

Summary
Background
The rs738409 C>G p.I148M variant in the patatin‐like phospholipase domain containing 3 (PNPLA3)‐gene promotes triglyceride accumulation in hepatocytes and hepatic stellate cell activation and has previously been linked to hepatic steatosis/liver fibrosis.
Aim
To investigate its impact on hepatic decompensation and (liver‐related) mortality in patients who had already developed portal hypertension. Moreover, we assessed its link with hepatic steatosis as evaluated by controlled attenuation parameter.
Methods
We performed a retrospective analysis in prospectively characterised patients with viral hepatitis/fatty liver disease‐induced portal hypertension (hepatic venous pressure gradient [HVPG] ≥ 6 mm Hg) diagnosed at the Medical University of Vienna who underwent HVPG measurement (until 2013; n = 372; longitudinal study) or simultaneous HVPG and controlled attenuation parameter measurement (2014‐2017; n = 153; cross‐sectional study).
Results
While survival was similar between PNPLA3‐C/C and ‐C/G patients, we observed substantially increased mortality in PNPLA3‐G/G patients. PNPLA3‐G/G had no impact on mortality in the subgroup of patients with viral hepatitis; however, we observed a strong independent association between PNPLA3‐G/G and hepatic decompensation (adjusted subdistribution hazard ratio [aSHR]: 2.1, 95% confidence interval [95% CI]: 1.1‐4; P = 0.024) as well as mortality (overall: aSHR: 2.2, 95% CI: 1.22‐3.98; P = 0.009; liver‐related: aSHR: 2.2, 95% CI: 1.08‐4.46; P = 0.029) in patients with fatty liver disease. Interestingly, even in the subgroup of patients who had already progressed to clinically significant portal hypertension (HVPG ≥ 10 mm Hg), PNPLA3‐G/G substantially increased mortality (aSHR: 2.33, 95% CI: 1.27‐4.29; P = 0.006). PNPLA3‐genotype had no influence on controlled attenuation parameter or the prevalence of values ≥248 dB/m.
Conclusion
PNPLA3‐G/G‐genotype seems to double the risks of hepatic decompensation and (liver‐related) mortality in patients with portal hypertension due to fatty liver disease. Further studies are warranted to investigate potential underlying pathophysiological mechanisms unrelated to hepatic steatosis.
1. INTRODUCTION
The patatin‐like phospholipase domain containing 3 (PNPLA3) protein is a lipase with activity towards triglycerides in hepatocytes1 as well as retinyl esters in hepatic stellate cells2 and experimental studies have shown that p.I148M substitution leads to a loss of function.3
The PNPLA3 rs738409 G allele encoding the I148M variant (Human Genom Variation Society Nomenclature: NC_000022.10:g.44324727C>G)4 has been linked to non‐alcoholic fatty liver disease (NAFLD)/non‐alcoholic steatohepatitis (NASH)1, 4 and disease severity,5 alcoholic liver disease (ALD)‐induced cirrhosis6, 7, 8, 9 and outcome after severe alcoholic hepatitis.10 Moreover, it has been linked to hepatic steatosis/liver fibrosis in hepatitis C virus‐monoinfected11 and HIV/hepatitis C virus‐coinfected12 patients, although its effects in viral hepatitis were less consistent when compared to patients with fatty liver disease.13 Finally, the PNPLA3 G allele has repeatedly been found to be associated with hepatocellular carcinoma (HCC) development.1, 13
Recent data indicates that the PNPLA3 G allele potentiates the proinflammatory and profibrogenic features of hepatic stellate cells.14, 15 Hepatic stellate cells are not only a key player in liver fibrogenesis but also play an essential role in the perpetuation of portal hypertension. Due to its impact on hepatic stellate cell activation, the PNPLA3 G allele might promote liver disease progression, even beyond the initiation of liver fibrosis/portal hypertension, and thus, have prognostic implications in advanced chronic liver disease (ie in patients who have already developed portal hypertension).
We aimed to investigate the impact of the PNPLA3 G allele on (liver‐related) mortality in our thoroughly characterised cohort of 372 patients who had already developed portal hypertension due to ALD/NAFLD or viral hepatitis (longitudinal study). To examine the underlying mechanism, we assessed whether PNPLA3 genotype is linked to hepatic steatosis, as evaluated by controlled attenuation parameter in a similar cohort of 153 consecutive patients (cross‐sectional study).
2. PATIENTS AND METHODS
2.1. Study population
To investigate the impact of the PNPLA3 G allele on mortality, we performed a retrospective analysis in 372 prospectively characterised patients with portal hypertension (hepatic venous pressure gradient [HVPG] ≥6 mm Hg) diagnosed at the Medical University of Vienna through 2013 (cohort I; longitudinal study).16 Since the effect of PNPLA3 might vary throughout different aetiologies, only patients with ALD/NAFLD‐ or viral hepatitis‐induced portal hypertension were included. Aetiology was determined based on medical history at the time of HVPG measurement.
The impact of PNPLA3 genotype on hepatic steatosis was evaluated in a cohort of 153 prospectively characterised patients who underwent both HVPG and controlled attenuation parameter measurement between 2014 and 2017 (similar inclusion criteria; cohort II; cross‐sectional study).
2.2. PNPLA3 genotyping
PNPLA3 rs738409 genotyping was performed by a StepOnePlus Real‐Time PCR System and a TaqMan SNP Genotyping Assay (Applied Biosystems, Foster City, CA, USA), using published sequences from the NCBI Entrez SNP Database ( http://www.ncbi.nlm.nih.gov/sites/entrez): 5′‐AAGGAGGGATAAGGCCACTGTA‐3′ as forward and 5′‐CTTTCACAGGCCTTGGTATGTTC‐3′ as reverse primer.
2.3. Hepatic venous pressure gradient measurement
The Vienna Hepatic Hemodynamic Laboratory at the Medical University of Vienna performed the HVPG measurements according to a standardised operating procedure.17, 18 HVPG measurements were performed in the absence of nonselective beta blockers and nitrates. Clinically significant portal hypertension was defined by HVPG values ≥10 mm Hg.19
2.4. Definition of hepatic decompensation
Patients’ medical records were reviewed for the following events which defined (further) hepatic decompensation: Requirement of paracentesis, admission for grade 3/4 hepatic encephalopathy, variceal bleeding and liver‐related death.
2.5. Liver stiffness and controlled attenuation parameter measurement
Measurement of controlled attenuation parameter was performed after an overnight fast by transient elastography using a FibroScan 502 Touch (Echosens, Paris, France).20 Hepatic steatosis was graded according to cut‐offs derived from a recent meta‐analysis based on individual patient data21: ≥248 dB × m−1 for ≥S1, ≥268 dB × m−1 for ≥S2 and ≥280 dB × m−1 for S3.
2.6. Statistical analyses
Statistical analyses were performed using IBM SPSS Statistics 24 (IBM, Armonk, NY, USA), GraphPad Prism 7 (GraphPad Software, La Jolla, CA, USA) and R 3.4.1 (R Core Team, R Foundation for Statistical Computing, Vienna, Austria).
Continuous variables were reported as mean ±standard error of the mean or median (interquartile range), while categorical variables were reported as number of patients with (proportion of patients with) the certain characteristic.
Student's t test and one‐way analysis of variance were used for group comparisons of continuous variables when applicable. Otherwise, Mann‐Whitney U test and Kruskal‐Wallis one‐way analysis of variance were applied. Group comparisons of categorical variables were performed using Chi‐squared or Fisher's Exact test.
The effect of PNPLA3 genotype on transplant‐free mortality was investigated using Kaplan‐Meier analysis, log‐rank test and Cox regression. Patients entered the survival analyses at the time of HVPG measurement. Patients who received a liver transplantation were censored at the day of surgery. Transplant‐free survival time was defined as the time to liver transplantation, death, or end of follow‐up.
To further investigate the impact of PNPLA3 G/G genotype on hepatic decompensation, liver‐related mortality as well as mortality, considering liver transplantation and, if applicable (nonliver‐related) death as competing risks, we used Fine and Gray competing risk regression models (cmprsk: Subdistribution Analysis of Competing Risks; https://CRAN.R-project.org/package=cmprsk).22
Variables that showed differences between PNPLA3 genotypes or those which we considered highly relevant for the endpoint of interest were included as covariates in multivariate analyses.
A P ≤0.05 was considered statistically significant.
2.7. Ethics
The study was conducted in accordance with the Declaration of Helsinki and approved by the ethics committee of the Medical University of Vienna (No. 1526/2017). All subjects were consented for genetic testing.
3. RESULTS
3.1. Characteristics of cohort I and comparison of PNPLA3 genotypes
A total of 372 patients had portal hypertension due to viral hepatitis (n = 231 [62%]) or fatty liver disease (n = 141 [38%]; ALD: 104 [28%]/NAFLD: 37 [10%]; Table S1). The median HVPG and model for end‐stage liver disease (MELD) score were 16 (10) mm Hg and 10 (4.7) points, respectively. Fifty‐six (15%) patients harboured the PNPLA3 G/G genotype, while 163 (44%) and 153 (41%) had the C/C and the C/G genotype, respectively.
PNPLA3 G allele carriers were more commonly male, when compared to C/C patients (Table S1). Moreover, ALD/NAFLD was overrepresented among patients harbouring a PNPLA3 G allele. PNPLA3 G allele carriers also showed more severe liver disease, as indicated by higher HVPG and MELD scores as well as lower serum albumin levels. A comparison between PNPLA3 G/G and non‐G/G patients yielded similar differences (Table 1). Stratifying patients by aetiology (viral hepatitis and fatty liver disease), we observed trends towards a higher proportion of males and higher HVPG values among PNPLA3 G/G patients of both aetiologies (Table 1).
Table 1.
Comparison of patient characteristics between patatin‐like phospholipase domain‐containing protein 3 (PNPLA3) G/G and non‐G/G in A the overall cohort, B among patients with viral hepatitis and C among patients with alcoholic (ALD)/nonalcoholic fatty liver disease (NAFLD) (cohort I)
| A | B | C | |||||||
|---|---|---|---|---|---|---|---|---|---|
| All patients, n = 372 | Viral hepatitis, n = 231 | ALD/NAFLD, n = 141 | |||||||
| Patient characteristics | Non‐G/G, n = 316 | G/G, n = 56 | P value | non‐G/G, n = 206 | G/G, n = 25 | P value | Non‐G/G, n = 110 | G/G, n = 31 | P value |
| Age, years | 52.5 (14.7) | 56.2 (16.6) | 0.131 | 51.8 ± 0.7 | 53.1 ± 2.1 | 0.529 | 58.6 (17.5) | 58.6 (17.9) | 0.594 |
| Sex | |||||||||
| Male | 240 (76%) | 50 (89%) | 0.026 | 155 (75%) | 22 (88%) | 0.155 | 85 (77%) | 28 (90%) | 0.108 |
| Female | 76 (24%) | 6 (11%) | 51 (25%) | 3 (12%) | 25 (23%) | 3 (10%) | |||
| Aetiology | |||||||||
| ALD/NAFLD | 110 (35%) | 31 (55%) | 0.003 | — | — | — | — | — | — |
| Viral | 206 (65%) | 25 (45%) | — | — | — | — | — | — | |
| HCC | 13 (4%) | 2 (4%) | 1 | 8 (4%) | 2 (8%) | 0.606 | 5 (5%) | 0 (0%) | 0.354 |
| HVPG, mm Hg | 15 (10) | 18 (8) | 0.003 | 13 (9) | 15 (7) | 0.154 | 18.4 ± 0.6 | 20.2 ± 5.7 | 0.155 |
| MELD, points | 10 (4) | 11 (6.8) | 0.029 | 9 (4) | 9 (3) | 0.481 | 11 (6.9) | 13 (6) | 0.284 |
| Albumin, g × L−1 | 37.2 (7.9) | 34.2 (8.4) | 0.028 | 37.7 (7.4) | 37 (7.6) | 0.227 | 35.8 (8.3) | 32.9 (9.8) | 0.236 |
BMI, body mass index; HCC, hepatocellular carcinoma; HVPG, hepatic venous pressure gradient; MELD, model for end‐stage liver disease.
3.2. Impact of PNPLA3 genotype on transplant‐free mortality in cohort I
Patients were followed for a median of 27.4 (37.6) months. Twenty‐five (7%) patients underwent liver transplantation, while 104 patients (28%) died.
While transplant‐free survival was similar between PNPLA3 C/C and C/G patients (at 5 years: 72% vs 66% P = 0.635; Figure S1), we observed substantially impaired transplant‐free survival in G/G patients (at 5 years: 47%; P = 0.026 vs C/C and P = 0.003 vs C/G). Thus, for further analyses, patients were grouped into PNPLA3 G/G and non‐G/G.
Overall, PNPLA3 G/G genotype (adjusted hazard ratio [aHR]: 1.42, 95% confidence interval [95% CI]: 0.879‐2.3; P = 0.151) tended to increase transplant‐free mortality in an analysis adjusted for age, sex, aetiology, HVPG, MELD and serum albumin levels (Table 2 and Figure 1).
Table 2.
Cox regression analyses on the influence of patatin‐like phospholipase domain‐containing protein 3 (PNPLA3) G/G genotype on transplant‐free survival in A the overall cohort, B among patients with viral hepatitis and C among patients with alcoholic (ALD)/non‐alcoholic fatty liver disease (NAFLD) (cohort I)
| A | B | C | |||||||
|---|---|---|---|---|---|---|---|---|---|
| All patients, n = 372 | Viral hepatitis, n = 231 | ALD/NAFLD, n = 141 | |||||||
| Patient characteristics | HR | 95% CI | P value | HR | 95% CI | P value | HR | 95% CI | P value |
| Age, years | 1.03 | 1.01‐1.05 | 0.004 | 1.03 | 1‐1.06 | 0.041 | 1.04 | 1.01‐1.08 | 0.008 |
| Sex, male vs female | 0.689 | 0.415‐1.14 | 0.15 | 0.648 | 0.332‐1.263 | 0.202 | 0.619 | 0.255‐1.5 | 0.288 |
| Aetiology, viral vs ALD/NAFLD | 0.651 | 0.404‐1.05 | 0.079 | — | — | — | — | — | — |
| HVPG, per mm Hg | 1.01 | 0.976‐1.05 | 0.498 | 1.05 | 0.994‐1.11 | 0.079 | 0.975 | 0.925‐1.03 | 0.356 |
| MELD, per point | 0.986 | 0.933‐1.04 | 0.611 | 0.974 | 0.891‐1.06 | 0.558 | 0.989 | 0.92‐1.06 | 0.768 |
| Albumin, per g x L−1 | 0.917 | 0.885‐0.952 | <0.001 | 0.907 | 0.851‐0.966 | 0.002 | 0.923 | 0.881‐0.966 | 0.001 |
| PNPLA3 G/G | 1.42 | 0.879‐2.3 | 0.151 | 0.845 | 0.358‐1.99 | 0.7 | 2.16 | 1.15‐4.06 | 0.017 |
HR, hazard ratio; 95% CI, 95% confidence interval; HVPG, hepatic venous pressure gradient; MELD, model for end‐stage liver disease.
Figure 1.
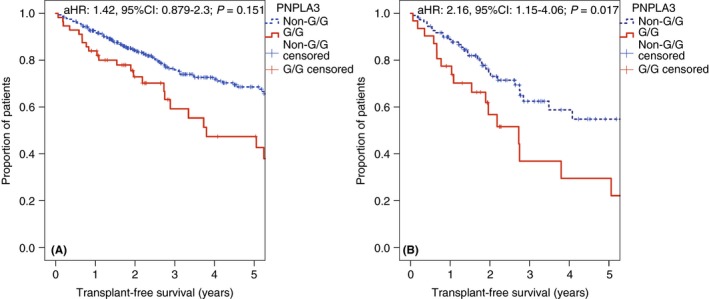
A, Transplant‐free survival in patients with the patatin‐like phospholipase domain‐containing protein 3 (PNPLA3) G/G genotype, or without (cohort I). B, Subgroup of patients with fatty liver disease (cohort I). aHR, adjusted hazard ratio; 95%CI, 95% confidence interval
Since previous studies on the impact of PNPLA3 on liver disease severity observed more consistent effects in fatty liver disease, as compared to viral hepatitis, we analysed these groups separately (Table 2 and Figure 1). While PNPLA3 showed no impact in patients with viral hepatitis (n = 231; aHR: 0.845, 95% CI: 0.358‐1.99; P = 0.7), harbouring the G/G genotype doubled the transplant‐free mortality risk among patients with fatty liver disease (n = 141; aHR: 2.16, 95% CI: 1.15‐4.06; P = 0.017), independently of potential confounding factors (age, sex, HVPG, MELD and serum albumin levels). Interestingly, even in the subgroup of patients who had already progressed to clinically significant portal hypertension, PNPLA3 G/G genotype substantially increased transplant‐free mortality (n = 129; aHR: 2.11, 95% CI: 1.09‐4.09; P = 0.027).
