mobility
Although available for many years as a stand‐alone technique, ion mobility coupled to MS (IM‐MS) is a recent development. Ion mobility is an analytical technique that separates gas‐phase ions based on their size and shape. Therefore, IM‐MS can be used to obtain complementary information about unknown analytes.
Several ion‐mobility techniques can be coupled to MS, and many papers describe these techniques and their different applications in more detail.78, 79, 80, 81 Although used mainly to analyze complex mixtures and the enhanced structural analysis of high molecular weight compounds such as peptides and proteins, IM‐MS has also been applied in the analysis of small molecules.82 In many cases, LC‐MS/MS will not discriminate among isomeric compounds. IM‐MS and IM‐MS/MS can sometimes overcome these limitations and identify small molecule positional isomers that cannot be differentiated based on MS fragmentation alone.83 Last, but not least, ion mobility can also simply be used to filter out ions from complex MS data based on their drift time26 or selectively remove background ions.84 This filtering dramatically reduces sample complexity and facilitates retrieval of the ions of interest.
4.3. Hydrogen/deuterium (H/D) exchange
Additional structural information can sometimes also be obtained from H/D exchange experiments, when the different structure proposals with the same elemental composition after mass spectrometry experiments contain a different number of exchangeable hydrogen atoms. Hydrogen atoms bound to heteroatoms such as oxygen, nitrogen, and sulfur are readily exchangeable with deuterium whereas those bound to carbon are not exchanged. The mass difference of approximately 1 Da per exchangeable hydrogen is easily detected with any mass spectrometer, and could exclude certain structure proposals. Next to decreasing the number of possible structure proposals for a given elemental composition, this technique can also be used to elucidate structures and fragmentation mechanisms through examination of the MS/MS spectrum after H/D exchange.
In general, two strategies exist for H/D exchange in solution. In the first one, D2O is added “post‐column” to the LC solvent flow or directly into the ESI source. In the second approach, D2O is used as mobile phase for the LC separation. In general and despite the higher cost due to the increased D2O consumption, the second option might be the most straightforward because this option ensures (almost) complete exchange, without any further intervention to the analytical setup. It is to be noted that when using methanol or other protic solvents as organic modifiers, these solvents should also be replaced with deuterated analogs to achieve a complete exchange. A comprehensive overview of the experimental setup and some applications are given by Liu et al.85, 86
4.4. Chemical derivatization
Chemical derivatization is often used to improve the physicochemical properties of a molecule for analysis. A chromophore can be added to an analyte to enable detection with UV/Vis spectrophotometry, or polar functional groups are derivatized to enhance retention on reversed phase LC or volatility and separation for GC(‐MS) analysis and even to protect thermolabile groups. In ESI and other API sources, derivatization often aims to improve the ionization characteristics for poorly ionizable compounds. An example of such chemical derivatization strategy was mentioned in section 3, because dansyl chloride is used to improve the ionization characteristics, chromatographic retention time, and also UV/Vis response of azide (see Figure 5 for the structure after derivatization).76 Such targeted approaches assume knowledge of the presence of a certain functional group that can be derivatized. On the other hand, chemical derivatization strategies can also be exploited in structural elucidation to prove the presence of a certain specific functional group. In fact, selective derivatization for the detection of functional groups is one of the oldest techniques in chemistry.87 Derivatization is particularly useful when several structure proposals are still possible for an unknown analyte after the elemental composition was determined with MS and tandem MS fragmentation study was performed. A comprehensive overview of the principles of chemical derivatization for mass spectrometry was given by Xu et al.88 Some interesting applications with regard to structural elucidation of drug metabolites were reviewed by Liu and Hop.86 An extensive overview of the application of chemical derivatization for mass spectrometry (not limited to structural elucidation) can be found in a book by Zaikin and Halket.89
5. INFORMATICS TO FACILITATE STRUCTURAL ELUCIDATION
Obviously, mass spectrometry yields primarily mass information through measuring m/z ratios. Unfortunately, even accurate mass alone is a poor discriminator to fully characterize a molecular structure. Especially when considering the mass accuracy of an instrument, numerous compounds and structural isoforms can fall within the same mass interval and cannot be discerned (see Table 2 for the example with celecoxib) by mass only. Luckily, the isotope pattern accompanies this mass information. Yet, given this additional information, it is still hard to distinguish among molecules with a similar mass and composition because the isotope distribution provides only marginal extra information; that is, it is connected to the atomic composition of a molecule and not to its structure. To unravel the identity of an unknown molecule, product‐ion information is required, as discussed in section 3. Computational methods can be a significant asset to improve capabilities and efficiency of this process. Unfortunately, the rule of “one size fits all” does not apply to mass spectrometry‐based compound identification. Depending on the application, different software‐aided strategies can be employed to pinpoint the molecular identify in a most optimal manner. These strategies are based on the amount and type of additional information that is known to the scientist who interprets the spectra. The types of additional information were already depicted in Table 1 (ie, presence of the spectrum or analyte structure in a knowledgebase, a priori expectations on the unknown analyte). Several comprehensive reviews have been published on the computational aspects of MS‐based structural elucidation.90, 91, 92, 93 Although it is not our intention to provide the reader with extensive lists of all published informatics tools, we mention a few of particular interest. Because this review is a tutorial, we focus on explanations of the general informatics strategies and concepts, instead of enumerating the characteristics of all available software tools. Figure 6 shows a general scheme of these strategies, and how they tie into each other. The strategies depicted in Figure 6 are further elaborated throughout this section, and are subdivided based on whether they use the MS or the MS/MS spectrum of the unknown as a starting point. It is important to note that these tools can all be used together to increase the odds of identification and that several of these strategies are actually combined in recent, more advanced, computational tools. All the tools mentioned in this section can aid the scientist in their quest to identify the molecular structure of a compound. However, caution should be applied with these tools. Knowledge about the underlying assumptions is important, and tools should not be considered as a black‐box. Experience and insight knowledge about molecular mass spectrometry remain essential to interpret the often long list of candidate molecules returned from software.
Figure 6.
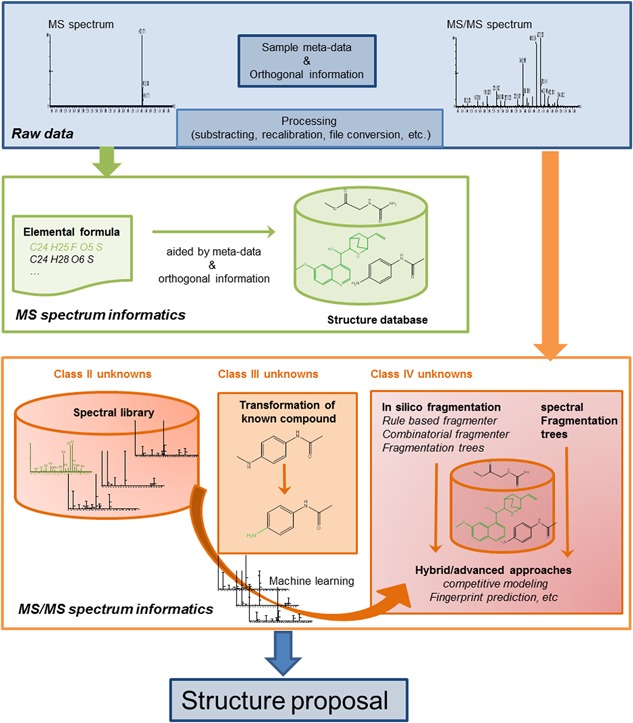
Scheme of the different informatics strategies discussed in section 5. It is important to note that these approaches are interwoven and that several approaches are often combined to obtain the structure of an unknown analyte. Some of the approaches lend themselves ideally to be used for certain classes of unknown compounds. In reality, all techniques can be and are used to gain structural information on the analyte that needs identification
5.1. The problem of mass spectrometry‐based compound identification
Computers have for a long time played a major role in various aspects of mass spectrometry‐based analysis of small molecules. Prior to identification, experimentalists rely on a range of signal‐processing steps (such as denoising, subtracting, calibrating, centroiding, etc), many of which are covered by the MS instrument vendor software or vendor‐independent workflow tools such as the XCMS software.94 A common hurdle is often the conversion of the raw data into a standard file format; for example, mzXML95 and mzML,96 that are suited as input for third‐party search algorithms. For data‐format conversion, we can refer the reader to, for example, the ProteomeWizard platform.97 Computers often also play a crucial role to match the experimentally obtained mass spectrometric evidence and available meta‐information to possible candidate compounds.90 Conceptually, identification is the task to effectively restrict an often large search space (ie, the collection of all theoretically possible structure candidates) to the most likely candidate. In this process, as much available evidence as possible that the scientist gathers from the sample origin, instrument method, and ions in the mass spectral data must be considered. Matching this evidence can be done against diverse types of information, from libraries of historical spectra to calculated molecular formulas. In compound identification, the available evidence is typically heterogeneous, and ranges from precursor‐ion spectra and product‐ion spectra to extra experimental features such as retention time and meta‐information such as the sample origin. To objectively compare experimental spectral data to candidate compounds, their similarity must be quantified with a score function. This comparison leads to a score from which the possible candidates can be prioritized and filtered. It is essential to understand that false positives and false negatives can arise in this process (outlined in detail by Ref.98). False negatives occur when a correct match is wrongfully discarded throughout this process. These false negatives typically originate from too‐large differences between the experimental spectrum and the library of other information against which it is matched. Many instrumental and experimental factors (eg, collision energy, collision gas, instrument design, etc) can introduce such extra variation in the observed spectra, especially in ESI and APCI workflows. On the contrary, false positives occur when a compound is positively matched against the wrong candidate. This incorrect matching happens mostly when the available spectral data are not able to distinguish among several structural features.98 These two potential error classes underline the importance of the incorporation of multiple types of evidence in the identification strategy. Even in cases where an exact identification of a compound is impossible, multiple candidates often belong to the same molecular class; therefore, a reliable identification up to the level of the structural class might be the best achievable.98
5.2. Information extracted from precursor‐ion spectra
The information that can be extracted from the full‐scan spectra essentially is the elemental composition or molecular formula of the unknown analyte (or, in most cases, a list of multiple possible elemental compositions). The way to arrive at this information is elaborated in section 2.2. The molecular formula(s) obtained for an unknown can be searched against numerous chemical structure databases such as PubChem (over 94 million compounds, February 2017), Chemspider (about 58 million structures, February 2017), and the CAS registry that can be searched with SciFinder (over 126 million substances, February 2017). Each database covers a range of compounds, but no database is complete nor is any of the databases comprehensive (ie, that contains all structures also present in the other databases). For most molecular formulas, multiple chemical structures will be found. As stated in the introduction to section 5 all additional meta‐information on the sample and analysis can be used to reduce the number of possibilities. For example, Lin et al discovered the presence of an extraneous contaminant with elemental composition C13H14N3 for the protonated ion during analysis of a pharmaceutical drug product.99 For this elemental formula, 66 molecules can be found in the CAS registry (through Scifinder) and 19 molecules are in Chemspider (searched in February 2017). One of the potential candidates, 1,3‐diphenylguanidine, is used as a rubber‐curing agent. Knowledge of the production and analysis protocols rendered the rubber safety filler of a pipette bulb used during the analysis as a potential source of contamination. The identity of 1,3‐diphenylguanidine was confirmed with a spiking experiment. Additional possibilities for a restriction of the search space are the use of organism‐specific databases and compound class‐specific database; for example, the Human Metabolite Database (41 993 metabolites, March 2017) and LipidMaps Structure database (40 360 lipids, March 2017), respectively. It should be noted that the accurate measured mass or the derived elemental composition that is extracted from the single MS spectrum, is a very effective way to restrict the number of possible candidates in strategies that mine chemical structure databases and spectral libraries based on product‐ion mass spectrometry data.
5.3. Information extracted from product ion spectra
In most cases, information about product ions is needed in order to elucidate the structure of small molecules. In contrast to accurate mass or isotope information, which are connected to atomic composition alone, product‐ion spectra can provide relevant structural insight for molecular identification, because they require the breaking of chemical bonds. In order to interpret product‐ion spectra, they are compared to potential candidate structures and ranked for fitness (similarity). This comparison process comprises a number of typical steps. First, the number of candidates can be restricted (“pruned”) with information complementary to the product‐ion spectrum (eg, precursor‐ion mass or elemental composition). The product‐ion spectrum can be compared with simulated or experimentally stored (in libraries) product‐ion spectra. This process involves scoring the similarity between the experimental spectrum and the candidate match. There are a few approaches to quantify this similarity; some of the most widely used are a normalized dot product100 or a probability based matching approach.101 These methods return a value that conveniently represents the similarity between experimental data and the candidate match.
5.3.1. Transformations from a known compound
In specific studies, it can be expected that unknown analytes are structurally related to a known compound. These are typical examples of class III unknowns as described in Table 1 and include, for example, degradation products and metabolites from pharmaceutical drugs. The identification of (drug) metabolites benefits from this relationship, because they share structural motifs with its “parent” compound. The pathways of biotransformations in metabolism are common, and can often be predicted. These in silico predictions are usually accompanied by large numbers of false positives; that is, biotransformations that are not observed experimentally, and are not able to give a quantitative estimation of metabolite abundances and metabolic rates. Therefore, they are unable to replace in vitro or in vivo experiments.102 In combination with mass spectral data, however, these tools can be very powerful. The localization of the exact site of a metabolic biotransformation with mass spectrometric analysis alone is in many cases challenging or impossible because regio‐isomeric metabolites are frequently not easily discriminated from product‐ion data. This is, for instance, the case for positional isomers with a site of metabolism on an aromatic ring. The biotransformation is often assigned to a part or substructure of the “parent” compound rather than on one particular position on the molecule. These partially described structures are also known as Markush structures. The assignment can be made with manual interpretation of the fragmentation data, or supported by automated localizations of biotransformations that are incorporated in most up‐to‐date mass spectrometry‐based metabolite identification software packages. All potential biotransformation bearing regio‐isomers are generated in silico and their fragmentation matched and scored against the detected unchanged fragments, fragments shifted with the biotransformation, and novel fragments relative to the parent drug fragmentation.103 Rather than blindly copying the position of the biotransformation suggested by the software, it is good practice to use these tools to improve efficiency by highlighting the most‐discriminating ions and to identify the more straightforward metabolites, and leave more time for the interpretation of more exceptional metabolites. In cases where the exact location of metabolism cannot be assigned from mass spectrometry analysis alone, one can opt for additional structure elucidation with orthogonal tools. In general, the combination of multidimensional NMR and (ultra)high‐resolution mass spectrometry will give you the highest information content for structural analysis of small molecules.104 Alternatively, one could apply in silico tools to predict the most‐likely site of metabolism within the mass spectrometry‐assigned substructure location. Especially in silico tools that apply quantum mechanical approaches to calculate the reactivity of the different sites in the molecule and/or the molecular interaction with the (phase I) metabolizing enzyme provides a high success rate in combination with MS data.105, 106 A comprehensive overview and pros and cons of different approaches to predict drug metabolism is described by Kirchmair et al.107
5.3.2. Spectral libraries
Spectral library searches are perhaps the oldest computational tool used for efficient and reliable identification of compounds. The idea is that mass spectra of confidently identified compounds (eg, recorded from reference material and measured in a particular fixed analytical setting); can be used to interpret new data that are produced in similar experimental conditions. In a comparison, the product ions from the trusted and confidently identified compound are used in similarity metrics to identify molecules from new experimental data. Spectral libraries for GC‐MS with EI have grown from thousands of compounds in the 70's to hundreds of thousands compounds (eg, The Wiley database with ∼600 000 compounds and the NIST/EPA/NIH mass spectral library for EI‐spectra containing ∼267 000 compounds). In contrast, spectral libraries for ESI and APCI LC‐MS spectra were developed more recently and at a much smaller scale.
An extensive review of all available ESI spectral libraries was recently published elsewhere.108 Nevertheless, we mention a few with some particular relevance in this context. MassBank109 contains EI MS and merged ESI MS/MS spectra of small‐molecule compounds, collected in over 20 contributing research laboratories. Massbank offers powerful spectral and substructure search functions. METLIN110 is a web‐based repository of MS and MS/MS data for endogenous metabolites, drugs, and drug metabolites, but it is not freely available to download. For targeted strategies that add compounds containing heavy isotopes (eg, 13C, 15N) to an in vivo test system to track their metabolic faith,111, 112 an analogous metabolite database, isoMETLIN, has been constructed.113 mzCloud is a commercial, highly curated database that contains multistage MS spectral trees for over 6000 compounds (concept based on Sheldon et al,101 database maintained by HighChem Ltd, Slovakia). Finally, NIST and Wiley also provide commercial MS/MS libraries.
Whereas spectral libraries have undoubtedly become an important component in the small‐molecule identification toolkit, the user should be aware that they are always incomplete. In an ideal world, spectral libraries allow one to discover an exact match between the experimental spectrum and the correct candidate library spectrum. In practice, even a partial match against a library can provide valuable information, because this partial match might allow interpretation of a subset of the data as a candidate substructure. The strength of spectral libraries is that assumptions on fragmentation behavior and theoretical models are not needed. Accurate information on masses and peak intensities is obtained from spectral libraries of trusted reference material. As such, spectral library searches can be performed with a higher sensitivity and specificity when compared to the classical interpretation by fragmentation models as discussed in section 5.3.3.
However, this advantage is ultimately also the weak point of spectral libraries. Spectra are recorded under certain conditions in an experimental setup. These fixed conditions make them inflexible and harder to exchange among different MS technologies, instruments, or even laboratories. This need for standardization is the main reason that the use of spectral libraries is still rather focused to volatile compounds that can be analyzed with GC‐MS with EI using standardized conditions (82% of analyses published in 2015, according to a recent overview114). The EI spectra are very reproducible when the “standard EI settings are used, regardless of the equipment. When ESI‐generated spectra are utilized in spectral libraries, many groups use home‐made libraries that often only contain specific compound classes of special interest to their research (eg, drugs of abuse115 or pesticides116). Recently, the Global Natural Products Social Molecular Networking base (GNPS) was released as a repository where researchers can share their MS and MS/MS data. The platform also contains a community‐curated set of MS/MS spectra of natural compounds.117
5.3.3. In silico fragmentation
There are many more structures known in data repositories (such as ChemSpider and PubChem) than there are compounds with mass spectra in spectral libraries. In 2010, Kind and Fiehn, estimated this ratio to approximate 50 million vs 1 million (for EI spectra).118 When experimental spectra are not available, in silico product‐ion spectra can be generated based on knowledge about the molecular structure of the compound and our accumulated experimental knowledge of fragmentation pathways. Doing so, in silico fragments are predicted that can be matched against experimental spectra. It is fairly straightforward for computers to compute all possible fragments that a molecule can generate. However, prediction of the most likely bond disconnection and assignment of intensity values that correspond to experimentally observed fragments is less trivial, especially, with the heterogeneity in molecules and the different ionization and fragmentation techniques. In general, in silico fragment generation can be based on two different paradigms or a combination thereof.
First, knowledge‐based and rule‐based fragmenters predict fragment ions based on cleavage rules learned from previous experimentation or from literature. In these approaches, although the information is often presented as fragmentation pathways, they are limited to certain types of molecular structures or instrument settings. Because fragment ions are predicted based on observations or deep insight about fragmentation mechanisms, one can regard the outcome of a rule‐based search approach highly reliable. Mass Frontier (HighChem Ltd, Slovakia) and ACD/MS Fragmenter (ACD Labs, Canada) are commercially available rule‐based fragment predictors. On the down‐side, rule‐based approaches have difficulties to handle spectra from novel ionization and/or fragmentation techniques because lessons learned in the past do not necessarily convey well to new technologies. These rule‐based fragmenters also tend to render false positive hits, which depend on the search‐settings.
A possible solution for this caveat comes from systematic bond‐disconnection tools also known as combinatorial fragmenters, the second paradigm to generate in silico fragments. Systematic bond disconnection enlists all possible fragment ions from a molecular structure tree. MetFrag from Wolf et al, EPIC from Hill et al, or FiD from Heinonen et al are such tools.119, 120, 121 The advantage of combinatorial fragmenters is that they do not rely on prior knowledge. A disadvantage is that all possible fragment combinations are produced, even the ones that are mechanistically impossible. As a consequence, good rule‐based scoring algorithms that rank the obtained results are crucial to reduce the number of false positives. To remediate the exhaustive listing of all possible fragmentation products, these tools often adopt simple heuristic rules to penalize bond disconnection. For example, the fragmentation of an aromatic system will be penalized more severely than an aliphatic system.
The rule‐based and combinatorial approaches are frequently embedded in data‐preprocessing and workflow managers like MetaboLyzer,122 Mass‐Metasite (Molecular Discovery) and MAGMa123 and Progenesis QI (Nonlinear Dynamics) that combine in silico‐calculated spectra from known compounds originating from public searchable databases such as PubChem; mostly preceded by search space reduction via accurate mass measurement on the MS level and other candidate preselection approaches. Excellent reviews on this topic are provided.91, 124 It is essential to note that none of the approaches used for fragmentation assignments can handle rearrangements that involve intramolecular reactions between two distinct parts of the molecule.
Recently, search algorithms were developed that combine information from in silico‐generated fragments and spectral libraries. These algorithms combine the best of both worlds, and can lead to a higher sensitivity and specificity in the identification of unknown analytes. An example of such an approach is embedded in the MetFusion tools.125
5.3.4. Advanced methods for de novo identification
As stated in the introduction unknown compounds that belong to class IV (ie, not present in a spectral database and not to be expected based on a priori information) are the hardest to identify. They require de novo identification, which usually necessitates a lot of experimental and interpretation work by skilled mass spectrometrists. In 2008, Böcker and Rasche published the concept of calculated fragmentation trees in an attempt to make this program computable.41 Basically, fragmentation trees are an automated way to draw a fragmentation scheme like those depicted in Figures 4B and 5B, but without prior knowledge of the molecular structure. Initially, this concept was introduced only as a way to determine the molecular formula of unknown analytes, but it was later turned into a method for structural elucidation.126, 127
During the last 5 years, novel approaches to assess MS/MS spectra were developed that take advantage of machine‐learning algorithms. Two technologies of particular interest in this regard are molecular‐fingerprint prediction, like the FingerID tool,128 and competitive‐fragmentation modeling.129 Just like for in silico‐fragmentation predictors, the results from these modeling techniques can be used to search chemical structure databases (like PubChem, Chemspider, etc). Recently, one of these techniques was combined with the spectral‐fragmentation tree concept,130 and this combination was further refined into the CSI:FingerID method.131 This advanced web‐based tool allows one to search the Pubchem database of chemical structures with a high resolving power MS and MS/MS spectrum. The technical details of the techniques described here are beyond the scope of this tutorial, but were the subject of a recent review.93 It is noteworthy that these machine‐learning approaches can only be developed with high‐quality training data, that can be retrieved from the spectral libraries discussed earlier. An overview that discusses the advances and pitfalls of these novel tools was given recently by Sebastian Böcker.132
6. LIMITATIONS IN MS‐BASED STRUCTURAL ELUCIDATION
The previous sections highlighted the virtue of ESI‐MS to identify small molecules. However, when embarking on the endeavor of MS‐based structural elucidation, it is important to know that there are also certain limitations and pitfalls that one must be aware of. Some of the most‐common ones are listed in Table 3. Good knowledge of these limitations is pivotal to raise the right expectations when one performs MS experiments.
