

Abstract
Mass spectrometry imaging (MSI) simultaneously detects and identifies the spatial distribution of numerous molecules throughout tissues. Currently, MSI is limited to providing a static and ex vivo snapshot of highly dynamic systems in which molecules are constantly synthesized and consumed. Herein, we demonstrate an innovative MSI methodology to study dynamic molecular changes of amino acids within biological tissues by measuring the dilution and conversion of stable isotopes in a mouse model. We evaluate the method specifically on hepatocellular metabolism of the essential amino acid l‐phenylalanine, associated with liver diseases. Crucially, the method reveals the localized dynamics of l‐phenylalanine metabolism, including its in vivo hydroxylation to l‐tyrosine and co‐localization with other liver metabolites in a time course of samples from different animals. This method thus enables the dynamics of localized biochemical synthesis to be studied directly from biological tissues.
Keywords: amino acids, dynamics, isotopic labeling, mass spectrometry imaging, metabolism
Mass spectrometry imaging (MSI) enables the study of molecular distributions and processes throughout biological tissues.1 Typically performed ex vivo on frozen sections, it provides only a static snapshot of inherently highly dynamic systems and cannot elucidate dynamic chemical conversions within the tissue. As MSI evolves to study localized biochemical pathways and their alteration in disease, spatially resolved dynamics of molecular synthesis and turnover becomes critical. Incorporation of stable isotopes into tissues, and thus their biosynthetic processes, provides a unique opportunity to study molecular kinetics in complex biological systems. Conventional MS modalities with the capability to analyze metabolic fluxes generally use stable‐isotope‐labeled precursor molecules.2 To date, such approaches have been widely employed using tissue/cell homogenates but are not able to detect spatial changes in heterogeneous tissues.3 Recent work has extended this approach to MSI to reveal localized incorporation throughout tissues. For example, nanoscale secondary ion mass spectrometry (SIMS) is employed to study localized turnover and isotopic incorporation in a variety of systems with subcellular resolution (ca. 50–100 nm).4 However, this approach suffers from the extensive fragmentation induced under dynamic SIMS conditions. Typically, such experiments monitor fragment signals of 13CH and 15N, making it difficult to assign incorporation to a specific biochemical precursor, especially if endogenous metabolism of the initially labeled compounds occurs. Louie et al. have developed similar approaches to study localized phospholipid synthesis following D2O administration using softer matrix‐assisted laser desorption ionization (MALDI).5 It was demonstrated that rates of lipid synthesis are altered in different tumor cell subpopulations, thus highlighting the need to study localized synthesis kinetics in tumor metabolism. In addition, MALDI‐MSI in combination with stable isotopes has been applied to study the formation of atherosclerotic plaques6 and the nitrogen cycle within plants.7
In this paper, we present a new method based on stable isotope‐labeling, which enables detection of localized amino acid metabolism over time for the first time. Stable isotopes of l‐phenylalanine (Phe) are commonly used in clinical studies to measure protein turnover.8 In liver, Phe is enzymatically hydroxylated into l‐tyrosine (Tyr), and altered levels are related to nonalcoholic steatohepatitis, nonalcoholic fatty liver disease, cirrhosis, and several cancer phenotypes.9 A bolus of ring‐13C6‐labeled l‐phenylalanine (13C6‐Phe) was injected into 11 mice and healthy liver tissue was collected at three time points (10, 30, and 60 min) after injection to track the incorporation of 13C6‐Phe into tissue using MALDI. As a control, four mice were injected with saline solution and sacrificed at 10 min (sham). All measurements were performed using four biological and two technical replicates.
To address the low ionization efficiency of many amino acids with MALDI, we performed on‐tissue derivatization using the reagent p‐N,N,N‐trimethylammonioanilyl N′‐hydroxysuccinimidyl carbamate iodide (TAHS).10 This enabled the sensitive detection of a variety of free amino acids and their labeled analogues as well as a suite of amine‐containing metabolites due to the addition of a quaternary nitrogen group. Tissues were imaged using MALDI‐Fourier transform ion cyclotron resonance (FTICR) mass spectrometry following on‐tissue derivatization. The experimental workflow is summarized in the Supporting Information, Figures S1 and S2. High resolution (25 μm) MSI data is provided in Figure S3 to demonstrate the preservation of tissue distributions during the sample preparation steps. Phe, Tyr, 13C6‐Phe, and 13C6‐Tyr could not be detected in nonderivatized tissue 10 minutes after tracer injection. In contrast, the derivatization enabled the detection throughout the tissues (Figure 1). Representative spectra for labeled and unlabeled Phe are shown in the Supporting Information, Figures S4 A and S3 B. In addition, TAHS derivatization enabled the simultaneous detection of approximately 60 amino metabolites in liver tissue (Supporting Information, Table S1 and S2). Using an accurate mass tolerance of <1 ppm, each m/z value could be assigned to a unique metabolite. Structural assignments of TAHS‐derivatized 13C6‐Phe and 13C6‐Tyr were also supported by tandem mass spectrometry, which revealed a characteristic TAHS fragment for derivatized metabolites (Figure S4). The detection of 13C6‐Tyr (Figure 1 B) indicated that 13C6‐labeled Phe was metabolized to 13C6‐Tyr through active hydroxylation in liver tissue.
Figure 1.
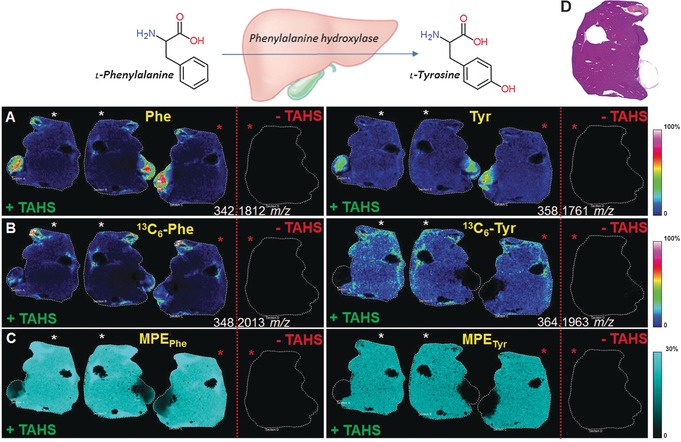
Representative MALDI‐FTICR‐MS images with (+TAHS) and without (−TAHS) derivatization showing the direct localization of the in vivo metabolism of Phe (left) and Tyr (right) in liver sections at 10 min after injection with 13C6‐Phe. (Top) l‐Phe is metabolized to l‐Tyr in the liver by phenylalanine hydroxylase. Consecutive sections with identical intersections, rotated 180°, are denoted by similarly colored asterisks. A) Molecular ion [M+TAHS]+ image of derivatized Phe and Tyr; B) molecular ion [M+TAHS]+ image of derivatized 13C6‐Phe and de novo synthesized 13C6‐Tyr; C) MPE of 13C6‐Phe and 13C6‐Tyr shows localized enrichment; D) H&E image of a representative tissue section.
Localized enrichment of 13C6‐Phe and 13C6‐Tyr in tissue was determined by calculating the ratio of labeled to total (labeled+unlabeled) peaks, also referred to as the molar percentage excess (MPE; Figure 1 C and Supporting Information). MPE was applied to measure the incorporation of 13C6‐Phe into liver and adjacent muscle and pancreas tissue. Computed for each location in 150‐μm increments, MPE values varied throughout the tissue (Figure 1 C). Although ionization efficiency and matrix effects can distort the absolute detection and quantification of metabolites (Supporting Information, Figures S5 A and S5 B),11 MPE values represent a robust measure (Figure S5C), since the tracer and target have the same chemical properties and are ionized in the same way.12 We obtained the same average MPE values for each biological replicate (n=4), measured in duplicate and in two randomized batches (standard error <1.2 %) (Supporting Information, Figure S6). Such reproducible and quantitative measurements are important to determine the kinetics of biologically altered amino acids, which are implicated in impairments in liver function.13
We measured the fraction of molecules isotopically labeled with 13C6 at 10, 30, and 60 min after bolus injection and compared these with control tissues to determine the kinetics of 13C6‐Phe. The MPE values for 13C6‐Phe and 13C6‐Tyr showed clear abundance throughout the liver at 10 min, indicating the transport and conversion of these labeled amino acids (Figure 2 B,C). At 30 min, 13C6‐Tyr levels were diminished, and 13C6‐Phe levels were cleared in liver tissue. By 60 min, both 13C6‐Phe and 13C6‐Tyr were cleared from liver tissue, indicating their predominant metabolism and incorporation into liver proteins (assessed by GC‐C‐IRMS in the Supporting Information, Figure S7). De novo hydroxylation was determined by the MPE ratio 13C6‐Tyr:13C6‐Phe to be approximately 1 (Figure 2 D). In addition, we measured enrichments in adjacent muscle and pancreas tissue (Figure S8). The MPE was lower for 13C6‐Tyr compared to 13C6‐Phe in the adjacent tissue at 10 min, which might be due to the absence of enzymes hydroxylating 13C6‐Phe into 13C6‐Tyr in non‐hepatic tissue.14 At 30 min, 13C6‐Phe and 13C6‐Tyr levels were elevated in adjacent pancreatic tissue (*, Figure S8A). These data suggest 13C6‐Phe and 13C6‐Tyr may have different uptake and diffusion mechanisms in pancreas compared to liver tissue.15
Figure 2.
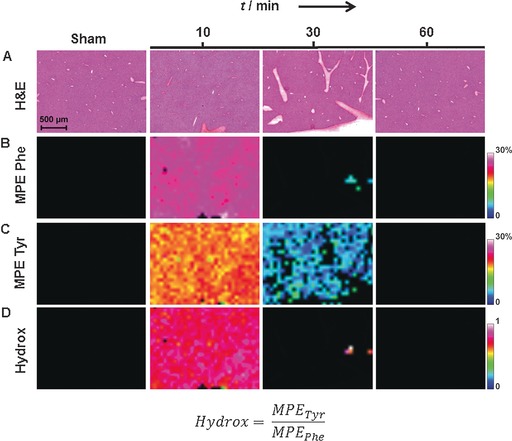
Detection of localized Phe and Tyr kinetics in liver tissue at three time points at a 150‐μm spatial resolution. Sections taken from mice 10 min after injection with saline (sham) are shown as a control. A) H&E stains; B–D) dynamic MS images for 13C6‐Phe MPE, 13C6‐Tyr MPE, and hepatic hydroxylation (Hydrox), respectively.
Spectra were generated from homogenized liver tissue using gas chromatography mass spectrometry (GC‐MS; Figure S6) and confirmed that 13C6‐Phe was successfully incorporated into tissue. Importantly, GC‐MS measurements gave similar enrichment trends across the time‐series as the MSI data, validating the accuracy of the MSI approach. These average enrichments may reflect the presence of other cells (muscle, pancreas, inflammatory cells) as the measurements were performed with homogenized tissues. This explains the slight differences in absolute enrichment between the methods.
The enrichment precision for MSI data was evaluated by comparing the detected natural enrichments of Phe and Tyr with their theoretical isotopic enrichments in tissues (M+1 vs. M+0, M+2 vs. M+0, and M+3 vs. M+0). In particular, the highly abundant Phe allowed more accurate reconstruction of low‐abundance natural 13C isotopes, whereas lower abundant isotopes (for example, Tyr M+3) may not be detected in all pixels due to a detection limit (Supporting Information, Table S3). Overall, an isotope ratio of more than 2 % is required to facilitate precise detection.
Principal component analysis combined with linear discriminant analysis (PCA‐LDA) was applied to MSI data on a pixel‐by‐pixel basis to identify the main metabolic differences across time points associated with Phe metabolism. The first discriminant function captures differences between matrix and tissue (data not shown). The second discriminant function (Supporting Information, Figure S9) demonstrated that the greatest metabolic differences were observed 10 min after tracer infusion, followed by 30 min, and almost reverted back to normal after 60 min, which is in agreement with the enrichments by MPE (Figure 2). The loadings of the negative discriminant (from the 10‐ and 30‐min samples) comprise the expected metabolites Phe, 13C6‐Phe, and Tyr, metabolites generated from Tyr, and polyamine synthesis (Table S4). The positive function (from the sham‐control and 60‐min samples) predominantly comprises other amino acids, pathways related to oxidative stress (both the substrates and product of glutathione synthase), and the urea cycle. We applied PCA to each pixel of each sample to gain further insight into local metabolic changes. As an example, the first principal component in the 30‐min sample correlates with different anatomical structures, liver tissue in the positive component and pancreatic tissue in the negative component (Supporting Information, Figure S10). This principal component is associated with taurine and glutamic acid in liver and Phe and 13C6‐Phe in pancreatic tissue. These findings may be explained by substrate competition for transporters of Phe with branched chain amino acids.16 Cellular import of aromatic amino acids is mainly mediated by l‐type amino acid transporters (LAT), which are found in most tissues. We found increased aromatic amino acid levels and diminished BCAA levels in liver sections after tracer infusion (Figure S9). Clinically, increased LAT transporter expression was shown to be of diagnostic and prognostic value.17
In conclusion, we present a novel and reproducible MSI method to explore the dynamic chemical changes of complex biological samples, allowing for the first time, the detection of co‐localization of hepatocellular 13C6‐Phe and amino acid kinetics. The method enabled the broad detection of over 60 endogenous metabolites, thus providing a visual map of liver function. This map enables translation of molecular kinetics to morphology for a wide variety of biological tissues. This allows differential kinetics between healthy and diseased tissue or altered amino acid metabolism in tumor cells and their microenvironment to be intimately followed.
Experimental Section
A bolus injection of 13C6‐Phe (n=11) or saline solution (n=4) was administered at the tail vein of tumor‐bearing immune‐compromised Crl:NU‐Foxn1nu nu/nu nude mice. The mice were sacrificed at 10, 30, or 60 min after injection, and the organs were snap‐frozen in liquid N2. Liver cryosections (10 μm) were covered with TAHS for on‐tissue derivatization and DHB as a matrix for high resolution MALDI‐FTICR‐MSI in positive mode (Solarix‐XR). A MATLAB algorithm was developed to calculate and visualize spatial tracer kinetics. After MSI analysis, liver sections were stained with hematoxylin and eosin, reviewed for tissue morphology, and co‐registered with the MALDI images. Additional experimental information is provided in the Supporting Information.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors thank Hang Nguyen for revising the manuscript, Rob Esser for the graphical design of the figures, Joan Senden for performing the GC‐MS analyses, and Marjon Gijbels for pathological review of the tissues. This work is financially supported by the Dutch Province of Limburg as part of the “LINK” program. L. D. Dubois is funded by a Worldwide Cancer Research (WCR), formerly known as AICR, project grant (15‐0345).
M. Arts, Z. Soons, S. R. Ellis, K. A. Pierzchalski, B. Balluff, G. B. Eijkel, L. J. Dubois, N. G. Lieuwes, S. M. Agten, T. M. Hackeng, L. J. C. van Loon, R. M. A. Heeren, S. W. M. Olde Damink, Angew. Chem. Int. Ed. 2017, 56, 7146.
References
- 1.
- 1a. Fujimura Y., Miura D., Metabolites 2014, 4, 319–346; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Chughtai K., Heeren R. M., Chem. Rev. 2010, 110, 3237–3277; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Cornett D. S., Reyzer M. L., Chaurand P., Caprioli R. M., Nat. Methods 2007, 4, 828–833. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. McIntosh T. S., Davis H. M., Matthews D. E., Anal. Biochem. 2002, 300, 163–169; [DOI] [PubMed] [Google Scholar]
- 2b. van Eijk H. M., Wijnands K. A., Bessems B. A., Olde Damink S. W., Dejong C. H., Poeze M., J. Chromatogr. B 2012, 905, 31–36; [DOI] [PubMed] [Google Scholar]
- 2c. Gogichaeva N. V., Alterman M. A., Methods Mol. Biol. 2012, 828, 121–135; [DOI] [PubMed] [Google Scholar]
- 2d. Buescher J. M., Antoniewicz M. R., Boros L. G., Burgess S. C., Brunengraber H., Clish C. B., DeBerardinis R. J., Feron O., Frezza C., Ghesquiere B., Gottlieb E., Hiller K., Jones R. G., Kamphorst J. J., Kibbey R. G., Kimmelman A. C., Locasale J. W., Lunt S. Y., Maddocks O. D., Malloy C., Metallo C. M., Meuillet E. J., Munger J., Noh K., Rabinowitz J. D., Ralser M., Sauer U., Stephanopoulos G., St-Pierre J., Tennant D. A., Wittmann C., Vander Heiden M. G., Vazquez A., Vousden K., Young J. D., Zamboni N., Fendt S. M., Curr. Opin. Biotechnol. 2015, 34, 189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Gogichaeva N. V., Williams T., Alterman M. A., J. Am. Soc. Mass Spectrom. 2007, 18, 279–284; [DOI] [PubMed] [Google Scholar]
- 3b. Piraud M., Vianey-Saban C., Petritis K., Elfakir C., Steghens J. P., Bouchu D., Rapid Commun. Mass Spectrom. 2005, 19, 1587–1602; [DOI] [PubMed] [Google Scholar]
- 3c. Kim I. Y., Suh S. H., Lee I. K., Wolfe R. R., Exp. Mol. Med. 2016, 48, e203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Steinhauser M. L., Bailey A. P., Senyo S. E., Guillermier C., Perlstein T. S., Gould A. P., Lee R. T., Lechene C. P., Nature 2012, 481, 516–519; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Senyo S. E., Steinhauser M. L., Pizzimenti C. L., Yang V. K., Cai L., Wang M., Wu T. D., Guerquin-Kern J. L., Lechene C. P., Lee R. T., Nature 2013, 493, 433–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Louie K. B., Bowen B. P., McAlhany S., Huang Y., Price J. C., Mao J. H., Hellerstein M., Northen T. R., Sci. Rep. 2013, 3, 1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Castro-Perez J., Hatcher N., Kofi Karikari N., Wang S. P., Mendoza V., Shion H., Millar A., Shockcor J., Towers M., McLaren D., Shah V., Previs S., Akinsanya K., Cleary M., Roddy T. P., Johns D. G., Rapid Commun. Mass Spectrom. 2014, 28, 2471–2479. [DOI] [PubMed] [Google Scholar]
- 7. Seaman C., Flinders B., Eijkel G., Heeren R. M., Bricklebank N., Clench M. R., Anal. Chem. 2014, 86, 10071–10077. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Olde Damink S. W., Jalan R., Deutz N. E., Dejong C. H., Redhead D. N., Hynd P., Hayes P. C., Soeters P. B., Hepatology 2007, 45, 560–568; [DOI] [PubMed] [Google Scholar]
- 8b. Tsun Z. Y., Possemato R., Semin. Cell Dev. Biol. 2015, 43, 22–32; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c. Matthews D. E., J. Nutr. 2007, 137, 1549S–1555S; discussion 1573S–1575S; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8d. Groen B. B., Horstman A. M., Hamer H. M., de Haan M., van Kranenburg J., Bierau J., Poeze M., Wodzig W. K., Rasmussen B. B., van Loon L. J., J. Clin. Endocrinol. Metab. 2016, 101, 3978–3988. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Wiggins T., Kumar S., Markar S. R., Antonowicz S., Hanna G. B., Cancer Epidemiol. Biomarkers Prev. 2015, 24, 32–38; [DOI] [PubMed] [Google Scholar]
- 9b. Safaei A., Arefi Oskouie A., Mohebbi S. R., Rezaei-Tavirani M., Mahboubi M., Peyvandi M., Okhovatian F., Zamanian-Azodi M., Gastroenterol. Hepatol. Bed Bench 2016, 9, 158–173. [PMC free article] [PubMed] [Google Scholar]
- 10. Toue S., Sugiura Y., Kubo A., Ohmura M., Karakawa S., Mizukoshi T., Yoneda J., Miyano H., Noguchi Y., Kobayashi T., Kabe Y., Suematsu M., Proteomics 2014, 14, 810–819. [DOI] [PubMed] [Google Scholar]
- 11. Ellis S. R., Bruinen A. L., Heeren R. M., Anal. Bioanal. Chem. 2014, 406, 1275–1289. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a.D. J. Wilkinson, Mass Spectrom. Rev 2016, 10.1002/mas.21507; [DOI] [PMC free article] [PubMed]
- 12b. Gregg C. T., Hutson J. Y., Prine J. R., Ott D. G., Furchner J. E., Life Sci. 1973, 13, 775–782. [DOI] [PubMed] [Google Scholar]
- 13. Dejong C. H., van de Poll M. C., Soeters P. B., Jalan R., Olde Damink S. W., J. Nutr. 2007, 137, 1579S–1585S; discussion 1597S–1598S. [DOI] [PubMed] [Google Scholar]
- 14. Lichter-Konecki U., Hipke C. M., Konecki D. S., Mol. Genet. Metab. 1999, 67, 308–316. [DOI] [PubMed] [Google Scholar]
- 15. Watanabe Y., Kurihara H., Itami J., Sasaki R., Arai Y., Sugimura K., Radiat. Oncol. 2017, 12, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.
- 16a. Kanai Y., Segawa H., Miyamoto K., Uchino H., Takeda E., Endou H., J. Biol. Chem. 1998, 273, 23629–23632; [DOI] [PubMed] [Google Scholar]
- 16b. Yanagida O., Kanai Y., Chairoungdua A., Kim D. K., Segawa H., Nii T., Cha S. H., Matsuo H., Fukushima J., Fukasawa Y., Tani Y., Taketani Y., Uchino H., Kim J. Y., Inatomi J., Okayasu I., Miyamoto K., Takeda E., Goya T., Endou H., Biochim. Biophys. Acta Biomembr. 2001, 1514, 291–302. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Glaudemans A. W., Enting R. H., Heesters M. A., Dierckx R. A., van Rheenen R. W., Walenkamp A. M., Slart R. H., Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 615–635; [DOI] [PubMed] [Google Scholar]
- 17b. Kaira K., Sunose Y., Ohshima Y., Ishioka N. S., Arakawa K., Ogawa T., Sunaga N., Shimizu K., Tominaga H., Oriuchi N., Itoh H., Nagamori S., Kanai Y., Yamaguchi A., Segawa A., Ide M., Mori M., Oyama T., Takeyoshi I., BMC Cancer 2013, 13, 482. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary