

Abstract
The adoption of hydrogen atom transfer (HAT) in a photocatalytic approach, in which an excited catalyst is responsible for substrate activation, offers unique opportunities in organic synthesis, enabling the straightforward activation of R–H (R = C, Si, S) bonds in desired reagents. Either a direct strategy, based on the intrinsic reactivity of a limited number of photocatalysts in the excited state, or an indirect one, in which a photocatalytic cycle is used for the generation of a thermal hydrogen abstractor, can be exploited. This microreview summarizes the most recent advances (mainly from the last two years) in this rapidly developing area of research, collecting the selected examples according to the nature of the species promoting the HAT process. From the synthetic point of view, this area has led to the development of a plethora of strategies for C–C, C–Si, C–N, C–S, and C–halogen (particularly, fluorine) bond formation, as well as for oxidation reactions.
Keywords: Photocatalysis, Hydrogen transfer, Radical reactions, C–H activation, Visible light, Photochemistry
1. Introduction
Hydrogen atom transfer (HAT) is a chemical transformation consisting of the concerted movement of two elementary particles – a proton and an electron – between two substrates in a single kinetic step.1, 2 The net reaction is given in Equation (1).
| (1) |
Conceptually, HAT can be considered a subclass of the larger family of proton‐coupled electron transfer (PCET) processes, in which the proton and the electron move together, sharing the starting and the final orbitals.3 Several research groups are active in the development of suitable models to explain HAT and to predict the rates at which these processes will occur. As an example, a conceptual framework for HAT through a Marcus theory approach has been developed.4 Furthermore, it has been demonstrated that the valence bond (VB) diagram model is capable of evaluating hydrogen‐abstraction barriers and deriving general trends from accessible raw data, such as bond energies.5 Significantly, HAT represents a key step in a wide variety of chemical reactions, including the combustion of hydrocarbons and aerobic oxidations, and it is involved in several atmospheric phenomena as well. In biology, several metalloenzymes are known to operate through a HAT step, and the role of such processes in the destructive effects of reactive oxygen species (ROS) in vivo and in the mechanism of action of antioxidants is studied in depth.1, 2, 6, 7, 8
On the other hand, HAT represents unique opportunities in organic synthesis, allowing the straightforward activation of (aliphatic) R–H bonds, often in a highly selective fashion. Accordingly, it is possible to skip the introduction of a directing moiety into a substrate, and the reactivity can be tuned by suitable choice of reaction conditions (e.g., hydrogen abstractor, solvent, etc.).9 Recently, photocatalysis10 has contributed significantly to the development of the field. In these reactions, an excited catalyst is responsible for the activation of the reaction partners (leading to the formation of reactive intermediates) and then reverts back to its original form, ready to start a new cycle. Upon light absorption, a photocatalyst is able to promote four different kinds of reactivity. The first is a direct HAT process,10, 11 in which the excited state of the photocatalyst (PC*) abstracts a hydrogen atom from a substrate R–H (pathway a in Scheme 1).
Scheme 1.
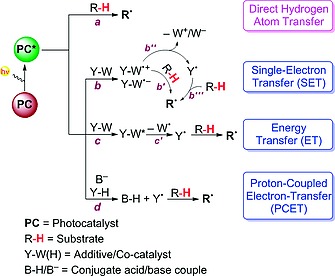
Substrate activation through a HAT step promoted through a direct (purple) or indirect (blue) photocatalytic approach.
Otherwise, PC* can be exploited to generate a thermal hydrogen abstractor by interaction with a suitable species Y–W(H), by three different mechanisms. The first possibility is to take advantage of the intrinsic capability of excited states to act as oxidants or reductants. Accordingly, PC* can promote a single‐electron‐transfer step (SET, pathway b), thus converting Y–W into the corresponding radical ion (either Y–W·+ or Y–W·–). This intermediate might be engaged directly in a HAT step with the substrate R–H (path b′) or might undergo the loss of a charged moiety (W+ or W–) to give a hydrogen‐abstractor species Y· (paths b′′ and b′′′). An alternative option involves an energy‐transfer step (ET, pathway c) between PC* and Y–W. The resulting excited species Y–W* then undergoes the homolytic cleavage of a labile bond, again generating a thermal hydrogen abstractor (Y·, path c′) prone to activate R–H through HAT. Finally, PC* can promote a PCET with an additive Y–H, also involving a suitable base (B–). As a result, radical Y· is formed, and is in turn able to promote the desired HAT step (pathway d).
In recent years, there has been an amazing development of photocatalytic approaches applied to organic synthesis, boosted by the exploitation of visible‐light‐absorbing transition‐metal complexes (e.g., based on Ru and Ir)12 and organic dyes,13 and this has led to a tremendous expansion of HAT‐based methodologies as well. Accordingly, the aim of this microreview is to provide an overview of the photocatalytic approaches available to activate an R–H bond through a HAT step. Thus, a classification to distinguish between direct and indirect methods is adopted, and the reported examples are collected according to the involved hydrogen abstractor. However, because many comprehensive reviews on photocatalysis have already been published,10, 12, 13, 14 the bias is on the most recent examples, mainly from 2014. Notwithstanding the very short period considered, more than 50 papers have been published in the field, clearly demonstrating the interest of photocatalysis practitioners in this topic.
Despite being, strictly speaking, related and often leading to the same net result, purely photocatalytic PCET strategies15 are not covered in this microreview. Similarly, examples in which the HAT event does not involve the actual substrate, but rather an intermediate, are not included. In that regard, here we mention the case of formation of iminium ions from amine radical cations16 or the recently reported example in which the HAT occurs within a photogenerated radical ion pair.17 Likewise, we do not consider processes in which a catalytic amount of a photoactive compound promotes a HAT step but is consumed during the reaction.18
2. Direct HAT Approaches
This approach is based on the capability of the excited state of a photocatalyst (PC*) to abstract a hydrogen atom directly from a substrate (Scheme 2).11 The catalytic cycle is then closed by means of a back‐HAT step (dashed arrow) to one of the intermediates formed during the process. Less commonly, a sequential electron/proton transfer operates.
Scheme 2.
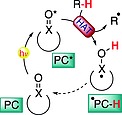
Typical reaction mechanism involving a direct HAT approach.
So far, the number of available photocatalysts able to perform this chemistry has been quite limited,11 being restricted to the families of aromatic ketones,19 polyoxometalates (in particular the decatungstate anion [W10O32]4–),20 and a few others, such as the uranyl cation [UO2]2+.21 In most cases, the states responsible for the observed reactivity share similar features, including the presence of a highly electrophilic oxygen site, mimicking the behavior of an alkoxy radical and enabling the desired HAT step.22
2.1. Activation by Aromatic Ketones
The capability of (aromatic) ketones to abstract a hydrogen atom upon irradiation has been known since the birth of photochemistry,23 and the first applications in the field of photocatalysis applied to organic synthesis date back to the mid‐1960s.[10b], [19a] The most recent examples involve the construction of C–C and C–N bonds, as well as oxidation and halogenation reactions.
In one instance, 4‐benzoylpyridine (4‐BzPy) was exploited to perform the conversion of a C(sp3)–H moiety into a C(sp3)–C(sp2) one through the introduction of an aldoxime function, resulting in a formal formylation procedure. This strategy has been successfully applied to ethers, thioethers, and protected amines. As an example, 1a–c were converted into adducts 3a–c in very good yields (>80 %; Scheme 3). Upon absorption of light, 4‐BzPy promoted a HAT to give radicals 4·a–c, which were trapped by 2, leading to adducts 5·a–c. The desired products 3a–c were then obtained upon elimination of sulfonyl radical 6·, which reacted with the reduced photocatalyst and gave sulfinic acid 7 as coupled product.24 The required amount of ketone, routinely employed in stoichiometric amount, is a significant limitation of this approach. However, it was shown that the system can also be optimized under catalytic conditions with use of 20 mol‐% of the aryl ketone.24
Scheme 3.
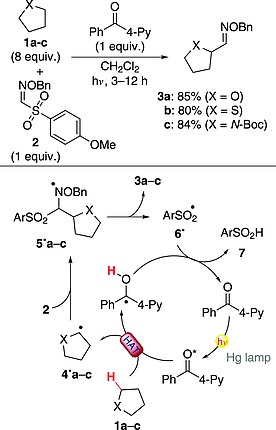
Introduction of an aldoxime function through HAT.
A similar approach was based on the use of 1,1‐bis(phenylsulfonyl)ethylene in the presence of 2‐chloroanthraquinone (10 mol‐%) as the photocatalyst. Thus, a variety of hydrogen donors, including hydrocarbons (alkanes, alkenes, and alkylaromatics), ethers, N‐Boc‐protected amines, thioethers, and alcohols, were smoothly functionalized. As a result, the final products each contained an active methine site CH(SO2Ph)2, highly suitable for further functionalization.25 In contrast, isomeric 1,2‐bis(phenylsulfonyl)ethylene enabled the direct alkenylation of C(sp3)–H bonds in ethers, alcohols, amides, and alkanes under irradiation in the presence of benzophenone (BP; 1 equiv.).26 A dual catalytic strategy for the carboxylation of allylic C–H bonds, promoted by a system composed of a xanthone derivative and a copper complex, has been reported. The key step involved a HAT from the allylic position by the excited xanthone, whereas the copper complex was involved in the carboxylation event.27
An elegant enantioselective alkynylation of C(sp3)–H bonds was performed by making recourse to a sulfoximine traceless chiral auxiliary group, lost at the end of the process. The reaction was carried out in the presence of BP under irradiation with a medium‐pressure lamp and followed a mechanism similar to that reported in Scheme 3. Thus, the α‐C–H bond in a protected amine (e.g., carbamate 8 in Scheme 4) was selectively functionalized through reaction with ethynylsulfoximine 9 to give 10 in 65 % yield with an enantiomeric ratio (er) of (S)/(R) = 84:16. The stereochemical output was interpreted on the basis of the preferential formation of transition state 11‡, in which the bulky groups adopted a pseudoequatorial arrangement, and a strong hydrogen bond between the S=N and the BocN–H moieties stabilized the structure. Also in this case the photocatalyst had to be used in large amounts (usually 3 equiv.), although the reaction worked to some extent (44 % yield) with a stoichiometric amount of BP.28 It is notable that a BP‐promoted stereoselective alkynylation strategy was adopted in the synthesis of (+)‐lactacystin, a potent inhibitor of the 20S proteasome.29
Scheme 4.
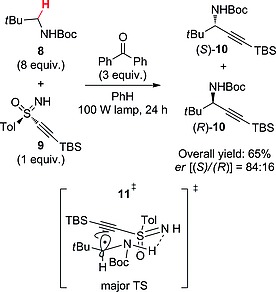
Enantioselective preparation of propargylamines (TBS = tert‐butyldimethylsilyl).
The hydrogen‐abstracting properties of excited benzophenone were likewise exploited to prepare a series of 2‐substituted benzoxazoles through C–C coupling between a hydrogen donor (e.g., an alcohol, an ether or a carbamate) and the chosen 2‐chlorobenzoxazole. Scheme 5 summarizes the reaction between THF (1a) and 2‐chlorobenzoxazole (12) to give 15. Thus, excited BP promoted the formation of 4·a, which was trapped by the heterocycle. Back‐HAT from ketyl radical to adduct 13· led to 14, which afforded the desired product 15 upon HCl elimination. The reaction was carried out with a 25 W energy‐saving UV‐A lamp under different reaction conditions. Indeed, 15 was obtained in 70 and 65 % yields when 100 and 25 mol‐%, respectively, of BP was used. Remarkably, the reaction can be scaled up to 18 mmol, and 2.48 g of 15 (73 % yield) were isolated after 60 h of irradiation (Scheme 5).30
Scheme 5.
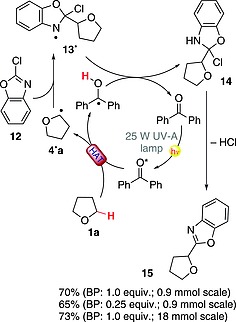
Metal‐free C–C coupling between 2‐chlorobenzoxazole and THF (BP = benzophenone).
Apart from C–C bond formation, HAT promoted by excited ketones can also be exploited to construct C–N bonds. A recent example involved the activation of formyl C(sp2)–H bonds in aliphatic (e.g., 16) and aromatic aldehydes by phenylglyoxylic acid (0.1 equiv.) and the ensuing addition to diisopropyl azodicarboxylate (17, DIAD; Scheme 6).31, 32 Interestingly, product 18 could be elaborated in a one‐pot fashion to prepare amide and hydroxamic acid derivatives by treatment with an amine31 or with hydroxylamine hydrochloride,32 respectively. As an example, products 19 31 and 20 32 were isolated in 95 and 89 % yields, respectively (Scheme 6). Remarkably, this chemistry has been successfully exploited for the synthesis of moclobemide31 (an antidepressant) and vorinostat32 (a compound employed for the treatment of cancer).
Scheme 6.
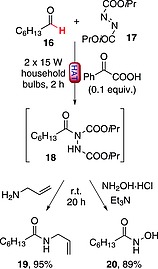
One‐pot approach to the synthesis of amides and hydroxamic acids from aldehydes.
In another instance, the preparation of alkyl azides through a C(sp3)–H to C–N bond conversion, making use of 4‐BzPy as the photocatalyst and tosyl azide in the role of an N3‐transfer agent, has been reported. The reaction was applied to tetrahydrofuran and pyrrolidine derivatives, as well as alkanes, and afforded the desired products in a single‐step fashion under mild reaction conditions.33
C–H activation in the α‐position of a primary alcohol offers an intriguing opportunity for selective oxidation. Indeed, it was shown that benzylic alcohols can be oxidized to the corresponding benzaldehydes under oxygen in the presence of sodium anthraquinone‐2‐sulfonate (AQNSS; 0.1 equiv.) in the role of a photocatalyst. Thus, excited AQNSS abstracted a hydrogen atom from each of 21a–c to give the corresponding oxidized derivatives 24a–c through the intermediacy of radicals 22·a–c and 23·a–c. Benzaldehydes 25a–c were then obtained through loss of hydrogen peroxide (Scheme 7).34 This reaction could be carried out in the presence of a chiral organocatalyst (e.g., hydroxyproline derivative 26) and a ketone (e.g., cyclohexanone), resulting in an overall photooxidative asymmetric aldol reaction. The process worked smoothly in water in the presence of acetic acid (0.5 equiv.), but required 48 h of irradiation with visible light. The final aldol adducts 27a–c were formed in good yields (ca. 60–70 %), with very good levels of diastereo‐ and enantioselectivity (Scheme 7).34
Scheme 7.
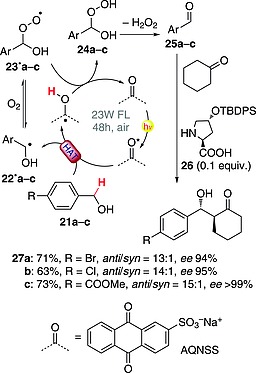
Photooxidative direct asymmetric aldol condensation starting from benzylic alcohols (FL = fluorescent lamp; TBDPS = tert‐butyldiphenylsilyl).
A related application involved the oxidation of cyclic acetals in the presence of anthraquinone‐2‐carboxylic acid under visible light irradiation conditions to give the corresponding hydroxyalkyl esters.35 The same photocatalyst could be also applied to the one‐pot epoxidation of alkenes. In detail, a peroxybenzoic acid was obtained in situ from the corresponding methyl‐substituted aromatic compound under aerobic photooxidative conditions and used directly in the desired epoxidation process.36 The photooxygenation of alkanes can be promoted by excited benzoquinone derivatives through a direct HAT in the presence of molecular oxygen. Indeed, this approach allowed the preparation of alkyl hydroperoxides, alcohols, and ketones under visible light irradiation with high quantum yields, although the starting alkane had to be used in large excess, in most cases as the solvent.37
Recently, the synthesis of α‐sulfonylated cyclic ethers has been achieved. This process involved a hydrogen atom abstraction from the ether promoted by excited benzophenone, whereas the sulfonyl unit was provided by sulfonyl chloride.38
Several research groups have recently been involved in the direct introduction of fluorine into unactivated derivatives. As an example, acetophenone was exploited to promote the fluorination of alkanes under irradiation with short‐violet light. The reaction occurred in acetonitrile with a low catalyst loading (5 mol‐%) and in the presence of the Selectfluor reagent as halogen source. As shown in Scheme 8, the fluorination of cyclooctane (28) proceeded readily (3 h irradiation) to afford the monofluorinated derivative 29 in a very good yield (82 %, NMR yield).39 A similar approach, based on the use of dibenzosuberenone (5 mol‐%), was adopted for C–H fluorination at the benzylic position in amino acids and short‐chain peptides, with a marked selectivity towards phenylalanine‐like residues.40 This chemistry could also easily be adapted to flow conditions and employed xanthone as an inexpensive photoorganocatalyst. Interestingly, in a case involving a biologically active molecule, the musk compound celestolide, a commonly utilized ingredient for fragrance compositions, was efficiently fluorinated at the benzylic position with a residence time of <10 min.41
Scheme 8.
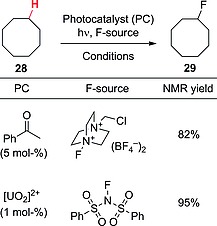
Photocatalytic fluorination of alkanes.
Apart from fluorination reactions, it has been shown that excited ketones (BP; 1 equiv.) can be exploited for the α‐chlorination of ethers. N,N‐Dimethylsulfamoyl chloride (Me2NSO2Cl) or CCl4 were conveniently used as halogen sources, and the resulting products could be used as synthetic intermediates for nucleophilic substitution processes, allowing the introduction of different oxygen‐, sulfur‐, and carbon‐based functionalities.42
2.2. Activation by Decatungstate Anion
Several polyoxometalates (POMs) – that is, inorganic cluster compounds containing oxygen and three or more metal centers – are known for their photochemical activity, and among them the decatungstate anion [W10O32]4– represents a unique case. It is often used as its tetrabutylammonium salt (TBADT) and, since the very first reports about its behavior under irradiation conditions,43 it has been exploited as photocatalyst in a number of synthetic procedures for the activation of organic substrates through HAT.20 Recent applications involve the formation of C–C and C–Si bonds, as well as dehydrogenation, oxidation, and fluorination reactions.
A library of γ‐lactones has been prepared with the aid of a flow apparatus made of PTFE tubing wrapped around a water‐cooled medium‐pressure Hg vapor lamp. This approach was based on a consecutive hydroacylation/reduction sequence starting from an aldehyde and an electron‐poor olefin in the presence of TBADT (1 mol‐%), with subsequent use of NaBH4 as reducing agent. Upon irradiation, the C(sp2)–H bond in heptanealdehyde (16) was cleaved by TBADT in its excited state to give acyl radical 30·. This reacted with diethyl maleate to give radical adduct 31· and then hydroacylated derivative 32. At the outlet of the photochemical reactor, the solution containing 32 was mixed with an NaBH4 solution in EtOH through a three‐way valve, promoting the reduction of the carbonyl group. Upon acidic workup, the resulting hydroxy ester underwent spontaneous cyclization to lactone 33 in an overall 65 % yield over two steps (Scheme 9).44 More recently, an upgraded version of this photochemical reactor has been reported, showing improved performance in terms of productivity and environmental sustainability, with a dramatic decrease in the process‐mass intensity (PMI) values (down to 10 kg kg–1) for the investigated processes with respect to the corresponding batch alternatives.45
Scheme 9.
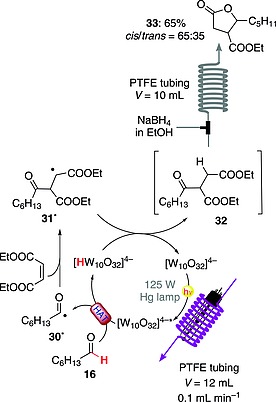
TBADT‐photocatalyzed synthesis of γ‐lactones.
Several reports have shown that excited decatungstate is particularly selective in the activation of C–H bonds, with a preference for nucleophilic hydrogen atoms. This behavior has recently been exploited in the functionalization of aliphatic nitriles, in which a reactivity trend following the general rule CH >> CH2 > CH3 has been observed. As an example, the reaction between isocapronitrile (34) and acrylonitrile in the presence of TBADT (4 mol‐%) led to the formation of adduct 35 as the only product (73 % yield; Scheme 10).46 Curiously, the decatungstate anion could be conveniently incorporated into the pores of a copper‐based metal‐organic framework (MOF). The resulting material functioned as a heterogeneous photocatalyst and afforded results comparable with those observed under homogeneous conditions (e.g., product 35 was isolated in 78 % yield; Scheme 10).47
Scheme 10.
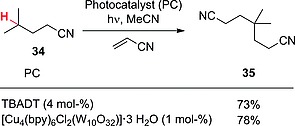
C–H to C–C conversion in aliphatic nitriles in the presence of decatungstate‐based photocatalysts (bpy = 4,4′‐bipyridine).
An asymmetric version of this chemistry based on the combined use of TBADT (5 mol‐%) and a chiral organocatalyst [(S,S)‐36, a primary amine containing a carbazolyl moiety, 20 mol‐%] has been reported. As detailed in Scheme 11, the process allowed the asymmetric construction of quaternary carbon systems from β‐substituted enones such as 37 and 1,3‐benzodioxole (38) through conjugate radical addition. Excited decatungstate allowed the generation of α,α‐dioxy radical 39·, which was in turn trapped by the iminium ion formed in situ through condensation of the organocatalyst (S,S)‐36 with 37. The thus‐formed intermediate 40·+ finally led to 41 upon restoration of the photocatalyst. Overall, the desired product 42 was formed with excellent enantiomeric excess (93 % ee) and in a good yield (75 %).48
Scheme 11.
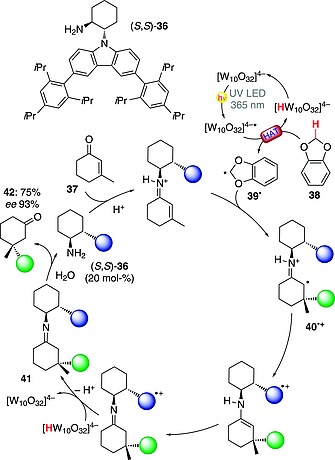
Asymmetric conjugate radical addition onto enones aided by iminium ion organocatalysis.
As well as in hydroalkylation reactions, the characteristic reactivity of electron‐poor olefins has also been exploited in hydrosilylation reactions. When TBADT (2 mol‐%) and trisubstituted silanes as hydrogen donors were used, clean activation of the Si–H bond resulted with aromatic derivatives, whereas competitive C–H versus Si–H functionalization was observed with trialkylsilanes. As an example, dimethyl(phenyl)silane (43) added to phenyl vinyl sulfone (44) to give adduct 45 in 80 % yield (Scheme 12; the mechanism is analogous to that depicted in Scheme 9). Interestingly, this protocol also worked under sunlight irradiation.49
Scheme 12.
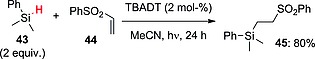
TBADT‐photocatalyzed hydrosilylation of electron‐poor olefins.
Alkanes were dehydrogenated through two different HAT steps by making use of a two‐catalyst system, in a reaction that has been defined as a cooperative HAT (cHAT) strategy. The system consisted of TBADT (0.4 mol‐%) and cobaloxime pyridine chloride {[Co(dmgH)2(pyr)Cl], COPC; 0.4 mol‐%} and worked under irradiation with UV light. Thus, excited TBADT was able to activate a strong C(sp3)–H bond in cyclooctane (28) to give radical 47·, in what has been called a “hard HAT” step. Subsequently, 47· underwent a second HAT (from the α‐position with respect to the radical site) promoted by COPC. The latter event has been termed an “easy HAT”, in view of the low BDE of the involved C–H bond. The catalytic system was regenerated through interaction between the reduced forms of the two catalysts, with release of a molecule of H2. Overall, the product cyclooctene (46) was obtained in 19 % yield upon irradiation for 48 h, demonstrating that the system worked in a catalytic fashion without the need for an added hydrogen acceptor (Scheme 13). Furthermore, the protocol was extended to (secondary) alcohols, which delivered the corresponding carbonyl derivatives in good preparative yields (up to 83 %).50
Scheme 13.
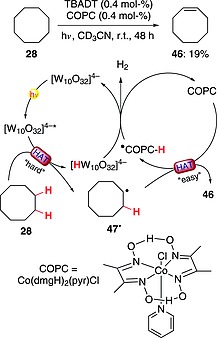
Dehydrogenation of alkanes in the presence of TBADT–cobaloxime pyridine chloride cooperative HAT strategy.
The decatungstate anion (1 mol‐%) was also exploited for the oxidation of different para‐substituted aryl alcohols to the corresponding acetophenones. The reaction proceeded smoothly with exceptionally high activity under UV/Vis irradiation and used molecular oxygen as a “green” oxidant. As an example, acetophenone derivative 49 was obtained with a very high selectivity (>99 %, 82 % conversion) from 48 after only 90 min (Scheme 14). Interestingly, even though a purely HAT mechanism has been proposed for TBADT in the activation of the benzyl C–H bond, a mixed mechanism involving both HAT and SET (with a predominance of the latter) was indicated when the decatungstate anion was supported over mesoporous TiO2 nanoparticles.51
Scheme 14.
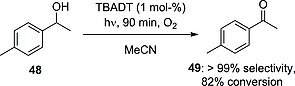
Oxidation of alcohols to ketones with O2 in the presence of TBADT.
When irradiated in the presence of N‐fluorobenzenesulfonimide (NFSI), decatungstate‐based salts proved also to be a convenient choice for replacing a hydrogen atom with a fluorine one. In particular, TBADT was exploited for the efficient fluorination of benzylic C–H bonds, and the protocol could readily be adapted to the use of flow conditions. An MeCN solution containing ibuprofen methyl ester (50), NaHCO3 (1 equiv.), NFSI (2 equiv.), and TBADT (2 mol‐%) was circulated in fluorinated ethylene‐propylene (FEP) tubing wrapped around a black‐light bulb (λ = 365 nm) to give fluorinated derivative 52 in 70 % yield upon irradiation for 5 h (a much shorter time than required under batch conditions). The process involved a HAT from 50 to give radical 51·, which in turn reacted with NFSI. The resulting nitrogen‐centered radical 53· was then involved in the restoration of the original photocatalyst (Scheme 15).52 A similar procedure (employing decatungstate sodium salt, Na4[W10O32]) was adopted in the synthesis of γ‐fluoroleucine methyl ester, a key intermediate in the route to odanacatib, a potent inhibitor of cathepsin K. Also in this case, the synthesis was carried out under flow conditions and could easily be scaled up to the preparation of ca. 45 g of product (90 % yield).53
Scheme 15.
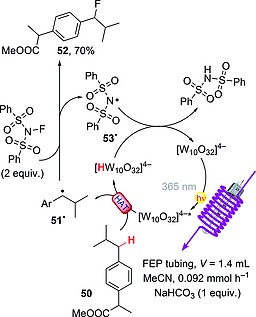
Fluorination of benzylic C–H bonds with NFSI aided by TBADT photocatalysis.
2.3. Activation by Uranyl Cation
Very recently, uranyl cation [UO2]2+ has been reported to act as a visible‐light‐absorbing photocatalyst for HAT processes, being applied to the fluorination of cyclooctane (28) in the presence of NFSI and allowing fluorocyclooctane (29) to be obtained in excellent yield (>95 %, NMR yield; see Scheme 8, above).21
3. Indirect HAT Approaches
In recent years, the limited number of photocatalysts able to perform direct HAT upon excitation has paved the way for the development of indirect strategies, in which a purposely added species Y–W(H) generates a thermal hydrogen abstractor upon activation by the excited photocatalyst (see Scheme 1 above). The Y–W(H) species may either be consumed during the process, thus acting as a stoichiometric additive, or behave catalytically, resulting in a dual‐catalytic approach.14
3.1. Activation by Carbon‐Based Abstractors
Carbon‐based abstractors have been generated through SET events promoted by (excited) photocatalysts. The use of additives of formula X3C–Br (with X = Cl or Br) is quite general, and the desired HAT is promoted by the corresponding X3C· species. A recent report highlighted the fact that hypervalent alkyl silicates can also be used for the generation of alkyl radicals, which are in turn capable of behaving as hydrogen abstractors. With regard to the applications of this methodology, carbon‐based abstractors have mainly been exploited for the construction of C–S bonds or for C–H to C–Br bond conversion.
A photocatalytic version of the thiol‐ene reaction, requiring the use of an RuII complex (1 mol‐%), Cl3CBr (5 mol‐%), and sodium ascorbate, has been proposed. The process involved reductive quenching of the excited RuII complex by sodium ascorbate, acting as electron donor. The resulting RuI species was then engaged in the monoelectronic reduction of Cl3CBr to give the trichloromethyl radical. The substrate methyl thioglycolate (54) was in turn activated through a HAT step promoted by Cl3C·, thus affording 57· and then 58· upon trapping by the olefin 55. The final product 56 (80 % yield) was then obtained through reaction with another molecule of 54, thus starting a radical chain (Scheme 16). Rigorously speaking, this reaction system should be considered a photocatalytic approach for the initiation of a thiol‐ene process. This is also confirmed by the use of Cl3CBr additive in substoichiometric quantity, despite it being consumed during the process.54 Recently, a variant of this approach involving the heterogeneous photocatalyst bismuth oxide (Bi2O3; 1–3 mol‐%) was adopted for the (gram‐scale) synthesis of different active pharmaceutical agents.55
Scheme 16.
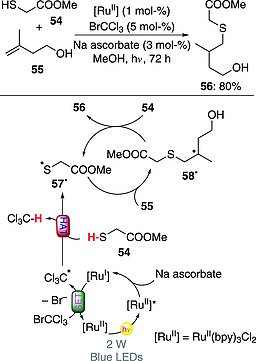
Photocatalytic initiation of a thiol‐ene reaction (bpy = 2,2′bipyridine).
The synthesis of aromatic thioethers from thiols and (hetero)aryl bromides has been carried out through a dual‐catalytic approach (based on an RuII photoredox catalyst and an NiII complex) and required the use of a hypervalent alkyl silicate. As depicted in Scheme 17, the process involved the monoelectronic oxidation of hypervalent alkyl silicate 61– [used as its (iPr)2NH2 + ammonium salt] to afford alkyl radical 63·. This abstracted a hydrogen atom from thiol 60, and the resulting sulfur‐centered radical 64· was intercepted by an Ni0 complex (formed in situ through reduction of the employed NiII salt). Oxidative addition of the thus‐formed NiI species onto aryl bromide 59 led to NiIII complex 65. The desired product 62 (94 % yield) was finally obtained by reductive elimination. The two catalytic cycles were finally closed through SET from RuI to NiI. Interestingly, an intramolecular version of this chemistry, based on the use of a thiol‐substituted alkyl silicate, was developed as well.56
Scheme 17.
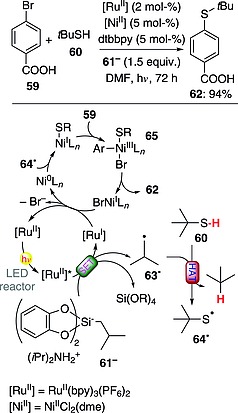
Dual‐catalytic synthesis of aromatic thioethers (dme = 1,2‐dimethoxyethane; dtbbpy = 4,4′‐di‐tert‐butyl‐2,2′‐bipyridine).
C–H to C–Br conversion at the benzylic position has recently been carried out with a photoredox catalyst (IrIII; 1 mol‐%) in the presence of CBr4 (1.5 equiv.). As shown in Scheme 18, the excited IrIII complex monoelectronically reduced CBr4, leading to the formation of the tribromomethyl radical. Hydrogen abstraction from 66 by Br3C· led to the formation of radical 69· and then of brominated derivative 67. The last step could occur either through the direct abstraction of a bromine atom from CBr4 or, less likely, through a two‐step mechanism involving an initial SET to the IrIV catalyst to give benzyl cation 69+, followed by trapping with Br–. Compound 67 was not isolated, but was treated instead with an excess of a nucleophile (e.g., morpholine, 20 equiv.) to give the corresponding benzylic amine 68 (80 % yield).57 A similar approach was exploited for the conversion of alkyl benzyl ethers into alkyl esters or alkyl alcohols.58
Scheme 18.
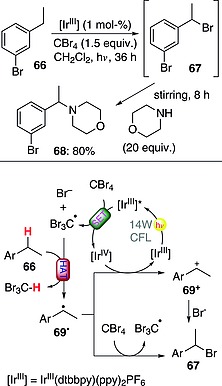
Photocatalytic bromination at the benzylic position (ppy = 2‐phenylpyridine).
3.2. Activation by Nitrogen‐Based Abstractors
Hydrogen abstraction can also be promoted by photocatalytically generated nitrogen‐centered radical cations or radicals. As in the former case, the generation of the desired abstracting species involves the monoelectronic oxidation of a tertiary amine. To drive the reactivity towards HAT and to prevent competitive pathways typical of amine radical cations (i.e., deprotonation from the α‐position),[19b] only selected types of precursors were employed, including triarylamines and bicyclic amines, such as quinuclidine derivatives. In the latter case, only one example has been reported so far and involved a PCET between an excited photocatalyst and a secondary amide. Nitrogen‐based abstractors have been exploited in different synthetic protocols for the construction of C–C (through cross‐coupling or Michael addition) and C–S (through a thiol‐ene process) bonds, whereas a single example involved the formation of a C–Br bond.
A triple catalytic system was employed to achieve C–H arylation of protected amines (carbamates), ureas, ethers, and alkylaromatics (e.g., p‐xylene). The protocol involved the use of a photoredox catalyst (an IrIII complex; 1 mol‐%), an organocatalyst (3‐acetoxyquinuclidine, 71; 1.1. equiv.), and an NiII complex (NiIIBr2 and ligand 4,7‐dimethoxy‐1,10‐phenanthroline, both 1 mol‐%, were used). The proposed mechanism involved absorption of a photon by the IrIII complex prone to activate 71 through a monoelectronic oxidation. The thus‐formed radical cation 71·+ abstracted a hydrogen atom from N‐Boc‐pyrrolidine (1c) to give radical 4·c. An active Ni0 species formed in situ underwent oxidative addition onto aryl bromide 70 and then gave alkyl(aryl)NiIII complex 73 upon interception of 4·c. The desired C–C coupling event occurred upon reductive elimination from 73 and led to the formation of product 72 (81 % yield) and of an NiI species. In the last step, the Ni0 species was regenerated through a SET with the reduced photoredox catalyst (Scheme 19).59
Scheme 19.
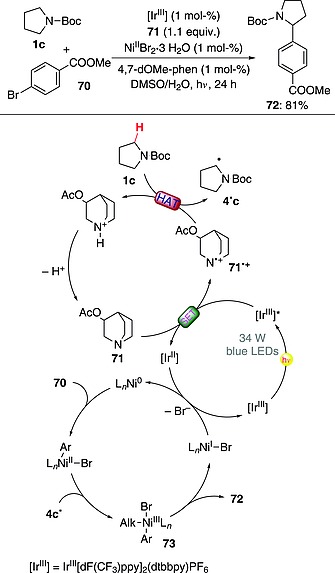
Photocatalytic C–H arylation in protected amines [dF(CF3)ppy = 2‐(2,4‐difluorophenyl)‐5‐(trifluoromethyl)pyridine; 4,7‐dOMe‐phen = 4,7‐dimethoxy‐1,10‐phenanthroline].
Similarly, C‐alkylation of alcohols with electron‐poor olefins (e.g., methyl acrylate) was performed by making recourse to a system based on a photoredox catalyst (an IrIII complex) and an organocatalyst (quinuclidine), in the presence of tetrabutylammonium phosphate. The key HAT activation step was promoted by quinuclidine radical cation (for a related example, see Scheme 19), that was able to activate the α‐C–H position of alcohols selectively, also thanks to the complexation of the O–H group by the phosphate anion.60
Very recently, it was demonstrated that the N–H bond in a secondary N‐alkyl amide can be activated through a concerted PCET mechanism involving an excited IrIII complex (2 mol‐%) and a base (tetrabutylammonium dibutyl phosphate; 5 mol‐%) to give the corresponding nitrogen‐centered radical. This operated as a hydrogen abstractor in intra‐ (resulting in a variant of Hofmann–Löffler–Freytag rearrangement) or intermolecular fashion to generate carbon‐centered radicals in alkanes, ethers, and protected amines. Scheme 20 shows an example in which this strategy was exploited for the activation of cyclohexane (74) and ensuing addition onto electron‐poor olefin 75. Thus, the phosphate anion interacted with N‐ethyl‐4‐methoxybenzamide (76; 1 equiv.), and the resulting hydrogen‐bonded complex reacted with the excited IrIII complex to give amidyl radical 78· through a concerted PCET process. Amidyl radical 78· was in turn able to abstract a hydrogen atom from 74 to give cyclohexyl radical and then adduct 79· upon trapping by 75. The starting catalytic system was restored through reduction of 79· and subsequent protonation by phosphoric acid to give the final product 77 (69 % yield; Scheme 20).61
Scheme 20.
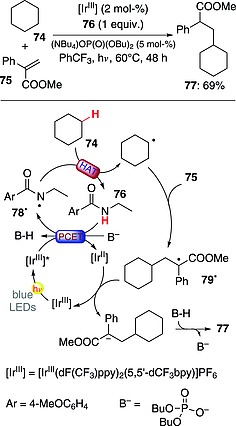
C–H activation in alkanes through the action of photogenerated amidyl radicals [5,5′‐dCF3bpy = 5,5′‐bis(trifluoromethyl)bipyridine].
A straightforward method for the construction of C–S bonds is through the thiol‐ene reaction. A recent report dealt with a photocatalytic variant of such approach. In particular, an RuII complex (0.25 mol‐%) was coupled with the use of an oxidative redox mediator, such as p‐toluidine (82; 0.5 equiv.). As depicted in Scheme 21, benzylmercaptan (80) added to cyclohexene (81) in 94 % yield. The mechanism involved an electron transfer between excited RuII and 82, leading to aniline radical cation 82·+ and then thiyl radical 84· upon HAT from 80 (a sequential electron‐proton transfer could not be completely ruled out, however). Sulfur‐centered radical 84· added to 81 to give a radical adduct that entered a chain reaction and finally led to the desired product 83. Restoration of the photoredox catalyst occurred thanks to the oxygen present in solution. Interestingly, the role of 82 as redox mediator was confirmed, because in its absence the reaction yield dropped to 16 %.62
Scheme 21.
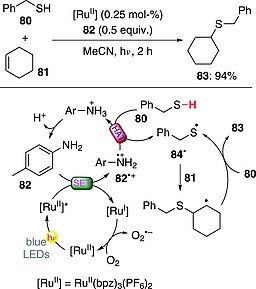
Photocatalytic thiol‐ene reaction (bpz = 2,2′‐bipyrazine).
Another application involved the selective bromination of C(sp3)–H bonds in aliphatic and benzylic derivatives. The reaction required the use of Eosin Y in the role of an inexpensive photocatalyst, CBr4 as the bromine source, and morpholine. The photogenerated N‐morpholino radical was responsible for the key hydrogen‐abstraction step, although the involvement of the Br3C· radical (formed by excited Eosin Y through monoelectronic reduction; see also Section 3.1, above) could not be excluded.63
3.3. Activation by Oxygen‐Based Abstractors
The generation of oxygen‐based abstractors usually requires an additive containing a labile O–X bond (with X = N, O). The activation step involves either an energy transfer or oxidative quenching of the excited state of the photocatalyst. Overall, the process leads to the formation of an oxygen‐centered radical, considered a model for the reactivity offered by excited photocatalysts operating through direct HAT chemistry (see Section 2, above). Indeed, oxygen‐based abstractors were exploited for the construction of C–C (through Michael‐type addition or radical aromatic substitution), C–S, and C–Cl bonds, as well as for oxidation reactions.
Some reports deal with the construction of C–C bonds through the combined use of a photocatalyst and tert‐butyl hydroperoxide (TBHP). This strategy has been exploited for the generation of different carbon‐centered radicals and their ensuing addition onto multiple bonds.64, 65, 66, 67, 68
One instance involved the activation of ethers and their addition onto alkynoates to give coumarin derivatives through a domino radical addition/cyclization sequence. The reaction mechanism is depicted in Scheme 22 and involved the activation of TBHP (4 equiv.) by the excited RuII photocatalyst (5 mol‐%) through a SET, leading to the formation of tert‐butoxyl radical. This was in turn responsible for the abstraction of a hydrogen atom from THF (1a) to give α‐oxy radical 4·a, which was trapped by alkynoate 85. The resulting adduct 87· underwent intramolecular cyclization and ensuing oxidation to give cation 88+. Product 86 (64 % yield) was finally obtained through deprotonation, helped by the hydroxide anion (Scheme 22).64 A variant of this approach involved the activation of the O–H bond in arylsulfinic acids, followed by addition onto phenyl propiolates, and resulted in the preparation of 3‐sulfonylated coumarins.65
Scheme 22.
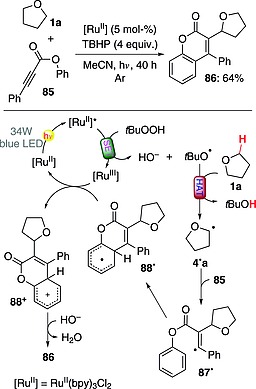
Photocatalytic synthesis of coumarins through a radical addition/cyclization sequence.
A similar strategy, based on Eosin Y (5 mol‐%) and TBHP (4 equiv.), was applied to the vinylation of THF with a series of alkynes. Interestingly, the proposed reaction mechanism involved an energy transfer between the excited photocatalyst and TBHP to generate the tert‐butoxyl radical, which was in turn responsible for the activation of THF.66 In another example, a library of α,β‐epoxy ketones was synthesized from styrenes and benzaldehydes in the presence of RuII(bpy)3Cl2 (2 mol‐%) and TBHP. The activation of the substrate is similar to that depicted in Scheme 22 and involved the cleavage of the formyl C–H bond in benzaldehydes promoted by the photogenerated tert‐butoxyl radical.67
An intramolecular asymmetric version of this C–H activation chemistry was also described. In particular, an IrIII complex (1 mol‐%) and an Rh‐based chiral Lewis acid catalyst (8 mol‐%) were adopted to promote a conjugate radical addition onto α,β‐unsaturated acylpyrazoles, leading to the desired products with high enantioselectivity (up to 97 % ee). As depicted in Scheme 23, the reaction required the use of an excess of Hantzsch ester (91; 1.5 equiv.), responsible for reductive quenching of the excited IrIII complex. The thus‐formed IrII species monoelectronically reduced N‐alkoxyphthalimide 89. The cleavage of the N–O bond in radical anion 89·– gave phthalimide (upon protonation by Hantzsch ester radical cation 91·+) and oxygen‐centered radical 94·, which in turn underwent an intramolecular 1,5‐HAT to give radical 95·. Trapping of this by a complex formed between α,β‐unsaturated acylpyrazole 90 and the Rh‐based chiral complex led, upon reaction with 93·, to the desired product 92 in good yield (81 %) and with excellent control of the stereochemistry (94 % ee; Scheme 23).68
Scheme 23.
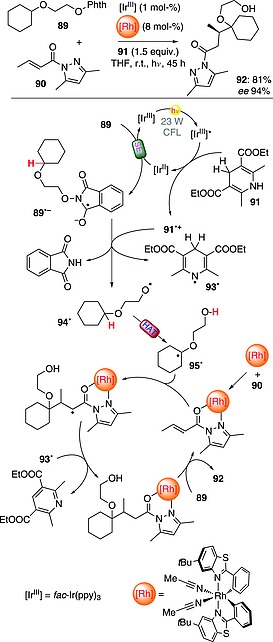
Catalytic asymmetric C–H functionalization through radical translocation.
Apart from addition onto multiple bonds, photocatalytically generated oxygen‐based abstractors were also exploited in the direct α‐arylation of ethers with heteroarenes in Minisci‐type coupling involving the construction of a C(sp3)–C(sp2) bond. In this case, an excited IrIII photoredox catalyst (2 mol‐%) monoelectronically reduced the persulfate anion (2 equiv. were required) to give a sulfate radical that was ultimately involved in the activation of the ether substrate.69
The metal‐free oxidation of benzylic alcohols to the corresponding carbonyl compounds under visible light irradiation conditions has recently been carried out. The proposed mechanism (Scheme 24) involved an energy transfer from excited Eosin Y to TBHP, which underwent a homolytic fragmentation. The thus‐formed tert‐butoxyl radical abstracted a hydrogen atom from the substrate (e.g., alcohol 96) to furnish the corresponding benzyl radical. The oxidized product 97 (95 % yield) was then formed through loss of a molecule of water aided by HO·. Notably, the reaction was applied both to primary and secondary alcohols and could also be adapted to a gram scale.70
Scheme 24.
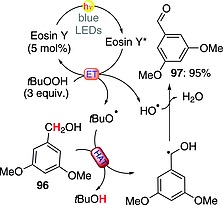
Metal‐free oxidation of benzylic alcohols.
A similar system was exploited for the preparation of α‐aryl thioethers from ethers and aromatic disulfides (Scheme 25). In this case, acridine red (2 mol‐%) under visible light irradiation conditions was responsible for the activation of TBHP, by a mechanism analogous to that shown in Scheme 24. Oxygen‐based abstractors tBuO· and HO· then allowed the activation of the chosen ether to give the corresponding α‐oxy radical, which was finally intercepted by the disulfide. As an example, 1,4‐dioxane (98) reacted with 99a–c to give adducts 100a–c in good yields (Scheme 25).71
Scheme 25.
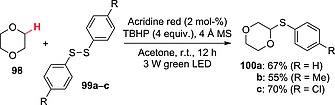
Photocatalytic synthesis of α‐aryl thioethers.
C–H to C–Cl conversion in aldehydes (e.g., 101a–c; Scheme 26) has been carried out by exploiting the reactivity of an RuII complex (1 mol‐%) under irradiation in the presence of TBHP (1.3 equiv.) and N‐chlorosuccinimide (3 equiv.) as Cl source. The reaction mechanism is similar to that reported in Scheme 22 and led, through the intermediacy of the corresponding benzoyl radicals, to the formation of acyl chlorides 102a–c. These were then converted into benzamides 103a–c in good to excellent yields by treatment with a suitable amine (e.g., benzylamine; Scheme 26). Interestingly, this procedure has been applied to the synthesis of the antidepressant moclobemide and of a D3 receptor intermediate.72
Scheme 26.
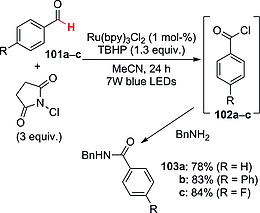
Synthesis of amides from aldehydes through in situ formation of acyl chlorides.
3.4. Activation by Sulfur‐Based Abstractors
The activation of an organic substrate with a sulfur‐based abstractor implies the use of a catalytic amount of a thiol. The activation of the S–H bond by the photocatalyst actually involves the formation of a thiyl radical through the formal removal of a hydrogen atom. According to the reports available in the literature, this may occur through a concerted PCET; otherwise the transfer of the electron and the proton may occur sequentially, depending on the reacting system considered. When the thus‐formed R–S· species promotes the desired HAT step, it is converted back into the original form so that it can be used catalytically. Indeed, this approach has been exploited for the introduction of a substituent onto a preformed aromatic ring and for radical addition onto multiple bonds (e.g., C=N), as well as for the construction of heterocycles.
One of the first examples of this chemistry involved the arylation of benzylic and allylic C–H bonds with cyanoaromatics in the presence of a photoredox catalyst and a thiol (methyl thioglycolate and triisopropylsilanethiol proved to be optimal choices).73, 74 An interesting example is reported in Scheme 27 and involves the arylation of 2,5‐dihydrofuran (104) by 1,4‐dicyanobenzene (105). The reaction system required the use of an IrIII photoredox catalyst (1 mol‐%), methyl thioglycolate (54; 20 mol‐%), octanal (1.2 equiv.), and a base (K2HPO4; 1 equiv.). The process involved oxidative quenching of the excited IrIII complex by 105 to give the corresponding radical anion. The resulting IrIV species was then involved in a PCET event (a stepwise electron‐proton transfer could be operative as well) with 54 to give 57·. This in turn abstracted a hydrogen atom from the allylic position in 104, affording radical 107· (in resonance with 107′·). The key C–C bond‐forming step occurred through a radical–radical anion coupling involving 107′· and 105· –, ultimately leading to the desired product 106 (82 % yield) as a single regioisomer through loss of a cyanide anion (scavenged by octanal).73 This chemistry was also exploited to perform the coupling of benzyl ethers with Schiff bases.75
Scheme 27.
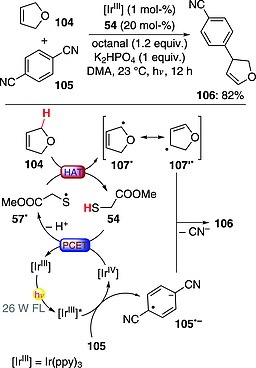
C–H arylation of 2,5‐dihydrofuran (DMA = N,N‐dimethylacetamide).
The use of thiols in combination with photoredox catalysis allowed the activation of the α‐position in alcohols and their use as alkylating agents in C–H functionalization in heteroarenes. Thus, methanol could be used as methylating agent for the functionalization of isoquinoline (108) in the presence of an IrIII complex (1 mol‐%) and an organocatalyst (ethyl 2‐mercaptopropionate, 109; 5 mol‐%). The process involved excitation of IrIII, which underwent a SET with a sacrificial amount of protonated 108. The resulting IrIV species was then engaged in the monoelectronic oxidation of 109 (followed by deprotonation), leading to thiyl radical 111·. Although the C–H bond in methanol is stronger than the S–H one in 109, 111· could activate CH3OH through a HAT step to give the hydroxymethyl radical, with regeneration of the starting thiol catalyst. Protonated isoquinoline (108‐H+) was then able to trap HOCH2 · to give the radical‐cationic adduct 112 ·+ and then intermediates 113· and 114· through a deprotonation and a spin center shift (accompanied by loss of a water molecule), respectively. The final product 110 (92 % yield) was then obtained by reduction (by excited IrIII) of benzyl radical 114· and protonation (Scheme 28).76
Scheme 28.
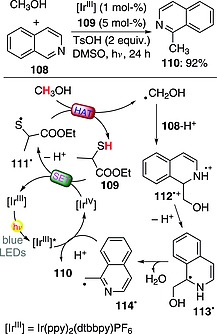
Methylation of isoquinoline by methanol.
A similar approach was adopted to promote the formation of nitrogen heterocycles of different sizes (four‐ to six‐membered), each starting from a substrate bearing a ketone and a tertiary amine moiety. The approach was again based on the use of an IrIII complex (1 mol‐%), a thiol (methyl thioglycolate, 54; usually 0.2 equiv.) and a base (K2HPO4; 0.2 equiv.). As mentioned above, this dual catalytic system allowed the formation of a thiyl radical, responsible for the activation of substrate 115 to give radical intermediate 116· and then the desired heterocycle 117 (83 % yield, dr = 10:1; Scheme 29).77
Scheme 29.
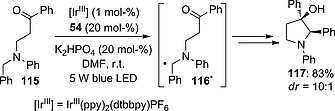
Construction of nitrogen heterocycles.
3.5. Activation by Halogen Atoms
In rare instances, a photocatalytically generated halogen atom has been exploited for substrate activation through a HAT step. In all cases chlorine or bromine atoms have been responsible for the desired hydrogen abstraction. This activation mode has been exploited for C–C and C–Cl bond‐forming reactions.
For instance, cross‐coupling between ethers (alkylaromatics and alkanes were suitable substrates as well) with aryl chlorides has been described. This involved the use of a dual catalytic system based on an IrIII photocatalyst (2 mol‐%) and an Ni0 complex (10 mol‐%) and also required the presence of a base (K3PO4; 2 equiv.) and a ligand (dtbbpy; 15 mol‐%). As shown in Scheme 30, the process involved the initial oxidative addition of the Ni0 complex onto aryl chloride 118 to give NiII intermediate 120. This was then oxidized to NiIII species 120+ through a SET promoted by the excited IrIII photoredox catalyst. NiIII species 120+ itself absorbed a photon, and this led to the formation of NiII species 121+ and of a chlorine atom, which in turn abstracted a hydrogen atom from 1a to give 4·a. The latter readily rebound to 121+, giving the new NiIII species 122+, which afforded the desired product 119 (80 % yield) upon reductive elimination. The two catalytic cycles were closed through a final SET event.78 In a related instance, cross‐coupling between aryl bromides and ethers was performed in the presence of an IrIII photocatalyst and an NiII salt. An alternative mechanistic scenario was proposed in this case, with the excited photocatalyst promoting an energy transfer step to an NiII complex, in turn leading to the formation of a bromine atom (involved in the key hydrogen‐abstraction step) through Ni–Br bond homolysis.79
Scheme 30.
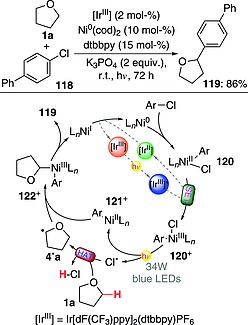
Cross‐coupling between ethers and aryl chlorides (cod = cycloocta‐1,5‐diene).
Chlorination of alkylarenes has been carried out in the presence of chloride anion and Ag nanoparticles. The reaction mechanism involved oxidation of the chloride anion to the corresponding radical, which was in turn able to abstract a hydrogen atom from the benzylic position of the chosen alkylaromatic. Overall, the process allowed benzyl chloride to be obtained from toluene with a very high selectivity (95 %) and with a good level of conversion (ca. 40 %).80
4. Conclusions
As can be seen in the reported examples, HAT processes promoted by photocatalytic approaches offer valuable opportunities in organic synthesis for the smooth activation of R–H (R = C, Si, S) bonds under mild conditions. The accessible intermediates according to the different strategies described in Sections 2 and 3 are collected in Table 1.
Table 1.
Radicals accessible through the proposed photocatalytic HAT steps
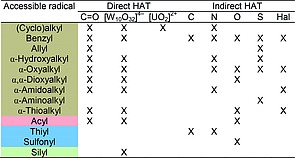
The direct approaches are long‐established methodologies and offer a large scope in terms of radical generation. With regard to the use of aromatic ketones, the amount of photocatalyst required, often (super)stoichiometric, is one of the main drawbacks. In contrast, the decatungstate anion can be used in small amounts (<5 mol‐%), and, most importantly, recent reports highlight that it can be heterogenized without loss of activity. Furthermore, this chemistry can also be adapted to flow conditions, allowing the desired processes to be performed on large scale. It should be noted that most of the reactions pertaining to the category of direct HAT processes are promoted by UV light. Nonetheless, recent trends involve the use of ketones that can work under visible light irradiation conditions and, in parallel, it has been demonstrated that solar light is a viable option for decatungstate photocatalysis. Due to the limited numbers of available alternatives, many efforts have been made to find new photocatalysts operating in this field, as demonstrated by the recent case of the uranyl cation.
In contrast, indirect methodologies have appeared in the literature only recently and, despite the fact that different types of hydrogen abstractors have been proposed, their scope is more limited than that of direct approaches in terms of substrate activation (see Table 1). The photocatalysts adopted to implement this methodology belong to the family of visible‐light‐absorbing derivatives, and most of the examples are based on the use of Ru or Ir complexes, as well as organic dyes. As a drawback, however, indirect approaches require the use of additives (sometimes used in catalytic amounts), thus making the overall protocol less appealing. Remarkably, different multicatalytic approaches in which thermal metal catalysts (e.g., based on Ni) are added to the reaction mixture have been developed, in turn giving access to a plethora of cross‐coupling processes.
On examination of the intermediates collected in Table 1, it appears that most of the radicals are nucleophilic in character (with the notable exception of a few sulfur‐based radicals). This is a general trend in photocatalytic HAT processes (either direct or indirect), and an interesting development in the future could be the exploitation of polarity‐reversal catalysis (PRC) principles, thus enabling the generation and use of electrophilic radicals.81 The development of HAT within the framework of photocatalytic strategies has seen impressive development in recent years, and this trend is expected to continue in the future, enabling synthesis practitioners to activate desired R–H bonds with high selectivity and efficiency (also in complex molecules; e.g., in a late‐stage functionalization process) through judicious choice of the optimal hydrogen‐abstraction protocol.
Acknowledgements
The authors thank Dr. Stefano Protti and Prof. Maurizio Fagnoni (University of Pavia) for fruitful discussions. D. R. thanks the Ministero dell'Università e della Ricerca (MIUR) for financial support (SIR Project “Organic Synthesis via Visible Light Photocatalytic Hydrogen Transfer”; Code: RBSI145Y9R).
Biographies
Luca Capaldo was born in Milano, Italy. He received his Master's degree in organic chemistry in 2015 under the guidance of Prof. Maurizio Fagnoni and Prof. Luisa De Cola, working on OLEDs, bioimaging, and superconductors. Later in the same year, he undertook his PhD project at the University of Pavia, focusing on the development of new photocatalytic approaches for sustainable synthesis.

Davide Ravelli obtained his PhD in 2012 from the University of Pavia with a thesis on decatungstate‐photocatalyzed processes applied to organic synthesis (Prof. A. Albini as supervisor). Since November 2015 he has been a fixed‐term researcher at the same university, and his main research interests focus on the generation of radical intermediates through photocatalyzed hydrogen atom transfer reactions and on the elucidation of organic reaction mechanisms by means of computational tools.

References
- 1. Hydrogen‐Transfer Reactions (Eds.: Hynes J. T., Klinman J. P., Limbach H.‐H. and Schowen R. L.), 2007. [Google Scholar]
- 2. Encyclopedia of Radicals in Chemistry, Biology and Materials (Eds.: Chatgilialoglu C. and Studer A.), John Wiley & Sons, Ltd, 2012. [Google Scholar]
- 3. Hammes‐Schiffer S. and Stuchebrukhov A. A., Chem. Rev., 2010, 110, 6939–6960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mayer J. M., Acc. Chem. Res., 2011, 44, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lai W., Li C., Chen H. and Shaik S., Angew. Chem. Int. Ed., 2012, 51, 5556–5578; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2012, 124, 5652–5676. [Google Scholar]
- 6. Ortiz de Montellano P. R., Chem. Rev., 2010, 110, 932–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Groves J. T. in: Cytochrome P450: Structure, Mechanism, and Biochemistry, 3rd ed. (Ed.: Ortiz de Montellano P. R.), Kluwer Academic/Plenum Publishers, New York, 2005, pp. 1–43; [Google Scholar]
- 8. Ingold K. U. and Pratt D. A., Chem. Rev., 2014, 114, 9022–9046. [DOI] [PubMed] [Google Scholar]
- 9. Salamone M. and Bietti M., Acc. Chem. Res., 2015, 48, 2895–2903. [DOI] [PubMed] [Google Scholar]
- 10.a) Ravelli D., Protti S. and Fagnoni M., Chem. Rev., 2016, 116, 9850–9913; [DOI] [PubMed] [Google Scholar]; b) Fagnoni M., Dondi D., Ravelli D. and Albini A., Chem. Rev., 2007, 107, 2725–2756. [DOI] [PubMed] [Google Scholar]
- 11. Protti S., Fagnoni M. and Ravelli D., ChemCatChem, 2015, 7, 1516–1523. [Google Scholar]
- 12.a) Prier C. K., Rankic D. A. and MacMillan D. W. C., Chem. Rev., 2013, 113, 5322–5363; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xi Y., Yi H. and Lei A., Org. Biomol. Chem., 2013, 11, 2387–2403; [DOI] [PubMed] [Google Scholar]; c) Reckenthäler M. and Griesbeck A. G., Adv. Synth. Catal., 2013, 355, 2727–2744; [Google Scholar]; d) Xuan J. and Xiao W.‐J., Angew. Chem. Int. Ed., 2012, 51, 6828–6838; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2012, 124, 6934–6944 [Google Scholar]; e) Narayanam J. M. R. and Stephenson C. R. J., Chem. Soc. Rev., 2011, 40, 102–113. [DOI] [PubMed] [Google Scholar]
- 13.a) Romero N. A. and Nicewicz D. A., Chem. Rev., 2016, 116, 10075–10166; [DOI] [PubMed] [Google Scholar]; b) Wei G., Basheer C., Tan C.‐H. and Jiang Z., Tetrahedron Lett., 2016, 57, 3801–3809. [Google Scholar]
- 14.a) Skubi K. L., Blum T. R. and Yoon T. P., Chem. Rev., 2016, 116, 10035–10074; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hopkinson M. N., Sahoo B., Li J.‐L. and Glorius F., Chem. Eur. J., 2014, 20, 3874–3886. [DOI] [PubMed] [Google Scholar]
- 15. Gentry E. C. and Knowles R. R., Acc. Chem. Res., 2016, 49, 1546–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a) Shi L. and Xia W., Chem. Soc. Rev., 2012, 41, 7687–7697; [DOI] [PubMed] [Google Scholar]; b) Bartling H., Eisenhofer A., König B. and Gschwind R. M., J. Am. Chem. Soc., 2016, 138, 11860–11871. [DOI] [PubMed] [Google Scholar]
- 17. Rusch F., Schober J.‐C. and Brasholz M., ChemCatChem, 2016, 8, 2881–2884. [Google Scholar]
- 18.a) Yadav V. K., Srivastava V. P. and Yadav L. D. S., Tetrahedron Lett., 2016, 57, 2502–2505; [Google Scholar]; b) Moteki S. A., Usui A., Selvakumar S., Zhang T. and Maruoka K., Angew. Chem. Int. Ed., 2014, 53, 11060–11064; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2014, 126, 11240–11244. [Google Scholar]
- 19.a) Ravelli D., Fagnoni M. and Albini A., Chem. Soc. Rev., 2013, 42, 97–113; [DOI] [PubMed] [Google Scholar]; b) Hoffmann N., Synthesis, 2016, 48, 1782–1802; [Google Scholar]; c) Oelgemöller M. and Hoffmann N., Pure Appl. Chem., 2015, 87, 569–582. [Google Scholar]
- 20.a) Ravelli D., Protti S. and Fagnoni M., Acc. Chem. Res., 2016, 49, 2232–2242; [DOI] [PubMed] [Google Scholar]; b) Tzirakis M. D., Lykakis I. N. and Orfanopoulos M., Chem. Soc. Rev., 2009, 38, 2609–2621. [DOI] [PubMed] [Google Scholar]
- 21. West J. G., Bedell T. A. and Sorensen E. J., Angew. Chem. Int. Ed., 2016, 55, 8923–8927; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem., 2016, 128, 9069–9073. [Google Scholar]
- 22. De Waele V., Poizat O., Fagnoni M., Bagno A. and Ravelli D., ACS Catal., 2016, 6, 7174–7182. [Google Scholar]
- 23. Albini A. and Dichiarante V., Photochem. Photobiol. Sci., 2009, 8, 248–254. [Google Scholar]
- 24. Kamijo S., Takao G., Kamijo K., Hirota M., Tao K. and Murafuji T., Angew. Chem. Int. Ed., 2016, 55, 9695–9699; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2016, 128, 9847–9851. [Google Scholar]
- 25. Kamijo S., Takao G., Kamijo K., Tsuno T., Ishiguro K. and Murafuji T., Org. Lett., 2016, 18, 4912–4915. [DOI] [PubMed] [Google Scholar]
- 26. Amaoka Y., Nagatomo M., Watanabe M., Tao K., Kamijo S. and Inoue M., Chem. Sci., 2014, 5, 4339–4345. [Google Scholar]
- 27. Ishida N., Masuda Y., Uemoto S. and Murakami M., Chem. Eur. J., 2016, 22, 6524–6527. [DOI] [PubMed] [Google Scholar]
- 28. Nagatomo M., Yoshioka S. and Inoue M., Chem. Asian J., 2015, 10, 120–123. [DOI] [PubMed] [Google Scholar]
- 29. Yoshioka S., Nagatomo M. and Inoue M., Org. Lett., 2015, 17, 90–93. [DOI] [PubMed] [Google Scholar]
- 30. Lipp A., Lahm G. and Opatz T., J. Org. Chem., 2016, 81, 4890–4897. [DOI] [PubMed] [Google Scholar]
- 31. Papadopoulos G. N. and Kokotos C. G., J. Org. Chem., 2016, 81, 7023–7028. [DOI] [PubMed] [Google Scholar]
- 32. Papadopoulos G. N. and Kokotos C. G., Chem. Eur. J., 2016, 22, 6964–6967. [DOI] [PubMed] [Google Scholar]
- 33. Kamijo S., Watanabe M., Kamijo K., Tao K. and Murafuji T., Synthesis, 2016, 48, 115–121. [Google Scholar]
- 34. Fujiya A., Nobuta T., Yamaguchi E., Tada N., Miura T. and Itoh A., RSC Adv., 2015, 5, 39539–39543. [Google Scholar]
- 35. Yamaguchi T., Kudo Y., Hirashima S.‐i., Yamaguchi E., Tada N., Miura T. and Itoh A., Tetrahedron Lett., 2015, 56, 1973–1975. [Google Scholar]
- 36. Taguchi M., Nagasawa Y., Yamaguchi E., Tada N., Miura T. and Itoh A., Tetrahedron Lett., 2016, 57, 230–232. [Google Scholar]
- 37. Ohkubo K., Hirose K. and Fukuzumi S., Photochem. Photobiol. Sci., 2016, 15, 731–734. [DOI] [PubMed] [Google Scholar]
- 38. Kamijo S., Hirota M., Tao K., Watanabe M. and Murafuji T., Tetrahedron Lett., 2014, 55, 5551–5554. [Google Scholar]
- 39. Xia J.‐B., Zhu C. and Chen C., Chem. Commun., 2014, 50, 11701–11704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bume D. D., Pitts C. R., Jokhai R. T. and Lectka T., Tetrahedron, 2016, 72, 6031–6036. [Google Scholar]
- 41. Cantillo D., de Frutos O., Rincón J. A., Mateos C. and Kappe C. O., J. Org. Chem., 2014, 79, 8486–8490. [DOI] [PubMed] [Google Scholar]
- 42. Kamijo S., Tao K., Takao G., Murooka H. and Murafuji T., Tetrahedron Lett., 2015, 56, 1904–1907. [Google Scholar]
- 43. Hill C. L., Synlett, 1995, 127–132. [Google Scholar]
- 44. Fagnoni M., Bonassi F., Palmieri A., Protti S., Ravelli D. and Ballini R., Adv. Synth. Catal., 2014, 356, 753–758. [Google Scholar]
- 45. Bonassi F., Ravelli D., Protti S. and Fagnoni M., Adv. Synth. Catal., 2015, 357, 3687–3695. [Google Scholar]
- 46. Yamada K., Okada M., Fukuyama T., Ravelli D., Fagnoni M. and Ryu I., Org. Lett., 2015, 17, 1292–1295. [DOI] [PubMed] [Google Scholar]
- 47. Shi D., He C., Sun W., Ming Z., Meng C. and Duan C., Chem. Commun., 2016, 52, 4714–4717. [DOI] [PubMed] [Google Scholar]
- 48. Murphy J. J., Bastida D., Paria S., Fagnoni M. and Melchiorre P., Nature, 2016, 532, 218–222. [DOI] [PubMed] [Google Scholar]
- 49. Qrareya H., Dondi D., Ravelli D. and Fagnoni M., ChemCatChem, 2015, 7, 3350–3357. [Google Scholar]
- 50. West J. G., Huang D. and Sorensen E. J., Nat. Commun., 2015, 6, 10093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Symeonidis T. S., Tamiolakis I., Armatas G. S. and Lykakis I. N., Photochem. Photobiol. Sci., 2015, 14, 563–568. [DOI] [PubMed] [Google Scholar]
- 52. Nodwell M. B., Bagai A., Halperin S. D., Martin R. E., Knust H. and Britton R., Chem. Commun., 2015, 51, 11783–11786. [DOI] [PubMed] [Google Scholar]
- 53. Halperin S. D., Kwon D., Holmes M., Regalado E. L., Campeau L.‐C., DiRocco D. A. and Britton R., Org. Lett., 2015, 17, 5200–5203. [DOI] [PubMed] [Google Scholar]
- 54. Keylor M. H., Park J. E., Wallentin C.‐J. and Stephenson C. R. J., Tetrahedron, 2014, 70, 4264–4269. [Google Scholar]
- 55. Fadeyi O. O., Mousseau J. J., Feng Y., Allais C., Nuhant P., Chen M. Z., Pierce B. and Robinson R., Org. Lett., 2015, 17, 5756–5759. [DOI] [PubMed] [Google Scholar]
- 56. Jouffroy M., Kelly C. B. and Molander G. A., Org. Lett., 2016, 18, 876–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hou T., Lu P. and Li P., Tetrahedron Lett., 2016, 57, 2273–2276. [Google Scholar]
- 58. Lu P., Hou T., Gu X. and Li P., Org. Lett., 2015, 17, 1954–1957. [DOI] [PubMed] [Google Scholar]
- 59. Shaw M. H., Shurtleff V. W., Terrett J. A., Cuthbertson J. D. and MacMillan D. W. C., Science, 2016, 352, 1304–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jeffrey J. L., Terrett J. A. and MacMillan D. W. C., Science, 2015, 349, 1532–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Choi G. J., Zhu Q., Miller D. C., Gu C. J. and Knowles R. R., Nature, 2016, 539, 268–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tyson E. L., Niemeyer Z. L. and Yoon T. P., J. Org. Chem., 2014, 79, 1427–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kee C. W., Chan K. M., Wong M. W. and Tan C.‐H., Asian J. Org. Chem., 2014, 3, 536–544. [Google Scholar]
- 64. Feng S., Xie X., Zhang W., Liu L., Zhong Z., Xu D. and She X., Org. Lett., 2016, 18, 3846–3849. [DOI] [PubMed] [Google Scholar]
- 65. Yang W., Yang S., Li P. and Wang L., Chem. Commun., 2015, 51, 7520–7523. [DOI] [PubMed] [Google Scholar]
- 66. Li J., Zhang J., Tan H. and Wang D. Z., Org. Lett., 2015, 17, 2522–2525. [DOI] [PubMed] [Google Scholar]
- 67. Li J. and Wang D. Z., Org. Lett., 2015, 17, 5260–5263. [DOI] [PubMed] [Google Scholar]
- 68. Wang C., Harms K. and Meggers E., Angew. Chem. Int. Ed., 2016, 55, 13495–13498; [DOI] [PubMed] [Google Scholar]; Angew. Chem., 2016, 128, 13693–13696. [Google Scholar]
- 69. Jin J. and MacMillan D. W. C., Angew. Chem. Int. Ed., 2015, 54, 1565–1569; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem., 2015, 127, 1585–1589. [Google Scholar]
- 70. Devari S., Rizvi M. A. and Shah B. A., Tetrahedron Lett., 2016, 57, 3294–3297. [Google Scholar]
- 71. Zhu X., Xie X., Li P., Guo J. and Wang L., Org. Lett., 2016, 18, 1546–1549. [DOI] [PubMed] [Google Scholar]
- 72. Iqbal N. and Cho E. J., J. Org. Chem., 2016, 81, 1905–1911. [DOI] [PubMed] [Google Scholar]
- 73. Qvortrup K., Rankic D. A. and MacMillan D. W. C., J. Am. Chem. Soc., 2014, 136, 626–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cuthbertson J. D. and MacMillan D. W. C., Nature, 2015, 519, 74–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hager D. and MacMillan D. W. C., J. Am. Chem. Soc., 2014, 136, 16986–16989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jin J. and MacMillan D. W. C., Nature, 2015, 525, 87–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Li W., Duan Y., Zhang M., Cheng J. and Zhu C., Chem. Commun., 2016, 52, 7596–7599. [DOI] [PubMed] [Google Scholar]
- 78. Shields B. J. and Doyle A. G., J. Am. Chem. Soc., 2016, 138, 12719–12722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Heitz D. R., Tellis J. C. and Molander G. A., J. Am. Chem. Soc., 2016, 138, 12715–12718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Liu S., Zhang Q., Li H., Yang Y., Tian X. and Whiting A., Chem. Eur. J., 2015, 21, 9671–9675. [DOI] [PubMed] [Google Scholar]
- 81. Roberts B. P., Chem. Soc. Rev., 1999, 28, 25–35. [Google Scholar]