Table 3.
Limitations of MS‐based structural elucidation
| Limitations and pitfalls in ESI‐MS based structural elucidation |
|---|
| 1. Not all compounds ionize by ESI and, thus, can be detected in MS (or only in positive or negative mode). |
| 2. The MS signal is spread over different isotopes, adducts, and/or fragments. |
| 3. The MS response is highly dependent on the chemical structure of the compound. |
| 4. The nature and abundance of the ions (adducts, fragments) is highly dependent on the sample solvent and matrix and the instrumentation hardware, settings and, overall, the experimental conditions used. |
| 5. Not all peaks in an MS spectrum are necessarily from the compound of interest: there can be interferences from chemical and non‐chemical (noise) background. |
| 6. Product ions that originate from a rearrangement or loss of a part of the molecule appearing as unchanged compared to a reference molecule, can be misleading. |
| 7. Isomers like enantiomers and rotamers cannot be differentiated with MS alone. |
| 8. Salt forms cannot be assigned with ESI‐MS. |
| 9. Structure elucidation with MS often leads to partially assigned structures (ie, Markush structures) rather than fully identified structures (eg, for regio‐isomers). |
7. CONCLUSION AND FUTURE PERSPECTIVES
ESI‐MS based structural elucidation of small molecules has experienced a tremendous evolution over the last two decades. Each manufacturer of MS equipment has its own flavor of ESI and APCI source; some have specific sources that are advantageous for specific analytes (eg, APPI) or for specific applications (eg, desorption electrospray ionization (DESI) of Direct Analysis in Real Time (DART) for ionization of analytes on a surface.133, 134 New ionization methods and sources are developed as well, such as UniSpray™135 that extend the range of analytes that can be ionized and analyzed with MS.
With the advent of (ultra) high‐resolution mass spectrometers combined with up to sub‐ppm accuracy and a sensitivity that has increased by orders of magnitude, identification of unknown analytes in extremely small samples has almost become routine. At the same time the complexity and shear amount of data increased to levels that are no longer manageable by a single operator or analyst. With these technical advancements, data analysis and spectral interpretation became the bottleneck in structural elucidation. Smart data processing will be required to de‐convolute the data (eg, M + H+, M + Na+, isotope pattern, fragmentation pattern, etc, that all lead to one analyte).When we consider the recent progress in informatics strategies for structural elucidation described in section 5 of this tutorial, especially in light of the application of machine‐learning techniques, we expect that this hurdle will be largely taken in the years to come.
Other advances in the field of MS‐based structural elucidation, next to informatics and the further improvements in MS technology, are likely to come from the combination of other types of separation strategies, ion activation methods for fragmentation and detectors. An interesting combination of multiple detectors is that of MS and infrared ion spectroscopy, that allow acquisition of IR spectra with MS selectivity and sensitivity. This was recently also demonstrated by the infrared ion spectroscopy (IR‐IS)—MS analysis of two hydroxylated drug metabolites from an in vitro hepatocyte sample.136 The revival of the combination capillary electrophoresis with mass spectrometry (CE‐MS) is a nice example of how additional separation techniques can broaden the scope of MS‐based structural elucidation of small molecules, especially in metabolomics.137 The further refinement of ion‐mobility technology (mainly towards resolving power) together with improvement and standardization of (in silico) collisional cross‐section (CCS) calculations for small molecules will also add to the toolbox of the structural elucidation community. Quantum mechanical approaches with density functional theory (DFT) can be a way to predict CCS ab initio.138 The same strategy can be used to predict product‐ion spectra139 but DFT calculation still tends to be consuming on computational resources, which hampers routine use, and the results are highly dependent on the assumptions the calculations are based on. Progress here will depend on further developments in computer hardware and software technology.
As stated in the introduction, this tutorial is primarily intended for researchers that are new in the field of MS‐based structural elucidation of small molecules and want to get familiar with the practical routines in this interesting and fundamental application of mass spectrometry. It is also meant to be a refresher for more‐seasoned mass spectrometrists or researchers who are active in other fields of research that use of MS‐based structural elucidation, like proteomics, lipidomics, and metabolomics. It is worth noting that merely reading a tutorial like this one will not make you a specialist in structural elucidation, and that practical experience will teach you more than theoretical knowledge does. As in every other aspect of science, the tips mentioned in this tutorial should not be applied without some critical thinking, and the (informatics) tools cited here should not be used as a “black box,” because that approach will lead to poor or even utterly wrong outcomes.
ACKNOWLEDGMENTS
Willy Janssens and Ronald de Vries (Janssen R&D, Belgium) are acknowledged for their critical review of the manuscript and their useful suggestions. The authors also wish to specially thank Guy Winderickx, Maurice Hermans, and Tom Vennekens for their expert advice on the fragmentation schemes in Figures 4 and 5. We are also grateful to the reviewers and the editor for their valuable comments and suggestions to improve this tutorial. TD, ER, KL, and DV are supported by the VlAIO (Vlaams Agentschap Innovatie & Ondernemen) grant HBC.2016.0890.
De Vijlder T, Valkenborg D, Lemière F, Romijn EP, Laukens K, Cuyckens F. A tutorial in small molecule identification via electrospray ionization‐mass spectrometry: The practical art of structural elucidation. Mass Spec Rev. 2018;37: 607–629. 10.1002/mas.21551
The article was updated on 20th November 2017, after first online publication.
REFERENCES
- 1. Onghena M, Van Hoeck E, Van Loco J, et al. Identification of substances migrating from plastic baby bottles using a combination of low‐resolving power and high‐resolving power mass spectrometric analysers coupled to gas and liquid chromatography. J Mass Spectrom. 2015; 50:1234–1244. [DOI] [PubMed] [Google Scholar]
- 2. Boschmans J, de Bruijn E, Van Schil P, Lemière F. Analysis of novel melphalan hydrolysis products formed under isolated lung perfusion conditions using liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom RCM. 2013; 27:835–841. [DOI] [PubMed] [Google Scholar]
- 3. Kind T, Tsugawa H, Cajka T, et al. Identification of small molecules using accurate mass MS/MS search. Mass Spectrom Rev. 2017; 36:581–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dallas DC, Guerrero A, Parker EA, et al. Current peptidomics: applications, purification, identification, quantification, and functional analysis. Proteomics. 2015; 15:1026–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shao W, Lam H. Tandem mass spectral libraries of peptides and their roles in proteomics research. Mass Spectrom Rev. 2017; 36:634–348. [DOI] [PubMed] [Google Scholar]
- 6.Verheggen K, Martens L, Berven FS, Barsnes H, Vaudel M. Database Search Engines: paradigms, Challenges and Solutions. Adv Exp Med Biol 2016; 919:147–156. [DOI] [PubMed] [Google Scholar]
- 7.Yang S, Rubin A, Eshghi ST, Zhang H. Chemoenzymatic method for glycomics: isolation, identification, and quantitation. Proteomics 2016; 16:241–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Blanksby SJ, Mitchell TW. Advances in mass spectrometry for lipidomics. Annu Rev Anal Chem Palo Alto Calif. 2010; 3:433–465. [DOI] [PubMed] [Google Scholar]
- 9. Hancock SE, Poad BLJ, Batarseh A, Abbott SK, Mitchell TW. Advances and unresolved challenges in the structural characterization of isomeric lipids. Anal Biochem. 2017; 524:45–55. [DOI] [PubMed] [Google Scholar]
- 10. Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray ionization for mass spectrometry of large biomolecules. Science. 1989; 246:64–71. [DOI] [PubMed] [Google Scholar]
- 11. Karas M, Bachmann D, Hillenkamp F. Influence of the wavelength in high‐irradiance ultraviolet laser desorption mass spectrometry of organic molecules. Anal Chem. 1985; 57:2935–2939. [Google Scholar]
- 12. Thurman EM, Ferrer I, Barceló D. Choosing between atmospheric pressure chemical ionization and electrospray ionization interfaces for the HPLC/MS analysis of pesticides. Anal Chem. 2001; 73:5441–5449. [DOI] [PubMed] [Google Scholar]
- 13. Schwudke D, Schuhmann K, Herzog R, Bornstein SR, Shevchenko A. Shotgun lipidomics on high resolution mass spectrometers. Cold Spring Harb Perspect Biol. 2011; 3:a004614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nagao T, Yukihira D, Fujimura Y, et al. Power of isotopic fine structure for unambiguous determination of metabolite elemental compositions: in silico evaluation and metabolomic application. Anal Chim Acta. 2014; 813:70–76. [DOI] [PubMed] [Google Scholar]
- 15. Johnson AR, Carlson EE. Collision‐induced dissociation mass spectrometry: a powerful tool for natural product structure elucidation. Anal Chem. 2015; 87:10668–10678. [DOI] [PubMed] [Google Scholar]
- 16. Qi Y, Volmer DA. Structural analysis of small to medium‐sized molecules by mass spectrometry after electron‐ion fragmentation (ExD) reactions. Analyst. 2016; 141:794–806. [DOI] [PubMed] [Google Scholar]
- 17. Keller BO, Sui J, Young AB, Whittal RM. Interferences and contaminants encountered in modern mass spectrometry. Anal Chim Acta. 2008; 627:71–81. [DOI] [PubMed] [Google Scholar]
- 18. Mathur R, O'Connor PB. Artifacts in Fourier transform mass spectrometry. Rapid Commun Mass Spectrom. 2009; 23:523–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xian F, Valeja SG, Beu SC, Hendrickson CL, Marshall AG. Artifacts induced by selective blanking of time‐domain data in Fourier transform mass spectrometry. J Am Soc Mass Spectrom. 2013; 24:1722–1726. [DOI] [PubMed] [Google Scholar]
- 20. Huang N, Siegel MM, Kruppa GH, Laukien FH. Automation of a Fourier transform ion cyclotron resonance mass spectrometer for acquisition, analysis, and E‐mailing of high‐resolving power exact‐mass electrospray ionization mass spectral data. J Am Soc Mass Spectrom. 1999; 10:1166–1173. [Google Scholar]
- 21. Furey A, Moriarty M, Bane V, Kinsella B, Lehane M. Ion suppression; a critical review on causes, evaluation, prevention and applications. Talanta. 2013; 115:104–122. [DOI] [PubMed] [Google Scholar]
- 22. Mortishire‐Smith RJ, O'Connor D, Castro‐Perez JM, Kirby J. Accelerated throughput metabolic route screening in early drug discovery using high‐resolving power liquid chromatography/quadrupole time‐of‐flight mass spectrometry and automated data analysis. Rapid Commun Mass Spectrom. 2005; 19:2659–2670. [DOI] [PubMed] [Google Scholar]
- 23. Cuyckens F, Balcaen LIL, De Wolf K, et al. Use of the bromine isotope ratio in HPLC‐ICP‐MS and HPLC‐ESI‐MS analysis of a new drug in development. Anal Bioanal Chem. 2008; 390:1717–1729. [DOI] [PubMed] [Google Scholar]
- 24. Rousu T, Pelkonen O, Tolonen A. Rapid detection and characterization of reactive drug metabolites in vitro using several isotope‐labeled trapping agents and ultra‐performance liquid chromatography/time‐of‐flight mass spectrometry. Rapid Commun Mass Spectrom. 2009; 23:843–855. [DOI] [PubMed] [Google Scholar]
- 25. Zhu P, Tong W, Alton K, Chowdhury S. An accurate‐mass‐based spectral‐averaging isotope‐pattern‐filtering algorithm for extraction of drug metabolites possessing a distinct isotope pattern from LC‐MS data. Anal Chem. 2009; 81:5910–5917. [DOI] [PubMed] [Google Scholar]
- 26. Cuyckens F, Dillen L, Cools W, et al. Identifying metabolite ions of peptide drugs in the presence of an in vivo matrix background. Bioanalysis. 2012; 4:595–604. [DOI] [PubMed] [Google Scholar]
- 27. Zhang H, Zhang D, Ray K. A software filter to remove interference ions from drug metabolites in accurate mass liquid chromatography/mass spectrometric analyses. J Mass Spectrom. 2003; 38:1110–1112. [DOI] [PubMed] [Google Scholar]
- 28. Zhu M, Ma L, Zhang D, et al. Detection and characterization of metabolites in biological matrices using mass defect filtering of liquid chromatography/high resolving power mass spectrometry data. Drug Metab Dispos Biol Fate Chem. 2006; 34:1722–1733. [DOI] [PubMed] [Google Scholar]
- 29. Mortishire‐Smith RJ, Castro‐Perez JM, Yu K, et al. Generic dealkylation: a tool for increasing the hit‐rate of metabolite rationalization, and automatic customization of mass defect filters. Rapid Commun Mass Spectrom. 2009; 23:939–948. [DOI] [PubMed] [Google Scholar]
- 30. Sleno L. The use of mass defect in modern mass spectrometry. J Mass Spectrom. 2012; 47:226–236. [DOI] [PubMed] [Google Scholar]
- 31. Wrona M, Mauriala T, Bateman KP, Mortishire‐Smith RJ, O'Connor D. “All‐in‐one” analysis for metabolite identification using liquid chromatography/hybrid quadrupole time‐of‐flight mass spectrometry with collision energy switching. Rapid Commun Mass Spectrom. 2005; 19:2597–2602. [DOI] [PubMed] [Google Scholar]
- 32. Hopfgartner G, Tonoli D, Varesio E. High‐resolving power mass spectrometry for integrated qualitative and quantitative analysis of pharmaceuticals in biological matrices. Anal Bioanal Chem. 2012; 402:2587–2596. [DOI] [PubMed] [Google Scholar]
- 33. Cuyckens F, Hurkmans R, Castro‐Perez JM, Leclercq L, Mortishire‐Smith RJ. Extracting metabolite ions out of a matrix background by combined mass defect, neutral loss and isotope filtration. Rapid Commun Mass Spectrom. 2009; 23:327–332. [DOI] [PubMed] [Google Scholar]
- 34. Böcker S, Letzel MC, Lipták Z, Pervukhin A. SIRIUS: decomposing isotope patterns for metabolite identification. Bioinforma Oxf Engl. 2009; 25:218–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Claesen J, Dittwald P, Burzykowski T, Valkenborg D. An efficient method to calculate the aggregated isotopic distribution and exact center‐masses. J Am Soc Mass Spectrom. 2012; 23:753–763. [DOI] [PubMed] [Google Scholar]
- 36. Kind T, Fiehn O. Metabolomic database annotations via query of elemental compositions: mass accuracy is insufficient even at less than 1 ppm. BMC Bioinformatics. 2006; 7:234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Suzuki S, Ishii T, Yasuhara A, Sakai S. Method for the elucidation of the elemental composition of low molecular mass chemicals using exact masses of product ions and neutral losses: application to environmental chemicals measured by liquid chromatography with hybrid quadrupole/time‐of‐flight mass spectrometry. Rapid Commun Mass Spectrom. 2005; 19:3500–3516. [DOI] [PubMed] [Google Scholar]
- 38. Kaufmann A. Determination of the elemental composition of trace analytes in complex matrices using exact masses of product ions and corresponding neutral losses. Rapid Commun Mass Spectrom. 2007; 21:2003–2013. [DOI] [PubMed] [Google Scholar]
- 39. Rojas‐Cherto M, Kasper PT, Willighagen EL, Vreeken RJ, Hankemeier T, Reijmers TH. Elemental composition determination based on MSn. Bioinformatics. 2011; 27:2376–2383. [DOI] [PubMed] [Google Scholar]
- 40. Pluskal T, Uehara T, Yanagida M. Highly accurate chemical formula prediction tool utilizing high‐resolving power mass spectra, MS/MS fragmentation, heuristic rules, and isotope pattern matching. Anal Chem. 2012; 84:4396–4403. [DOI] [PubMed] [Google Scholar]
- 41. Böcker S, Rasche F. Towards de novo identification of metabolites by analyzing tandem mass spectra. Bioinformatics. 2008; 24:49–55. [DOI] [PubMed] [Google Scholar]
- 42. Budzikiewicz H, Grigsby RD. Mass spectrometry and isotopes: a century of research and discussion. Mass Spectrom Rev. 2006; 25:146–157. [DOI] [PubMed] [Google Scholar]
- 43. Meija J, Coplen TB, Berglund M, et al. Isotopic compositions of the elements 2013 (IUPAC technical report). Pure Appl Chem. 2016; 88:293–306. [Google Scholar]
- 44. Wang Y, Gu M. The concept of spectral accuracy for MS. Anal Chem. 2010; 82:7055–7062. [DOI] [PubMed] [Google Scholar]
- 45. Valkenborg D, Mertens I, Lemière F, Witters E, Burzykowski T. The isotopic distribution conundrum. Mass Spectrom Rev. 2012; 31:96–109. [DOI] [PubMed] [Google Scholar]
- 46. Rockwood AL, Palmblad M. Isotopic distributions. Methods Mol Biol. 2013; 1007:65–99. [DOI] [PubMed] [Google Scholar]
- 47. Knolhoff AM, Callahan JH, Croley TR. Mass accuracy and isotopic abundance measurements for HR‐MS instrumentation: capabilities for non‐targeted analyses. J Am Soc Mass Spectrom. 2014; 25:1285–1294. [DOI] [PubMed] [Google Scholar]
- 48. Soffer MD. The molecular formula generalized in terms of cyclic elements of structure. Science. 1958; 127:880. [DOI] [PubMed] [Google Scholar]
- 49. Karni M, Mandelbaum A. The even‐electron rule. Org Mass Spectrom. 1980; 15:53–64. [Google Scholar]
- 50. Kind T, Fiehn O. Seven Golden Rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinformatics. 2007; 8:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sleno L, Volmer DA. Ion activation methods for tandem mass spectrometry. J Mass Spectrom. 2004; 39:1091–1112. [DOI] [PubMed] [Google Scholar]
- 52. Qi Y, Volmer DA. Electron‐based fragmentation methods in mass spectrometry: an overview: ExD IN MS. Mass Spectrom Rev. 2017; 36:4–15. [DOI] [PubMed] [Google Scholar]
- 53. Nikolskiy I, Mahieu NG, Chen Y‐J, Tautenhahn R, Patti GJ. An untargeted metabolomic workflow to improve structural characterization of metabolites. Anal Chem. 2013; 85:7713–7719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bateman KP, Castro‐Perez J, Wrona M, et al. MSE with mass defect filtering for in vitro and in vivo metabolite identification. Rapid Commun Mass Spectrom. 2007; 21:1485–1496. [DOI] [PubMed] [Google Scholar]
- 55. Simons B, Kauhanen D, Sylvänne T, Tarasov K, Duchoslav E, Ekroos K. Shotgun lipidomics by sequential precursor ion fragmentation on a hybrid quadrupole time‐of‐flight mass spectrometer. Metabolites. 2012; 2:195–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Berendsen BJA, Wegh RS, Meijer T, Nielen MWF. The assessment of selectivity in different quadrupole‐orbitrap mass spectrometry acquisition modes. J Am Soc Mass Spectrom. 2015; 26:337–346. [DOI] [PubMed] [Google Scholar]
- 57. Scheidweiler KB, Jarvis MJY, Huestis MA. Nontargeted SWATH acquisition for identifying 47 synthetic cannabinoid metabolites in human urine by liquid chromatography‐high‐resolving power tandem mass spectrometry. Anal Bioanal Chem. 2015; 407:883–897. [DOI] [PubMed] [Google Scholar]
- 58. Sheldon MT, Mistrik R, Croley TR. Determination of ion structures in structurally related compounds using precursor ion fingerprinting. J Am Soc Mass Spectrom. 2009; 20:370–376. [DOI] [PubMed] [Google Scholar]
- 59. Rojas‐Cherto M, Peironcely JE, Kasper PT, et al. Metabolite identification using automated comparison of high‐resolving power multistage mass spectral trees. Anal Chem. 2012; 84:5524–5534. [DOI] [PubMed] [Google Scholar]
- 60. Smith R, Loo J, Barinaga C, Edmonds C, Udseth H. Collisional activation and collision‐activated dissociation of large multiply charged polypeptides and proteins produced by electrospray ionization. J Am Soc Mass Spectrom. 1990; 1:53–65. [DOI] [PubMed] [Google Scholar]
- 61. Siegel MM, Huang J, Lin B, Tsao R, Edmonds CG. Structures of bacitracin A and isolated congeners: sequencing of cyclic peptides with blocked linear side chains by electrospray ionization mass spectrometry. Biol Mass Spectrom. 1994; 23:186–204. [DOI] [PubMed] [Google Scholar]
- 62.Koppen V, Mortishire‐Smith R, Cuyckens F. Multi‐stage mass Sspectrometry up to MS4 on a QTof system. Waters application note 2014. APNT134814078.
- 63. McLafferty FW, Turecek F. Interpretation of Mass Spectra. Herndon VA: University Science Books; 1993. p. 371. [Google Scholar]
- 64. Levsen K, Schiebel H‐M, Terlouw JK, et al. Even‐electron ions: a systematic study of the neutral species lost in the dissociation of quasi‐molecular ions. J Mass Spectrom. 2007; 42:1024–1044. [DOI] [PubMed] [Google Scholar]
- 65. Holcapek M, Jirásko R, Lísa M. Basic rules for the interpretation of atmospheric pressure ionization mass spectra of small molecules. J Chromatogr A. 2010; 1217:3908–3921. [DOI] [PubMed] [Google Scholar]
- 66. Weissberg A, Dagan S. Interpretation of ESI(+)‐MS‐MS spectra—towards the identification of “unknowns.” Int J Mass Spectrom. 2011; 299:158–168. [Google Scholar]
- 67. Niessen WMA. Fragmentation of toxicologically relevant drugs in negative‐ion liquid chromatography‐tandem mass spectrometry. Mass Spectrom Rev. 2012; 31:626–665. [DOI] [PubMed] [Google Scholar]
- 68. Zhou SL, Cook KD. Protonation in electrospray mass spectrometry: wrong‐way‐round or right‐way‐round? J Am Soc Mass Spectrom. 2000; 11:961–966. [DOI] [PubMed] [Google Scholar]
- 69. Ehrmann BM, Henriksen T, Cech NB. Relative importance of basicity in the gas phase and in solution for determining selectivity in electrospray ionization mass spectrometry. J Am Soc Mass Spectrom. 2008; 19:719–728. [DOI] [PubMed] [Google Scholar]
- 70. Cheng C, Gross ML. Applications and mechanisms of charge‐remote fragmentation. Mass Spectrom Rev. 2000; 19:398–420. [DOI] [PubMed] [Google Scholar]
- 71. Thurman EM, Ferrer I, Pozo OJ, Sancho JV, Hernandez F. The even‐electron rule in electrospray mass spectra of pesticides. Rapid Commun Mass Spectrom. 2007; 21:3855–3868. [DOI] [PubMed] [Google Scholar]
- 72. Niessen WMA. Fragmentation of toxicologically relevant drugs in positive‐ion liquid chromatography‐tandem mass spectrometry. Mass Spectrom Rev. 2011; 30:626–663. [DOI] [PubMed] [Google Scholar]
- 73. Tsugawa H, Kind T, Nakabayashi R, et al. Hydrogen rearrangement rules: computational MS/MS fragmentation and structure elucidation using MS‐FINDER software. Anal Chem. 2016; 88:7946–7958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bijlsma L, Sancho JV, Hernández F, Niessen WMA. Fragmentation pathways of drugs of abuse and their metabolites based on QTOF MS/MS and MS(E) accurate‐mass spectra. J Mass Spectrom JMS. 2011; 46:865–875. [DOI] [PubMed] [Google Scholar]
- 75. Niessen WMA. Group‐specific fragmentation of pesticides and related compounds in liquid chromatography‐tandem mass spectrometry. J Chromatogr A. 2010; 1217:4061–4070. [DOI] [PubMed] [Google Scholar]
- 76. Dillen L, Sips L, de Vries R, et al. Quantitative LC‐MS/MS analysis of azide and azidoalanine in in vitro samples following derivatisation with dansyl chloride. Anal Methods. 2013; 5:3136–3141. [Google Scholar]
- 77. Wang Z, Hop CECA, Kim M‐S, Huskey S‐EW, Baillie TA, Guan Z. The unanticipated loss of SO2 from sulfonamides in collision‐induced dissociation. Rapid Commun Mass Spectrom. 2003; 17:81–86. [DOI] [PubMed] [Google Scholar]
- 78. Ewing MA, Glover MS, Clemmer DE. Hybrid ion mobility and mass spectrometry as a separation tool. J Chromatogr A. 2016; 1439:3–25. [DOI] [PubMed] [Google Scholar]
- 79. Kanu AB, Dwivedi P, Tam M, Matz L, Hill HH. Ion mobility‐mass spectrometry. J Mass Spectrom. 2008; 43:1–22. [DOI] [PubMed] [Google Scholar]
- 80. May JC, Goodwin CR, McLean JA. Ion mobility‐mass spectrometry strategies for untargeted systems, synthetic, and chemical biology. Curr Opin Biotechnol. 2015; 31:117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhong Y, Hyung S‐J, Ruotolo BT. Ion mobility‐mass spectrometry for structural proteomics. Expert Rev Proteomics. 2012; 9:47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lapthorn C, Pullen F, Chowdhry BZ. Ion mobility spectrometry‐mass spectrometry (IMS‐MS) of small molecules: separating and assigning structures to ions. Mass Spectrom Rev. 2013; 32:43–71. [DOI] [PubMed] [Google Scholar]
- 83. Cuyckens F, Wassvik C, Mortishire‐Smith RJ, Tresadern G, Campuzano I, Claereboudt J. Product ion mobility as a promising tool for assignment of positional isomers of drug metabolites. Rapid Commun Mass Spectrom. 2011; 25:3497–3503. [DOI] [PubMed] [Google Scholar]
- 84. Eckers C, Laures AM‐F, Giles K, Major H, Pringle S. Evaluating the utility of ion mobility separation in combination with high‐pressure liquid chromatography/mass spectrometry to facilitate detection of trace impurities in formulated drug products. Rapid Commun Mass Spectrom. 2007; 21:1255–1263. [DOI] [PubMed] [Google Scholar]
- 85. Liu DQ, Wu L, Sun M, MacGregor PA. On‐line H/D exchange LC‐MS strategy for structural elucidation of pharmaceutical impurities. J Pharm Biomed Anal. 2007; 44:320–329. [DOI] [PubMed] [Google Scholar]
- 86. Liu DQ, Hop C. Strategies for characterization of drug metabolites using liquid chromatography‐tandem mass spectrometry in conjunction with chemical derivatization and on‐line H/D exchange approaches. J Pharm Biomed Anal. 2005; 37:1–18. [DOI] [PubMed] [Google Scholar]
- 87. Halket JM, Zaikin VG. Review: derivatization in mass spectrometry‐5. Specific derivatization of monofunctional compounds. Eur J Mass Spectrom. 2005; 11:127–160. [DOI] [PubMed] [Google Scholar]
- 88. Xu F, Zou L, Liu Y, Zhang Z, Ong CN. Enhancement of the capabilities of liquid chromatography‐mass spectrometry with derivatization: general principles and applications. Mass Spectrom Rev. 2011; 30:1143–1172. [DOI] [PubMed] [Google Scholar]
- 89. Zaikin V, Halket J. A Handbook of Derivatives for Mass Spectrometry. Chichester: IM Publications LLP; 2009. p. 543. [Google Scholar]
- 90. Neumann S, Böcker S. Computational mass spectrometry for metabolomics: identification of metabolites and small molecules. Anal Bioanal Chem. 2010; 398:2779–2788. [DOI] [PubMed] [Google Scholar]
- 91. Scheubert K, Hufsky F, Böcker S. Computational mass spectrometry for small molecules. J Cheminformatics. 2013; 5:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Hufsky F, Scheubert K, Böcker S. Computational mass spectrometry for small‐molecule fragmentation. TrAC Trends Anal Chem. 2014; 53:41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Hufsky F, Böcker S. Mining molecular structure databases: identification of small molecules based on fragmentation mass spectrometry data. Mass Spectrom Rev. 2017; 36:624–633. [DOI] [PubMed] [Google Scholar]
- 94. Smith CA, Want EJ, O'Maille G, Abagyan R, Siuzdak G. XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal Chem. 2006; 78:779–787. [DOI] [PubMed] [Google Scholar]
- 95. Pedrioli PG, Eng JK, Hubley R, et al. A common open representation of mass spectrometry data and its application to proteomics research. Nat Biotechnol. 2004; 22:1459–1466. [DOI] [PubMed] [Google Scholar]
- 96. Martens L, Chambers M, Sturm M, et al. MzML‐a community standard for mass spectrometry data. Mol Cell Proteomics. 2011; 10:R110.000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Chambers MC, Maclean B, Burke R, et al. A cross‐platform toolkit for mass spectrometry and proteomics. Nat Biotechnol. 2012; 30:918–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Stein S. Mass spectral reference libraries: an ever‐expanding resource for chemical identification. Anal Chem. 2012; 84:7274–7282. [DOI] [PubMed] [Google Scholar]
- 99. Lin M, Li M, Rustum A. Identification of an unknown extraneous contaminant in pharmaceutical product analysis. J Pharm Biomed Anal. 2007; 45:747–755. [DOI] [PubMed] [Google Scholar]
- 100. Stein SE, Scott DR. Optimization and testing of mass spectral library search algorithms for compound identification. J Am Soc Mass Spectrom. 1994; 5:859–866. [DOI] [PubMed] [Google Scholar]
- 101. McLafferty FW, Hertel RH, Villwock RD. Probability based matching of mass spectra. Rapid identification of specific compounds in mixtures. Org Mass Spectrom. 1974; 9:690–702. [Google Scholar]
- 102. T'jollyn H, Boussery K, Mortishire‐Smith RJ, et al. Evaluation of three state‐of‐the‐art metabolite prediction software packages (Meteor, MetaSite, and StarDrop) through independent and synergistic use. Drug Metab Dispos Biol Fate Chem. 2011; 39:2066–2075. [DOI] [PubMed] [Google Scholar]
- 103. Leclercq L, Mortishire‐Smith RJ, Huisman M, Cuyckens F, Hartshorn MJ, Hill A. IsoScore: automated localization of biotransformations by mass spectrometry using product ion scoring of virtual regioisomers. Rapid Commun Mass Spectrom. 2009; 23:39–50. [DOI] [PubMed] [Google Scholar]
- 104. Wang C, He L, Li D, Bruschweiler‐Li L, Marshall AG, Bruschweiler R. Accurate identification of unknown and known metabolic mixture components by combining 3D NMR with FT‐ICR MS/MS. J Proteome Res. 2016; 16:3774–3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Trunzer M, Faller B, Zimmerlin A. Metabolic soft spot identification and compound optimization in early discovery phases using MetaSite and LC‐MS/MS validation. J Med Chem. 2009; 52:329–335. [DOI] [PubMed] [Google Scholar]
- 106. Pähler A, Brink A. Software aided approaches to structure‐based metabolite identification in drug discovery and development. Drug Discov Today Technol. 2013; 10:e207–e217. [DOI] [PubMed] [Google Scholar]
- 107. Kirchmair J, Göller AH, Lang D, et al. Predicting drug metabolism: experiment and/or computation? Nat Rev Drug Discov. 2015; 14:387–404. [DOI] [PubMed] [Google Scholar]
- 108. Vinaixa M, Schymanski EL, Neumann S, Navarro M, Salek RM, Yanes O. Mass spectral databases for LC/MS‐ and GC/MS‐based metabolomics: state of the field and future prospects. Trac‐Trends Anal Chem. 2016; 78:23–35. [Google Scholar]
- 109. Horai H, Arita M, Kanaya S, et al. MassBank: a public repository for sharing mass spectral data for life sciences. J Mass Spectrom. 2010; 45:703–714. [DOI] [PubMed] [Google Scholar]
- 110. Smith CA, O'Maille G, Want EJ, et al. METLIN: a metabolite mass spectral database. Ther Drug Monit. 2005; 27:747–751. [DOI] [PubMed] [Google Scholar]
- 111. Huang X, Chen Y‐J, Cho K, Nikolskiy I, Crawford PA, Patti GJ. X13CMS: global tracking of isotopic labels in untargeted metabolomics. Anal Chem. 2014; 86:1632–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Chen Y‐J, Mahieu NG, Huang X, et al. Lactate metabolism is associated with mammalian mitochondria. Nat Chem Biol. 2016; 12:937–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Cho K, Mahieu N, Ivanisevic J, et al. IsoMETLIN: a database for isotope‐based metabolomics. Anal Chem. 2014; 86:9358–9361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Milman BL, Zhurkovich IK. Mass spectral libraries: a statistical review of the visible use. Trac‐Trends Anal Chem. 2016; 80:636–640. [Google Scholar]
- 115. Oberacher H, Schubert B, Libiseller K, Schweissgut A. Detection and identification of drugs and toxicants in human body fluids by liquid chromatography‐tandem mass spectrometry under data‐dependent acquisition control and automated database search. Anal Chim Acta. 2013; 770:121–131. [DOI] [PubMed] [Google Scholar]
- 116. Zhang K, Wong JW, Yang P, et al. Protocol for an electrospray ionization tandem mass spectral product ion library: development and application for identification of 240 pesticides in foods. Anal Chem. 2012; 84:5677–5684. [DOI] [PubMed] [Google Scholar]
- 117. Wang M, Carver JJ, Phelan VV, et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat Biotechnol. 2016; 34:828–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Kind T, Fiehn O. Advances in structure elucidation of small molecules using mass spectrometry. Bioanal Rev. 2010; 2:23–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Wolf S, Schmidt S, Müller‐Hannemann M, Neumann S. In silico fragmentation for computer assisted identification of metabolite mass spectra. BMC Bioinformatics. 2010; 11:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Hill DW, Kertesz TM, Fontaine D, Friedman R, Grant DF. Mass spectral metabonomics beyond elemental formula: chemical database querying by matching experimental with computational fragmentation spectra. Anal Chem. 2008; 80:5574–5582. [DOI] [PubMed] [Google Scholar]
- 121. Heinonen M, Rantanen A, Mielikäinen T, et al. FiD: a software for ab initio structural identification of product ions from tandem mass spectrometric data. Rapid Commun Mass Spectrom. 2008; 22:3043–3052. [DOI] [PubMed] [Google Scholar]
- 122. Mak TD, Laiakis EC, Goudarzi M, Fornace AJ. MetaboLyzer: a novel statistical workflow for analyzing Postprocessed LC‐MS metabolomics data. Anal Chem. 2014; 86:506–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Ridder L, van der Hooft JJJ, Verhoeven S, de Vos RCH, van Schaik R, Vervoort J. Substructure‐based annotation of high‐resolving power multistage MS(n) spectral trees. Rapid Commun Mass Spectrom. 2012; 26:2461–2471. [DOI] [PubMed] [Google Scholar]
- 124. Dunn WB, Erban A, Weber RJM, et al. Mass appeal: metabolite identification in mass spectrometry‐focused untargeted metabolomics. Metabolomics. 2013; 9:44–66. [Google Scholar]
- 125. Gerlich M, Neumann S. MetFusion: integration of compound identification strategies. J Mass Spectrom. 2013; 48:291–298. [DOI] [PubMed] [Google Scholar]
- 126. Rasche F, Svatoš A, Maddula RK, Böttcher C, Böcker S. Computing fragmentation trees from tandem mass spectrometry data. Anal Chem. 2011; 83:1243–1251. [DOI] [PubMed] [Google Scholar]
- 127. Rasche F, Scheubert K, Hufsky F, et al. Identifying the unknowns by aligning fragmentation trees. Anal Chem. 2012; 84:3417–3426. [DOI] [PubMed] [Google Scholar]
- 128. Heinonen M, Shen H, Zamboni N, Rousu J. Metabolite identification and molecular fingerprint prediction through machine learning. Bioinformatics. 2012; 28:2333–2341. [DOI] [PubMed] [Google Scholar]
- 129. Allen F, Pon A, Wilson M, Greiner R, Wishart D. CFM‐ID: a web server for annotation, spectrum prediction and metabolite identification from tandem mass spectra. Nucleic Acids Res. 2014; 42:W94–W99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Shen H, Dührkop K, Böcker S, Rousu J. Metabolite identification through multiple kernel learning on fragmentation trees. Bioinformatics. 2014; 30:157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Dührkop K, Shen H, Meusel M, Rousu J, Böcker S. Searching molecular structure databases with tandem mass spectra using CSI:FingerID. Proc Natl Acad Sci USA. 2015; 112:12580–12585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Böcker S. Searching molecular structure databases using tandem MS data: are we there yet? Curr Opin Chem Biol. 2017; 36:1–6. [DOI] [PubMed] [Google Scholar]
- 133. Takáts Z, Wiseman JM, Gologan B, Cooks RG. Mass spectrometry sampling under ambient conditions with desorption electrospray ionization. Science. 2004; 306:471–473. [DOI] [PubMed] [Google Scholar]
- 134. Cody RB, Laramée JA, Durst HD. Versatile new ion source for the analysis of materials in open air under ambient conditions. Anal Chem. 2005; 77:2297–2302. [DOI] [PubMed] [Google Scholar]
- 135. Lubin A, Bajic S, Cabooter D, Augustijns P, Cuyckens F. Atmospheric pressure ionization using a high voltage target compared to electrospray ionization. J Am Soc Mass Spectrom. 2017; 28:286–293. [DOI] [PubMed] [Google Scholar]
- 136. Martens J, Koppen V, Berden G, Cuyckens F, Oomens J. Combined liquid chromatography‐infrared ion spectroscopy for identification of regioisomeric drug metabolites. Anal Chem. 2017; 89:4359–4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Ramautar R, Somsen GW, de Jong GJ. CE‐MS for metabolomics: developments and applications in the period 2014–2016. Electrophoresis. 2017; 38:190–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Boschmans J, Jacobs S, Williams JP, et al. Combining density functional theory (DFT) and collision cross‐section (CCS) calculations to analyze the gas‐phase behaviour of small molecules and their protonation site isomers. Analyst. 2016; 141:4044–4054. [DOI] [PubMed] [Google Scholar]
- 139. Alex A, Harvey S, Parsons T, Pullen FS, Wright P, Riley J‐A. Can density functional theory (DFT) be used as an aid to a deeper understanding of tandem mass spectrometric fragmentation pathways? Rapid Commun Mass Spectrom. 2009; 23:2619–2627. [DOI] [PubMed] [Google Scholar]
- 140. Tautenhahn R, Patti GJ, Rinehart D, Siuzdak G. XCMS online: a web‐based platform to process untargeted metabolomic data. Anal Chem. 2012; 84:5035–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]