

Abstract
Objective
To determine the effect of selexipag, an oral, selective IP prostacyclin receptor agonist, on the frequency of attacks of Raynaud's phenomenon (RP) in patients with systemic sclerosis (SSc).
Methods
Patients with SSc‐related RP were randomized 1:1 to placebo (n = 38) or selexipag (n = 36) in individualized doses (maximum of 1,600 μg twice daily) during a 3‐week titration period. The primary end point was the weekly average number of RP attacks during the study maintenance period, analyzed using a Bayesian approach with a negative binomial model adjusted for baseline number of RP attacks. Other outcome measures included Raynaud's Condition Score (RCS), RP attack duration, and treatment‐emergent adverse events (AEs).
Results
Baseline characteristics were comparable between treatment groups. For 83.3% of patients, the individualized maintenance dosage of selexipag was ≤800 μg twice daily. No significant difference was observed between placebo and selexipag in weekly average number of electronic diary (eDiary)–recorded RP attacks during the maintenance period (14.2 attacks during the maintenance period and 21.5 attacks during the baseline week in the placebo group [n = 32] versus 18.0 attacks during the maintenance period and 22.4 attacks during the baseline week in the selexipag group [n = 27]; adjusted mean treatment difference of 3.4 in favor of placebo). No significant treatment effect was observed on RCS or RP attack duration. In the double‐blind period, 86.8% of placebo‐treated patients and 100% of selexipag‐treated patients reported ≥1 AE; 55.3% and 91.7%, respectively, reported ≥1 prostacyclin‐associated AE.
Conclusion
Treatment with selexipag did not reduce the number of RP attacks compared with placebo. The safety profile of selexipag was similar to that previously reported. This study provides important information about the feasibility of eDiary reporting of RP attacks in clinical trials.
Raynaud's phenomenon (RP) is experienced by >90% of patients with systemic sclerosis (SSc), often as the first symptom of the disease 1, 2, 3. RP is part of the spectrum of vasculopathy associated with SSc, which also includes digital ulceration and critical digital ischemia 2. It is an important clinical manifestation of the disease, as it is thought that vasculopathy may play a key role in the early pathogenesis of SSc 4. RP occurs due to episodic, reversible vasospasm of the small arteries and arterioles, usually in the fingers and toes, and is mainly triggered by cold or emotional stress 5, 6. In addition, RP secondary to SSc is linked with structural changes of the vasculature, resulting in blood vessel narrowing and impairment of blood flow 5. Because RP is burdensome, improvements in RP have been linked to better quality of life 7, 8.
Management of RP is challenging and requires a multifaceted approach, including risk factor avoidance and targeted drug therapy 2, such as calcium‐channel blockers 9 and, more recently, at least in patients with severe SSc‐related RP, phosphodiesterase V inhibitors 10, 11, 12. Angiotensin receptor blockers are sometimes recommended, but there is little evidence to support their efficacy 13. Intravenous prostanoids, particularly iloprost infusions, are recommended for patients with severe RP when treatment with other agents has failed 2, 9. Although intravenous iloprost has demonstrated efficacy in decreasing severity, frequency, and duration of RP attacks in patients with SSc 14, 15, 16, 17, 18, intravenous administration is burdensome. Currently, there is limited evidence for the benefit of oral prostacyclin analogs in patients with RP 8. Therefore, there is a need to identify oral therapies that act on the prostacyclin receptor for the management of RP secondary to SSc.
Selexipag is an oral, selective IP prostacyclin receptor agonist that has recently been approved for the long‐term treatment of pulmonary arterial hypertension (PAH) in adults with World Health Organization functional class II/III symptoms 19, 20. The present study aimed to determine the effect of selexipag on the frequency of RP attacks in patients with RP secondary to SSc.
Patients and Methods
Study design. This was a multicenter, double‐blind, randomized, placebo‐controlled, parallel‐group, phase II study comprising a 2–4‐week single‐blind placebo run‐in period, an 8‐week treatment period (3‐week titration, 5‐week maintenance), and a 30‐day posttreatment safety follow‐up period. The baseline week was the last 7 days before randomization during the run‐in period. Patients were randomized in a 1:1 ratio to placebo or selexipag, stratified by the presence or absence of digital ulcers at baseline. Data on RP attacks were collected using an electronic diary (eDiary).
During the single‐blind run‐in period, patients received placebo twice daily. This run‐in period was designed primarily to determine eligibility with respect to RP attack frequency. In the 3‐week titration period, selexipag or matching placebo was initiated at a dosage of 200 μg twice daily and was increased every 3 days in increments of 200 μg until unmanageable adverse effects associated with prostacyclin use (e.g., headache or diarrhea) developed. The dose was then either continued or decreased by 200 μg in both daily dosages, and this was considered to be the individualized highest tolerated dosage. The maximum dosage allowed was 1,600 μg twice daily. During the maintenance period, dose increases were not permitted; however, dose reductions for tolerability reasons and subsequent titration to the dose previously reached were allowed. The individualized maintenance dose was defined as the dose that the patient was exposed to for the longest duration during the maintenance period.
The study was conducted during the winter months in the Northern Hemisphere to minimize seasonal variability. At screening, patients were trained by the investigator in how to recognize an attack as well as the information to be recorded in the eDiary (number of RP attacks per day; attack duration in minutes). An RP attack was defined as an episode of at least a 2‐phase color change in the fingers in response to cold exposure or emotion, consisting of pallor and/or cyanosis and reactive hyperemia associated with finger discomfort. Written informed consent was provided by all patients. Ethical approval was received from the independent ethics committee or institutional review board of all participating centers prior to study commencement. The study was conducted in accordance with the principles of the Declaration of Helsinki. A list of the Raynaud Study Investigators is provided in Appendix A.
Patient selection. Eligible patients were age ≥18 years with a diagnosis of SSc according to the American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR) 2013 classification criteria (total score of ≥9, including a score of 3 for the RP item) 21. Patients were required to have had ≥7 RP attacks on ≥5 different days during the baseline week and ≥80% eDiary compliance during the run‐in period. We excluded patients with a history of other conditions that can affect RP evaluation (for example, surgery [cervicothoracic sympathectomy, recent amputation, debridement] or recent treatment with botulinum toxin). Patients who received prostacyclin or prostacyclin analogs within 3 months of the screening visit were not eligible. Patients were permitted to take calcium‐channel blockers, nitrates or nitric oxide donors, endothelin receptor antagonists, alpha‐blockers, antithrombotic agents, nonsteroidal antiinflammatory agents, angiotensin‐converting enzyme inhibitors, beta‐blockers, clonidine, systemic corticosteroids, and fluoxetine during the study, provided that the dose had been stable in the month prior to screening and remained stable during the treatment period. Complete inclusion/exclusion criteria are provided in Supplementary Table 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40242/abstract.
Study outcome measures. The primary efficacy end point was the weekly average number of RP attacks during the maintenance period. Other prespecified efficacy end points included number and proportion of patients with weekly average number of RP attacks in categories of improved (change from baseline week of at least –15%), stable (change from baseline week of between –15% and 15%), and worsened (change from baseline week of >15%) during the maintenance period; change from baseline week to week 8 in the weekly average RP attack duration following randomization; change from baseline week to each postbaseline week in the weekly average Raynaud's Condition Score (RCS) 22 following randomization; number of new digital ulcers and number of baseline digital ulcers completely healed at week 8, and changes from baseline to week 8 in quality of life, as measured by the overall Scleroderma Health Assessment Questionnaire 23, the Health Assessment Questionnaire disability index (HAQ DI) 24, and the hand components of the HAQ DI. Safety end points included treatment‐emergent adverse events (AEs) and laboratory assessments.
Statistical analysis. Efficacy end points were analyzed on the per‐protocol set, which included all patients who had ≥7 RP attacks on ≥5 days during the baseline week, did not receive forbidden concomitant medication from the start of the run‐in period until end of treatment, did not prematurely discontinue treatment before day 30, and completed ≥70% of the eDiary RP assessments during the maintenance period. The primary efficacy end point was analyzed using a negative binomial model adjusted for the baseline number of RP attacks to assess the following joint proof‐of‐concept criteria in a Bayesian framework: statistical significance was achieved if there was a high probability (≥0.95) that the difference in the mean weekly average number of RP attacks (selexipag minus placebo) was <0 during the maintenance period (i.e., the probability of a difference of <0 was ≥0.95); clinical significance was achieved if the probability of a difference of <–4 was ≥0.5. Missing data were minimized by using the per‐protocol set for the primary analysis. Weekly rates of RP attacks were standardized based on each patient's follow‐up time in the maintenance period, to account for different follow‐up times and/or missing days of RP attacks.
Power and sample size were determined using simulations based on the assumed total number of RP attacks at baseline and during the maintenance period. With 25 patients per arm qualifying for the per‐protocol set, the operating characteristics of the Bayesian approach were a true‐positive probability of >85% to fulfill both proof‐of‐concept criteria if the true difference between the means of the weekly average number of RP attacks during the maintenance period was at least 5.25, and a false‐positive probability of <1% to fulfill both proof‐of‐concept criteria if the true difference between the means was 0, assuming at least a 30% reduction from the baseline week with ≥17.5 RP attacks. Based on this, it was determined to randomize 35 patients per treatment arm. A prespecified subgroup analysis of the primary efficacy variable was conducted based on the presence/absence of digital ulcers at baseline, number of RP attacks during the baseline week (≤17, >17), smoking status at screening (smoker, nonsmoker/former smoker), and use/no use of calcium‐channel blockers at baseline.
Descriptive statistics, counts and percentages (categorical variables), and means and SDs (continuous variables) were provided without imputation for missing data. Between‐group changes from baseline to week 8 in the RCS were compared using a nonparametric analysis of covariance adjusted for the baseline score. Safety analyses were performed on the safety analysis set, which included all patients who received ≥1 dose of study treatment.
Results
Patient disposition and baseline characteristics. Ninety‐two patients were screened between November 2014 and February 2015 from 16 centers in France, Germany, and the UK. Seventy‐four patients were randomized to placebo (n = 38) or selexipag (n = 36), of whom 59 (placebo n = 32; selexipag n = 27) formed the per‐protocol set (Figure 1).
Figure 1.
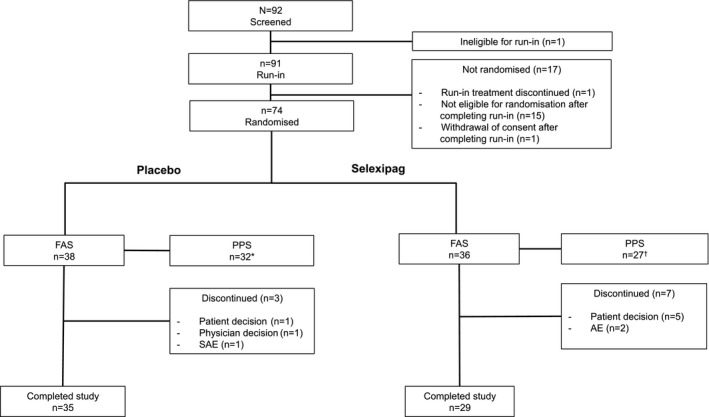
Patient disposition. * = Six patients were excluded from the placebo per‐protocol set (PPS), due to premature study treatment discontinuation (before day 30) (n = 2), ≥30% missing data for assessment of Raynaud's phenomenon (RP) during the maintenance period (n = 2), and the concomitant use of forbidden medication (n = 2). † = Nine patients were excluded from the selexipag per‐protocol set, due to premature study treatment discontinuation (before day 30) (n = 7), ≥30% missing data for RP assessment during the maintenance period (n = 1), and <7 RP attacks/RP attacks not experienced on ≥5 different days prior to randomization (n = 1). FAS = full analysis set (all randomized patients); SAE = serious adverse event.
Baseline demographics and clinical characteristics were similar between all randomized patients (Table 1) and the per‐protocol set (see Supplementary Table 2, http://onlinelibrary.wiley.com/doi/10.1002/art.40242/abstract). The treatment arms were generally similar, although more patients in the placebo group had a history of digital ulcers (71.1%) compared with the selexipag group (44.4%). Baseline use of calcium‐channel blockers in the per‐protocol set was greater in placebo‐treated patients (71.9%) compared with selexipag‐treated patients (33.3%) (see Supplementary Table 2).
Table 1.
Baseline demographic and clinical characteristics of all of the randomized patientsa
| Placebo (n = 38) | Selexipag (n = 36) | All patients (n = 74) | |
|---|---|---|---|
| Sex, no. (%) | |||
| Male | 7 (18.4) | 7 (19.4) | 14 (18.9) |
| Female | 31 (81.6) | 29 (80.6) | 60 (81.1) |
| Age, mean ± SD years | 52.6 ± 11.9 | 52.7 ± 12.2 | 52.6 ± 12.0 |
| Race, no. (%) | |||
| White | 34 (89.5) | 35 (97.2) | 69 (93.2) |
| Asian | 3 (7.9) | – | 3 (4.1) |
| Other | 1 (2.6) | 1 (2.8) | 2 (2.7) |
| SSc subset, no. (%) | |||
| Limited cutaneous | 22 (57.9) | 22 (61.1) | 44 (59.5) |
| Diffuse cutaneous | 14 (36.8) | 12 (33.3) | 26 (35.1) |
| Other | 2 (5.3) | 2 (5.6) | 4 (5.4) |
| Time since SSc diagnosis, mean ± SD yearsb | 7.4 ± 6.3 | 7.3 ± 7.2 | 7.3 ± 6.7 |
| Time since first non‐RP symptom, mean ± SD yearsb | 8.5 ± 6.4 | 9.5 ± 6.8 | 9.0 ± 6.6 |
| Time since first RP symptom, mean ± SD yearsb | 13.4 ± 10.7 | 14.9 ± 10.7 | 14.1 ± 10.7 |
| PAH and/or ILD, no. (%)c, d | |||
| PAH | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| ILD | 10 (26.3) | 4 (11.1) | 14 (18.9) |
| Unknown/not answered | 3 (7.9) | 3 (8.3) | 6 (8.1) |
| SSc‐related antibodies, no. (%)c | |||
| Anticentromere | 19 (50.0) | 16 (44.4) | 35 (47.3) |
| Anti–topoisomerase I | 12 (31.6) | 6 (16.7) | 18 (24.3) |
| Anti–RNA polymerase III | 5 (13.2) | 4 (11.1) | 9 (12.2) |
| Unknown | 1 (2.6) | 2 (5.6) | 3 (4.1) |
| RP attacks in the baseline week, mean ± SD | 21.6 ± 14.7 | 22.1 ± 16.1 | 21.8 ± 15.3 |
| History of digital ulcers, no. (%) | 27 (71.1) | 16 (44.4) | 43 (58.1) |
| Digital ulcers present at baseline, no. (%) | 7 (18.4) | 4 (11.1) | 11 (14.9) |
| Smoking status, no. (%) | |||
| Current smoker | 6 (15.8) | 6 (16.7) | 12 (16.2) |
| Former smoker | 9 (23.7) | 11 (30.6) | 20 (27.0) |
| Nonsmoker | 23 (60.5) | 19 (52.8) | 42 (56.8) |
| Baseline use of CCBs, no. (%) | 24 (63.2) | 15 (41.7) | 39 (52.7) |
SSc = systemic sclerosis; RP = Raynaud's phenomenon; CCBs = calcium‐channel blockers.
Calculated from date of randomization.
Classes not mutually exclusive.
Data on pulmonary arterial hypertension (PAH)/interstitial lung disease (ILD) collected as part of the American College of Rheumatology/European League Against Rheumatism criteria 21.
Dosing and exposure. Of all randomized patients, 71.1% (27 of 38) in the placebo group had an individualized maintenance dose corresponding to 1,600 μg twice daily, while 83.3% (30 of 36) of patients receiving selexipag had an individualized maintenance dose of ≤800 μg twice daily (median 600 [interquartile range 200–800]) (see Supplementary Table 3, http://onlinelibrary.wiley.com/doi/10.1002/art.40242/abstract). The median duration of exposure to study drug for all randomized patients in the double‐blind period was 55.5 days (interquartile range 54.0–57.0 days) in the placebo group and 55.5 days (interquartile range 50.5–56.0 days) in the selexipag group.
Primary efficacy end point. During the maintenance period, there was a decrease from the baseline week in the weekly average number of RP attacks for both the placebo and selexipag groups (Table 2). As the probabilities of observing a difference (selexipag minus placebo) of <0 (statistical significance) and of <–4 (clinical efficacy) in the mean weekly average number of RP attacks were below the proof‐of‐concept criteria of ≥0.95 and ≥0.5, respectively (observed probabilities 0.03 and 0.00, respectively), the primary objective was not met (Table 2 and Figure 2). Similar results were observed in the prespecified subgroups (Figure 3).
Table 2.
Summary of weekly RP attacksa
| Placebo (n = 32) | Selexipag (n = 27) | |
|---|---|---|
| Summary statistics | ||
| Average number of RP attacks during baseline week, mean ± SD | 21.5 ± 13.5 | 22.4 ± 15.9 |
| Weekly average number of RP attacks during maintenance period, mean ± SD | 14.2 ± 10.3 | 18.0 ± 14.1 |
| Statistical inference | ||
| Posterior weekly average number of RP attacks during the maintenance phase, mean ± SDb | 12.5 ± 1.1 | 15.9 ± 1.5 |
| Adjusted treatment difference, mean (90% CI)b, c | 3.4 (0.4–6.6) | |
| P for difference <0b | 0.03 | |
| P for difference <–4b | 0.00 | |
Per‐protocol set. 90% CI = 90% confidence interval.
Statistics from negative binomial model in Bayesian framework.
Selexipag minus placebo, adjusted for the average number of attacks of Raynaud's phenomenon (RP) during the baseline week.
Figure 2.
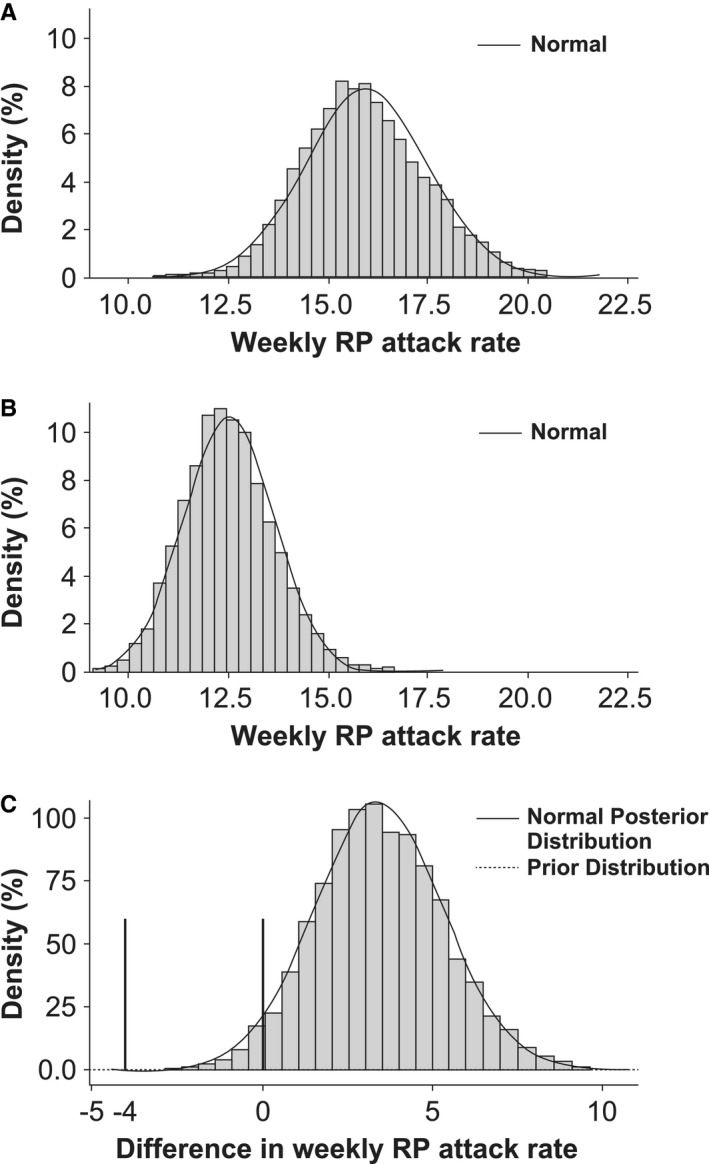
Posterior distribution of weekly Raynaud's phenomenon (RP) attack rate in selexipag‐treated patients (A) and placebo‐treated patients (B), and difference in weekly RP attack rate between treatment arms (per‐protocol set) (C). The probabilities of observing a difference (selexipag minus placebo) of <0 (statistical significance; right vertical bar) and of <–4 (clinical efficacy; left vertical bar) in the mean weekly average number of RP attacks were below the proof‐of‐concept criteria of ≥0.95 and ≥0.5, respectively (observed probabilities 0.03 and 0.00, respectively).
Figure 3.
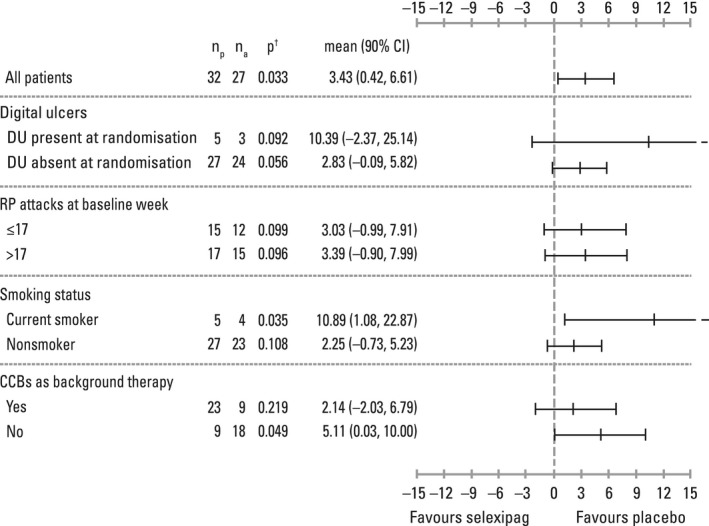
Forest plot of summary statistics of weekly attacks of Raynaud's phenomenon (RP) from posterior distribution of negative binomial Bayesian model (subgroup analyses; per‐protocol set). np = number of patients receiving placebo; na = number of patients receiving active treatment; P† = probability that the difference between the treatment means (selexipag minus placebo) for weekly average number of RP attacks in the maintenance period is <0; 90% CI = 90% confidence interval; DU = digital ulcer; CCBs = calcium‐channel blockers.
Other end points. The weekly average number of RP attacks during the maintenance period improved in 81.3% of placebo‐treated patients and in 63.0% of selexipag‐treated patients; it remained stable in 12.5% and 22.2% of patients, respectively, and worsened in 6.3% and 14.8% of patients, respectively. During the baseline week, the average RP attack duration was 21.5 minutes in the placebo group and 24.2 minutes in the selexipag group; the mean ± SD change in weekly average RP attack duration at week 8 was 4.6 ± 26.5 minutes in the placebo group and 2.7 ± 17.0 minutes in the selexipag group (n = 19 for both groups). The mean RCS at baseline was 3.3 in placebo‐treated patients (n = 30) and 4.0 in selexipag‐treated patients (n = 25). No difference was observed between placebo and selexipag in changes from baseline in RCS at any time during the study (data not shown).
At baseline, 5 placebo‐treated patients (15.6%) had a total of 8 digital ulcers, and 3 selexipag‐treated patients (11.1%) had a total of 3 digital ulcers (see Supplementary Table 2, http://onlinelibrary.wiley.com/doi/10.1002/art.40242/abstract). The number of new digital ulcers reported during the double‐blind period was low in both groups (0.2 per patient in the placebo group and 0.4 per patient in the selexipag group). At the end of treatment, 5 of the 8 baseline digital ulcers were healed in the placebo group and all baseline digital ulcers were healed in the selexipag group. There were no differences between treatment groups in the quality‐of‐life assessments (data not shown).
Safety and tolerability. Overall, 86.8% of placebo‐treated patients and 100% of selexipag‐treated patients reported ≥1 AE (Table 3). Most AEs were reported as mild or moderate in intensity (34.2% and 44.7%, respectively, with placebo; 16.7% and 61.1%, respectively, with selexipag). Serious AEs (SAEs) reported in the placebo group were RP worsening (2 patients), bronchitis (1 patient), and skin ulcer (1 patient); SAEs reported in the selexipag group were musculoskeletal chest pain (1 patient) and pulmonary hypertension (1 patient). In the placebo group, AEs leading to study drug discontinuation were bronchitis and RP worsening. In the selexipag group, all but 1 AE (syncope) leading to study drug discontinuation were AEs typically associated with therapies targeting the prostacyclin pathway. At least 1 AE typically associated with therapies targeting the prostacyclin pathway occurred in 55.3% of placebo‐treated patients and 91.7% of selexipag‐treated patients, of which headache was the most frequently reported (36.8% in the placebo group, 63.9% in the selexipag group). Most of these AEs were reported as mild or moderate in intensity (61.9% and 33.3%, respectively, with placebo; 36.4% and 48.5%, respectively, with selexipag). There were no deaths during the study.
Table 3.
Summary of AEsa
| Placebo (n = 38) | Selexipag (n = 36) | |
|---|---|---|
| Patients with AEs | 33 (86.8) | 36 (100.0) |
| Patients with SAEs | 4 (10.5) | 2 (5.6) |
| Patients with AEs leading to study drug discontinuation | 2 (5.3) | 6 (16.7)b |
| AEs occurring in ≥10% of patients in either treatment group | ||
| Headache | 14 (36.8) | 23 (63.9) |
| Nausea | 4 (10.5) | 13 (36.1) |
| Diarrhea | 5 (13.2) | 10 (27.8) |
| Dizziness | 2 (5.3) | 8 (22.2) |
| Pain in extremity | 2 (5.3) | 8 (22.2) |
| Pain in jaw | 0 (0.0) | 8 (22.2) |
| Fatigue | 3 (7.9) | 6 (16.7) |
| Myalgia | 2 (5.3) | 5 (13.9) |
| Arthralgia | 1 (2.6) | 5 (13.9) |
| Nasopharyngitis | 6 (15.8) | 4 (11.1) |
| Flushing | 1 (2.6) | 4 (11.1) |
| Back pain | 0 (0.0) | 4 (11.1) |
| Raynaud's phenomenon worsening | 4 (10.5) | 2 (5.6) |
| Abdominal pain, upper | 4 (10.5) | 1 (2.8) |
| Skin ulcer | 5 (13.2) | 0 (0.0) |
Safety analysis set for the double‐blind treatment period. Values are the number (%). SAEs = serious adverse events.
Includes 1 patient who discontinued due to an AE (headache) with onset during the run‐in period.
Discussion
The primary objective of the study was to evaluate the effect of selexipag on the frequency of RP attacks in patients with RP secondary to SSc. The rationale for this evaluation included the observation that other drugs targeting the prostacyclin pathway (e.g., intravenous iloprost) have shown some efficacy in RP secondary to SSc 18. However, selexipag did not reduce the number of RP attacks compared with placebo, and therefore the study did not meet its primary objective. The safety profile of selexipag was consistent with that observed previously in studies of patients with PAH 19, 25, with no new safety events identified.
Recent systematic reviews have noted that there have been few randomized controlled trials in RP, including in SSc‐related RP 26, 27, 28, 29. This dearth of studies is related to the complexities of study design in RP, which includes the need to run trials during the winter months to minimize the effects of seasonality 30. Despite its negative findings, our study is important because it draws attention to a number of learning points that will help to optimize clinical trial design. One particular point of note for future trial design is the issue of a placebo response in studies of RP. The placebo effect is often a confounder in the evaluation of RP in a clinical trial setting 12, 31. In our study, the placebo effect was notable, with many placebo‐treated patients reporting good outcomes. Patient‐reported outcomes may be particularly sensitive to the placebo effect 30; it may be that placebo‐treated patients experience fewer AEs and subsequently report better outcomes compared with patients receiving active treatment who are subject to side effects. Another potential contributing factor may be the difference between groups in the number of tablets taken; a greater proportion of placebo‐treated patients reached a higher placebo‐equivalent dose and therefore received more tablets. Taking more tablets may be associated with an increased placebo effect 32. Also, by specifically recruiting patients who report a high number of RP attacks, we may have selected a population in which the placebo effect is particularly apparent.
The timing and time period of the study may have imposed certain limitations. As stated earlier, seasonal variability is a potential confounding factor in studies that evaluate RP 30. In this study, the observation period was limited to the winter season to avoid seasonal variability. This restriction affected the time allowed to titrate selexipag up to the individualized highest tolerated dose, and further increases were not permitted during the maintenance period. In the Prostacyclin Receptor Agonist In Pulmonary Arterial Hypertension study, the titration period for selexipag (up to 1,600 μg twice daily) was 12 weeks 19, while in our study, for the same maximum allowed dose, selexipag was titrated to an individualized highest tolerated dose over 3 weeks. The short titration period in the present study meant that patients had a limited amount of time to adjust to AEs associated with selexipag treatment and, as a result, may not have reached their efficacious dose.
Structural vasculopathy and vasospasm are features of SSc 33, and it is possible that treatment acting to restore vasoreactivity could lead to greater awareness of RP attacks, thereby masking a potential treatment effect. This potential confounding factor is likely to be more acute over a short observation period, and a longer observation period may be warranted to discern any potential treatment effect.
The number of new digital ulcers reported during the double‐blind period was low in both treatment groups and lower than that in previous studies with double‐blind or open‐label treatment that specifically focused on digital ulcers 34, 35, 36, 37, 38. However, this study was not designed or powered to assess the impact of selexipag on digital ulcers.
Although not fully understood, the pathogenesis of RP secondary to SSc is linked to structural and functional changes in the vasculature leading to impaired blood flow and an imbalance in the levels of neurotransmitters controlling vasodilation and vasoconstriction, and SSc‐related RP has been associated with smoking, hormonal changes, and genetic factors 39. As there is evidence that selexipag is efficacious in other forms of vasculopathy and some evidence that other therapies targeting the prostacyclin pathway can have a positive effect on RP, differences in efficacy due to the route of administration also need to be considered. Intravenous iloprost has shown efficacy in reducing the number, severity, and duration of RP attacks 14, 15, 16, 17, 40, 41; however, consistent benefits have not been seen in trials evaluating oral iloprost 42, 43, 44. This raises the question of whether the route of administration of selexipag may have an impact on the potential for a treatment response. One might consider targeting SSc vasculopathy with IP prostacyclin receptor agonists, such as selexipag, but via a different mode of administration. Furthermore, future studies may include the use of objective measurements to assess clinically relevant end points in SSc vasculopathy.
There are a number of additional points to note about the design and conduct of our study. First, patients used an eDiary to record the frequency and duration of their RP attacks. Compared with a paper diary, this electronic tool was expected to facilitate better compliance and accuracy 45. Indeed, 95.9% of patients were compliant in completing the eDiary; only 2 of 38 placebo‐treated patients and 1 of 36 selexipag‐treated patients were excluded from the per‐protocol set due to lack of eDiary compliance, a key finding that benchmarks this novel method of recording RP attacks for future studies. Second, despite the short enrollment period, it was feasible to recruit a good number of patients who were representative of the patients with SSc seen in daily clinical practice; the enrolled population was comparable to that reported in large cohorts such as the EULAR Scleroderma Trials and Research group database 1. Third, a precise definition of RP attacks, based on the ACR/EULAR criteria 21, was used in this study, while earlier studies of intravenous or oral treatments for RP used less precise and inconsistent definitions of RP attacks.
In conclusion, treatment with selexipag in the present study did not reduce the number of RP attacks compared with placebo in patients with RP secondary to SSc. The safety profile of selexipag was consistent with that previously observed in studies of patients with PAH, with no new safety events identified. Some aspects of this study may offer a potentially robust template for future studies in RP secondary to SSc, including the use of the eDiary as an innovative tool in disease monitoring.
Author Contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Denton had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Denton, Frenoux, Frey, Herrick.
Acquisition of data
Denton, Hachulla, Riemekasten, Schwarting, Herrick.
Analysis and interpretation of data
Denton, Hachulla, Riemekasten, Schwarting, Frenoux, Frey, Le Brun, Herrick.
Role of the Study Sponsor
Actelion Pharmaceuticals was involved in all aspects of the study design, the collection, analysis, and interpretation of the data, and the decision to submit the manuscript for publication, with full involvement of the authors and investigators. Actelion Pharmaceuticals funded medical writing assistance provided by ApotheCom, London, UK. Publication of this article was not contingent upon approval by Actelion Pharmaceuticals.
Supporting information
Acknowledgment
We thank Lynda McEvoy, PhD, ApotheCom, London, UK, for medical writing assistance, which was funded by Actelion Pharmaceuticals.
Appendix A. The Raynaud Study Investigators
The Raynaud Study Investigators are as follows: C. Agard (CHU de Nantes–Hôpital Hôtel Dieu, PHUS–Service de Médecine Interne, Nantes, France), A. Ambach (Otto‐von‐Guericke‐Universität Magdeburg, Universitätsklinik für Dermatologie und Venerologie, Magdeburg, Germany), M. Anderson (University Hospital Aintree–Rheumatology Department, Liverpool, UK), M. Buch (Leeds Institute of Rheumatic and Musculoskeletal Medicine, Leeds, UK), P. Carpentier (CHU Grenoble–Hôpital Michallon, Service de médecine vasculaire, Grenoble, France), C. P. Denton (The Royal Free Hospital, Department of Rheumatology, Royal Free London NHS Foundation Trust, London, UK), J. Distler (Friedrich‐Alexander‐Universität Erlangen‐Nürnberg, Medizinische Klinik 3, Rheumatologie und Immunologie, Erlangen, Germany), E. Feist (Universitätsmedizin Berlin, Medizinische Klinik mit Schwerpunkt, Rheumatologie und Klinische Immunologie, Berlin, Germany), É. Hachulla (CHRU de Lille–Hôpital Claude Huriez, Service de Médecine Interne, Lille, France), A. Herrick (Salford Royal NHS Foundation Trust, Rheumatic Diseases Centre, Clinical Research Facility, Salford, UK), R. König (Kerckhoff‐Klinik GmbH, Abteilung für Rheumatologie und Klinische Immunologie, Bad Nauheim, Germany), P. Moinzadeh (Uniklinik Köln, Klinik und Poliklinik für Dermatologie und Venerologie, Köln, Germany), L. Mouthon (Hôpital Cochin, Service de Médecine Interne, Paris, France), J. Pauling (Royal National Hospital for Rheumatic Diseases, Department of Rheumatology, Bath, UK), G. Riemekasten (Universitätsmedizin Berlin, Medizinische Klinik mit Schwerpunkt, Rheumatologie und Klinische Immunologie, Berlin, Germany), A. Schwarting (Universitätsmedizin der Johannes Gutenberg‐Universität Mainz, I. Medizinische Klinik und Poliklinik, Rheumatologie und klinische Immunologie, Mainz, Germany), J. Sibilia (Hôpital de Hautepierre, Service de Rhumatologie, Strasbourg, France).
ClinicalTrials.gov identifier: NCT02260557.
Supported by Actelion Pharmaceuticals.
Dr. Denton has received consulting fees and/or speaking fees from Actelion Pharmaceuticals, GlaxoSmithKline, Bayer, Inventiva, and Takeda (less than $10,000 each) and research support from Actelion Pharmaceuticals, CSL Behring, and Novartis. Dr. Hachulla has received consulting fees and/or speaking fees from Actelion Pharmaceuticals, GlaxoSmithKline, Bayer, and Pfizer (less than $10,000 each) and research support from Actelion Pharmaceuticals and GlaxoSmithKline. Dr. Riemekasten has received consulting fees, speaking fees, and/or honoraria from Bayer, Schering/Bayer, and Actelion Pharmaceuticals. (less than $10,000 each) and research support from Actelion Pharmaceuticals. Dr. Schwarting has received consulting fees and/or speaking fees from GlaxoSmithKline (less than $10,000) and research support from Actelion Pharmaceuticals. Drs. Frenoux and Frey and Mr. Le Brun own stock or stock options in Actelion Pharmaceuticals. Dr. Herrick has received consulting fees and/or speaking fees from Actelion Pharmaceuticals, Apricus, and GlaxoSmithKline (less than $10,000 each) and research support from Actelion Pharmaceuticals.
[The copyright line for this article was changed on August 4, 2018 after original online publication.]
References
- 1. Meier FM, Frommer KW, Dinser R, Walker UA, Czirjak L, Denton CP, et al. Update on the profile of the EUSTAR cohort: an analysis of the EULAR Scleroderma Trials and Research group database. Ann Rheum Dis 2012;71:1355–60. [DOI] [PubMed] [Google Scholar]
- 2. Hughes M, Ong VH, Anderson ME, Hall F, Moinzadeh P, Griffiths B, et al. Consensus best practice pathway of the UK Scleroderma Study Group: digital vasculopathy in systemic sclerosis. Rheumatology (Oxford) 2015;54:2015–24. [DOI] [PubMed] [Google Scholar]
- 3. Cappelli L, Wigley FM. Management of Raynaud Phenomenon and digital ulcers in scleroderma. Rheum Dis Clin North Am 2015;41:419–38. [DOI] [PubMed] [Google Scholar]
- 4. Matucci‐Cerinic M, Kahaleh B, Wigley FM. Evidence that systemic sclerosis is a vascular disease [review]. Arthritis Rheum 2013;65:1953–62. [DOI] [PubMed] [Google Scholar]
- 5. Sunderkotter C, Riemekasten G. Pathophysiology and clinical consequences of Raynaud's phenomenon related to systemic sclerosis. Rheumatology (Oxford) 2006;45 Suppl 3:iii33–5. [DOI] [PubMed] [Google Scholar]
- 6. Flavahan NA. A vascular mechanistic approach to understanding Raynaud phenomenon. Nat Rev Rheumatol 2015;11:146–58. [DOI] [PubMed] [Google Scholar]
- 7. Milio G, Corrado E, Genova C, Amato C, Raimondi F, Almasio PL, et al. Iloprost treatment in patients with Raynaud's phenomenon secondary to systemic sclerosis and the quality of life: a new therapeutic protocol. Rheumatology (Oxford) 2006;45:999–1004. [DOI] [PubMed] [Google Scholar]
- 8. Wigley FM, Flavahan NA. Raynaud's phenomenon. N Engl J Med 2016;375:556–65. [DOI] [PubMed] [Google Scholar]
- 9. Kowal‐Bielecka O, Fransen J, Avouac J, Becker M, Kulak A, Allanore A, et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann Rheum Dis 2017;76:1327–39. [DOI] [PubMed] [Google Scholar]
- 10. Herrick AL, van den Hoogen F, Gabrielli A, Tamimi N, Reid C, O'Connell D, et al. Modified‐release sildenafil reduces Raynaud's phenomenon attack frequency in limited cutaneous systemic sclerosis. Arthritis Rheum 2011;63:775–82. [DOI] [PubMed] [Google Scholar]
- 11. Hachulla E, Hatron PY, Carpentier P, Agard C, Chatelus E, Jego P, et al. Efficacy of sildenafil on ischaemic digital ulcer healing in systemic sclerosis: the placebo‐controlled SEDUCE study. Ann Rheum Dis 2016;75:1009–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Roustit M, Blaise S, Allanore Y, Carpentier PH, Caglayan E, Cracowski JL. Phosphodiesterase‐5 inhibitors for the treatment of secondary Raynaud's phenomenon: systematic review and meta‐analysis of randomised trials. Ann Rheum Dis 2013;72:1696–9. [DOI] [PubMed] [Google Scholar]
- 13. Goundry B, Bell L, Langtree M, Moorthy A. Diagnosis and management of Raynaud's phenomenon. BMJ 2012;344:e289. [DOI] [PubMed] [Google Scholar]
- 14. Rademaker M, Cooke ED, Almond NE, Beacham JA, Smith RE, Mant TG, et al. Comparison of intravenous infusions of iloprost and oral nifedipine in treatment of Raynaud's phenomenon in patients with systemic sclerosis: a double blind randomised study. BMJ 1989;298:561–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Scorza R, Caronni M, Mascagni B, Berruti V, Bazzi S, Micallef E, et al. Effects of long‐term cyclic iloprost therapy in systemic sclerosis with Raynaud's phenomenon: a randomized, controlled study. Clin Exp Rheumatol 2001;19:503–8. [PubMed] [Google Scholar]
- 16. Wigley FM, Seibold JR, Wise RA, McCloskey DA, Dole WP. Intravenous iloprost treatment of Raynaud's phenomenon and ischemic ulcers secondary to systemic sclerosis. J Rheumatol 1992;19:8. [PubMed] [Google Scholar]
- 17. Wigley FM, Wise RA, Seibold JR, McCloskey DA, Kujala G, Medsger TA Jr, et al. Intravenous iloprost infusion in patients with Raynaud phenomenon secondary to systemic sclerosis: a multicenter, placebo‐controlled, double‐blind study. Ann Intern Med 1994;120:199–206. [DOI] [PubMed] [Google Scholar]
- 18. Pope J, Fenlon D, Thompson A, Shea B, Furst D, Wells G, et al. Iloprost and cisaprost for Raynaud's phenomenon in progressive systemic sclerosis. Cochrane Database Syst Rev 2000:CD000953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sitbon O, Channick R, Chin KM, Frey A, Gaine S, Galie N, et al. Selexipag for the treatment of pulmonary arterial hypertension. N Engl J Med 2015;373:2522–33. [DOI] [PubMed] [Google Scholar]
- 20. European Medicines Agency . Uptravi summary of product characteristics. 2016. URL: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/003774/WC500207173.pdf.
- 21. Van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 2013;65:2737–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Merkel PA, Herlyn K, Martin RW, Anderson JJ, Mayes MD, Bell P, et al. Measuring disease activity and functional status in patients with scleroderma and Raynaud's phenomenon. Arthritis Rheum 2002;46:2410–20. [DOI] [PubMed] [Google Scholar]
- 23. Steen VD, Medsger TA Jr. The value of the Health Assessment Questionnaire and special patient‐generated scales to demonstrate change in systemic sclerosis patients over time. Arthritis Rheum 1997;40:1984–91. [DOI] [PubMed] [Google Scholar]
- 24. Fries JF, Spitz P, Kraines RG, Holman HR. Measurement of patient outcome in arthritis. Arthritis Rheum 1980;23:137–45. [DOI] [PubMed] [Google Scholar]
- 25. Simonneau G, Torbicki A, Hoeper MM, Delcroix M, Karlocai K, Galie N, et al. Selexipag: an oral, selective prostacyclin receptor agonist for the treatment of pulmonary arterial hypertension. Eur Respir J 2012;40:874–80. [DOI] [PubMed] [Google Scholar]
- 26. Herrick A. Raynaud's phenomenon (secondary). BMJ Clin Evid 2008;2008:1125. [PMC free article] [PubMed] [Google Scholar]
- 27. Stewart M, Morling JR. Oral vasodilators for primary Raynaud's phenomenon. Cochrane Database Syst Rev 2012:CD006687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Garcia de la Pena Lefebvre P, Nishishinya MB, Pereda CA, Loza E, Sifuentes Giraldo WA, Roman Ivorra JA, et al. Efficacy of Raynaud's phenomenon and digital ulcer pharmacological treatment in systemic sclerosis patients: a systematic literature review. Rheumatol Int 2015;35:1447–59. [DOI] [PubMed] [Google Scholar]
- 29. Ennis H, Hughes M, Anderson ME, Wilkinson J, Herrick AL. Calcium channel blockers for primary Raynaud's phenomenon. Cochrane Database Syst Rev 2016;2:CD002069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wilkinson J. Design and reporting of randomised controlled trials for Raynaud's phenomenon In: Wigley FM, Herrick AL, Flavahan NA, editors. Raynaud's phenomenon: a guide to pathogenesis and treatment. New York: Springer‐Verlag; 2015. p. 287–97. [Google Scholar]
- 31. Khanna D, Gladue H, Seibold JR. Clinical outcome measures in Raynaud's phenomenon In: Wigley FM, Herrick AL, Flavahan NA, editors. Raynaud's phenomenon: a guide to pathogenesis and treatment. New York: Springer; 2015, p. 279–86. [Google Scholar]
- 32. De Craen AJ, Moerman DE, Heisterkamp SH, Tytgat GN, Tijssen JG, Kleijnen J. Placebo effect in the treatment of duodenal ulcer. Br J Clin Pharmacol 1999;48:853–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Guiducci S, Giacomelli R, Cerinic MM. Vascular complications of scleroderma. Autoimmun Rev 2007;6:520–3. [DOI] [PubMed] [Google Scholar]
- 34. Matucci‐Cerinic M, Denton CP, Furst DE, Mayes MD, Hsu VM, Carpentier P, et al. Bosentan treatment of digital ulcers related to systemic sclerosis: results from the RAPIDS‐2 randomised, double‐blind, placebo‐controlled trial. Ann Rheum Dis 2011;70:32–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chung L, Ball K, Yaqub A, Lingala B, Fiorentino D. Effect of the endothelin type A‐selective endothelin receptor antagonist ambrisentan on digital ulcers in patients with systemic sclerosis: results of a prospective pilot study. J Am Acad Dermatol 2014;71:400–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Khanna D, Denton CP, Merkel PA, Krieg T, Le Brun FO, Marr A, et al. Effect of macitentan on the development of new ischemic digital ulcers in patients with systemic sclerosis: DUAL‐1 and DUAL‐2 randomized clinical trials. JAMA 2016;315:1975–88. [DOI] [PubMed] [Google Scholar]
- 37. Korn JH, Mayes M, Matucci Cerinic M, Rainisio M, Pope J, Hachulla E, et al. Digital ulcers in systemic sclerosis: prevention by treatment with bosentan, an oral endothelin receptor antagonist. Arthritis Rheum 2004;50:3985–93. [DOI] [PubMed] [Google Scholar]
- 38. Brueckner CS, Becker MO, Kroencke T, Huscher D, Scherer HU, Worm M, et al. Effect of sildenafil on digital ulcers in systemic sclerosis: analysis from a single centre pilot study. Ann Rheum Dis 2010;69:1475–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Herrick AL. Pathogenesis of Raynaud's phenomenon. Rheumatology (Oxford) 2005;44:587–96. [DOI] [PubMed] [Google Scholar]
- 40. McHugh NJ, Csuka M, Watson H, Belcher G, Amadi A, Ring EF, et al. Infusion of iloprost, a prostacyclin analogue, for treatment of Raynaud's phenomenon in systemic sclerosis. Ann Rheum Dis 1988;47:43–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yardumian DA, Isenberg DA, Rustin M, Belcher G, Snaith ML, Dowd PM, et al. Successful treatment of Raynaud's syndrome with iloprost, a chemically stable prostacyclin analogue. Br J Rheumatol 1988;27:220–6. [DOI] [PubMed] [Google Scholar]
- 42. Belch JJ, Capell HA, Cooke ED, Kirby JD, Lau CS, Madhok R, et al. Oral iloprost as a treatment for Raynaud's syndrome: a double blind multicentre placebo controlled study. Ann Rheum Dis 1995;54:197–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Black CM, Halkier‐Sørensen L, Belch JJ, Ullman S, Madhok R, Smit AJ, et al. Oral iloprost in Raynaud's phenomenon secondary to systemic sclerosis: a multicentre, placebo‐controlled, dose‐comparison study. Br J Rheumatol 1998;37:952–60. [DOI] [PubMed] [Google Scholar]
- 44. Wigley FM, Korn JH, Csuka ME, Medsger TA Jr, Rothfield NF, Ellman M, et al. Oral iloprost treatment in patients with Raynaud's phenomenon secondary to systemic sclerosis: a multicenter, placebo‐controlled, double‐blind study. Arthritis Rheum 1998;41:670–7. [DOI] [PubMed] [Google Scholar]
- 45. Lauritsen K, Degl’ Innocenti A, Hendel L, Praest J, Lytje MF, Clemmensen‐Rotne K, et al. Symptom recording in a randomised clinical trial: paper diaries vs. electronic or telephone data capture. Control Clin Trials 2004;25:585–97. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials