

Abstract
There is strong interest in the production of bispecific monoclonal antibodies that can simultaneously bind two distinct targets or epitopes to achieve novel mechanisms of action and efficacy. Regeneron's bispecific technology, based upon a standard IgG, consists of a heterodimer of two different heavy chains, and a common light chain. Coexpression of two heavy chains leads to the formation of two parental IgG impurities, the removal of which is facilitated by a dipeptide substitution in the Fc portion of one of the heavy chains that ablates Fc Protein A binding. Therefore, the affinity capture (Protein A) step of the purification process must perform both bulk capture and high resolution of these mAb impurities, a task current commercially available resins are not designed for. Resolution can be further impaired by the ability of Protein A to bind some antibodies in the variable region of the heavy chain (VH). This article details development of a novel Protein A resin. This resin combines an alkali stable ligand with a base matrix exhibiting excellent mass transfer properties to allow high capacity single step capture and resolution of bispecific antibodies (bsAbs) with high yields. The developed resin, named MabSelect SuRe™ pcc, is implemented in GMP production processes for several bsAbs. © 2018 The Authors Biotechnology Progress published by Wiley Periodicals, Inc. on behalf of American Institute of Chemical Engineers Biotechnol. Prog., 34:650–658, 2018
Keywords: bispecific antibody, protein A chromatography, MabSelect SuRe™, affinity chromatography, z‐domain
Introduction
Bispecific antibodies (bsAbs) are antibody‐derived proteins with the ability to bind to two different epitopes on the same or different antigens.1 As such, they combine specificities of two antibodies: an attractive therapeutic concept, especially in the fields of immuno‐oncology and immune disease.2, 3, 4, 5, 6 A variety of novel molecular mechanisms have been proposed, including: recruitment of immune cells to target cells, simultaneous inhibition of two cell surface receptors, simultaneous blocking of two ligands, and receptor cross‐linking.2 The combination of two binding specificities on a single molecule could also be attractive without a mechanistic necessity, as it would avoid the complicated and costly development of combination therapies.7
There is no one common molecular structure of a bsAb. On the contrary, over the past two decades over 60 different formats of bsAbs have been proposed, with varying size, valency, flexibility, half‐life, biodistribution, ease of manufacture, options for Fc‐mediated effector functions, and immunogenic potential.8, 9, 10 Generally, formats can be divided into two major classes, those bearing an Fc region and those lacking an Fc region.8 Bispecifics from both classes have achieved marketing approval. Catumaxomab, an Fc‐containing bsAb for the treatment of patients with malignant ascites was approved in 2009.11 Blinatumomomab was approved in 2014 for the treatment of Philadelphia chromosome‐negative precursor B cell ALL.12, 13, 14 Blinatumomomab, a bispecific T‐cell engager (BiTE®) is an example of a format lacking an Fc region. It is based on the two single‐chain variable fragments joined via a flexible linker.
Regeneron's bsAb format was designed as a fully human IgG antibody.15 It consists of a heterodimer of two different heavy chains, which confer binding specificity, and a common light chain. Therefore, light‐heavy chain pairing issues are addressed by choosing heavy chains that can retain their different specificities but employ identical light chains. Coexpression of two different heavy chains does however lead to the formation of two parental IgG impurities. The removal of these impurities is facilitated by a dipeptide substitution in the Fc portion of one of the heavy chains (then named Fc*) that ablates Protein A binding (Figure 1). This allows the isolation of the bispecific dimer via selective elution from a Protein A column. The Fc*‐containing parental will flow through the column, while the bispecific can be separated from the Protein A binding parental by using the decreased avidity with which the bispecific binds to Protein A. The two amino acid residues substituted are taken from the equivalent region in the IgG3 isotype; therefore, no new non‐human potentially immunogenic sites are introduced. While the exact residues replaced can vary depending on the IgG isotype and individual antibody, in all cases the substitutions, replace the histidine residue critical for Fc‐Protein A binding.15
Figure 1.
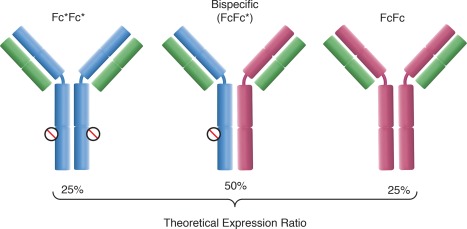
Bispecific (FcFc*) and novel product‐related parental antibody impurities (FcFc, Fc*Fc*) expressed in the production bioreactor. The theoretical expression ratio assumes equal production of either heavy chain and no thermodynamic preference for the formation of either quaternary structure. The star substitution present on the Fc* heavy chain (blue) is indicated via the circle. The Protein A binding Fc heavy chain lacking the star substitution is shown as red, and the common light chain is green.
The drawback of this approach is that, assuming that Fc* and the unmodified heavy chain (Fc) are produced in equal amounts and formation of the bispecific is thermodynamically equivalent to formation of the FcFc and Fc*Fc* parental impurities, the theoretical expression ratio will be 1:2:1 (FcFc:bsAb:Fc*Fc*). This means cell culture productivity is intrinsically half that of a standard monoclonal antibody. The advantage over other Fc‐containing bispecific formats is that the fully human nature of the antibody minimizes immunogenicity while maximizing molecule stability and pharmacokinetics. An additional advantage over similar full length antibody formats, such as the knobs‐into‐holes design,16, 17, 18 is that if parental antibodies or other variants do form with these techniques, their removal can be highly challenging. In contrast, the star substitution eases purification development by providing a well characterized, platformable mechanism of separation of parental antibody impurities.
The classical interaction between Staphylococcal Protein A (SpA) and the Fc region has been studied in detail using X‐ray crystallography and exploited by the Fc* substitution, which ablates the interaction.19, 20 However, a subset of antibodies bind Protein A in the variable region of the heavy chain (Figure 2).21, 22, 23, 24 In particular, IgGs that contain heavy chains derived from the VH3 gene family have been shown to exhibit this behavior. Nearly half of human VH germline genes belong to this family.25, 26 Because of this, on SpA‐based resins, many bsAbs exhibit poor resolution of the two binding species as well as retention of the non‐binding Fc*Fc* parental antibody. This is because VH binding reduces the avidity difference between the bispecific and the FcFc parental, and Fc*Fc* parental is also retained by VH binding. This phenomenon can be resolved through the use of the alkali stabilized MabSelect SuRe ligand. This is because while binding studies between SpA and antibodies have shown that all five domains of SpA (E, D, A, B, and C) bind antibodies via the Fc region, only domains D and E exhibit significant Fab binding.23, 27, 28 As the MabSelect SuRe ligand is a tetramer of the Z‐domain, a protein‐engineered version of the native SpA B‐domain, it would be expected to lack significant Fab binding, and this has been demonstrated in multiple studies.23, 27, 29, 30
Figure 2.
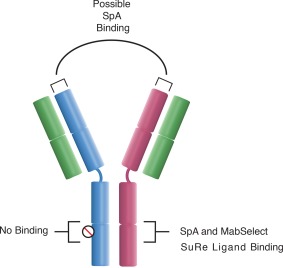
Binding sites of Staphylococcal Protein A (SpA) and the MabSelect SuRe ligand to IgG1, IgG2, and IgG4 antibodies. While both ligands bind in the CH2‐CH3 interfacial region, only SpA is capable of strongly binding antibodies of the VH3 gene family in the VH region.
This finding is also supported by previous work where it was shown that use of MabSelect SuRe allows for flow‐through removal of the Fc*Fc* parental antibody during column loading.31 However, commercially available resins functionalized with the MabSelect SuRe ligand did not provide adequate separation of the bispecific from the FcFc parental. This necessitated insertion of another Protein A step after primary capture to remove this SpA binding impurity. Resolution was achieved via the use of a commercially available resin (POROS™ MabCapture™ A, Thermo Scientific) exhibiting superior mass transfer and resolution characteristics based on (i) large through pores that give an element of perfusive flow in the base matrix, (ii) a large mean pore diameter of 1,100 Angstroms, and (iii) a smaller average particle size of 45 µm (c.f. to MabSelect SuRe resin average particle size of 85 µm).32, 33
Although this resin used SpA, the Fab‐binding was minimized via the use of chaotropic salts in the elution buffer. A salt tolerant multimodal resin was then used in a positive mode as both a polishing step, and to achieve buffer exchange. The resultant process was broadly applicable, with bispecific purities (Eq. 1) >95% and a robust operating space. However, the requirement of extra chromatographic step was compounded by the increased cost of Protein A resins compared to nonproteinaceous ligands.34, 35 This led to the desire to create a resin that could combine the mass transfer benefits of the POROS MabCapture A base matrix with the MabSelect SuRe ligand to allow single step affinity chromatography for all star substitution‐based bsAbs.
Materials and Methods
Materials
All bsAbs (C, D, and F) used in this study were expressed in CHO cells and produced at Regeneron Pharmaceuticals. Commercially available chromatographic resins used were obtained from their manufacturers: MabSelect SuRe and MabSelect SuRe pcc (GE Healthcare), and POROS MabCapture A (Thermo Scientific). All chemicals used were supplied by J.T. Baker. Developmental resins were supplied by GE Healthcare Custom Design Resin Group.
Equipment
Laboratory scale chromatographic separations were performed using an ÄKTA™ avant 25 chromatographic system (GE Healthcare) and 1.0 cm inner diameter (ID) Omnifit® Benchmark chromatography columns (Omnifit Ltd.). Pilot‐ and production‐scale chromatography was conducted on a ÄKTAprocess™ chromatography skid using either 20 cm ID INdEX™ chromatography columns or ReadyToProcess™ MabSelect SuRe pcc 20 columns (cat#29–1875‐64; GE Healthcare), with the exception of pilot scale pressure‐flow data that used a Axichrom™ 200 (GE Healthcare). UPLC analysis leveraged an ACQUITY® UPLC system from Waters Corporation. All material was derived from cell culture using either a 500 L or 2,000 L single use bioreactor (Thermo Scientific).
Pilot‐scale preparation of affinity capture pools for chromatographic development
When clarified cell culture fluid was not used directly, load material for affinity resolving development was produced by affinity capture chromatography using 20 ± 1 cm bed height MabSelect SuRe columns to remove the Fc*Fc* parental. After equilibration with 2 CV of 20 mM sodium phosphate pH 7.2, the columns were loaded to 10–40 g FcFc+ bsAb per L of packed bed with clarified cell culture fluid. Columns were washed and all bound protein eluted in 3 CVs using a proprietary buffer system at pH 3.0. After elution a 2 CV column strip is performed to regenerate the column. The entire elution peak containing FcFc and bispecific was collected and neutralized to pH 7.0 ± 0.2 with 2 M Tris base.
Resin prototype screening using gradient elution methodology
Chromatography resins screened were packed to 20 ± 1.0 cm bed height in 1.0 cm ID columns. Columns were equilibrated with 2 CVs of 20 mM sodium phosphate, pH 7.2 before load application of bsAb C to 10 g FcFc + bsAb/L. Load material had been previously subjected to affinity capture as described above, and was 77% bispecific purity as per Eq. 1. Following loading, columns were washed with a proprietary wash buffer system and eluted in 40 mM acetate, 500 mM calcium chloride, pH 6–3 over 20 CV. All steps were performed at a 4 min residence time. UNICORN™ 6.1 software (GE Healthcare) was used for chromatographic analysis including calculation of chromatographic peak resolution (Rs see Eq. 2) assuming Gaussian peaks using the width at half height method (resolution algorithm 3).
Affinity resolving chromatography using isocratic elution approach
All affinity resolving chromatography runs were performed using 20 ± 1 cm bed height columns. Columns were equilibrated with 2 CVs of 20 mM sodium phosphate, pH 7.2 before load application. Following loading, columns were washed with a proprietary wash buffer system and eluted using 40 mM acetate, 500 mM calcium chloride at a specified pH and elution volume. Pool collection began 0.5 CV into the elution volume and was terminated at the end of the elution step. Following elution, columns were stripped with 2 CV of buffer. Column clean in place (CIP) was performed with 0.5 N NaOH at a 15 min contact time. Statistical design of experiments (DoE), analysis, and modeling were performed using JMP® 11.1.1 (SAS Institute Inc). Small‐scale pools were neutralized to pH 7–8 using 2 M Tris base. Pilot‐ and production‐scale pools were buffer exchanged by standard flat sheet ultrafiltration techniques into low conductivity buffer, subjected to viral inactivation by low pH hold, neutralized, and filtered before process‐ and product‐related impurity measurement. Yield was calculated over the affinity resolving step only.
Process‐ and product‐related impurity measurement
Soluble aggregate quantification was achieved by two ACQUITY UPLC PrST SEC Columns, 200 Å, 1.7 µm, 4.6 × 150 mm2 cat#186005225 in series in a 10 mM sodium phosphate, 500 mM sodium chloride, pH 7.0 mobile phase. Bispecific purity was measured using three prepacked POROS A 20 µm columns (2.1 × 30 mm2, 0.1 mL) cat#2‐1001‐00 in series and an isocratic elution buffer system. Bispecific and FcFc titers were measured using a POROS A 20 µm column (2.1 × 30 mm2, 0.1 mL) cat#2–1001‐00, and Fc*Fc* titers were measured by loading the Protein A flowthrough over a POROS G 20 µm column (2.1 × 30 mm2, 0.1 mL) cat#2‐1002‐00 (Thermo Scientific). HCP quantification was performed using a commercially available ELISA kit cat#F550 (Cygnus Technologies). Leached affinity ligand was quantified using the commercially available ELISA kit cat#F400 (Cygnus Technologies).
Results and Discussion
Base matrix selection and screening
The aim of this study was to couple the MabSelect SuRe ligand to a base matrix capable of resolving the bispecific from the FcFc parental, while maintaining acceptable flow properties for large‐scale commercial protein production for pharmaceutical applications. Three agarose‐based prototype resins with a range of mean particle sizes and mean pore diameters were provided (Table 1). For base matrix evaluation, ligand density was fixed at an equivalent level to the commercially available MabSelect SuRe. MabSelect SuRe resin was used as a control.
Table 1.
Properties of Chromatographic Base Matrices Studied
| Base Matrix | Average Particle Size (d50V) | Relative Mean Pore Diameter |
|---|---|---|
| MabSelect SuRe (control) | 85 | Large |
| MabSelect Xtra™ | 75 | Extra large |
| Capto ImpRes | 40 | Medium |
| Capto ImpAct | 50 | Large |
Bispecific antibody C (bsAb C) was used as a model bsAb for resin screening. Table 2 details attributes of this and other bsAbs used in this study. Resins were loaded to 10 g FcFc + bsAb/L resin with load bispecific purity (Eq. 1) of 77%. For the purposes of screening the prototype resins, elution was achieved with a pH 6‐3 pH gradient over 20 column volumes (CV) rather than by isocratic step elution. This allowed measurement of the chromatographic peak resolution obtained between bsAb C and FcFc peaks as calculated by Eq. 2. Resolutions varied from 1.06 to 1.66 between the four resins studied (Figure 3).
Table 2.
Attributes of the bsAbs Studied
| BsAb | Isoelectric Point | Subtype | Member of VH3 Gene Family? |
|---|---|---|---|
| C | 9.2 | IgG1 | Y |
| D | 8.8 | IgG4 | Y |
| F | 9.3 | IgG4 | Y |
Figure 3.
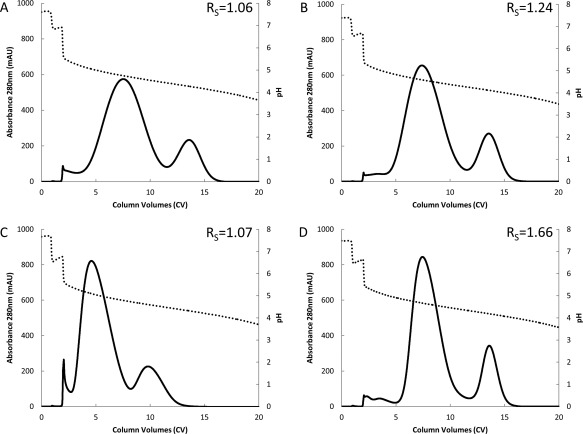
Chromatograms illustrating pH (dotted) and absorbance at 280 nm (solid) during elution step for purification of bsAb C using either MabSelect SuRe (A), or prototype resins with the MabSelect SuRe ligand attached to MabSelect Xtra base matrix (B), Capto ImpRes base matrix (C), or Capto ImpAct base matrix (D). Resolution (Rs) obtained between bsAb and FcFc parental are displayed in the top right of either pane.
All three prototype resins demonstrated improved resolution (Rs = 1.07 – 1.66) from MabSelect SuRe control (Rs = 1.06). As might be expected for separation of a large protein such as an antibody, resolution seems to be related to both average particle size and mean pore diameter. Specifically, the Capto™ ImpAct base matrix combined the second smallest (50 μm) particle size with a larger mean pore diameter and was found to provide the best resolution of tested resins. Based on these data, Capto ImpAct was chosen as the base matrix for the new resin.
Influence of ligand density upon dynamic binding capacity and resolution
The Capto ImpAct base matrix was functionalized with MabSelect SuRe ligand at a range of ligand densities (2‐11 mg ligand per mL of resin) and the dynamic binding capacity (g FcFc + bsAb/L) at 5% breakthrough of bsAb (DBC5%) determined at a four minute residence time with bsAb C (see Figure 4). It should be noted the bsAb breaks through before the FcFc parental via a displacement mechanism likely caused by the avidity difference between the two molecules (data not shown). A higher ligand density on the chromatography medium directly translates into an increased binding capacity, but diminishing results are observed at higher ligand densities. Increasing Protein A ligand density leading to a situation of diminishing returns for binding capacity due to increased steric hindrance on the ligand surface is studied in a previous work.34
Figure 4.
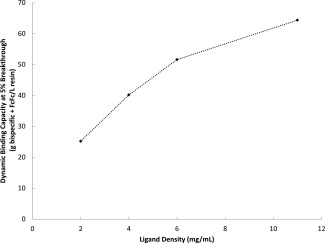
Dynamic binding capacity observed at 5% breakthrough for BsAb C in relation to the MabSelect SuRe ligand density attached to the Capto ImpAct base matrix.
Following this initial univariate screening experiment, a 31‐run D‐optimal custom DoE study was performed with the aim of determining an optimal ligand density that could give the most robust separation between the bispecific and the parental antibodies (FcFc and Fc*Fc*). An isocratic elution was chosen to simplify possible plant fit and technology transfer while also reducing buffer consumption and processing time. Other factors known to be critical to the bispecific separation from a previous study (elution pH and length [CV], and column loading) were also included as factors (Table 3).31 This allowed possible interactions between ligand density and other process parameters to be determined in order to make the most informed ligand density choice. Residence time for all chromatographic steps, including loading, wash, and elution, was held constant at 4 min (300 cm/h) as this was previously observed not to be a significant factor for bispecific yield or bispecific purity with isocratic elution. Yield and pool bispecific purity were studied as responses. The load material for the study was bsAb C that had been previously subjected to standard positive mode affinity chromatography using MabSelect SuRe resin. This resulted in a load bispecific purity for the DoE of 77%, with all parental antibody present being FcFc.
Table 3.
Factors Studied using Custom D‐Optimal DoEs Study to Determine Optimal Ligand Density for Prototype Resin
| Factor | Range Studied | Levels |
|---|---|---|
| Elution pH | 4.3–4.9 | 3 |
| Loading | 50–100% of determined DBC for ligand density | 3 |
| Ligand Density | 2–11 mg/mL | 4 |
| Step Elution Length | 4–6 CV | 3 |
Good models were obtained for both bispecific yield (R 2 = 0.97, = 0.96, RMSE = 4.2%, P < 0.001) and bispecific purity (R 2 = 0.97, = 0.97, RMSE = 1.4%, P < 0.001) using a standard least squares fit algorithm. Using a 95% confidence interval, ligand density was found be a significant parameter for both yield and purity (P < 0.0001 for both responses), and several interactions and quadratic effects were also identified (Table 4).
Table 4.
Significant Factors Determined using Custom D‐Optimal DoEs Study
| Factor | Yield | Bispecific Purity | ||
|---|---|---|---|---|
| Prob > |t| | Effect Size (%) | Prob > |t| | Effect Size (%) | |
| Elution pH | <0.0001 | −33.6 | <0.0001 | 8.2 |
| Loading | 0.0002 | 8.4 | 0.0550 | −1.1 |
| Ligand Density | <0.0001 | 11.5 | <0.0001 | 8.5 |
| Step Elution Length | – | – | – | – |
| Elution pH × Loading | <0.0001 | 14.4 | 0.0114 | 1.5 |
| Elution pH × Ligand Density | <0.0001 | −16.1 | <0.0001 | −9.8 |
| Elution pH × Step Elution Length | 0.0290 | 4.4 | – | – |
| Loading × Ligand Density | <0.0001 | 10.4 | 0.0006 | 2.4 |
| Loading × Step Elution Length | 0.0275 | −3.9 | – | – |
| Elution pH × Elution pH | 0.0009 | −7.7 | <0.0001 | −4.6 |
| Loading × Loading | <0.0001 | −6.4 | – | – |
| Ligand Density × Ligand Density | – | – | 0.037 | −2.1 |
The model predicted that attainable bispecific purities are optimized at increased ligand densities (Figure 5). A recent work observed competitive binding of antibody species on ProA resins despite the tight binding affinity.36 The speed of this displacement was shown to vary between different commercial SpA based resins, attributed to ligand attachment chemistry. We believe this competitive binding mechanism may explain this increase in purity with higher ligand densities. A higher ligand density may result in more frequent multiple binding events of the same antibody per ligand for the FcFc parental, which would not be possible with the bispecific because one of the two Fc binding sites has been ablated. Thus, a higher ligand density enhances the mechanism of separation via facilitating multiple binding to the FcFc parental.
Figure 5.
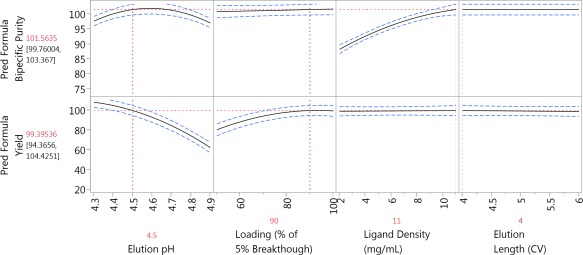
Prediction profiler depicting changes in bispecific purity and yield as a function of the factors studied in a custom D‐optimal DoE study (N = 31); 95% confidence intervals are shown in blue. Optimized conditions and predicted outputs are shown in red.
Optimizing the model for both yield and purity suggested conditions predicted to give 100% purity with >99% bispecific yield, as well as the highest DBC5%, at the highest ligand density studied (11 mg/mL). Therefore, a MabSelect SuRe ligand density of 11 mg/mL was targeted for the new resin, using the Capto ImpAct base matrix, and this new resin was named MabSelect SuRe pcc.
Sweet spot analysis was conducted using the models for bispecific yield and purity obtained. CaCl2 concentration in the elution buffer was held constant at 500 mM and elution volume fixed at 4 CVs. As shown in (Figure 6), the desirable operating window, or sweet spot, is indicated by the white area when imposing constraints of bispecific yield >80% (blue contour lines) and bispecific purities >95% (red contour lines).
Figure 6.
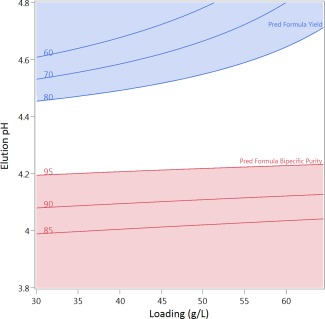
Sweet spot analysis utilizing models obtained from DoE evaluation of the design space for isocratic affinity chromatography for resolution of bsAb C from the parental (FcFc and Fc*Fc*) using the newly developed MabSelect SuRe pcc. The sweet spot is the white region where bispecific yield >80% and pool bispecific purity >95%.
In a previous work, similar studies were performed to evaluate both MabSelect SuRe and POROS MabCapture A resins for ability to separate bsAb C from the parental antibodies (FcFc and Fc*Fc*) in Protein A pool (i.e., affinity resolving chromatography step).31 Here, load material had also been previously subjected to standard positive mode affinity chromatography using MabSelect SuRe resin, resulting in 64% bispecific purity. The MabSelect SuRe pcc resin seems to exhibit a larger sweet spot, indicating increased process robustness, with the same bsAb when compared with the two resins used in the previous study. Further data gained directly comparing MabSelect SuRe pcc to MabSelect SuRe and POROS MabCapture A resin using other Regeneron bsAbs further supports this claim (data not shown). Moreover, the use of the MabSelect SuRe ligand, as compared with the SpA used by POROS MabCapture, alleviates the need for a prior affinity capture step to resolve the Fc*Fc* parental antibodies (FcFc and Fc*Fc*) decreasing cost of goods and increasing productivity of the purification process.
As studied in the previous work, the unit operation is sensitive to column loading and therefore also the bispecific purity of the load material.31 Thus, column loading, and the effect of resin cycling upon resin degradation, need to be carefully controlled as part of the manufacturing strategy. Furthermore, the reproducibility of the upstream process to provide material of consistent bispecific purity must be assessed as part of the development strategy.
Alkaline stability and pressure flow characteristics
The MabSelect SuRe ligand was originally developed to withstand stronger alkaline conditions than SpA, thereby facilitating CIP protocols with cleaning agents such as sodium hydroxide.37 As the developed MabSelect SuRe pcc resin utilizes this affinity ligand, it was expected to demonstrate similar alkaline stability to other commercially available resins produced with this ligand (e.g., MabSelect SuRe and MabSelect SuRe LX, [GE Healthcare]).
Alkaline stability was assessed at small‐scale (20 cm BH, 1.0 cm I.D.). Dynamic binding capacity was measured at a six minute residence time using bsAb D before (naïve resin) and after soaking in 0.1–0.5 M sodium hydroxide for up to 12 h. MabSelect SuRe pcc was found to retain >95% binding capacity after a 12 h exposure to up to 0.3 N sodium hydroxide. Furthermore, ∼90% binding capacity was retained with 12 h exposure of up to 0.5 M sodium hydroxide (Figure 7). Assuming a 15 min exposure time per CIP step, with CIP performed every four cycles, 12 h would correspond to ∼200 production cycles.
Figure 7.
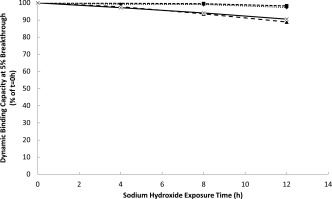
Dynamic binding capacity of MabSelect SuRe pcc as a function of exposure time to sodium hydroxide at 0.1 (♦), 0.3 (▪), 0.4(▲), and 0.5 (x) N concentrations.
Pressure flow characteristics of the new resin were assessed at pilot‐ (19.3 cm BH, 20 cm I.D.) and production‐scale (20.0 cm BH, 35.7 cm I.D.). These column diameters were deliberately chosen for evaluation as more representative of production batches, which would require larger columns with less wall support.38 Pressure drop over packed columns was measured at a range of linear velocities at 22°C using water as the mobile phase. The nonlinear profile observed is characteristic of compressible media (Figure 8). Based on these data a maximum linear velocity of 200 cm/h was recommended with 20 cm bed height columns to avoid pressure limitations with scale‐up.
Figure 8.
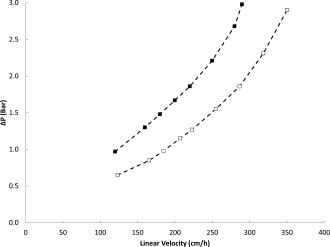
Pressure‐flow profiles for MabSelect SuRe pcc media packed to either 19.3 cm BH, 20 cm I.D. (□), or 20.0 cm BH, 35.7 cm I.D. (▪). Dashed lines connect the data points and are not model predictions.
Process performance and scale‐up
The performance of MabSelect SuRe pcc was assessed in the context of a complete downstream process for clinical manufacturing. As part of determining operating conditions for this process, a custom D‐optimal DoE (N = 12) was performed using a small‐scale column (20 cm BH, 1.0 cm I.D.) and clarified cell culture fluid containing bsAb F, a bsAbs, of the IgG4 subtype (Table 2). Elution pH (range of 0.6 pH units), elution length (4–6 CV), and column loading (35–65 g FcFc + bsAb/L resin), and all primary interactions and quadratic effects were considered as factors, based on the prior study (Table 4). It should be noted that 4–6 CV are used for elution to maximize yield, as the use of the higher elution pH to allow selective elution of the bispecific causes elution peak broadening. Residence time was held constant for all chromatographic steps at 6 min, the maximal recommended flow at production‐scale due to loss of wall support in larger columns. Good models were obtained for both responses of yield (R 2 = 0.99, = 0.97, RMSE = 4.4%, P < 0.001) and bispecific purity (R 2 = 0.98, = 0.97, RMSE = 0.62%, P < 0.001) using a least squares fit algorithm. Using the model, a sweet spot analysis was performed, with the desirable area defined as >80% yield and >95% bispecific purity (%; Figure 9). Based on this and further analysis of the predictive models, isocratic elution over 5 CV at pH 4.4 ± 0.1 was shown to predict robust operation, with >95% bispecific purity and >80% yields over a wide range of column loadings (35–65 g/L) throughout the entire pH range.
Figure 9.
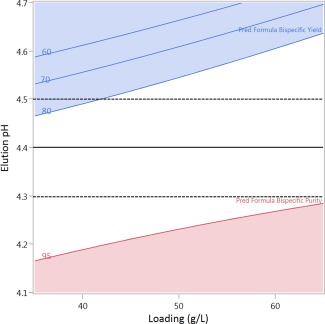
Sweet spot analysis using models obtained from DoE evaluation of the design space for isocratic affinity chromatography for resolution of bsAb F from the parentals (FcFc and Fc*Fc*) using MabSelect SuRe pcc (C). Elution volume is held constant at 5 CV. The sweet spot is the white region where bispecific yield >80% and pool bispecific purity >95%. The recommended elution pH of 4.4 is indicated by the solid line, with dotted lines at ±0.1 pH units to illustrate possible variation in the buffer during manufacturing. Expected column loading at pilot‐ and production‐scale is 50–60 g FcFc + bsAb/L.
The purification scheme developed for bsAb F was scaled up and process performance and product quality analyzed at pilot‐scale (500 L bioreactor) and during GMP production (2 kL bioreactor, N = 2). Load material was cell culture fluid clarified by direct depth filtration and all runs were performed at a constant residence time of 6 min. The results are shown in Table 5 and show a high level of reproducibility across scale and site. The goals of >80% yield and >95% bispecific purity were met for both scales of production. High molecular weight (HMW) species, leached Protein A and HCP levels observed in resulting filtered viral inactivated Protein A pool were comparable to typical values obtained at Regeneron Pharmaceuticals for a traditional mAb process using commercially available agarose base matrix Protein A resins.
Table 5.
Quantitative Purification Performance of Single Step Affinity Resolving Chromatography using MabSelect SuRe pcc at Pilot‐ and Production‐Scale
| Scale | Pilot Scale | Production Scale Batch 1 | Production Scale Batch 2 |
|---|---|---|---|
| Parameter | |||
| Bioreactor Size (L) | 500 | 2,000 | 2,000 |
| MabSelect SuRe pcc CV (L) | 6.3 | 20 | 20 |
| Column Loading (g FcFc + bsAb/L resin) | 60 | 56 | 53 |
| Output | |||
| Final Bispecific Purity (%) | 98.7 | 98.1 | 98.5 |
| HCP (ppm) | 113 | 229 | 210 |
| HMW (%) | 6.0 | 4.3 | 5.2 |
| Yield (%) | 92.3 | 92.4 | 83.0 |
| Leached Protein A (ppm) | 2.9 | 0.8 | 0.5 |
Conclusions
A previous study detailed an approach for purification of a fully human bsAbs format. This process was limited by the need for two Protein A resins when applied to bsAbs of the VH3 gene family. A novel Protein A resin, MabSelect SuRe pcc, has been developed that combines an alkali stabilized, non VH‐region binding ligand with a base matrix exhibiting excellent mass transfer properties. Thus, high capacity single step capture and resolution of native format bsAbs with high yield is achieved, and demonstrated at pilot‐ and production‐scale. This allows the consolidation of the two Protein A columns used in the prior approach, increasing productivity while decreasing cost of goods. A downstream processing platform has therefore been established to facilitate the manufacturing of fully human bsAbs.
Although MabSelect SuRe pcc was developed for the purification of bsAbs, the high DBC (>60 g/L) at moderate residence times, and the alkaline stability (0.5 N NaOH) compare favorably with other commercially available Protein A resins.39 The resin may be especially appropriate for use in continuous capture by periodic counter‐current chromatography (PCC).40 The ability of PCC to use shorter column bed heights could allow use of shorter residence times without pressure restrictions, important because of the relatively small average particle size (50 µm) of the MabSelect SuRe pcc resin.
Conflicts of Interest
The authors are currently or have been employees of either Regeneron Pharmaceuticals Inc. or GE Healthcare
Equations
Equation 1: Definition of bispecific purity
Equation 2: Equation used to determine peak resolution (Rs) using peak widths at half height (W 1/2) and their retention times (t R)
Notation
- BH
bed height
- bsAb
bispecific antibody
- CIP
clean in place
- CV
column volume
- DBC
dynamic binding capacity
- DoE
statistical design of experiments
- Fc*Fc*
parental antibody containing star substitution on both heavy chains
- FcFc
parental antibody containing native heavy chains
- FcFc*
bispecific antibody containing a single chain with the star substitution
- HCP
CHO host cell protein
- ID
inner diameter
- Rs
chromatographic peak resolution
- SpA
Staphylococcal Protein A
Acknowledgments
The authors thank Michelle LaFond, Mark Czeterko, Anthony DeBiase, Shey Ng, and Matthew Klaas' group for carrying out antibody productions, Benjamin Adams for column packing, and Michael Perrone, Audrey Rodriquez, and Kathir Muthusamy for impurity quantification. They acknowledge Scott Staton for invaluable help with figures.
Literature Cited
- 1. Byrne H, Conroy PJ, Whisstock JC, O'Kennedy RJ. A tale of two specificities: bispecific antibodies for therapeutic and diagnostic applications. Trends Biotechnol. 2013;31:621–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. May C, Sapra P, Gerber HP. Advances in bispecific biotherapeutics for the treatment of cancer. Biochem Pharmacol. 2012;84:1105–1112. [DOI] [PubMed] [Google Scholar]
- 3. Kontermann R. Dual targeting strategies with bispecific antibodies. mAbs 2012;4:182–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Demarest SJ, Hariharan K, Dong J. Emerging antibody combinations in oncology. mAbs 2011;3:338–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Muller D, Kontermann RE. Bispecific antibodies for cancer immunotherapy: current perspectives. BioDrugs 2010;24:89–98. [DOI] [PubMed] [Google Scholar]
- 6. Weidle UH, Kontermann RE, Brinkmann U. Tumor‐antigen‐binding bispecific antibodies for cancer treatment. Semin Oncol. 2014;41:653–660. [DOI] [PubMed] [Google Scholar]
- 7. Holmes D. Buy buy bispecific antibodies. Nat Rev Drug Discov. 2011;10:798–800. [DOI] [PubMed] [Google Scholar]
- 8. Kontermann RE, Brinkmann U. Bispecific antibodies. Drug Discov Today. 2015;20:838–847. [DOI] [PubMed] [Google Scholar]
- 9. Spiess C, Zhai Q, Carter PJ. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol Immunol. 2015;67:95–106. [DOI] [PubMed] [Google Scholar]
- 10. KleinSustmann C, Thomas C, Stubenrauch M, Croasdale K, Schanzer R, Brinkmann J, Kettenberger UH, Regula J, Schaefer TW. Progress in overcoming the chain association issue in bispecific heterodimeric IgG antibodies. mAbs 2012;4:653–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Linke R, Klein A, Seimetz D. Catumaxomab: clinical development and future directions. mAbs 2010;2:129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wolf E, Hofmeister R, Kufer P, Schlereth B, Baeuerle PA. BiTEs: bispecific antibody constructs with unique anti‐tumor activity. Drug Discov Today 2005;10:1237–1244. [DOI] [PubMed] [Google Scholar]
- 13. Baeuerle P, Kufer A, Bargou PR. BiTE: teaching antibodies to engage T‐cells for cancer therapy. Curr Opin Mol Ther. 2009;11:22–30. [PubMed] [Google Scholar]
- 14. Kufer P, Lutterbuse R, Baeuerle PA. A revival of bispecific antibodies. Trends Biotechnol. 2004;22:238–244. [DOI] [PubMed] [Google Scholar]
- 15. Smith EJ, Olson K, Haber LJ, Varghese B, Duramad P, Tustian AD, Oyejide A, Kirshner JR, Canova L, Menon J, Principio J, MacDonald D, Kantrowitz J, Papadopoulos N, Stahl N, Yancopoulos GD, Thurston G, Davis S. A novel, native‐format bispecific antibody triggering T‐cell killing of B‐cells is robustly active in mouse tumor models and cynomolgus monkeys. Sci Rep. 2016;5:17943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Merchant AM, Zhu Z, Yuan JQ, Goddard A, Adams CW, Presta LG, Carter P. An efficient route to human bispecific IgG. Nat Biotechnol. 1998;16:677–681. [DOI] [PubMed] [Google Scholar]
- 17. Jackman J, Chen Y, Huang A, Moffat B, Scheer JM, Leong SR, Lee WP, Zhang J, Sharma N, Lu Y, Iyer S, Shields RL, Chiang N, Bauer MC, Wadley D, Roose‐Girma M, Vandlen R, Yansura DG, Wu Y, Wu LC. Development of a two‐part strategy to identify a therapeutic human bispecific antibody that inhibits IgE receptor signaling. J Biol Chem. 2010;285:20850–20859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Spiess C, Bevers J III, Jackman J, Chiang N, Nakamura G, Dillon M, Liu H, Molina P, Elliott JM, Shatz W, Scheer JM, Giese G, Persson J, Zhang Y, Dennis MS, Giulianotti J, Gupta P, Reilly D, Palma E, Wang J, Stefanich E, Scheerens H, Fuh G, Wu LC. Development of a human IgG4 bispecific antibody for dual targeting of interleukin‐4 (IL‐4) and interleukin‐13 (IL‐13) cytokines. J Biol Chem. 2013;288:26583–26593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Deisenhofer J. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9‐ and 2.8‐A resolution. Biochemistry 1981;20:2361–2370. [PubMed] [Google Scholar]
- 20. Deisenhofer J, Jones TA, Huber R, Sjodahl J, Sjoquist J. Crystallization, crystal structure analysis and atomic model of the complex formed by a human Fc fragment and fragment B of protein A from Staphylococcus aureus. Hoppe Seylers Z Physiol Chem. 1978;359:975–985. [DOI] [PubMed] [Google Scholar]
- 21. Akerstrom B, Nilson BH, Hoogenboom H, Bjorck RL. On the interaction between single chain Fv antibodies and bacterial immunoglobulin‐binding proteins. J Immunol Methods. 1994;177:151–163. [DOI] [PubMed] [Google Scholar]
- 22. Inganas M. Comparison of mechanisms of interaction between protein A from Staphylococcus aureus and human monoclonal IgG, IgA and IgM in relation to the classical FC gamma and the alternative F(ab')2 epsilon protein A interactions. Scand J Immunol. 1981;13:343–352. [DOI] [PubMed] [Google Scholar]
- 23. Starovasnik MA, O'Connell MP, Fairbrother WJ, Kelley RF. Antibody variable region binding by Staphylococcal protein A: thermodynamic analysis and location of the Fv binding site on E‐domain. Protein Sci. 1999;8:1423–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vidal MA, Conde FP. Alternative mechanism of protein A‐immunoglobulin interaction the VH‐associated reactivity of a monoclonal human IgM. J Immunol. 1985;135:1232–1238. [PubMed] [Google Scholar]
- 25. Schroeder HW Jr., Hillson JL, Perlmutter RM. Structure and evolution of mammalian VH families. Int Immunol. 1990;2:41–50. [DOI] [PubMed] [Google Scholar]
- 26. Walter MA, Cox DW. Analysis of genetic variation reveals human immunoglobulin VH‐region gene organization. Am J Hum Genet. 1988;42:446–451. [PMC free article] [PubMed] [Google Scholar]
- 27. Ljungberg UK, Jansson B, Niss U, Nilsson R, Sandberg BE, Nilsson B. The interaction between different domains of staphylococcal protein A and human polyclonal IgG, IgA, IgM and F(ab')2: separation of affinity from specificity. Mol Immunol. 1993;30:1279–1285. [DOI] [PubMed] [Google Scholar]
- 28. Roben PW, Salem AN, Silverman GJ. VH3 family antibodies bind domain D of staphylococcal protein A. J Immunol. 1995;154:6437–6445. [PubMed] [Google Scholar]
- 29. Cedergren L, Andersson R, Jansson B, Uhlen M, Nilsson B. Mutational analysis of the interaction between staphylococcal protein A and human IgG1. Protein Eng. 1993;6:441–448. [DOI] [PubMed] [Google Scholar]
- 30. Ghose S, Allen M, Hubbard B, Brooks C, Cramer SM. Antibody variable region interactions with Protein A: implications for the development of generic purification processes. Biotechnol Bioeng. 2005;92:665–673. [DOI] [PubMed] [Google Scholar]
- 31. Tustian AD, Endicott C, Adams B, Mattila J, Bak H. Development of purification processes for fully human bispecific antibodies based upon modification of protein A binding avidity. mAbs 2016;8:828–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Afeyan NB, Gordon NF, Mazsaroff I, Varady L, Fulton SP, Yang YB, Regnier FE. Flow‐through particles for the high‐performance liquid chromatographic separation of biomolecules: perfusion chromatography. J Chromatogr. 1990;519:1–29. [DOI] [PubMed] [Google Scholar]
- 33. Whitney D, McCoy M, Gordon N, Afeyan N. Characterization of large‐pore polymeric supports for use in perfusion biochromatography. J Chromatogr A. 1998;807:165–184. [DOI] [PubMed] [Google Scholar]
- 34. Ghose S, Hubbard B, Cramer SM. Binding capacity differences for antibodies and Fc‐fusion proteins on protein A chromatographic materials. Biotechnol Bioeng. 2007;96:768–779. [DOI] [PubMed] [Google Scholar]
- 35. Follman DK, Fahrner RL. Factorial screening of antibody purification processes using three chromatography steps without protein A. J Chromatogr A 2004;1024:79–85. [DOI] [PubMed] [Google Scholar]
- 36. WeinbergZhang J, Crews S, Healy G, Carta E, Przybycien GT. Polyclonal and monoclonal IgG binding on protein A resins‐Evidence of competitive binding effects. Biotechnol Bioeng. 2017;114:1803–1812. [DOI] [PubMed] [Google Scholar]
- 37. Hober S, Nord K, Linhult M. Protein A chromatography for antibody purification. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;848:40–47. [DOI] [PubMed] [Google Scholar]
- 38. Stickel JJ, Fotopoulos A. Pressure‐flow relationships for packed beds of compressible chromatography media at laboratory and production scale. Biotechnol Prog. 2001;17:744–751. [DOI] [PubMed] [Google Scholar]
- 39. Bolton GR, Mehta KK. The role of more than 40 years of improvement in protein A chromatography in the growth of the therapeutic antibody industry. Biotechnol Prog. 2016;32:1193–1202. [DOI] [PubMed] [Google Scholar]
- 40. Godawat R, Brower K, Jain S, Konstantinov K, Riske F, Warikoo V. Periodic counter‐current chromatography – design and operational considerations for integrated and continuous purification of proteins. Biotechnol J. 2012;7:1496–1508. [DOI] [PubMed] [Google Scholar]