

Abstract
Drebrin is a major F‐actin binding protein in dendritic spines that is critically involved in the regulation of dendritic spine morphogenesis, pathology, and plasticity. In this study, we aimed to identify a novel drebrin‐binding protein involved in spine morphogenesis and synaptic plasticity. We confirmed the beta subunit of Ca2+/calmodulin‐dependent protein kinase II (CaMKIIβ) as a drebrin‐binding protein using a yeast two‐hybrid system, and investigated the drebrin–CaMKIIβ relationship in dendritic spines using rat hippocampal neurons. Drebrin knockdown resulted in diffuse localization of CaMKIIβ in dendrites during the resting state, suggesting that drebrin is involved in the accumulation of CaMKIIβ in dendritic spines. Fluorescence recovery after photobleaching analysis showed that drebrin knockdown increased the stable fraction of CaMKIIβ, indicating the presence of drebrin‐independent, more stable CaMKIIβ. NMDA receptor activation also increased the stable fraction in parallel with drebrin exodus from dendritic spines. These findings suggest that CaMKIIβ can be classified into distinct pools: CaMKIIβ associated with drebrin, CaMKIIβ associated with post‐synaptic density (PSD), and CaMKIIβ free from PSD and drebrin. CaMKIIβ appears to be anchored to a protein complex composed of drebrin‐binding F‐actin during the resting state. NMDA receptor activation releases CaMKIIβ from drebrin resulting in CaMKIIβ association with PSD.
Keywords: drebrin, CaMKIIβ, drebrin‐binding protein, dendritic spine
Abbreviations used
- BSA
bovine serum albumin
- CaMKIIα
alpha subunit of Ca2+/calmodulin‐dependent protein kinase II
- CaMKIIβ
beta subunit of Ca2+/calmodulin‐dependent protein kinase II
- DIV
days in vitro
- F‐actin
filamentous actin
- FRAP
fluorescence recovery after photobleaching
- irDrebrin
RNAi‐resistant drebrin
- KD
knockdown
- MEM
minimum essential medium
- NMDAR
NMDA receptor
- PBS
phosphate‐buffered saline
- PSD
post‐synaptic density
- RNAi
RNA interference
- RRIDs
research resource identifier
Drebrin is a filamentous actin (F‐actin)‐binding protein consisting of two isoforms, drebrin E and drebrin A (Shirao 1995), which are highly expressed in the brain and are localized mainly in dendritic spines of mature neurons (Hayashi et al. 1996). It has been proposed that drebrin regulates local organization of the actin network and modifies cell morphology (for review, see Shirao et al. 2017). Over‐expression of the neuron‐specific isoform drebrin A in mature neurons leads to abnormal enlargement of dendritic spines (Hayashi and Shirao 1999) and augments the amplitude and frequency of glutamatergic mEPSC (Ivanov et al. 2009). Knockdown (KD) of drebrin A during neuronal development suppresses the morphological maturation of dendritic spines and accumulation of post‐synaptic density (PSD)‐95 at dendritic spines (Takahashi et al. 2003). Furthermore, activation of NMDA receptor (NMDAR) induces the exodus of drebrin from dendritic spines (Mizui et al. 2014). Dendritic spine morphology is strongly linked to neuropsychiatric disorders, including Alzheimer's disease, Down's syndrome, fragile X syndrome, and autism spectrum disorder (Penzes et al. 2011), and loss of drebrin in dendritic spines is observed in patients with Alzheimer's disease (Harigaya et al. 1996; Hatanpaa et al. 1999; Counts et al. 2006) and Down's syndrome (Shim and Lubec 2002). This evidence demonstrates a critical role for drebrin in the regulation of dendritic spine morphogenesis, pathology, and plasticity.
Drebrin binds to F‐actin in vitro without severing, capping, or nucleating activities (Ishikawa et al. 1994), and competes with tropomyosin, α‐actinin, fascin, and myosin V for actin binding (Ishikawa et al. 1994, 2007; Sasaki et al. 1996; Sekino et al. 2007). In addition, drebrin binds to the actin‐monomer‐binding protein and stimulator of actin polymerization, profilin (Mammoto et al. 1998). These data suggest that drebrin can regulate F‐actin organization by interacting with other actin‐associated proteins. Aside from F‐actin and actin‐regulators, drebrin also interacts with the synaptic proteins Homer (Shiraishi‐Yamaguchi et al. 2009) and spikar (Yamazaki et al. 2014). Homer regulates dendritic spine morphology in cooperation with Shank (Sala et al. 2001). Spikar is stabilized in dendritic spines by drebrin and is involved in spine formation (Yamazaki et al. 2014). Moreover, drebrin binds to the microtubule tip‐tracking protein EB3, which is involved in spine morphology through interaction with p140Cap and cortactin (Jaworski et al. 2009). These findings suggest that drebrin acts as a central hub for spine‐regulating proteins, in addition to its involvement in actin organization. However, although increasing evidence indicates a pivotal role in synapses, the molecular mechanisms of the drebrin complex have not been fully elucidated.
In this study, we aimed to identify a novel drebrin‐binding protein localized in dendritic spines with roles in spine morphogenesis and synaptic plasticity. In the course of screening for drebrin‐binding proteins, we isolated beta subunit of Ca2+/calmodulin‐dependent protein kinase II (CaMKIIβ) as a novel drebrin‐binding partner. CaMKIIβ exists in large quantity in neuron as heteromeric complexes with CaMKIIα. Although CaMKII is known to be a pivotal Ca2+ transducer in neurons, they function not only as a protein kinase but also as a structural component in dendritic spines (Okamoto et al. 2007). CaMKIIβ binds to and bundles F‐actin in neurons, and the interaction between CaMKIIβ and F‐actin is negatively regulated by synaptic activity (Kim et al. 2015). When long‐term potentiation is induced by Ca2+ influx, active CaMKIIβ is released from F‐actin and then translocates into post‐synaptic density (PSD). On the other hand, inactive CaMKIIβ under basal condition is attached to F‐actin and localized in a wide range of a dendritic spine (Lu et al. 2014). However, an upstream protein governing the localization of CaMKIIβ in a dendritic spine is unknown. We supposed that drebrin is a candidate protein that is responsible for spine localization of CaMKIIβ at resting state. In this study, we show the change in CaMKIIβ localization and dynamics in dendritic spines and shafts in drebrin‐knockdown (KD) neurons. The results suggest the existence of both drebrin‐dependent and drebrin‐independent pools of CaMKIIβ in a dendritic spine.
Materials and Methods
Materials
The following materials were used: mouse anti‐drebrin monoclonal antibody (clone M2F6, Shirao and Obata 1986; from our laboratory; RRID:AB_2532045), mouse anti‐CaMKIIβ monoclonal antibody (Thermo Fisher Scientific, Waltham, MA, USA; Cat.# 13‐9800, RRID:AB_2533045), mouse anti‐CaMKIIα monoclonal antibody (Thermo Fisher Scientific; Cat.# MA1‐048, RRID:AB_325403), mouse anti‐c‐Myc monoclonal antibody (Santa Cruz, Santa Cruz, CA, USA; Cat.# sc‐40, RRID:AB_627268), mouse anti‐β‐actin monoclonal antibody (Sigma, St. Louis, MO, USA; Cat.# F3022, RRID:AB_476970), mouse anti‐FLAG monoclonal antibody (Sigma; Cat.# F3165, RRID:AB_259529), rat anti‐HA monoclonal antibody (Roche, Basel, Switzerland; Cat.# 11867431001, RRID:AB_390919) and EDTA‐free protease inhibitor cocktail (Roche; Cat.# 11 873 580 001), anti‐rat and anti‐mouse Horseradish Peroxidase (HRP)‐conjugated IgG (Cappel, West Chester, PA, USA; Cat.# 55770 and # 55550), Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA; Cat. # 11668‐027), minimum essential medium (MEM; Thermo Fisher Scientific; Cat. # 1095080), Dulbecco's MEM (Wako, Tokyo, Japan; Cat. # 041‐29775) and B‐27 supplement (Thermo Fisher Scientific; Cat. # 17504044), and p3XFLAG‐CMV‐14 vector (Sigma; Cat. # E7908), pACT2 (Clontech, Palo Alto, CA, USA; Cat. # 638822), pEGFP‐C1 (Clontech; Cat. # 6084‐1), pCMV‐Myc (Clontech; Cat. # 631604), and pCMV‐HA vectors (Clontech; Cat. # K6003‐1). Rabbit antiserum against drebrin was described previously (Hayashi et al. 1996). Other chemicals were purchased from Wako (Tokyo, Japan).
Animals
The study was not pre‐registered. Pregnant Wistar rats (fetuses at 13 days of gestation; Strain Crlj:WI, Cat# 2312504, RRID:RGD_2312504) were obtained from Charles River Japan Inc. (Tokyo, Japan). The animals had ad libitum access to food and tap water and were maintained under conditions of controlled temperature (23 ± 1°C) and lighting (light/dark cycle: lights on, 06 : 00; lights off, 18 : 00). All animal experiments were carried out in accordance with the guidelines of the Animal Care and Experimentation Committee of Gunma University, Showa Campus. All efforts were made to minimize the number of animals used and to minimize suffering (protocol #17‐024).
DNA constructs
Rat CaMKIIβ constructs were generated by PCR using Pfu turbo polymerase (Stratagene, La Jolla, CA, USA; Cat. # 600250). The expression vectors of c‐Myc‐tagged (QKLISEEDL) full‐length rat CaMKIIβ (Myc‐CaMKIIβ) cDNA and c‐Myc‐tagged full‐length rat CaMKIIβe (Myc‐CaMKIIβe) cDNA (Okuno et al. 2012) were constructed by subcloning the EcoRI/XhoI fragments into the pCMV‐Myc vector. HA‐tagged (YPYDVPDYA) full‐length rat drebrin A (HA‐Drebrin) cDNA was constructed by subcloning an XhoI fragment into the pCMV‐HA vector. HA‐CaMKIIβ 1‐280 and 281‐542 were constructed by subcloning the EcoRI/BglII fragments into the pCMV‐HA vector. The cDNA of enhanced GFP‐tagged rat CaMKIIβ (GFP‐CaMKIIβ) cDNA was constructed by subcloning the XhoI/BamHI fragment into the pEGFP‐C1 vector. The GFP‐CaMKIIβe cDNA was constructed by subcloning the XhoI/EcoRI fragment into the pEGFP‐C1 vector. The FLAG‐tagged (DYKDDDDK) full‐length rat drebrin A (Drebrin‐FLAG) was constructed by subcloning the BglII/KpnI fragment into the p3XFLAG‐CMV‐14 vector (Sigma). For RNA interference (RNAi) KD, we used a pSUPERneo+gfp vector (OligoEngine, Seattle, WA, USA; Cat. # VEC‐PBS‐0002) and pSUPERneo vector (OligoEngine; Cat. # VEC‐PBS‐0006). The shRNA sequences were: drebrin shRNA#1; sense 5′‐GATCCCCGTGATGTGTGGTTTCTGTATTCAAGAGATGCAGAAACCGTACATCACTTTTTGGAAA‐3′ and antisense 5′‐AGCTTTTCCAAAAAGTGATGTACGGTTTCTGCATCTCTTGAATACAGAAACCACACATCACGGG‐3′; drebrin shRNA#2; sense 5′‐GATCCCCCCAGAAAGTGATGTACGGTTTCAAGAGAACCGTACATCACTTTCTGGTTTTTA‐3′ and antisense 5′‐AGCTTAAAAACCAGAAAGTGATGTACGGTTCTCTTGAAACCGTACATCACTTTCTGGGGG‐3′; CaMKIIβ shRNA; sense 5′‐GATCCCCGAGTATGCGGCTAGGATTATTCAAGAGATGATCTTAGCTGCATACTCTTTTTGGAAA‐3′ and antisense 5′‐AGCTTTTCCAAAAAGAGTATGCAGCTAAGATCATCTCTTGAATAATCCTAGCCGCATACTCGGG‐3′; and non‐target negative shRNA; sense 5′‐GATCCCCATCCGCGCGATAGTACGTATTCAAGAGATACGTACTATCGCGCGGATTTTTTA‐3′ and antisense 5′‐AGCTTAAAAAATCCGCGCGATAGTACGTATCTCTTGAATACGTACTATCGCGCGGATGGG‐3′. The efficiency of drebrin shRNA was confirmed in a previous study (Kato et al. 2012). The details of CaMKIIβ shRNA, non‐target negative shRNA, and RNAi‐resistant drebrin used in this study have been described in previous papers (CaMKIIβ, Okamoto et al. 2007; non‐target negative shRNA, Okuno et al. 2012; RNAi‐resistant drebrin, Yamazaki et al. 2014). The nucleotide sequences in each construct were verified by DNA sequencing.
Yeast two‐hybrid system
Saccharomyces cerevisiae transformation and two‐hybrid screening were carried out using the Y190 strain (MATa gal4 gal80 his3 trp1‐901 ade2‐101 ura3‐52 leu2‐3, ‐112+URA3::GAL‐lacZ, LYS2::GAL(UAS)‐HIS3 cyk r) as previously described (Yamazaki et al. 2014). To obtain the constructs for the yeast two‐hybrid system, rat drebrin A cDNA (Accession No. X59267) was used as a template. The N‐terminal region of rat drebrin (amino acid residues 1–233) was subcloned into the pAS404 vector derived from pAS1 (Sekiguchi et al. 2001). The bait plasmid pAS404‐Dr1‐233 was transformed into Y190 cells using the conventional lithium acetate–polyethylene glycol method. Cells were grown on tryptophan minus SD medium (SD−Trp) and further characterized by testing for protein expression and self‐activation of the bait before screening. Large‐scale transformation was performed using a rat brain cDNA library constructed in pACT2 vector (Clontech), and cells were grown on a 3‐aminotriazole (25 mM) plus SD−Trp, Leu, and His plate and consequently assayed for activation of the HIS3 gene. For two‐hybrid binding assay, the 5′‐untranslated regions of CaMKIIβ and CaMKIIα (Brickey et al. 1990) were subcloned into pACT2 vector.
Cell culture and transfection
COS7 monkey kidney epithelial cells (RRID: CVCL_0224) were seeded at a density of 106 cells in a 35‐mm plastic dish (Falcon, Franklin Lakes, NJ, USA) and grown in Dulbecco's MEM supplemented with 10% fetal bovine serum at 37°C in a 5% CO2 atmosphere. Cells were allowed to adhere for 12–16 h and then transfected with appropriate plasmids using Lipofectamine 2000 reagent, following the manufacturer's instructions.
Low‐density cultures of hippocampal neurons were prepared by Banker's methods (Takahashi et al. 2003). Pregnant Wistar rats were killed by cervical dislocation. Hippocampi were dissected from embryonic 18‐day‐old Wistar rats, dissociated by trypsin treatment, and triturated through a Pasteur pipette. The rat embryos were not divided by the sex. The neurons were plated at a density of 5000 cells/cm2 on coverslips coated with poly‐L‐lysine in MEM supplemented with 10% fetal bovine serum. After cell attachment, coverslips were transferred into a dish containing a glial monolayer sheet and maintained in serum‐free MEM with a B‐27 supplement. Cytosine β‐D‐arabinofuranoside (5 μM) was added to the cultures 4 days after plating to inhibit glial proliferation. Neurons were transfected at 7–8 days in vitro (DIV) using Lipofectamine 2000 reagent, and assayed at 16–17 DIV (KD and FRAP) or 20 DIV (NMDAR activation).
Immunoprecipitation and western blotting
Immunoprecipitation was performed using the ProFound Mammalian Myc Tag IP/Co‐IP kit (Pierce, Rockford, IL, USA) according to the manufacturer's instructions. In brief, after 48 h of transfection, COS7 cells were washed three times with ice‐cold 25 mM Tris‐buffered saline (pH 7.2) and lysed in M‐PER buffer (Pierce) containing protease inhibitor cocktail. The cell lysates were centrifuged at 16 000 g for 20 min at 4°C, and supernatants were incubated with immobilized anti‐Myc antibody agarose for 12–16 h at 4°C with gentle agitation. The immune complexes were precipitated, washed three times with Tris‐buffered saline containing 0.05% Tween‐20, and eluted by sample buffer at 95°C. For immunoprecipitation of drebrin and CaMKIIβe or short CaMKIIβ fragments, HEK293 cells (RRID: CVCL_0045) were harvested in lysis buffer (150 mM NaCl, 50 mM HEPES pH 7.4, 1% Triton X‐100, plus inhibitors) and centrifuged at 16 000 g for 20 min at 4°C. The supernatants were pre‐cleared with Protein A/G agarose (Pierce) for 1h at 4°C. The pre‐cleared lysates were then incubated for 12–16 h at 4°C with 3 μg anti‐FLAG monoclonal antibody. After addition of Protein A/G agarose, the immunoprecipitates were incubated for 1 h at 4°C. The Protein A/G agarose with immunoprecipitates were washed three times with lysis buffer, and eluted by sample buffer at 95°C. Samples were separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and the proteins were then transferred onto an Immobilon Transfer Membrane (Millipore, Bedford, MA, USA). After transfer, the membrane was blocked in 10% non‐fat milk/phosphate‐buffered saline (PBS) and incubated for 12–16 h at 4°C in 3% bovine serum albumin (BSA)/PBS with the appropriate antibody. After washing in PBS, the membrane was incubated for 1 h at 24°C with the appropriate Horseradish Peroxidase‐conjugated IgG in 3% BSA/PBS. Immunopositive signals were visualized with an image analyzer (LAS‐3000, Fuji?lm, Tokyo, Japan). using ECL detection reagents (GE Healthcare, Piscataway, NJ, USA).
Immunocytochemistry and glutamate treatment
Hippocampal neurons were fixed with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.2) and then permeated with 0.1% Triton X‐100 in PBS. The cells were then incubated with 3% BSA/PBS and with appropriate antibodies for 12–16 h at 4°C. After washing with PBS, cells were incubated for 1 h at 24°C in 3% BSA/PBS with an appropriate fluorescence‐labeled anti‐IgG antibody. For NMDAR activation, cultured neurons were pre‐incubated in Tyrode's solution (119 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 25 mM HEPES pH 7.4, 30 mM glucose) for 10 min, and then treated with glutamate (50 μM)/glycine (10 μM) for 10 min (Shen and Meyer 1999).
Image analysis and quantification
All experiments were repeated at least three times using three different cultures. Each fluorescent image was obtained using a Zeiss Axioplan microscope (Carl Zeiss, Oberkochen, Germany) with a 63× objective lens (NA, 1.4) and a Photometrics series Cool Snap fx cooled CCD camera (Photometrics, Tucson, AZ, USA). The microscopic images (1300 × 1030 pixels) were captured at a spatial sampling of 106 nm/pixel. To evaluate the spine:dendrite ratio for each protein (CaMKIIβ, CaMKIIβe, CaMKIIα, and drebrin), 8–10 mushroom‐type spines were selected from two dendrites of GFP‐positive neurons for each condition. The immunofluorescence intensities were measured in circles (0.26 μm2) of the spines and the adjacent dendritic shaft. Line scans were performed in MetaMorph software (Molecular Devices, Sunnyvale, CA, USA). For colocalization analysis, binary images were created in Fiji (Schindelin et al. 2012) from manual tracings of the dendritic spines and dendritic shaft (20–30 μm) for each neuron after auto threshold. Manders’ colocalization coefficients were determined in Fiji using the Coloc2 plug‐in. The samples were coded, and image capture and analysis were done by an experimenter blind to treatment groups. Sample size calculation nor power analysis was conducted to predetermine the size.
FRAP analysis
Cultured neurons were transfected with GFP‐CaMKIIβ (or GFP‐CaMKIIβe) alone, GFP‐CaMKIIβ (or GFP‐CaMKIIβe) and drebrin shRNA or negative shRNA at 8 DIV and imaged at 15–16 DIV. The neurons were imaged with a Zeiss LSM510 confocal microscope (Carl Zeiss) and a 40× water‐immersion objective lens (NA, 1.2) in Tyrode's solution. The GFP fluorescence in a region of interest was bleached 10 times with a 488‐nm laser at 100% output. During fluorescence recovery after photobleaching (FRAP) imaging, the GFP fluorescence was captured before and after photobleaching with 5‐s intervals for 300 s for GFP‐CaMKIIβ and GFP‐CaMKIIβe, and 1‐s intervals for 120 s for GFP alone. Raw data were quantified using MetaMorph software (Molecular Devices). Background intensity was subtracted from the intensity of the region of interest. The intensity data were normalized to the intensity measured at unbleached spines to correct for fluorescence loss during FRAP imaging. Finally, all intensity data were normalized to the average fluorescence intensity of five scans acquired just before bleaching. The FRAP curve was fitted with KaleidaGraph software (Synergy Software, Reading, PA, USA) to the equation: F(t) = 1−fs−fm × exp(−t/τ) derived by Star et al. (2002), where fs, fm, t and τ are the stable fraction, dynamic fraction, time (min) and time constant, respectively.
Statistical analysis
Statistical analyses were performed using SPSS software (SPSS, Chicago, IL, USA) and Statcel3 software (OMS, Tokorozawa, Japan). We used Welch's t‐tests or Mann–Whitney U‐test for single comparisons and anova followed by Tukey–Kramer or Steel–Dwass post hoc test for multiple comparisons. Bar graphs present means ± SEM. Box plots show the median, interquartile, and range. p values of < 0.05 were considered significant.
Results
Drebrin binds to CaMKIIβ
We performed a yeast two‐hybrid screen to identify novel binding partners for drebrin. The N‐terminal region of drebrin (amino acids 1–233), which is common between drebrin isoforms, was used as bait to screen a rat brain cDNA library (Fig. 1a). We isolated 89 positive clones, two of which were CaMKIIβ gene products. One clone encoded full‐length CaMKIIβ protein (Fig. 1b, Clone 1), while the other lacked part of the nucleic acid sequence (Fig. 1b, Clone 2). The clone 2 is identical to CaMKIIβ isoform X11 (accession number XP_017454561). These clones had conserved identical sequences in the non‐coding regions at the 3′ and 5′ ends. However, we did not obtain any clones encoding the alpha subunit of Ca2+/calmodulin‐dependent protein kinase II (CaMKIIα), which is another major subtype of CaMKII in neurons (Erondu and Kennedy 1985). To clarify if drebrin could bind to CaMKIIα, we performed a yeast two‐hybrid assay using both CaMKIIα and CaMKIIβ with the N‐terminal region of drebrin. CaMKIIβ clone 1 showed a positive result in the growth test, but neither CaMKIIα, the 5′‐untranslated region of CaMKIIβ, nor the empty vector gave a positive result (Fig. 1c and d). These findings indicate that CaMKIIβ, but not CaMKIIα, selectively interacts with drebrin.
Figure 1.
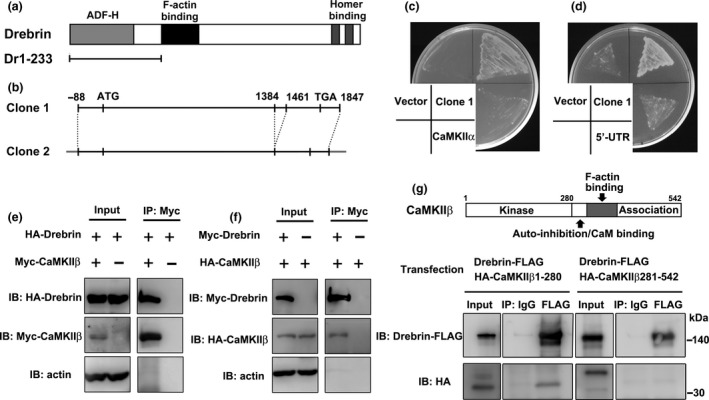
CaMKIIβ binds to drebrin. (a) Domain structure of drebrin. The N‐terminal region of drebrin (Dr1‐233) was used as bait for yeast two‐hybrid screening. (b) We isolated two CaMKIIβ clones by screening. One clone encoded full‐length CaMKIIβ (Clone 1) and the other clone lacked 78 nucleotide residues in the association domain (Clone 2). (c and d) Yeast two‐hybrid assay between Dr1‐233 and CaMKIIβ (Clone 1), CaMKIIα, or 5′‐UTR of CaMKIIβ. The yeast grew when Dr1‐233 was coexpressed with CaMKIIβ but not with CaMKIIα and 5′‐UTR of CaMKIIβ. (e and f) Coimmunoprecipitation between CaMKIIβ and drebrin. COS7 cells were transfected with HA‐drebrin (or Myc‐drebrin) and Myc‐CaMKIIβ (or HA‐CaMKIIβ). Myc‐CaMKIIβ (or HA‐CaMKIIβ) single transfection was performed for control experiments. Cell lysates were immunoprecipitated with anti‐Myc‐agarose gel. (g) Coimmunoprecipitation between CaMKIIβ fragments and drebrin. HEK293 cells were transfected with Drebrin‐FLAG and HA‐CaMKIIβ 1‐280 or HA‐CaMKIIβ 281‐542. Cell lysates were immunoprecipitated with anti‐FLAG antibody.
We then analyzed the interaction between drebrin and CaMKIIβ by immunoprecipitation. HA‐drebrin and Myc‐CaMKIIβ were coexpressed in COS7 cells. In control experiments, only HA‐drebrin was expressed in COS7 cells. The supernatants of the cell lysates were immunoprecipitated with anti‐Myc antibody and the precipitated proteins were then analyzed by western blotting. The band for HA‐drebrin was observed in the immunoprecipitation sample at an equivalent level as that in the input sample (Fig. 1e). In contrast, the band for HA‐drebrin was not detected in the control immunoprecipitation without Myc‐CaMKIIβ (Fig. 1e). A similar set of experiments using Myc‐drebrin and HA‐CaMKIIβ showed that HA‐CaMKIIβ was immunoprecipitated by anti‐Myc beads from a cell lysate containing Myc‐drebrin and HA‐CaMKIIβ (Fig. 1f). Moreover, we analyzed the drebrin‐binding region of CaMKIIβ by immunoprecipitation from HEK293 cells transfected with Drebrin‐FLAG and HA‐CaMKIIβ 1‐280 or 281‐542. Anti‐FLAG immunoprecipitations showed that drebrin interacts with CaMKIIβ 1‐280, but not CaMKIIβ 281‐542 (Fig. 1g).
Drebrin colocalizes with CaMKIIβ in filopodia and mature dendritic spines
Drebrin and CaMKIIβ are localized in dendritic spines of mature neurons (Hayashi et al. 1996; Shen et al. 1998). We studied the localization of drebrin and CaMKIIβ within dendritic spines/filopodia in developing cultured neurons. Cultured neurons were transfected with GFP and immunolabeled with drebrin and CaMKIIβ at two neuronal developmental stages. Drebrin and CaMKIIβ colocalized diffusely in dendritic filopodia (Fig. 2a, line scan image of 7 DIV) and at the submembranous region of dendritic shafts in immature neurons, although CaMKIIβ was also observed in the center region of dendritic shafts (Fig. 2a). On the other hand, CaMKIIβ colocalized with drebrin in almost all dendritic spines in mature neurons (Fig. 2a). In a dendritic spine, CaMKIIβ was closer to plasma membrane than drebrin (Fig. 2a, line scan image of 21 DIV). Manders’ colocalization coefficients confirmed that CaMKIIβ was colocalized with drebrin in dendritic filopodia and spines (Fig. 2b). These suggest that CaMKIIβ is partly colocalized with drebrin from an early developmental stage.
Figure 2.
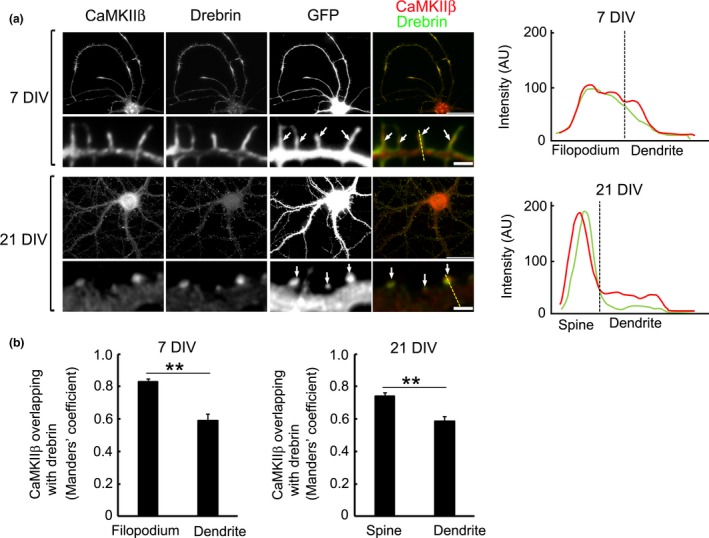
CaMKIIβ colocalizes with drebrin in cultured hippocampal neurons. (a) CaMKIIβ was localized in dendrites of immature neurons. CaMKIIβ colocalized with drebrin in dendritic filopodia [arrows, 7 days in vitro (DIV)] and dendritic spines of a mature neuron (arrows, 21 DIV). Dashed yellow lines in the merged images of 7 DIV and 21 DIV indicate the positions of the line scan graphs shown right. The line scans show CaMKIIβ (red) and drebrin (green) pixel intensities across the region of interest. AU, Arbitrary units. Scale bars, 30, 2 μm (dendrite images). (b) Quantification of Mander's coefficients for CaMKIIβ overlap with drebrin. Data are presented as means ± SEM (n = 86 filopodia for 7 DIV neuron, n = 29 dendrite regions for 7 DIV neuron, n = 74 spines for 21 DIV neuron, n = 35 dendrite regions for 21 DIV neuron, **p < 0.01, Welch's t‐test).
Drebrin is responsible for dendritic spine localization of CaMKIIβ
We showed that drebrin governs synaptic targeting of PSD‐95 through clustering F‐actin in proto‐spine (Takahashi et al. 2003). Based on the above results, we considered that drebrin may govern spine localization of CaMKIIβ. We investigated the hypothesis that CaMKIIβ was localized in dendritic spines in a drebrin‐dependent manner by measuring the spine:dendrite ratio of CaMKIIβ immunofluorescence intensity in drebrin‐KD neurons. We transfected cultured neurons with drebrin shRNA, control shRNA, or drebrin shRNA and RNAi‐resistant drebrin, and imaged the CaMKIIβ immunoreactivity of these neurons (Fig. 3a). CaMKIIβ was localized diffusely in dendrites of drebrin‐KD neurons compared with control neurons (Fig. 3b, line scan). Moreover, the spine:dendrite ratio of CaMKIIβ was decreased in drebrin‐KD neurons (Fig. 3c), and this ratio decrease was rescued by coexpression of RNAi‐resistant drebrin (Fig. 3c). Actually, the immunofluorescence intensity was decreased in spines but not in dendritic shafts in drebrin‐KD neurons (Figure S1). We also examined CaMKIIα localization in drebrin‐KD neurons and found that CaMKIIα was diffusely localized in dendritic shafts (Fig. 3d–f), consistent with the fact that CaMKIIβ forms a holoenzyme with CaMKIIα (Bennett et al. 1983). Similar results were obtained in neurons treated with latrunculin A, an F‐actin‐disrupting reagent. Latrunculin A treatment nearly abolished drebrin cluster from dendritic spines (Figure S2a and b). In parallel, the immunoreactivity of CaMKIIβ were decreased in dendritic spines but not in dendritic shafts (Figure S2c–e). In contrast, drebrin immunofluorescence intensity was unchanged in CaMKIIβ‐KD neurons (Fig. 3g–i). These results suggest that drebrin contributes to the localization of CaMKIIβ in dendritic spines, but not vice versa.
Figure 3.
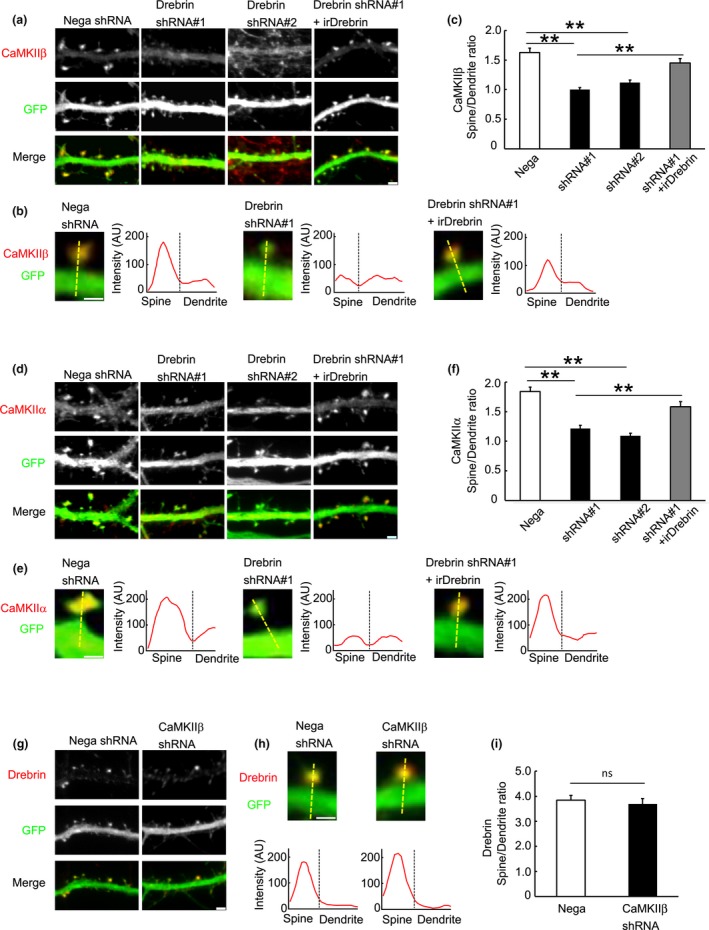
Spine localization of CaMKIIβ depends on drebrin. (a) Cultured neurons were transfected with negative shRNA (control), drebrin shRNA#1, drebrin shRNA#2, or drebrin shRNA#1 + RNAi‐resistant drebrin (irDrebrin). Immunofluorescence intensities of CaMKIIβ were measured in spines and parent dendrites. Scale bars, 2 μm. (b) Enlarged images of the dendritic spine of each condition. Dashed yellow lines indicate the positions of the line scan graph. They show CaMKIIβ pixel intensities across the spine and dendrite. Scale bar, 1 μm. (c) Quantifications of the intensity ratio (spine:dendrite) of CaMKIIβ. Data are presented as means ± SEM (n = 26 neurons for negative shRNA, n = 56 neurons for drebrin shRNA#1, n = 26 neurons for drebrin shRNA#2, and n = 36 neurons for drebrin shRNA#1+irDrebrin, **p < 0.01, one‐way anova with Tukey–Kramer post hoc tests). (d–f) Immunofluorescence intensities of CaMKIIα were also measured in drebrin‐KD neurons. (e) The line scans show CaMKIIα pixel intensities across the spine and dendrite. Scale bar, 1 μm. (f) Quantifications of the intensity ratio of CaMKIIα. Data are presented as means ± SEM (n = 25 neurons for negative shRNA, n = 22 neurons for drebrin shRNA#1, n = 25 neurons for drebrin shRNA#2, and n = 20 neurons for drebrinshRNA#1+irDrebrin, **p < 0.01, one‐way anova with Tukey–Kramer post hoc tests). (g) Cultured neurons were transfected with negative shRNA or CaMKIIβ shRNA. Drebrin immunoreactivities in spines of CaMKIIβ‐KD neurons were similar to those in control neurons. Scale bar, 2 μm. (h) The line scans show drebrin pixel intensities across the spine and dendrite. Scale bar, 1 μm. (i) Quantifications of the intensity ratio of drebrin. Data are presented as means ± SEM (n = 13 neurons for negative shRNA and n = 16 neurons for CaMKIIβ shRNA, Welch's t‐test).
Drebrin knockdown increases the stable fraction of CaMKIIβ
We analyzed the drebrin‐dependence of CaMKIIβ stability in dendritic spines by FRAP analysis of individual dendritic spines (Fig. 4a). As a control for this analysis, we imaged GFP FRAP from neurons transfected with GFP only (Figure S3a). The fluorescence of GFP in dendritic spines recovered rapidly after photobleaching to nearly the pre‐bleaching level (Figure S3b), indicating that this protocol hardly damaged dendritic spines. We determined the fluorescence intensities of GFP‐CaMKIIβ before and after photobleaching and quantified the stable fraction (Fig. 4b and c). Unexpectedly, the stable fraction of GFP‐CaMKIIβ was greater in drebrin‐KD compared with control neurons, and this increase was canceled by cotransfection with RNAi‐resistant drebrin (Fig. 4c). Since both drebrin and CaMKIIβ are F‐actin binding proteins, it raises a possibility that the changes in CaMKIIβ observed in drebrin‐KD neurons are because of changes in F‐actin. To test the specificity of drebrin‐KD, we analyzed a non‐F‐actin binding CaMKIIβ isoform CaMKIIβe in drebrin‐KD neurons. We first confirmed the interaction between CaMKIIβe and drebrin by an immunoprecipitation (Fig. 5a). This indicates that CaMKIIβ binds to drebrin, but not through F‐actin. In drebrin‐KD neurons, the spine accumulation of Myc‐CaMKIIβe was weak compared with that in control neurons (Fig. 5b and c). Indeed, the spine:dendrite ratio of Myc‐CaMKIIβe was decreased in drebrin‐KD neurons (Fig. 5d). The FRAP analysis of GFP‐CaMKIIβe showed similar data to those obtained from GFP‐CaMKIIβ (Fig. 5e–g). These results suggest that the CaMKIIβ changes in drebrin‐KD neurons depend on specific interaction between CaMKIIβ and drebrin.
Figure 4.
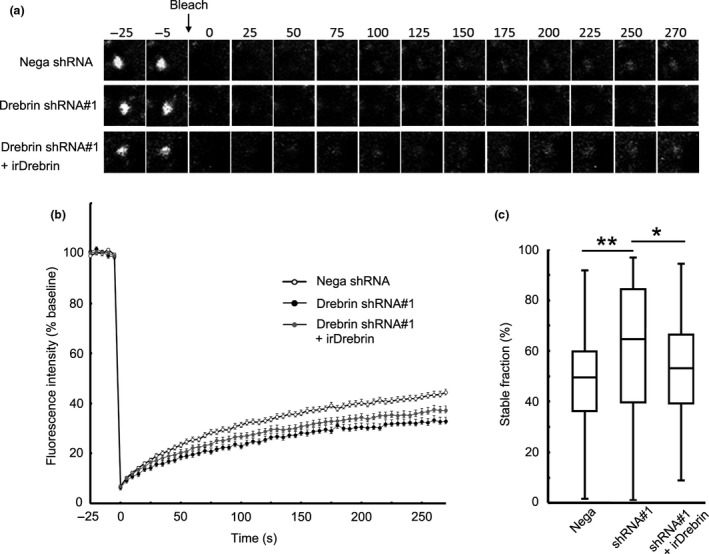
Ratio of stable CaMKIIβ is increased in drebrin‐KD neurons. (a) Cultured neurons were transfected with GFP‐CaMKIIβ + negative shRNA, drebrin shRNA#1, or drebrin shRNA#1 + RNAi‐resistant drebrin (irDrebrin). Live confocal images of GFP‐CaMKIIβ were captured every 5 s for 300 s. Photobleaching was performed at the time point before 0 s. (b) FRAP curves of GFP‐CaMKIIβ in each condition. (c) Quantification of the stable fractions. Box plots show the median, interquartiles, and range (n = 149 spines for negative shRNA, n = 134 spines for drebrin shRNA#1, and n = 126 spines for drebrin shRNA#1+ irDrebrin, *p < 0.05, **p < 0.01, one‐way anova with Steel–Dwass post hoc test).
Figure 5.
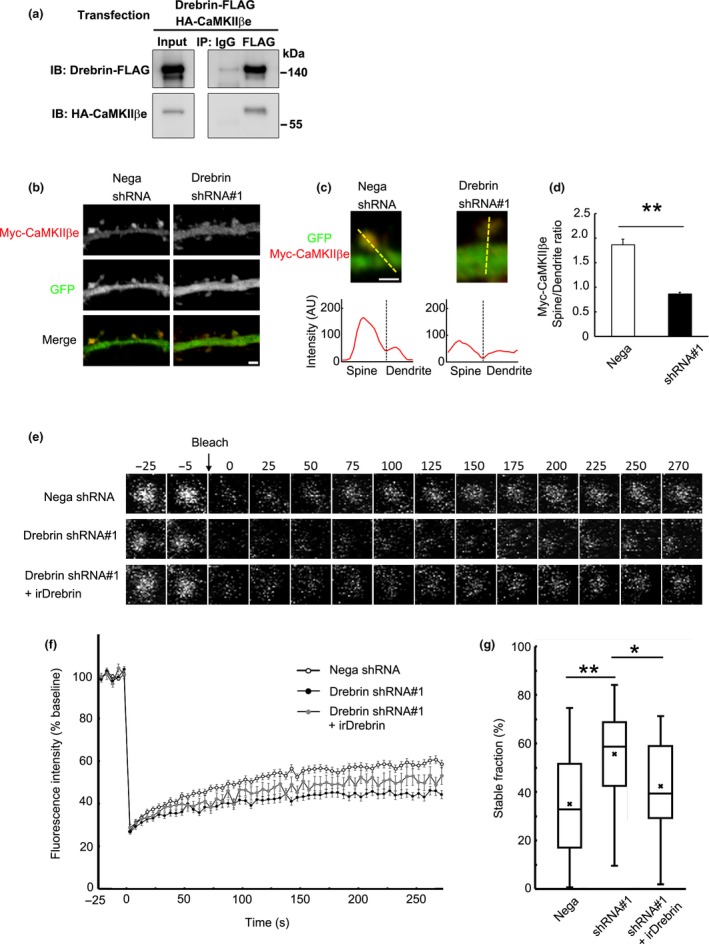
Spine localization of non‐F‐actin binding isoform CaMKIIβe depends on drebrin. (a) Coimmunoprecipitation between CaMKIIβe and drebrin. HEK293 cells were transfected with Drebrin‐FLAG and HA‐CaMKIIβe. Cell lysates were immunoprecipitated with anti‐FLAG antibody. (b) Cultured neurons were transfected with Myc‐CaMKIIβe + negative shRNA (control) or drebrin shRNA#1. Immunofluorescence intensities of Myc‐CaMKIIβe were measured in spines and parent dendrites. Scale bars, 2 μm. (c) Enlarged images of the dendritic spine of control and drebrin‐KD neurons. The line scans show Myc‐CaMKIIβe pixel intensities across the spine and dendrite. Scale bar, 1 μm. (d) Quantifications of the intensity ratio (spine:dendrite) of CaMKIIβe. Data are presented as means ± SEM (n = 24 neurons for negative shRNA, and n = 29 neurons for drebrin shRNA#1, **p < 0.01, Welch's t‐test). (e) FRAP analysis of GFP‐CaMKIIβe. Cultured neurons were transfected with GFP‐CaMKIIβe + negative shRNA, drebrin shRNA#1, or drebrin shRNA#1 + RNAi‐resistant drebrin (irDrebrin). Live confocal images of GFP‐CaMKIIβe were captured every 5 s for 300 s. (f) FRAP curves of GFP‐CaMKIIβe in each condition. (g) Quantification of the stable fractions. Box plots show the mean, median, interquartiles, and range (n = 80 spines for negative shRNA, n = 34 spines for drebrin shRNA#1, and n = 22 spines for drebrin shRNA#1+ irDrebrin, *p < 0.05, **p < 0.01, one‐way anova with Steel–Dwass post hoc test).
Our FRAP data indicate that CaMKIIβ was more stable in dendritic spines in the absence of drebrin, suggesting the existence of a pool of more stable CaMKIIβ, in addition to drebrin‐binding CaMKIIβ. To validate this hypothesis, we focused on the fact that NMDAR activation induces CaMKIIβ accumulation (Shen and Meyer 1999) and drebrin exodus (Sekino et al. 2006; Mizui et al. 2014) in parallel. Similar to a previous study (Shen and Meyer 1999), in control neurons, NMDAR activation with 50 μM glutamate with 10 μM glycine (Glu/Gly) for 10 min enhanced spine localization of CaMKIIβ (Fig. 6a–c) and decreased spine localization of drebrin (Fig. 6d–f). In drebrin‐KD neurons, NMDAR activation similarly increased CaMKIIβ intensity in spines (Fig. 6c, right column). These indicate that the activity‐dependent accumulation of CaMKIIβ into spines is not dependent on drebrin.
Figure 6.
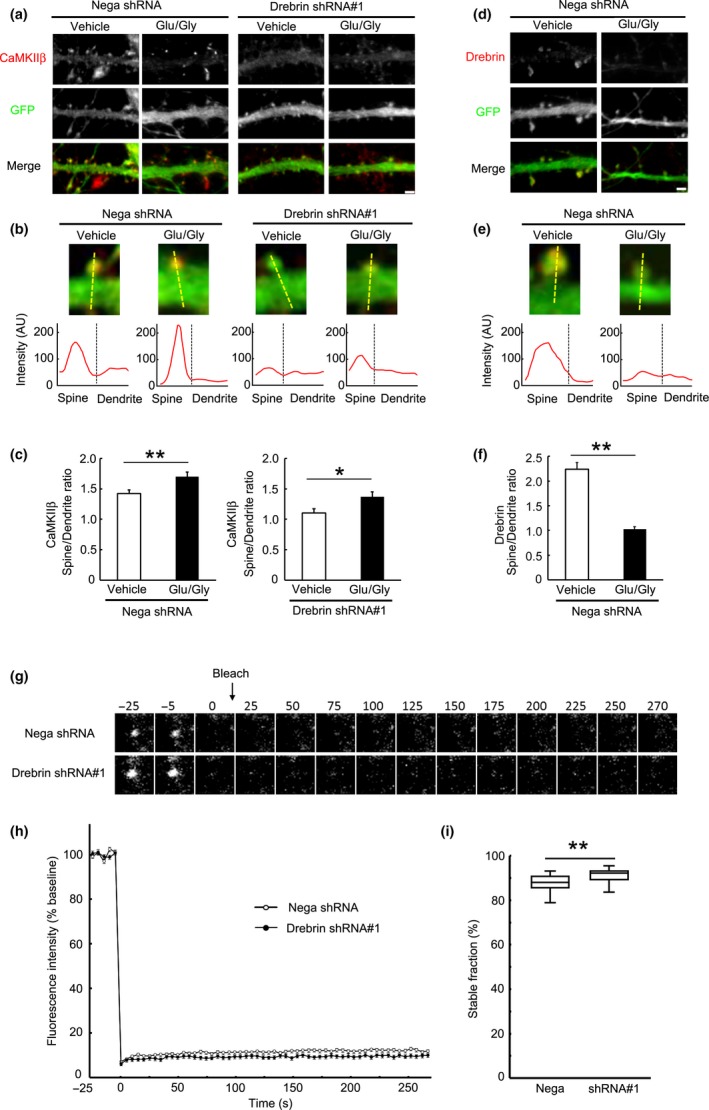
CaMKIIβ is stably localized in dendritic spines after NMDAR activation. (a) Activity‐induced CaMKIIβ localization in spines does not depend on drebrin. Cultured neurons were transfected with negative shRNA or drebrin shRNA#1. Neurons were treated with Glu (50 μM)/Gly (10 μM) or vehicle for 10 min. Scale bars, 2 μm. (b) Enlarged images of the dendritic spine of each condition. The line scans show CaMKIIβ pixel intensities across the spine and dendrite. Scale bar, 1 μm. (c) Quantification of the intensity ratio of CaMKIIβ. Data are presented as means ± SEM (n = 14 neurons for negative shRNA/vehicle, n = 23 neurons for negative shRNA/Glu/Gly, n = 18 neurons for drebrin shRNA#1/vehicle, and n = 18 neurons for drebrin shRNA#1/Glu/Gly, *p < 0.05, **p < 0.01, Welch's t‐test). (d) The spine localization of drebrin was decreased by Glu/Gly treatment. (e) Enlarged images of the dendritic spine of each condition. The line scans show drebrin pixel intensities across the spine and dendrite. Scale bar, 1 μm. (f) Quantification of the intensity ratio of drebrin. Data are presented as means ± SEM (n = 22 neurons for vehicle, n = 29 neurons for Glu/Gly, **p < 0.01, Welch's t‐test). (g) FRAP analysis of GFP‐CaMKIIβ in Glu/Gly‐treated neurons. Cultured neurons were transfected with GFP‐CaMKIIβ + negative shRNA or drebrin shRNA#1, and then treated with Glu/Gly followed by FRAP imaging. (h) FRAP curves of GFP‐CaMKIIβ. NMDAR activation immobilized CaMKIIβ in dendritic spines in each condition. However, the stable fraction of CaMKIIβ was slightly larger in drebrin‐KD neurons. (i) Quantification of the stable fractions. Box plots show the median, interquartiles, and range (n = 67 spines for negative shRNA and n = 79 spines for drebrin shRNA#1, **p < 0.01, Mann–Whitney U‐test).
We further analyzed the stable fraction of synaptic CaMKIIβ accumulated by NMDAR activation. Cultured neurons expressing GFP‐CaMKIIβ were treated with Glu/Gly, followed by washing for 10 min, photobleaching, and imaging (Fig. 6g). FRAP analysis showed that NMDAR activation markedly increased the apparent stable fraction of CaMKIIβ in control neurons (Fig. 6h and i) compared with non‐activated neurons (Fig. 4c). Interestingly, the stable fraction after NMDAR activation was slightly but significantly larger in drebrin‐KD neurons than control neurons (Fig. 6h and i).
Collectively these indicate that NMDAR activation results in the accumulation of drebrin‐independent CaMKIIβ, which forms a large stable fraction in dendritic spines. This is consistent with an increase in the stable fraction of CaMKIIβ in drebrin‐KD neurons in the resting state (Fig. 4c), despite low levels of CaMKIIβ in dendritic spines (Fig. 3a–c). Furthermore, it is suggested that drebrin‐dependent and drebrin‐independent pools of CaMKIIβ are present in dendritic spines, and that synaptic activity regulates the accumulation of drebrin‐independent CaMKIIβ.
Discussion
This study provides the first evidence to show that CaMKIIβ interacts with drebrin in dendritic spines. Drebrin‐silencing caused diffuse localization of CaMKIIβ in cultured neurons, suggesting that drebrin acts as a scaffold for CaMKIIβ in dendritic spines. We propose that drebrin‐dependent stable CaMKIIβ is localized in the central area of the spines, because drebrin levels are low in the PSD area compared with the cytoplasmic area (Kobayashi et al. 2007). CaMKIIα localization in the spines was also impaired in drebrin‐KD neurons although CaMKIIα does not bind to drebrin. The mislocalization of CaMKIIα is likely mediated by a decrease in drebrin‐dependent CaMKIIβ, because CaMKIIβ knockout causes mislocalization of CaMKIIα in cultured neurons (Borgesius et al. 2011). F‐actin disruption by latrunculin A treatment also caused diffuse localization of CaMKIIβ. Since cluster like drebrin‐immunoreactivity was almost abolished in the latrunculin A treated neurons, we consider that the CaMKIIβ decrease in dendritic spines is caused by disappearance of drebrin cluster. Interestingly, the latrunculin A treatment did not decrease the immunofluorescence intensity of CaMKIIβ in dendritic shafts. This suggests that CaMKIIβ localization in dendritic shafts is not dependent on actin cytoskeleton.
Both drebrin (Ishikawa et al. 1994) and CaMKIIβ (Shen et al. 1998; Okamoto et al. 2007) bind to F‐actin, raising the possibility that these molecules interact with each other via F‐actin. Given that one drebrin molecule binds stoichiometrically to five actin molecules (Ishikawa et al. 1994), more actin than drebrin would thus be expected in the immunoprecipitated complex; however, the present immunoprecipitation experiments showed very little actin in the immunoprecipitate, indicating that CaMKIIβ directly interacts with drebrin, rather than indirectly via F‐actin. Moreover, our immunoprecipitation data indicate that CaMKIIβe, a non‐F‐actin binding isoform, can interact with drebrin.
CaMKIIβ binds to F‐actin, resulting in the stabilization of actin cytoskeleton structure in dendritic spines via preventing the interaction between F‐actin and other actin modulating proteins such as cofilin, gelsolin, and Arp2/3 (Kim et al. 2015). The binding of drebrin to F‐actin also stabilizes actin cytoskeleton structure by preventing the interaction between F‐actin and other actin modulating proteins, such as cofilin, fascin, and α‐actinin. (Ishikawa et al. 1994; Sasaki et al. 1996; Grintsevich and Reisler 2014). This study shows that the N‐terminal region of drebrin interacts with the kinase domain of CaMKIIβ. These regions do not overlap with the F‐actin binding regions of both proteins. This raises a possibility that a tripartite interaction among drebrin, CaMKIIβ, and F‐actin contributes to the stabilization of actin cytoskeletons. Thus, we propose that the binding of drebrin‐CaMKIIβ complex to actin cytoskeleton plays a role in the stabilization of dendritic spine under basal condition.
Drebrin has several conserved consensus sites for phosphorylation in its amino acid sequence (Kojima et al. 1993) and is significantly enriched in phosphoaffinity column eluates (Chew et al. 2005). Mass spectrometry‐based analysis has shown that amino acid residues 140–147 of drebrin are phosphorylated in vivo, and one serine residue (Ser142) is a potential CaMKII phosphorylation site (Chew et al. 2005). Given that the N‐terminal sequence of drebrin (amino acids 1–233) used in the present yeast two‐hybrid experiment contained this putative phosphorylation site, CaMKIIβ may phosphorylate drebrin at Ser142 after association of these two proteins. Actually, our data showed that drebrin binds to kinase domain of CaMKIIβ. Meanwhile, the Ser142 is identified as a phosphorylation site of CDK5 (Worth et al. 2013; Tanabe et al. 2014). The modification of drebrin Ser142 might thus be regulated temporally and spatially by a number of kinase including CaMKII and CDK5.
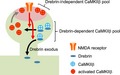
CaMKIIβ is necessary for maintaining spine structure by bundling of F‐actin (Okamoto et al. 2007). As is the case with CaMKIIβ, drebrin is involved in morphological changes in the dendritic spines (Hayashi and Shirao 1999; Mizui et al. 2005; Takahashi et al. 2006). Drebrin translocates from the spines to the parent dendrite following a Ca2+ influx through NMDARs, leading to morphological changes (Sekino et al. 2006; Mizui et al. 2014). This study demonstrated that, while the interaction between drebrin and CaMKIIβ was maintained during the resting state in dendritic spines, the interaction was broken by robust Ca2+ influx from NMDARs, leading to CaMKIIβ release from drebrin and accumulation with PSD, where activated CaMKIIβ functions as a protein kinase. This is coordinated with previous studies showing that CaMKIIβ activation led to a loss of actin‐binding activity (O'Leary et al. 2006; Okamoto et al. 2007; Kim et al. 2015). The interaction between drebrin and CaMKIIβ, which may be regulated by Ca2+ influx via NMDARs, may thus play a pivotal role in morphological synaptic plasticity. Our findings suggest a three‐pool model of CaMKIIβ in dendritic spines (Fig. 7). The first pool includes CaMKIIβ associated with drebrin, which contributes to CaMKIIβ accumulation in dendritic spines during the resting state, but disappears in parallel with the NMDAR‐mediated drebrin exodus in the activated state (Mizui et al. 2014). In the first pool, CaMKIIβ mainly functions as a structural, and can be under standby condition for activation by Ca2+ influx. We suppose that this pool contributes immediate PSD‐translocation for CaMKIIβ. The second pool includes CaMKIIβ associated with PSD, which forms the major population in dendritic spines during the activated state. FRAP analysis showed that this pool contributes strongly to the increase in the stable fraction of CaMKIIβ induced by NMDAR activation, consistent with a recent study showing that CaMKII activated by NMDAR activation is localized at the PSD (Lu et al. 2014). In the second pool, CaMKIIβ is separated from drebrin and would be autophosphorylated. It may function as a protein kinase for several PSD proteins, and is localized in dendritic spines more stable than first pool CaMKIIβ. The third pool includes CaMKIIβ free from both drebrin and PSD, but which probably binds to F‐actin. Because CaMKIIβ is not accumulated in dendritic spines of drebrin‐KD neurons, this pool may not contribute to the accumulation of CaMKIIβ in dendritic spines. CaMKIIβ in the third pool would function as a structural protein like the first pool.
Figure 7.
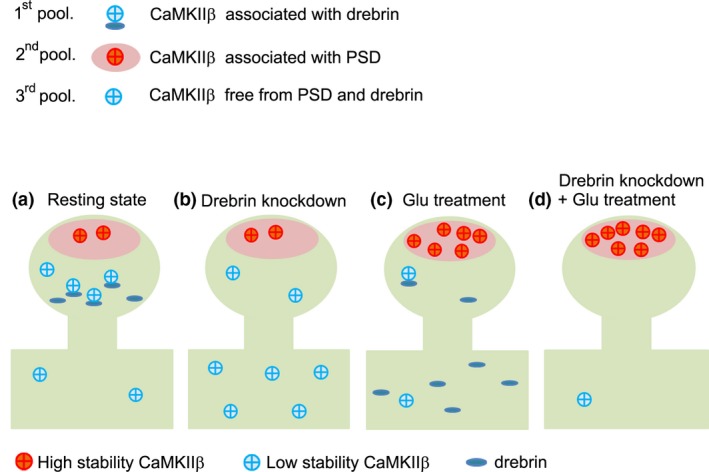
Three‐pool model of CaMKIIβ in dendritic spines. CaMKIIβ is classified into three distinct pools: CaMKIIβ associated with drebrin, CaMKIIβ associated with post‐synaptic density (PSD), and CaMKIIβ free from PSD and drebrin (probably associated with F‐actin). (a) In the resting state, CaMKIIβ associated with drebrin predominates in dendritic spines, and maintains the spine structure by cooperating with drebrin in the core of the spines. (b) CaMKIIβ does not accumulate in dendritic spines in drebrin‐knockdown neurons because of the lack of drebrin in dendrites. Interestingly, the relative increase in CaMKIIβ associated with PSD, caused by a decrease in drebrin‐associated CaMKIIβ following drebrin knockdown, means that the stable fraction is larger than the control, even though CaMKIIβ is not accumulated in dendritic spines. (c) Glutamate stimulation activates CaMKIIβ, resulting in dissociation of CaMKIIβ from drebrin in parallel with drebrin exodus and association of CaMKIIβ with PSD. CaMKIIβ associated with PSD consequently predominates in the activated state in the dendritic spine. Stability of CaMKIIβ in this pool contributes the increase in the stable fraction of CaMKIIβ in the activated state. (d) Glutamate stimulation in drebrin‐knockdown neurons increases second pool of CaMKIIβ more than glutamate stimulation in normal neurons.
In conclusion, we report the relationship between drebrin and CaMKIIβ in dendritic spines. CaMKIIβ binds to drebrin, and the protein complex is anchored on actin cytoskeleton in core region of a dendritic spine. This interaction is regulated by NMDAR activation. When NMDAR is activated, CaMKIIβ is released from drebrin‐F‐actin complex, which consequently accumulates at PSD.
Supporting information
Figure S1. The immunofluorescence intensity of CaMKIIβ was decreased in dendritic spines but not in dendritic shafts in drebrin‐KD neurons.
Figure S2. F‐actin disruption decreased drebrin and CaMKIIβ from dendritic spines.
Figure S3. FRAP analysis of GFP in dendritic spines.
Acknowledgments and conflict of interest disclosure
This work was partly supported by JSPS KAKENHI Grant Number 26430063 to HY, 17H06312 to HB, 19200029 and 15K14344 to TS, and AMED under Grant Number JP17bk0104077. Authors have no conflict of interest related to this research.
All experiments were conducted in compliance with the ARRIVE guidelines.
References
- Bennett M. K., Erondu N. E. and Kennedy M. B. (1983) Purification and characterization of a calmodulin‐dependent protein kinase that is highly concentrated in brain. J. Biol. Chem. 258, 12735–12744. [PubMed] [Google Scholar]
- Borgesius N. Z., van Woerden G. M., Buitendijk G. H., Keijzer N., Jaarsma D., Hoogenraad C. C. and Elgersma Y. (2011) betaCaMKII plays a nonenzymatic role in hippocampal synaptic plasticity and learning by targeting alphaCaMKII to synapses. J. Neurosci. 31, 10141–10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickey D. A., Colbran R. J., Fong Y. L. and Soderling T. R. (1990) Expression and characterization of the alpha‐subunit of Ca2+/calmodulin‐dependent protein kinase II using the baculovirus expression system. Biochem. Biophys. Res. Commun. 173, 578–584. [DOI] [PubMed] [Google Scholar]
- Chew C. S., Okamoto C. T., Chen X. and Thomas R. (2005) Drebrin E2 is differentially expressed and phosphorylated in parietal cells in the gastric mucosa. Am. J. Physiol. Gastrointest. Liver Physiol. 289, G320–G331. [DOI] [PubMed] [Google Scholar]
- Counts S. E., Nadeem M., Lad S. P., Wuu J. and Mufson E. J. (2006) Differential expression of synaptic proteins in the frontal and temporal cortex of elderly subjects with mild cognitive impairment. J. Neuropathol. Exp. Neurol. 65, 592–601. [DOI] [PubMed] [Google Scholar]
- Erondu N. E. and Kennedy M. B. (1985) Regional distribution of type II Ca2+/calmodulin‐dependent protein kinase in rat brain. J. Neurosci. 5, 3270–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grintsevich E. E. and Reisler E. (2014) Drebrin inhibits cofilin‐induced severing of F‐actin. Cytoskeleton (Hoboken) 71, 472–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harigaya Y., Shoji M., Shirao T. and Hirai S. (1996) Disappearance of actin‐binding protein, drebrin, from hippocampal synapses in Alzheimer's disease. J. Neurosci. Res. 43, 87–92. [DOI] [PubMed] [Google Scholar]
- Hatanpaa K., Isaacs K. R., Shirao T., Brady D. R. and Rapoport S. I. (1999) Loss of proteins regulating synaptic plasticity in normal aging of the human brain and in Alzheimer disease. J. Neuropathol. Exp. Neurol. 58, 637–643. [DOI] [PubMed] [Google Scholar]
- Hayashi K. and Shirao T. (1999) Change in the shape of dendritic spines caused by overexpression of drebrin in cultured cortical neurons. J. Neurosci. 19, 3918–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K., Ishikawa R., Ye L. H., He X. L., Takata K., Kohama K. and Shirao T. (1996) Modulatory role of drebrin on the cytoskeleton within dendritic spines in the rat cerebral cortex. J. Neurosci. 16, 7161–7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa R., Hayashi K., Shirao T., Xue Y., Takagi T., Sasaki Y. and Kohama K. (1994) Drebrin, a development‐associated brain protein from rat embryo, causes the dissociation of tropomyosin from actin filaments. J. Biol. Chem. 269, 29928–29933. [PubMed] [Google Scholar]
- Ishikawa R., Katoh K., Takahashi A., Xie C., Oseki K., Watanabe M., Igarashi M., Nakamura A. and Kohama K. (2007) Drebrin attenuates the interaction between actin and myosin‐V. Biochem. Biophys. Res. Commun. 359, 398–401. [DOI] [PubMed] [Google Scholar]
- Ivanov A., Esclapez M., Pellegrino C., Shirao T. and Ferhat L. (2009) Drebrin A regulates dendritic spine plasticity and synaptic function in mature cultured hippocampal neurons. J. Cell Sci. 122, 524–534. [DOI] [PubMed] [Google Scholar]
- Jaworski J., Kapitein L. C., Gouveia S. M. et al (2009) Dynamic microtubules regulate dendritic spine morphology and synaptic plasticity. Neuron 61, 85–100. [DOI] [PubMed] [Google Scholar]
- Kato K., Shirao T., Yamazaki H., Imamura K. and Sekino Y. (2012) Regulation of AMPA receptor recruitment by the actin binding protein drebrin in cultured hippocampal neurons. JNSNE 1, 153–160. [Google Scholar]
- Kim K., Lakhanpal G., Lu H. E. et al (2015) A temporary gating of actin remodeling DURING synaptic plasticity consists of the interplay between the kinase and structural functions of CaMKII. Neuron 87, 813–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi C., Aoki C., Kojima N., Yamazaki H. and Shirao T. (2007) Drebrin a content correlates with spine head size in the adult mouse cerebral cortex. J. Comp. Neurol. 503, 618–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima N., Shirao T. and Obata K. (1993) Molecular cloning of a developmentally regulated brain protein, chicken drebrin A and its expression by alternative splicing of the drebrin gene. Brain Res. Mol. Brain Res. 19, 101–114. [DOI] [PubMed] [Google Scholar]
- Lu H. E., MacGillavry H. D., Frost N. A. and Blanpied T. A. (2014) Multiple spatial and kinetic subpopulations of CaMKII in spines and dendrites as resolved by single‐molecule tracking PALM. J. Neurosci. 34, 7600–7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammoto A., Sasaki T., Asakura T., Hotta I., Imamura H., Takahashi K., Matsuura Y., Shirao T. and Takai Y. (1998) Interactions of drebrin and gephyrin with profilin. Biochem. Biophys. Res. Commun. 243, 86–89. [DOI] [PubMed] [Google Scholar]
- Mizui T., Takahashi H., Sekino Y. and Shirao T. (2005) Overexpression of drebrin A in immature neurons induces the accumulation of F‐actin and PSD‐95 into dendritic filopodia, and the formation of large abnormal protrusions. Mol. Cell Neurosci. 30, 630–638. [PubMed] [Google Scholar]
- Mizui T., Sekino Y., Yamazaki H., Ishizuka Y., Takahashi H., Kojima N., Kojima M. and Shirao T. (2014) Myosin II ATPase activity mediates the long‐term potentiation‐induced exodus of stable F‐actin bound by drebrin A from dendritic spines. PLoS ONE 9, e85367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K., Narayanan R., Lee S. H., Murata K. and Hayashi Y. (2007) The role of CaMKII as an F‐actin‐bundling protein crucial for maintenance of dendritic spine structure. Proc. Natl Acad. Sci. USA 104, 6418–6423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuno H., Akashi K., Ishii Y. et al (2012) Inverse synaptic tagging of inactive synapses via dynamic interaction of Arc/Arg3.1 with CaMKIIbeta. Cell 149, 886–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary H., Lasda E. and Bayer K. U. (2006) CaMKIIbeta association with the actin cytoskeleton is regulated by alternative splicing. Mol. Biol. Cell 17, 4656–4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P., Cahill M. E., Jones K. A., VanLeeuwen J. E. and Woolfrey K. M. (2011) Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 14, 285–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala C., Piech V., Wilson N. R., Passafaro M., Liu G. and Sheng M. (2001) Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron 31, 115–130. [DOI] [PubMed] [Google Scholar]
- Sasaki Y., Hayashi K., Shirao T., Ishikawa R. and Kohama K. (1996) Inhibition by drebrin of the actin‐bundling activity of brain fascin, a protein localized in filopodia of growth cones. J. Neurochem. 66, 980–988. [DOI] [PubMed] [Google Scholar]
- Schindelin J., Arganda‐Carreras I., Frise E. et al (2012) Fiji: an open‐source platform for biological‐image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiguchi T., Hirose E., Nakashima N., Ii M. and Nishimoto T. (2001) Novel G proteins, Rag C and Rag D, interact with GTP‐binding proteins, Rag A and Rag B. J. Biol. Chem. 276, 7246–7257. [DOI] [PubMed] [Google Scholar]
- Sekino Y., Tanaka S., Hanamura K., Yamazaki H., Sasagawa Y., Xue Y., Hayashi K. and Shirao T. (2006) Activation of N‐methyl‐D‐aspartate receptor induces a shift of drebrin distribution: disappearance from dendritic spines and appearance in dendritic shafts. Mol. Cell Neurosci. 31, 493–504. [DOI] [PubMed] [Google Scholar]
- Sekino Y., Kojima N. and Shirao T. (2007) Role of actin cytoskeleton in dendritic spine morphogenesis. Neurochem. Int. 51, 92–104. [DOI] [PubMed] [Google Scholar]
- Shen K. and Meyer T. (1999) Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science 284, 162–166. [DOI] [PubMed] [Google Scholar]
- Shen K., Teruel M. N., Subramanian K. and Meyer T. (1998) CaMKIIbeta functions as an F‐actin targeting module that localizes CaMKIIalpha/beta heterooligomers to dendritic spines. Neuron 21, 593–606. [DOI] [PubMed] [Google Scholar]
- Shim K. S. and Lubec G. (2002) Drebrin, a dendritic spine protein, is manifold decreased in brains of patients with Alzheimer's disease and Down syndrome. Neurosci. Lett. 324, 209–212. [DOI] [PubMed] [Google Scholar]
- Shiraishi‐Yamaguchi Y., Sato Y., Sakai R., Mizutani A., Knopfel T., Mori N., Mikoshiba K. and Furuichi T. (2009) Interaction of Cupidin/Homer2 with two actin cytoskeletal regulators, Cdc42 small GTPase and Drebrin, in dendritic spines. BMC Neurosci. 10, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirao T. (1995) The roles of microfilament‐associated proteins, drebrins, in brain morphogenesis: a review. J. Biochem. 117, 231–236. [DOI] [PubMed] [Google Scholar]
- Shirao T. and Obata K. (1986) Immunochemical homology of 3 developmentally regulated brain proteins and their developmental change in neuronal distribution. Brain Res. 394, 233–244. [DOI] [PubMed] [Google Scholar]
- Shirao T., Hanamura K., Koganezawa N., Ishizuka Y., Yamazaki H. and Sekino Y. (2017) The role of drebrin in neurons. J. Neurochem. 141, 819–834. [DOI] [PubMed] [Google Scholar]
- Star E. N., Kwiatkowski D. J. and Murthy V. N. (2002) Rapid turnover of actin in dendritic spines and its regulation by activity. Nat. Neurosci. 5, 239–246. [DOI] [PubMed] [Google Scholar]
- Takahashi H., Sekino Y., Tanaka S., Mizui T., Kishi S. and Shirao T. (2003) Drebrin‐dependent actin clustering in dendritic filopodia governs synaptic targeting of postsynaptic density‐95 and dendritic spine morphogenesis. J. Neurosci. 23, 6586–6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H., Mizui T. and Shirao T. (2006) Down‐regulation of drebrin A expression suppresses synaptic targeting of NMDA receptors in developing hippocampal neurones. J. Neurochem. 97(Suppl 1), 110–115. [DOI] [PubMed] [Google Scholar]
- Tanabe K., Yamazaki H., Inaguma Y. et al (2014) Phosphorylation of drebrin by cyclin‐dependent kinase 5 and its role in neuronal migration. PLoS ONE 9, e92291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worth D. C., Daly C. N., Geraldo S., Oozeer F. and Gordon‐Weeks P. R. (2013) Drebrin contains a cryptic F‐actin‐bundling activity regulated by Cdk5 phosphorylation. J. Cell Biol. 202, 793–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki H., Kojima N., Kato K. et al (2014) Spikar, a novel drebrin‐binding protein, regulates the formation and stabilization of dendritic spines. J. Neurochem. 128, 507–522. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The immunofluorescence intensity of CaMKIIβ was decreased in dendritic spines but not in dendritic shafts in drebrin‐KD neurons.
Figure S2. F‐actin disruption decreased drebrin and CaMKIIβ from dendritic spines.
Figure S3. FRAP analysis of GFP in dendritic spines.