3.3. Influence of PNPLA3 G/G genotype on hepatocellular carcinoma development, hepatic decompensation and liver‐related mortality in patients with fatty liver disease
Among patients without a history of HCC, 5 (16%) and 9 (9%) patients were diagnosed with HCC during follow‐up in the subgroup of patients with and without the PNPLA3 G/G genotype, respectively. HCC incidence rates were numerically higher among patients harbouring the PNPLA3 G/G genotype (8% per person‐year; 95% CI: 3‐17) when compared to non‐G/G patients (4% per person‐year; 95% CI: 2‐7).
To further investigate the impact of PNPLA3 G/G genotype on mortality, we conducted a competing risk regression analysis adjusted for age, sex, aetiology, HVPG, MELD, and serum albumin levels, considering liver transplantation as a competing risk (Table 3). The mortality risk among patients harbouring the PNPLA3 G/G genotype was about twice as high when compared to non‐G/G patients (adjusted subdistribution hazard ratio [aSHR]: 2.2, 95% CI: 1.22‐3.98; P = 0.009).
Table 3.
Competing risk regression analyses on the influence of patatin‐like phospholipase domain‐containing protein 3 (PNPLA3) G/G genotype on A mortality, B liver‐related mortality and C hepatic decompensation in patients with alcoholic (ALD)/non‐alcoholic fatty liver disease (NAFLD) (cohort I)
| A | B | C | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Mortalitya | Liver‐related mortalityb | Hepatic decompensationb | |||||||
| Patient characteristics | SHR | 95% CI | P value | SHR | 95% CI | P value | HR | 95% CI | P value |
| Age, years | 1.04 | 1.01‐1.08 | 0.011 | 1.05 | 1.01‐1.1 | 0.008 | 1.03 | 0.998‐1.05 | 0.068 |
| Sex, male vs female | 0.713 | 0.344‐1.48 | 0.36 | 0.76 | 0.335‐1.73 | 0.51 | 1.83 | 0.943‐3.57 | 0.074 |
| HVPG, per mmHg | 0.972 | 0.918‐1.03 | 0.34 | 0.961 | 0.901‐1.03 | 0.23 | 1.01 | 0.956‐1.07 | 0.66 |
| MELD, per point | 0.976 | 0.897‐1.06 | 0.58 | 1 | 0.921‐1.09 | 0.98 | 0.978 | 0.915‐1.04 | 0.5 |
| Albumin, per g x L−1 | 0.922 | 0.876‐0.97 | 0.002 | 0.925 | 0.876‐0.978 | 0.006 | 0.951 | 0.911‐0.944 | 0.025 |
| PNPLA3 G/G | 2.2 | 1.22‐3.98 | 0.009 | 2.2 | 1.08‐4.46 | 0.029 | 2.1 | 1.1‐4 | 0.024 |
Considering liver transplantation as a competing risk.
Considering liver transplantation and nonliver‐related death as competing risks.
SHR, subdistribution hazard ratio; 95% CI, 95% confidence interval; HVPG, hepatic venous pressure gradient; MELD, model for end‐stage liver disease.
Among the 50 deaths in patients with fatty liver disease, 10 were nonliver‐related (G/G: 18% [3/17]; non‐G/G: 21% [6/33]). Harbouring the PNPLA3 G/G genotype doubled the risk of liver‐related mortality among patients with fatty liver disease (aSHR: 2.2, 95% CI: 1.08‐4.46; P = 0.029), considering liver transplantation and nonliver‐related death as competing risks and adjusting for potential confounding factors (Table 3 and Figure 2).
Figure 2.
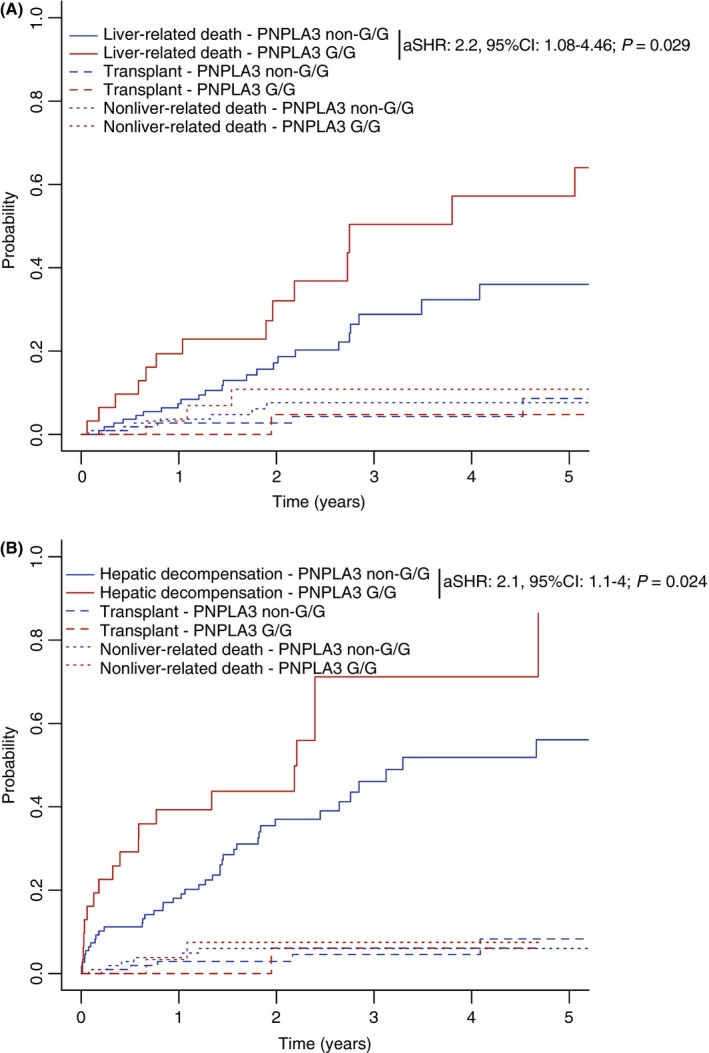
A, Liver‐related mortality and B, hepatic decompensation in fatty liver disease patients with the patatin‐like phospholipase domain‐containing protein 3 (PNPLA3) G/G genotype, or without, considering liver transplantation and nonliver‐related death as competing risks (cohort I). aSHR, adjusted subdistribution hazard ratio; 95% CI, 95% confidence interval
In an analysis considering liver transplantation and nonliver‐related death as competing risks, PNPLA3 G/G genotype was associated with a twofold increase in risk of hepatic decompensation (aSHR: 2.1, 95% CI: 1.1‐4; P = 0.024), after adjusting for potential confounding factors (Table 3 and Figure 2).
Finally, another competing risk regression analysis (considering liver transplantation as a competing risk) adjusted for age, sex, aetiology, HVPG, MELD and serum albumin levels restricted to patients who had already progressed to clinically significant portal hypertension, confirmed that harbouring the PNPLA3 G/G genotype substantially increased mortality (aSHR: 2.33, 95% CI: 1.27‐4.29; P = 0.006) in this subgroup.
3.4. Impact of PNPLA3 genotype on hepatic steatosis in cohort II
Cohort II comprised 153 patients and was comparable to cohort I in terms of liver disease severity (median HVPG: 18 (10) mm Hg; median MELD: 10 (5) points; Table S2). The mean body mass index was 25.7 ± 0.4 kg × m−2, with 85 (56%) patients being overweight/obese (body mass index [BMI] ≥25 kg × m−2). Twenty‐four (16%) patients were obese (BMI ≥30 kg × m−2) and 33 (22%) had diabetes mellitus type 2.
Similar to cohort I, we observed a trend towards an overrepresentation of fatty liver disease among patients harbouring a PNPLA3 G allele. Except for HCC prevalence, all other relevant patient characteristics (eg BMI) were comparable between PNPLA3 genotype groups.
Interestingly, PNPLA3 genotype had no influence on controlled attenuation parameter (C/C: 242 ± 7 vs C/G: 252 ± 7 vs G/G: 223 ± 13 dB × m−1; P = 0.639; Table S2 and Figure S2) or the prevalence of values ≥248 dB × m−1 (C/C: 28 [44%] vs C/G: 33 [54%] vs G/G: 12 [41%]; P = 0.443) in this population of patients with portal hypertension due to fatty liver disease or viral hepatitis.
4. DISCUSSION
Previous studies established firm associations between the PNPLA3 G allele and NAFLD/NASH4 and disease severity.5 More recently, the PNPLA3 G allele has also been linked to ALD‐induced cirrhosis.6, 7, 8, 9 As opposed to this, its effects in viral hepatitis were less consistent.13
We observed an overrepresentation of ALD/NAFLD among carriers of the PNPLA3 G allele, supporting its particular relevance in these aetiologies (cohort I; trend in cohort II). This is in line with a study by Fatelli et al23 which also found a higher frequency of the PNPLA3 G allele in patients with cirrhosis due to fatty liver disease, when compared to viral cirrhosis. Similarly, Friedrich and co‐workers24 observed an increased prevalence of the PNPLA3 G/G genotype (vs a reference population) in ALD patients listed for liver transplantation. Of note, the highest frequency of the PNPLA3 G/G genotype was observed in patients with cryptogenic cirrhosis, which might have comprised a considerable proportion of patients with NAFLD.25
While liver disease severity at the time of HVPG measurement showed a stepwise increase with each PNPLA3 G allele in cohort I, no such association was observed in cohort II. In our opinion, the cross‐sectional findings on liver disease severity at the time of HVPG measurement should not be overinterpreted, since the duration of disease/time to cirrhosis are unknown. Furthermore, the numbers of patients with HCC at the time of HVPG measurement were small in both cohorts, which substantially limits the significance of our findings on HCC prevalence.
Although there is a broad body of evidence derived from cross‐sectional analyses, comparatively few longitudinal studies assessed the impact of PNPLA3 genotype on clinical endpoints other than HCC development. Thus, the results of our study provide valuable information on the prognostic impact of the PNPLA3 G allele in patients who have already developed portal hypertension.
Friedrich and colleagues24 investigated the effect of PNPLA3 genotype in patients listed for liver transplantation and reported increased risks of (further) hepatic decompensation (ie ascites and hepatic encephalopathy) and mortality in carriers of the G allele. However, since the proportion of patients who underwent liver transplantation differed substantially throughout the PNPLA3 genotypes, this study does not allow to draw conclusions on the impact of PNPLA3 genotype on mortality. Furthermore, due to the transplant setting, patients with ongoing alcohol consumption were excluded from this study, which might limit the generalisability of its findings. Following alcohol withdrawal, Rausch and colleagues26 observed a delayed regression of liver stiffness measured by transient elastography in PNPLA3 G allele carriers. Moreover, an analysis based on data of the Steroids or Pentoxifylline for Alcoholic Hepatitis (STOPAH) trial27 revealed an association between PNPLA3 genotype and severe alcoholic hepatitis development as well as medium‐term mortality. This effect was limited to patients who were abstinent from drinking. However, the proportion of patients with underlying advanced chronic liver disease included in the STOPAH trial remains unclear. In our study, harbouring the PNPLA3 G/G genotype doubled the mortality risk of patients with portal hypertension due to (predominantly alcoholic) fatty liver disease (cohort I). Interestingly, this effect was also observed in the subgroup of patients with clinically significant portal hypertension, in whom splanchnic vasodilatation/hyperdynamic circulation, rather than “intrahepatic” factors such as PNPLA3, is considered as the main determinant of portal pressure.28, 29 However, this paradigm has already been challenged by improvements in portal pressure after achieving sustained virologic response to hepatitis C treatments, which were observed throughout all strata of portal hypertension severity.30, 31, 32
Although the numerically increased rates of HCC development among PNPLA3 G/G patients might have contributed to the observed association between PNPLA3 G/G and mortality (cohort I), considering the small number of patients who were diagnosed with HCC, differences in (liver‐related) mortality cannot solely explained by HCC development.
To further explore the mechanism by which the PNPLA3 G/G genotype might promote hepatic decompensation and increase (liver‐related) mortality in patients who have already developed portal hypertension, we analysed controlled attenuation parameter (possibly indicating hepatic steatosis) in a similar patient population (cohort II). Notably, PNPLA3 genotype had no impact on controlled attenuation parameter, which would suggest that its effect on hepatic steatosis is less pronounced in patients who have already progressed to advanced chronic liver disease. Importantly, previous studies have demonstrated the disappearance of hepatic steatosis with liver fibrosis progression in NAFLD patients (ie “burnt‐out NASH”),33 a phenomenon which might also occur in ALD and might have attenuated the effect of PNPLA3 genotype on controlled attenuation parameter in our study, although the proportion of patients with controlled attenuation parameter values indicative of hepatic steatosis was still 48%. Moreover, there is only limited data on the performance of controlled attenuation parameter for diagnosing hepatic steatosis in patients with advanced chronic liver disease; however, it was not impacted by the presence of cirrhosis in a recent meta‐analysis of individual patient data.21 In addition, its diagnostic performance seemed to be similar among ALD patients with advanced liver fibrosis or without.34 Considering these potential limitations as well as the small sample size of cohort II, our findings should be interpreted with caution and require confirmation.
Until recently, the impact of PNPLA3 genotype on hepatic lipid metabolism, and thus, hepatic steatosis, was considered as the primary explanation for its association with liver disease and its progression.3 Notably, Pingitore et al14 as well as Bruschi et al15 demonstrated that the PNPLA3 G allele enhances proinflammatory and profibrogenic features of hepatic stellate cells. Although the association between the PNPLA3 G/G genotype and increased transplant‐free mortality in patients with fatty liver disease was independent of baseline HVPG in our study (cohort I), harbouring the PNPLA3 G/G genotype might have potentiated liver disease progression and aggravated portal hypertension during follow‐up.
The main limitation of this study is its retrospective design; however, patients were prospectively characterised at the time of HVPG measurement. Moreover, the number of patients with fatty liver disease included in cohort I was limited and our study did not include a validation cohort. Aetiology was determined based on medical history and the diagnostic work up did not include a liver biopsy in most cases. In addition, alcohol abstinence was not systematically assessed. Finally, the impact of PNPLA3 genotype on clinical events and controlled attenuation parameter was not investigated in the same patient cohort, since controlled attenuation parameter became available in 2014 at our institution. However, inclusion criteria and study setting were exactly the same for both cohorts, which resulted in similar patient characteristics. Analysing clinical events in patients evaluated before 2014 allowed for a longer follow‐up duration, a higher number of clinical events, and thus, more robust estimates of the impact of PNPLA3 genotype.
In summary, PNPLA3 G/G genotype seems to double the risks of liver‐related morbidity and mortality in patients with portal hypertension due to fatty liver disease, even after the development of clinically significant portal hypertension. Further studies are warranted to investigate potential underlying pathophysiological mechanisms unrelated to hepatic steatosis.
AUTHORSHIP
Guarantor of the article: Arnulf Ferlitsch.
Author contributions: All authors contributed either to research design (M.M., Bernhard Scheiner, and A.F.), and/or the acquisition (genotyping: A.F.S. and P.F.; phenotyping: all authors), analysis (M.M., Bernhard Scheiner, and D.B.) or interpretation (all authors) of data. M.M. and Bernhard Scheiner drafted the manuscript, which was then critically revised by all other authors. All authors gave approval of the submitted version of the manuscript.
Supporting information
ACKNOWLEDGEMENTS
Declaration of personal interests: The authors have nothing to disclose regarding the work under consideration for publication. However, the authors disclose the following relevant financial activities outside the submitted work: M.M. has served as a speaker and/or consultant and/or advisory board member for AbbVie, Bristol‐Myers Squibb, Gilead, W. L. Gore & Associates, and Janssen. Bernhard Scheiner has nothing to disclose. A.F.S. has served as a speaker and/or consultant and/or advisory board member for Boehringer Ingelheim, Gilead, Janssen, MSD and Roche. P.S. has served as a speaker and/or consultant and/or advisory board member for Boehringer Ingelheim. R.P. has nothing to disclose. D.B. has nothing to disclose. Benedikt Schaefer has nothing to disclose. H.Z. has served as a speaker and/or consultant and/or advisory board member for AbbVie, Bristol Myer‐Squibb, Gilead, Janssen and MSD. M.P. has served as a speaker and/or consultant and/or advisory board member for Abbott, AbbVie, Bayer, Boehringer Ingelheim, Bristol‐Myers Squibb, Gilead, Janssen, Lilly and MSD and received research funding from AbbVie, ArQule, Bayer, Daiichi Sankyo, Gilead and MSD. M.T. has served as a speaker and/or consultant and/or advisory board member for Albireo, Bristol‐Myers Squibb, Dr. Falk Pharma, Gilead, Intercept, MSD, Novartis, and Phenex Pharmaceuticals and has received research funding from Albireo, Dr. Falk Pharma, Gilead, Intercept, MSD and Takeda. M.T. owns patents on the use of nor‐ursodeoxycholic acid for the treatment and/or prevention of cholestatic liver disease and alcoholic/nonalcoholic steatohepatitis. T.R. has served as a speaker and/or consultant and/or advisory board member for AbbVie, Bayer, Boehringer Ingelheim, Gilead, W. L. Gore & Associates and MSD and has received research funding from AbbVie, Gilead, Boehringer Ingelheim, Phenex Pharmaceuticals and Philips. P.F. has served as a speaker and/or consultant and/or advisory board member for AbbVie, Bristol Myer‐Squibb, Gilead, MSD and Roche and has received research funding from Gilead and Roche. A.F. has served as a speaker and/or consultant and/or advisory board member for AbbVie, Gilead and Intercept. A.F. owns a patent on a catheter for the measurement of hepatic venous pressure gradient.
Declaration of funding interests: No financial support specific to this study was received.
Mandorfer M, Scheiner B, Stättermayer AF, et al. Impact of patatin‐like phospholipase domain containing 3 rs738409 G/G genotype on hepatic decompensation and mortality in patients with portal hypertension. Aliment Pharmacol Ther. 2018;48:451–459. 10.1111/apt.14856
The Handling Editor for this article was Professor Stephen Harrison, and it was accepted for publication after full peer‐review.
M.M. and Bernhard Scheiner contributed equally to this manuscript.
[Correction added on 12 July 2018, after first online publication: The 7th author’s surname Schäfer was incorrect. This is now corrected as Schaefer in this version.]
REFERENCES
- 1. Dongiovanni P, Romeo S, Valenti L. Genetic factors in the pathogenesis of nonalcoholic fatty liver and steatohepatitis. Biomed Res Int. 2015;2015:460190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pirazzi C, Valenti L, Motta BM, et al. PNPLA3 has retinyl‐palmitate lipase activity in human hepatic stellate cells. Hum Mol Genet. 2014;23:4077‐4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Trepo E, Romeo S, Zucman‐Rossi J, Nahon P. PNPLA3 gene in liver diseases. J Hepatol. 2016;65:399‐412. [DOI] [PubMed] [Google Scholar]
- 4. Romeo S, Kozlitina J, Xing C, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461‐1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stattermayer AF, Traussnigg S, Aigner E, et al. Low hepatic copper content and PNPLA3 polymorphism in non‐alcoholic fatty liver disease in patients without metabolic syndrome. J Trace Elem Med Biol. 2017;39:100‐107. [DOI] [PubMed] [Google Scholar]
- 6. Stickel F, Buch S, Lau K, et al. Genetic variation in the PNPLA3 gene is associated with alcoholic liver injury in caucasians. Hepatology. 2011;53:86‐95. [DOI] [PubMed] [Google Scholar]
- 7. Trepo E, Gustot T, Degre D, et al. Common polymorphism in the PNPLA3/adiponutrin gene confers higher risk of cirrhosis and liver damage in alcoholic liver disease. J Hepatol. 2011;55:906‐912. [DOI] [PubMed] [Google Scholar]
- 8. Salameh H, Raff E, Erwin A, et al. PNPLA3 gene polymorphism is associated with predisposition to and severity of alcoholic liver disease. Am J Gastroenterol. 2015;110:846‐856. [DOI] [PubMed] [Google Scholar]
- 9. Buch S, Stickel F, Trepo E, et al. A genome‐wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol‐related cirrhosis. Nat Genet. 2015;47:1443‐1448. [DOI] [PubMed] [Google Scholar]
- 10. Atkinson SR, Way MJ, McQuillin A, Morgan MY, Thursz MR. Homozygosity for rs738409: G in PNPLA3 is associated with increased mortality following an episode of severe alcoholic hepatitis. J Hepatol. 2017;67:120‐127. [DOI] [PubMed] [Google Scholar]
- 11. Stattermayer AF, Scherzer T, Beinhardt S, Rutter K, Hofer H, Ferenci P. Review article: genetic factors that modify the outcome of viral hepatitis. Aliment Pharmacol Ther. 2014;39:1059‐1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mandorfer M, Payer BA, Schwabl P, et al. Revisiting liver disease progression in HIV/HCV‐coinfected patients: the influence of vitamin D, insulin resistance, immune status, IL28B and PNPLA3. Liver Int. 2015;35:876‐885. [DOI] [PubMed] [Google Scholar]
- 13. Scheiner B, Mandorfer M, Schwabl P, et al. The impact of PNPLA3 rs738409 SNP on liver fibrosis progression, portal hypertension and hepatic steatosis in HIV/HCV coinfection. PLoS ONE. 2015;10:e0143429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pingitore P, Dongiovanni P, Motta BM, et al. PNPLA3 overexpression results in reduction of proteins predisposing to fibrosis. Hum Mol Genet. 2016;25:5212‐5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bruschi FV, Claudel T, Tardelli M, et al. The PNPLA3 I148M variant modulates the fibrogenic phenotype of human hepatic stellate cells. Hepatology. 2017;65:1875‐1890. [DOI] [PubMed] [Google Scholar]
- 16. Mandorfer M, Schwabl P, Paternostro R, et al. Von Willebrand factor indicates bacterial translocation, inflammation, and procoagulant imbalance and predicts complications independently of portal hypertension severity. Aliment Pharmacol Ther. 2018;47:980‐988. [DOI] [PubMed] [Google Scholar]
- 17. Ferlitsch A, Bota S, Paternostro R, et al. Evaluation of a new balloon occlusion catheter specifically designed for measurement of hepatic venous pressure gradient. Liver Int. 2015;35:2115‐2120. [DOI] [PubMed] [Google Scholar]
- 18. Schwarzer R, Kivaranovic D, Mandorfer M, et al. Randomised clinical study: the effects of oral taurine 6 g/day vs placebo on portal hypertension. Aliment Pharmacol Ther. 2018;47:86‐94. [DOI] [PubMed] [Google Scholar]
- 19. Reiberger T, Püspök A, Schoder M, et al. Austrian consensus guidelines on the management and treatment of portal hypertension (Billroth III). Wien Klin Wochenschr. 2017;129(Suppl 3):135‐158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schwabl P, Bota S, Salzl P, et al. New reliability criteria for transient elastography increase the number of accurate measurements for screening of cirrhosis and portal hypertension. Liver Int. 2015;35:381‐390. [DOI] [PubMed] [Google Scholar]
- 21. Karlas T, Petroff D, Sasso M, et al. Individual patient data meta‐analysis of controlled attenuation parameter (CAP) technology for assessing steatosis. J Hepatol. 2017;66:1022‐1030. [DOI] [PubMed] [Google Scholar]
- 22. Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. Journal of the American Statistical Association. 1999;94:496‐509. [Google Scholar]
- 23. Falleti E, Fabris C, Cmet S, et al. PNPLA3 rs738409C/G polymorphism in cirrhosis: relationship with the aetiology of liver disease and hepatocellular carcinoma occurrence. Liver Int. 2011;31:1137‐1143. [DOI] [PubMed] [Google Scholar]
- 24. Friedrich K, Wannhoff A, Kattner S, et al. PNPLA3 in end‐stage liver disease: alcohol consumption, hepatocellular carcinoma development, and transplantation‐free survival. J Gastroenterol Hepatol. 2014;29:1477‐1484. [DOI] [PubMed] [Google Scholar]
- 25. Maheshwari A, Thuluvath PJ. Cryptogenic cirrhosis and NAFLD: are they related? Am J Gastroenterol. 2006;101:664‐668. [DOI] [PubMed] [Google Scholar]
- 26. Rausch V, Peccerella T, Lackner C, et al. Primary liver injury and delayed resolution of liver stiffness after alcohol detoxification in heavy drinkers with the PNPLA3 variant I148M. World J Hepatol. 2016;8:1547‐1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Atkinson SR, Way MJ, McQuillin A, Morgan MY, Thursz MR. Homozygosity for rs738409: G in PNPLA3 is associated with increased mortality following an episode of severe alcoholic hepatitis. J Hepatol. 2017;67(1):120‐127. [DOI] [PubMed] [Google Scholar]
- 28. Mandorfer M, Reiberger T. Beta blockers and cirrhosis, 2016. Dig Liver Dis. 2017;49:3‐10. [DOI] [PubMed] [Google Scholar]
- 29. Reiberger T, Mandorfer M. Beta adrenergic blockade and decompensated cirrhosis. J Hepatol. 2017;66:849‐859. [DOI] [PubMed] [Google Scholar]
- 30. Mandorfer M, Kozbial K, Schwabl P, et al. Sustained virologic response to interferon‐free therapies ameliorates HCV‐induced portal hypertension. J Hepatol. 2016;65:692‐699. [DOI] [PubMed] [Google Scholar]
- 31. Schwabl P, Mandorfer M, Steiner S, et al. Interferon‐free regimens improve portal hypertension and histological necroinflammation in HIV/HCV patients with advanced liver disease. Aliment Pharmacol Ther. 2017;45:139‐149. [DOI] [PubMed] [Google Scholar]
- 32. Lens S, Alvarado‐Tapias E, Marino Z, et al. Effects of all‐oral anti‐viral therapy on HVPG and systemic hemodynamics in patients with hepatitis C virus‐associated cirrhosis. Gastroenterology. 2017;153:1273‐1283; e1. [DOI] [PubMed] [Google Scholar]
- 33. Powell EE, Cooksley WG, Hanson R, Searle J, Halliday JW, Powell LW. The natural history of nonalcoholic steatohepatitis: a follow‐up study of forty‐two patients for up to 21 years. Hepatology. 1990;11:74‐80. [DOI] [PubMed] [Google Scholar]
- 34. Thiele M, Rausch V, Fluhr G, et al. Controlled attenuation parameter and alcoholic hepatic steatosis: diagnostic accuracy and role of alcohol detoxification. J Hepatol 2018;68:1025‐1032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials