

Abstract
Objective
To investigate the efficacy and safety of belimumab, a human immunoglobulin monoclonal antibody against B lymphocyte stimulator, in a subset of patients with systemic lupus erythematosus (SLE) who were hypocomplementemic (C3 <90 mg/dl and/or C4 <10 mg/dl) and anti–double‐stranded DNA (anti‐dsDNA) positive (≥30 IU/ml) at baseline.
Methods
In this phase III, double‐blind, placebo‐controlled study (BEL112341; ClinicalTrials.gov identifier: NCT01484496), patients with moderate to severe SLE (Safety of Estrogens in Lupus Erythematosus National Assessment version of the Systemic Lupus Erythematosus Disease Activity Index [SELENA–SLEDAI] score ≥8) were randomized (2:1) to receive weekly subcutaneous (SC) belimumab 200 mg or placebo, plus standard SLE therapy, for 52 weeks. The primary end point was SLE Responder Index 4 (SRI‐4) response rate at week 52. Secondary end points were time to severe flare and reduction in corticosteroid dose (weeks 40–52). Safety was assessed throughout.
Results
Of the 836 patients in the intent‐to‐treat (ITT) population, 356 were hypocomplementemic and anti‐dsDNA positive at baseline (108 in the placebo group and 248 in the SC belimumab 200 mg group). Compared with placebo, the belimumab group contained more SRI‐4 responders (47.2% versus 64.6%; P = 0.0014), had a lower incidence of severe flare according to the SELENA‐SLEDAI flare index (31.5% versus 14.1%), and had a greater percentage of patients who reduced corticosteroid dosage by ≥25% to ≤7.5 mg/day during weeks 40–52 (11.4% versus 20.7%; P = 0.0844). Adverse events (AEs) were similar between treatment groups.
Conclusion
Our findings indicate that in hypocomplementemic, anti‐dsDNA–positive SLE patients, weekly SC belimumab 200 mg significantly improves SRI‐4 response, decreases severe flare incidence, and reduces corticosteroid use versus placebo; a trend toward greater benefit compared with the overall ITT population was observed. AEs were consistent with the known safety profile of belimumab.
Belimumab is a human immunoglobulin monoclonal antibody against B lymphocyte stimulator 1, 2, a potent B cell survival factor associated with human systemic lupus erythematosus (SLE) 3, 4, 5. The safety and efficacy of intravenous (IV) belimumab 10 mg/kg plus standard SLE therapy in patients with SLE have been demonstrated in 2 large phase III trials (the Study of Belimumab in Subjects with SLE 52‐week trial [BLISS‐52; ClinicalTrials.gov identifier: NCT00424476] and 76‐week trial [BLISS‐76; ClinicalTrials.gov identifier: NCT00410384]) 6, 7. Following these studies, IV administration of belimumab was approved by the US Food and Drug Administration and the European Medicines Agency for the treatment of patients with active, autoantibody‐positive SLE who are receiving standard therapy, including corticosteroids, antimalarials, immunosuppressive drugs, and nonsteroidal antiinflammatory drugs 8, 9. The effectiveness and safety of belimumab were subsequently reaffirmed in uncontrolled studies carried out in a clinical practice setting 10, 11.
The development of a novel liquid formulation of belimumab, along with a prefilled syringe and autoinjector device, enables patients to self‐administer subcutaneous (SC) belimumab, an option demonstrated as preferable to IV administration by the majority of individuals 12. Single‐ and multi‐dose studies of self‐administered SC belimumab 200 mg have demonstrated that both the prefilled syringe and autoinjector devices show good usability, reliability, and safety 13, 14. Furthermore, administration of weekly SC belimumab 200 mg achieved a target belimumab steady‐state exposure similar to that obtained with IV belimumab 10 mg/kg every 4 weeks 15, 16.
Post hoc analyses of the BLISS IV studies demonstrated that patients with SLE who had low complement (C3 or C4) levels and anti–double‐stranded DNA (anti‐dsDNA) positivity at baseline demonstrated greater benefits in response to IV belimumab treatment versus placebo than patients without these characteristics 17. The identification of factors able to predict patient response to belimumab may represent a first step toward personalized medicine in SLE, matching subsets of patients to the most appropriate therapy. The efficacy, safety, and tolerability of SC belimumab administered via prefilled syringe in patients with active, autoantibody‐positive SLE has been demonstrated (61.4% of the patients receiving belimumab were SLE Responder Index 4 [SRI‐4] responders at week 52 compared with 48.4% of the patients receiving placebo) (odds ratio [OR] 1.68 [95% confidence interval (95% CI) 1.25, 2.25]; P = 0.0006) 18. Here we report a prespecified analysis for a subset of patients who were hypocomplementemic (C3 level <90 mg/dl and/or C4 level <10 mg/dl) and anti‐dsDNA positive (≥30 IU/ml) at baseline.
Patients and Methods
Study design and patients. We conducted a prespecified analysis of a randomized, double‐blind, placebo‐controlled phase III trial (BEL112341; ClinicalTrials.gov identifier: NCT01484496), the study design for which has been described previously 18. Briefly, patients stratified by screening Safety of Estrogens in Lupus Erythematosus National Assessment (SELENA) version of the Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) score (8–9 versus ≥10), baseline complement level (low C3 and/or C4 level versus other), and race (black versus other) were randomized 2:1 to receive weekly SC belimumab 200 mg or placebo administered using a prefilled syringe, in addition to stable standard therapy, for 52 weeks. The first and second SC administrations (week 0 and week 1) were carried out at the study site under supervision. Patients or caregivers were then allowed to administer subsequent doses at home, albeit at the investigator's discretion. Patients recorded the following information in a log book: date, injection site (rotated between the abdomen and thighs), and dose administered.
Patient inclusion and exclusion criteria for the study have been described previously 18. Briefly, inclusion criteria consisted of a SELENA–SLEDAI score of ≥8 at screening and antinuclear antibody and/or anti‐dsDNA positivity. Exclusion criteria included severe lupus kidney disease (proteinuria >6 gm/24 hours or equivalent using spot urine protein‐to‐creatinine ratio, or serum creatinine >2.5 mg/dl) and severe central nervous system (CNS) lupus. All patients provided written informed consent prior to enrollment. The main study site (Clinical Trials Unit, Los Angeles, CA) received Institutional Review Board (IRB) approval from the University of Southern California Health Sciences Campus Institutional Review Board (approval number IRB00002880). The study and all protocols were approved by the relevant IRBs for all 207 study sites (see Supplementary Table 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40511/abstract) and conducted in accordance with the Declaration of Helsinki 2008 19.
Changes in the total dose of systemic steroids were permitted up to week 24, but the total systemic dose must have returned to ~25% or 5 mg over the baseline dose (whichever was higher) by week 24, or the patient was considered to have experienced a treatment failure. Within the 8 weeks before week 52, no new increase over the baseline or week 44 steroid dose (whichever was higher) was allowed. After week 24, an increase of 25% or 5 mg over the baseline dose (whichever was higher) necessary to combat SLE activity was considered a treatment failure.
End points and assessments. The primary end point was the SRI‐4 response rate at week 52 20, a composite index of ≥4‐point reduction in SELENA–SLEDAI score, increase of <0.3 in physician's global assessment of disease activity, and no new British Isles Lupus Assessment Group (BILAG) A organ domain scores or no more than 1 new BILAG B organ domain score at week 52 compared with baseline. End points supporting the primary end point were SRI‐4 and SRI‐5 through SRI‐8 response over time 18.
Key secondary end points included time to first severe flare (SELENA–SLEDAI flare index modified to exclude the single criterion of an increase in SELENA–SLEDAI to >12) 21, 22, 23, reduction in corticosteroid dosage (percentage of patients with mean corticosteroid [prednisone or equivalent] dosage reduced by ≥25% from baseline to ≤7.5 mg/day during weeks 40–52 in patients receiving >7.5 mg/day at baseline). The mean change from baseline in Functional Assessment of Chronic Illness Therapy–Fatigue (FACIT‐Fatigue) score and the percentage of patients with an improved FACIT‐Fatigue score of ≥4 (minimal clinically important difference) from baseline to week 52 were analyzed. Safety evaluations included reporting of adverse events (AEs) and analyzing laboratory parameters.
Data analysis. The prespecified analyses reported here included all patients from the intent‐to‐treat (ITT) population (defined as all patients who were randomized and treated with at least 1 dose of study treatment) who had low C3 levels (<90 mg/dl) and/or low C4 levels (<10 mg/dl) and who were anti‐dsDNA positive (≥30 IU/ml) at baseline. A step‐down sequential testing procedure was used for the primary and key secondary end points to control the overall Type I error rate (the incorrect rejection of a true null hypothesis). The prespecified sequence for assessing statistical significance (2‐sided alpha level of 0.05) was: 1) SRI‐4 response rate at week 52; 2) time to first severe SLE flare; and 3) percentage of patients with a reduction in corticosteroid dosage. End points in the sequence above could only be interpreted as statistically significant if statistical significance was achieved by all prior tests. The proportion of patients with an SRI‐4 response at week 52 was compared between treatment groups using a logistic regression model. Analyses of other efficacy end points (all 2‐sided with a significance level of 0.05) were not subject to a multiple comparison procedure. Patients who withdrew or who were deemed to have experienced treatment failure were analyzed as nonresponders. An additional post hoc analysis of SRI‐5 through SRI‐8 response was conducted.
Results
Patient population. Overall, 356 of 836 patients in the ITT population had low C3 levels and/or low C4 levels and were anti‐dsDNA positive at baseline. Of these, 108 patients received placebo and 248 received SC belimumab 200 mg (Figure 1), with a total of 77 patients (71.3%) and 207 patients (83.5%), respectively, completing through week 52 of the study. The majority of the patients were women (98.1% in the placebo group and 95.2% in the belimumab group). The mean age was 34.6 years in both treatment groups, and the mean ± SD SELENA–SLEDAI score was 11.7 ± 3.14 in the placebo group and 11.5 ± 3.31 in the belimumab group (Table 1). The majority of the patients were receiving corticosteroids at baseline (91.7% in the placebo group and 93.1% in the belimumab group) with a mean ± SD daily dosage (among all patients) of 11.4 ± 7.39 mg/day in the placebo group and 12.2 ± 8.34 mg/day in the belimumab group (Table 1).
Figure 1.

Disposition of the patients. ITT = intent‐to‐treat; SC = subcutaneous; anti‐dsDNA = anti–double‐stranded DNA; AEs = adverse events.
Table 1.
Baseline characteristics of the patients with SLE treated with placebo or belimumaba
| Placebo (n = 108) | SC belimumab 200 mg (n = 248) | |
|---|---|---|
| Women, no. (%) | 106 (98.1) | 236 (95.2) |
| Age, mean ± SD years | 34.6 ± 10.38 | 34.6 ± 10.96 |
| Weight, mean ± SD kg | 64.9 ± 17.86 | 64.5 ± 16.30 |
| Enrollment by region, no. (%) | ||
| US | 24 (22.2) | 55 (22.2) |
| Americas excluding US | 21 (19.4) | 52 (21.0) |
| Western Europe/Australia/Israel | 6 (5.6) | 24 (9.7) |
| Eastern Europe | 24 (22.2) | 58 (23.4) |
| Asia | 33 (30.6) | 59 (23.8) |
| Ethnicity, no. (%) | ||
| Hispanic or Latino | 33 (30.6) | 70 (28.2) |
| Not Hispanic or Latino | 75 (69.4) | 178 (71.8) |
| Race, no. (%) | ||
| White | 58 (53.7) | 140 (56.5) |
| Asian | 32 (29.6) | 63 (25.4) |
| African American/African heritage | 7 (6.5) | 26 (10.5) |
| Other | 13 (12.0) | 23 (9.3) |
| Disease duration, median (range) years | 4.0 (0–32) | 5.1 (0–35) |
| SELENA–SLEDAI score, mean ± SDb | 11.7 ± 3.14 | 11.5 ± 3.31 |
| SELENA–SLEDAI score, no. (%)b | ||
| ≤9 | 30 (27.8) | 62 (25.0) |
| ≥10 | 78 (72.2) | 186 (75.0) |
| SELENA–SLEDAI organ involvement, no. (%) | ||
| Mucocutaneous | 91 (84.3) | 205 (82.7) |
| Musculoskeletal | 72 (66.7) | 184 (74.2) |
| Immunologic | 108 (100.0) | 248 (100.0) |
| Renal | 26 (24.1) | 38 (15.3) |
| Hematologic | 13 (12.0) | 23 (9.3) |
| Vascular | 4 (3.7) | 14 (5.6) |
| Cardiovascular and respiratory | 6 (5.6) | 16 (6.5) |
| ≥1 flare, no. (%)c | 24 (22.2) | 48 (19.4) |
| ≥1 severe flare, no. (%)c | 1 (0.9) | 5 (2.0) |
| PGA, mean ± SD | 1.57 ± 0.457 | 1.59 ± 0.434 |
| FACIT‐Fatigue, mean ± SD | 33.4 ± 10.82 | 34.0 ± 11.75 |
| Medications | ||
| Any corticosteroid, no. (%) | 99 (91.7) | 231 (93.1) |
| Corticosteroid dosage, mean ± SD mg/dayd | 11.4 ± 7.39 | 12.2 ± 8.34 |
| Corticosteroid dosage, no. (%) | ||
| 0 mg/day | 9 (8.3) | 17 (6.9) |
| >0 to ≤7.5 mg/day | 29 (26.9) | 67 (27.0) |
| >7.5 mg/day | 70 (64.8) | 164 (66.1) |
| Any antimalarial, no. (%) | 68 (63.0) | 177 (71.4) |
| Any immunosuppressant, no. (%) | 59 (54.6) | 117 (47.2) |
| Azathioprine | 26 (24.1) | 48 (19.4) |
| Cyclosporine | 2 (1.9) | 4 (1.6) |
| Cyclophosphamide | 1 (0.9) | 1 (0.4) |
| Leflunomide | 0 | 1 (0.4) |
| Methotrexate | 12 (11.1) | 24 (9.7) |
| Mizoribine | 4 (3.7) | 6 (2.4) |
| Mycophenolate | 16 (14.8) | 35 (14.1) |
| Tacrolimus | 5 (4.6) | 5 (2.0) |
SLE = systemic lupus erythematosus; SC = subcutaneous; PGA = physician's global assessment of disease activity; FACIT‐Fatigue = Functional Assessment of Chronic Illness Therapy–Fatigue.
Patients were required to have a score of ≥8 on the Safety of Estrogens in Lupus Erythematosus National Assessment (SELENA) version of the Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) at screening (occurring within 35 days prior to baseline).
During the screening period (day −35 to day 0).
Prednisone equivalent.
Primary end point of SRI‐4 response. At week 52, among patients who were hypocomplementemic and anti‐dsDNA positive at baseline, 64.6% of the patients who received SC belimumab 200 mg were SRI‐4 responders compared with 47.2% of those who received placebo (OR 2.23 [95% CI 1.36, 3.64]; P = 0.0014) (Table 2). Among patients who were not hypocomplementemic and anti‐dsDNA positive at baseline, 58.8% of the patients who received SC belimumab 200 mg were SRI‐4 responders compared with 49.1% of those who received placebo (OR 1.46 [95% CI 1.00, 2.14]; P = 0.0488; interaction P value for treatment by subgroup interaction = 0.2393). All components of the SRI‐4 were significantly higher for the SC belimumab 200 mg group than for the placebo group at week 52 (Table 2). The observed treatment difference in the subpopulation studied here (17.4%) was greater than that observed in the overall ITT population (13.0% treatment difference; 61.4% of the patients receiving belimumab were responders versus 48.4% of the patients receiving placebo) (OR 1.68 [95% CI 1.25, 2.25]; P = 0.0006) 18. Exclusion of the anti‐dsDNA and complement components of the SELENA–SLEDAI (post hoc analysis) resulted in SRI‐4 responses at week 52 of 60.6% for the belimumab group and 44.4% for the placebo group (treatment difference 16.1%; OR 2.13 [95% CI 1.30, 3.51]; P = 0.0027).
Table 2.
SRI‐4 response rate and component scores at week 52a
| Placebo (n = 108) | SC belimumab 200 mg (n = 248) | OR (95% CI) | P | |
|---|---|---|---|---|
| SRI‐4 response rate, % | 47.2 | 64.6 | 2.23 (1.36, 3.64) | 0.0014 |
| 4‐point reduction in SELENA–SLEDAI score, % | 47.2 | 65.7 | 2.40 (1.46, 3.92) | 0.0005 |
| No worsening in PGA, % | 68.5 | 82.5 | 2.16 (1.28, 3.65) | 0.0040 |
| No new A or and no more than 1 new BILAG B domain score, % | 70.4 | 81.9 | 1.90 (1.12, 3.21) | 0.0165 |
SRI‐4 = Systemic Lupus Erythematosus Responder Index 4; SC = subcutaneous; OR = odds ratio; 95% CI = 95% confidence interval; SELENA–SLEDAI = Safety of Estrogens in Lupus Erythematosus National Assessment version of the Systemic Lupus Erythematosus Disease Activity Index; PGA = physician's global assessment of disease activity; BILAG = British Isles Lupus Assessment Group.
Significantly higher increases in complement levels from baseline (post hoc analysis) were observed in patients receiving belimumab compared with those receiving placebo (P < 0.05) at weeks 12–20 and 28–52 (for C3 level) and weeks 12–52 (for C4 level) (see Supplementary Figure 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.40511/abstract). In contrast, in both groups, changes from baseline in anti‐dsDNA antibody levels were found not to be significant at any time point (data not shown).
Post hoc analysis revealed durable SRI‐4 response between weeks 44 and 52, for patients who were hypocomplementemic and anti‐dsDNA positive at baseline, to be 56.9% (in the belimumab group) and 36.1% (in the placebo group), with an observed treatment difference of 20.8% (OR 2.50 [95% CI 1.53, 4.08]; P = 0.0002).
Additional efficacy end points. SRI‐4 response over time was significantly greater from week 16 through week 52 in the belimumab group than in the placebo group (Figure 2A). The median time to first SRI‐4 response, which was maintained through week 52, was 225.0 days (interquartile range [IQR] 86.0–not calculable) for belimumab and 364.0 days (IQR 171.0–not calculable) for placebo (hazard ratio [HR] 1.73 [95% CI 1.26, 2.38]; P = 0.0007). Compared with the placebo group, SRI‐5, SRI‐6, SRI‐7, and SRI‐8 responses were also significantly greater in the belimumab group from as early as week 16 (Figure 2B).
Figure 2.
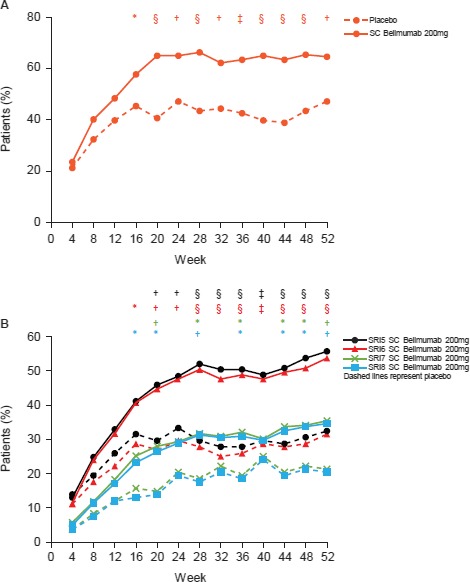
A, Systemic Lupus Erythematosus Responder Index 4 (SRI‐4) response over time in the patients treated with placebo (n = 108) and those treated with subcutaneous (SC) belimumab 200 mg (n = 248). B, Post hoc analysis of SRI‐5 through SRI‐8 response over time in the patients treated with placebo (n = 108) and those treated with SC belimumab 200 mg (n = 246). The definitions of SRI‐5 through SRI‐8 were the same as that of SRI‐4, except with increasingly higher thresholds for Safety of Estrogens in Lupus Erythematosus National Assessment version of the Systemic Lupus Erythematosus Disease Activity Index score reduction (5–8 points). * = P ≤ 0.05; † = P ≤ 0.01; ‡ = P ≤ 0.001; § = P ≤ 0.0001, versus placebo.
Severe flares . The incidence of severe flares was 31.5% in the placebo group and 14.1% in the belimumab group; patients receiving belimumab were 62% less likely to experience a severe flare than those receiving placebo (HR 0.38 [95% CI 0.24, 0.61]; P < 0.0001). The median time to first severe flare, among patients who experienced a severe flare, was 126.5 days (IQR 57.0–243.0) for placebo‐treated patients and 90.0 days (IQR 39.0–204.0) for belimumab‐treated patients (Figure 3).
Figure 3.
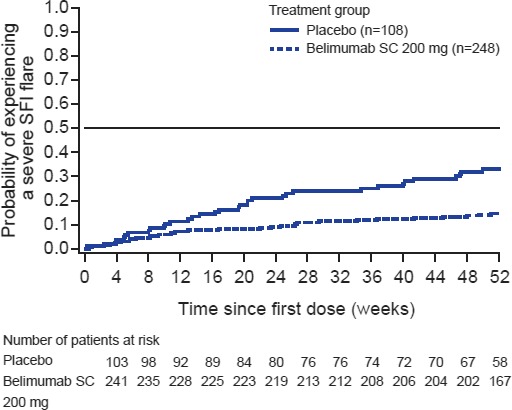
Time to first severe flare according to the Safety of Estrogens in Lupus Erythematosus National Assessment version of the Systemic Lupus Erythematosus Disease Activity Index flare index (SFI) in the placebo group and belimumab group. SC = subcutaneous.
Corticosteroid dosage. Of patients who received a corticosteroid dosage of >7.5 mg/day at baseline (164 [66.1%] of the patients receiving belimumab and 70 [64.8%] of the patients receiving placebo) (Table 1), a greater proportion of those in the belimumab group were able to reduce corticosteroid dosage by ≥25% to ≤7.5 mg/day during weeks 40–52 compared with those in the placebo group, although the difference was not statistically significant (20.7% versus 11.4%) (OR 2.08 [95% CI 0.91, 4.77]; P = 0.0844).
FACIT‐Fatigue. A significantly higher proportion of patients in the belimumab group had an improvement in FACIT‐Fatigue score of ≥4 at week 52 compared with patients in the placebo group (44.8% versus 33.3%, respectively; OR 1.82 [95% CI 1.10, 3.01]; P = 0.0199). The mean change from baseline in FACIT‐Fatigue score at week 52 was 5.4 in the belimumab group and 3.6 in the placebo group (treatment difference 2.1 [95% CI 0.2, 4.1]; P = 0.0324).
Proteinuria. At baseline, 66 (26.6%) of the 248 patients in the belimumab group had high (>0.5 gm/24 hours) proteinuria (mean ± SD 1.47 ± 1.07 gm/24 hours) compared with 31 (28.7%) of the 108 patients in the placebo group (mean ± SD 2.07 ± 1.43 gm/24 hours). At week 52, proteinuria was reduced to ≤0.5 gm/24 hours in 24 (54.5%) of 44 patients in the belimumab group compared with 4 (25.0%) of 16 patients in the placebo group. The percentage change in proteinuria (least squares mean ± SEM), any time after baseline, was 56.1 ± 54.74% for the belimumab group (n = 65 with data available) compared with 184.5 ± 65.03% for the placebo group (n = 30 with data available) (treatment difference −128.4 [95% CI −247.2, −9.76]; P = 0.0344).
Safety . Overall, 88 (81.5%) of the patients in the placebo group and 194 (78.2%) of the patients in the belimumab group experienced a treatment‐emergent AE, 29 (26.9%) and 79 (31.9%) of which, respectively, were considered to be treatment related. Serious AEs were reported for 25 (23.1%) of the patients in the placebo group and 33 (13.3%) of the patients in the belimumab group (Table 3). The most commonly reported AEs were found within the system organ class infections and infestations. The percentages of patients reporting an opportunistic infection or herpes zoster infection, both considered AEs of special interest, were higher in the placebo group than in the SC belimumab 200 mg group (0.9% versus 0.4% and 6.5% versus 2.8%, respectively). The AE of special interest sepsis was reported by 0.9% of patients who received placebo compared with 2.0% of patients who received belimumab (Table 3). Twenty‐one (8.5%) of the patients receiving SC belimumab 200 mg and 13 (12.0%) of the patients receiving placebo reported a post‐injection systemic reaction, none of which were considered serious. Three (2.8%) of the patients receiving placebo and 11 (4.4%) of the patients receiving belimumab experienced depression; none of these episodes were serious. One case of serious suicidal ideation (0.4% of patients in the belimumab group), adjudicated by GlaxoSmithKline physicians, and no cases of suicidal behavior were reported. Two deaths in the placebo group (1 of vascular causes and 1 related to SLE), and 3 deaths in the belimumab group (all due to infections; 1 of bacterial sepsis, 1 of urosepsis, and 1 of tuberculosis of the CNS) were reported (Table 3).
Table 3.
Summary of AEsa
| Placebo (n = 108) | SC belimumab 200 mg (n = 248) | |
|---|---|---|
| Any treatment‐emergent AEb | 88 (81.5) | 194 (78.2) |
| Infections and infestations | 59 (54.6) | 137 (55.2) |
| Gastrointestinal disorders | 28 (25.9) | 56 (22.6) |
| Musculoskeletal and connective tissue disorders | 22 (20.4) | 55 (22.2) |
| Skin and subcutaneous tissue disorders | 22 (20.4) | 45 (18.1) |
| Nervous system disorders | 19 (17.6) | 46 (18.5) |
| Respiratory, thoracic, and mediastinal disorders | 13 (12.0) | 43 (17.3) |
| General disorders and administration site conditions | 14 (13.0) | 35 (14.1) |
| Investigations | 10 (9.3) | 31 (12.5) |
| Injury, poisoning, and procedural complications | 13 (12.0) | 24 (9.7) |
| Blood and lymphatic system disorders | 13 (12.0) | 22 (8.9) |
| Psychiatric disorders | 14 (13.0) | 17 (6.9) |
| Renal and urinary disorders | 13 (12.0) | 18 (7.3) |
| Treatment‐related AEs | 29 (26.9) | 79 (31.9) |
| SAEsc | 25 (23.1) | 33 (13.3) |
| Infections and infestations | 8 (7.4) | 15 (6.0) |
| Renal and urinary disorders | 6 (5.6) | 6 (2.4) |
| Blood and lymphatic system disorders | 4 (3.7) | 3 (1.2) |
| Vascular disorders | 3 (2.8) | 3 (1.2) |
| AEs of special interest | ||
| Malignancies (including nonmelanoma skin cancer) | 1 (0.9) | 0 (0.0) |
| Post‐injection systemic reactionsd | 13 (12.0) | 21 (8.5) |
| Serious delayed non‐acute hypersensitivity reactionse | 0 (0.0) | 0 (0.0) |
| Opportunistic infectionse | 1 (0.9) | 1 (0.4) |
| Herpes zoster | 7 (6.5) | 7 (2.8) |
| Serious | 0 (0.0) | 1 (0.4) |
| Sepsis | 1 (0.9) | 5 (2.0) |
| Serious | 0 (0.0) | 4 (1.6) |
| Depression | 3 (2.8) | 11 (4.4) |
| Serious | 0 (0.0) | 0 (0.0) |
| Serious suicidal ideatione | 0 (0.0) | 1 (0.4) |
| Suicidal behaviore | 0 (0.0) | 0 (0.0) |
| Deaths | 2 (1.9) | 3 (1.2) |
Values are the number (%). SC = subcutaneous.
Adverse events (AEs) by system organ class that occurred in ≥10% of patients in either treatment group are listed.
Serious AEs (SAEs) by system organ class that occurred in >2% of patients in either treatment group are listed.
Defined based on a query for anaphylactic in the Medical Dictionary for Regulatory Activities.
Per adjudication by GlaxoSmithKline.
Discussion
This study evaluated both the efficacy and safety of belimumab in a subset of patients with SLE who were hypocomplementemic and anti‐dsDNA positive at baseline. The results demonstrate that in this SLE subpopulation, the primary end point, SRI‐4 response rate at week 52, was significantly greater among belimumab‐treated patients than placebo‐treated patients (P = 0.0014). The SRI‐4 response rate of 47.2% among patients receiving placebo plus standard therapy is, however, high and likely due to several factors, including administration of standard therapy; an increased chance of receiving active treatment due to the unbalanced randomization schedule (2:1 belimumab:placebo), thereby resulting in a psychological benefit; and the high frequency of visits and patient satisfaction associated with clinical trials 24, 25, 26. A comparable SRI‐4 response (44%) to placebo plus standard therapy was also observed in the BLISS‐52 study, perhaps for similar reasons 7. The greater treatment difference among hypocomplementemic and anti‐dsDNA–positive patients with SLE, compared with the overall ITT population (17.4% versus 13.0%, respectively) 18, can be attributed to both the modestly increased response rate in the belimumab group (64.6% versus 61.4%, respectively) and the modestly decreased response rate in the placebo group (47.2% versus 48.4%, respectively), suggesting that this subpopulation may be more difficult to treat with standard therapies. This subset of patients comprises a large proportion of the overall population and may substantially affect overall outcomes, potentially masking a larger difference from patients who were not hypocomplementemic and dsDNA positive at baseline; however, there is no significant difference in the effect of treatment between these 2 groups (interaction P value 0.2393).
This study and the analyses of the BLISS IV studies have demonstrated that irrespective of the formulation used, belimumab is effective in patients with SLE who are hypocomplementemic and anti‐dsDNA positive 17. The SRI‐4 treatment differences (belimumab compared with placebo) for the complete population and subpopulation of the BLISS IV trials were 11.8% and 19.8%, respectively, and are similar to those observed in this study with SC belimumab (13.0% and 17.4%, respectively) 17. This reduction in SRI‐4 response was associated with a trend toward reduced steroid dosage (discussed further below); however, this was not statistically significant owing to a small sample size, less power for such analyses, and no protocol‐mandated steroid taper. The study populations for the IV and SC belimumab studies were similar; however, the present study included patients with a SELENA–SLEDAI score of ≥8 and 43% of the patients were hypocomplementemic and/or anti‐dsDNA positive, whereas the BLISS‐52 and BLISS‐76 studies recruited patients with SELENA–SLEDAI scores of ≥6 and 52% of the patients were hypocomplementemic and/or anti‐dsDNA positive 6, 7, 17. It would be of interest to see whether a greater treatment effect is observed in the subpopulation studied here for patients with a reduced level of disease severity (i.e., with a SELENA–SLEDAI score of <8).
Of the patients who were hypocomplementemic and anti‐dsDNA positive and received corticosteroid dosages of >7.5 mg/day at baseline, 20.7% reduced steroid dosage by ≥25% to ≤7.5 mg/day during weeks 40–52 of belimumab treatment; a slightly lower reduction of 18.2% was seen in the overall ITT population 18. However, among patients receiving belimumab, there was a higher incidence of severe flares in this subgroup compared with the overall ITT population (14.1% and 10.6%, respectively). Nevertheless, the incidence of severe flares was significantly reduced with belimumab compared with placebo in both the complete ITT population and the hypocomplementemic and anti‐dsDNA–positive subpopulation 18. Among patients receiving belimumab, the median time to severe flare was also shorter in this subgroup compared with the overall population (90 days and 171 days, respectively). Furthermore, in the subgroup analysis the median time to severe flare was in fact shorter for the belimumab group compared with the placebo group (126.5 days). Previous studies have demonstrated that the incidence of flares decreases as the duration of belimumab use increases 27; therefore, this finding may be a reflection of later‐occurring flares being prevented in the belimumab group while early flares were unaffected, resulting in a shorter median time to first flare. In placebo‐treated patients, the occurrence of neither early nor late flares was affected; therefore, the median time was greater.
Significant separation from the placebo group was observed as early as week 16 for SRI‐4 in the belimumab group, and median time to first SRI‐4 response maintained through to week 52 was significantly shorter in the belimumab group compared with the placebo group. Furthermore, significant differences from placebo were also observed as early as week 16 in the belimumab group following analysis of the more stringent SRI‐5 through SRI‐8 measures. The proportion of patients with improved FACIT‐Fatigue scores of ≥4 at week 52 was also found to be significantly greater in the belimumab group compared with the placebo group. The mean change from baseline in FACIT‐Fatigue scores was also greater following belimumab treatment (5.4) than placebo treatment (3.6). Such results, along with those of the primary study 18, provide substantial support for the introduction of SC belimumab, especially in patients with SLE who are hypocomplementemic and anti‐dsDNA positive.
For some patients, the requirement of a regular clinic visit to receive belimumab incurs substantial costs and impact on time; a recent survey of patients with SLE found that >50% of those receiving IV belimumab would prefer to self‐administer their treatment at home 28. The introduction of SC belimumab may enable physicians to offer a therapy that would better suit the patient's preferences in a real‐world setting.
The safety of SC belimumab 200 mg plus standard therapy was similar to that of placebo in this population. Furthermore, the incidence of AEs was similar to that in the overall ITT population (78.2% versus 80.8%, respectively) 18, and was consistent with the known safety profile of belimumab.
Some aspects of this study were identified as potential limitations. Within the hypocomplementemic and anti‐dsDNA–positive subset population, only 65.7% of the patients received steroid dosages of >7.5 mg/day at baseline; thus (as in the overall population), this end point was not powered for statistical significance. In addition, this study excluded patients with a SELENA–SLEDAI score of <8, active nephritis, or active CNS disease at screening. Accordingly, the conclusions drawn from this study may not necessarily pertain to these patients. A separate study investigating patients with lupus nephritis is currently ongoing (ClinicalTrials.gov identifier: NCT01639339).
In conclusion, fixed‐dose weekly SC belimumab 200 mg, in a subgroup of patients with SLE who were hypocomplementemic and anti‐dsDNA positive, reduced SLE disease activity and improved patient‐reported levels of fatigue compared with placebo. Furthermore, the reduced likelihood of response of these patients to standard therapy (placebo group), compared with the ITT population, suggests a benefit of belimumab.
Author Contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Doria had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Stohl, Hammer, Bass, Fox, Roth, Gordon.
Acquisition of data
Doria, Stohl, Schwarting, Okada, Scheinberg, Hammer, Groark, Bass, Fox, Roth, Gordon.
Analysis and interpretation of data
Doria, Stohl, Schwarting, Okada, Scheinberg, van Vollenhoven, Hammer, Groark, Bass, Fox, Roth, Gordon.
Role of the Study Sponsor
GlaxoSmithKline (GSK) designed, conducted, and funded this study. GSK contributed to collection, analysis, and interpretation of the data, and supported the authors in the development of the manuscript. All authors, including those employed by GSK, approved the content of the submitted manuscript. GSK is committed to publicly disclosing the results of GSK‐sponsored clinical research that evaluates GSK medicines, and as such was involved in the decision to submit. Medical writing and editorial assistance were provided by Katie White, PhD, and Sam Halliwell, PhD, of Fishawack Indicia Ltd, UK, which was funded by GSK, but they did not contribute to the study design, or acquisition, analysis, or interpretation of data.
Supporting information
ClinicalTrials.gov identifier: NCT01484496.
Supported by GlaxoSmithKline.
Dr. Doria has received consulting fees and/or honoraria from GlaxoSmithKline, Pfizer, AstraZeneca, Celgene, Eli Lilly and Company, Baxalta, Bristol‐Myers Squibb, and Roche (less than $10,000 each). Dr. Stohl has received consulting fees from Akros Pharma, Janssen, and Sanofi (less than $10,000 each) and research support from GlaxoSmithKline and Pfizer. Dr. Schwarting has received speaking fees (less than $10,000) and research support from GlaxoSmithKline. Dr. Okada has received speaking fees and/or honoraria from Ayumi Pharmaceutical, Mitsubishi Tanabe Pharma, Pfizer, and Abbott Japan (less than $10,000 each). Dr. Scheinberg has received consulting fees from Pfizer, GlaxoSmithKline, Epirus, Samsung Bioepis, and Janssen Pharmaceutica Products (less than $10,000 each). Dr. van Vollenhoven has received consulting fees and/or honoraria from AbbVie, Biotest, Bristol‐Myers Squibb, Crescendo, GlaxoSmithKline, Janssen, Eli Lilly and Company, Merck, Pfizer, Roche, UCB, and Vertex (less than $10,000 each) and research grants from AbbVie, Bristol‐Myers Squibb, GlaxoSmithKline, Pfizer, Roche, and UCB. Ms Hammer and Drs. Groark, Bass, Fox, Roth, and Gordon own stock or stock options in GlaxoSmithKline.
References
- 1. Baker KP, Edwards BM, Main SH, Choi GH, Wager RE, Halpern WG, et al. Generation and characterization of LymphoStat‐B, a human monoclonal antibody that antagonizes the bioactivities of B lymphocyte stimulator. Arthritis Rheum 2003;48:3253–65. [DOI] [PubMed] [Google Scholar]
- 2. Benlysta (belimumab) prescribing information. Rockville (MD): GlaxoSmithKline; 2017. [Google Scholar]
- 3. Cheema GS, Roschke V, Hilbert DM, Stohl W. Elevated serum B lymphocyte stimulator levels in patients with systemic immune–based rheumatic diseases. Arthritis Rheum 2001;44:1313–9. [DOI] [PubMed] [Google Scholar]
- 4. Stohl W, Metyas S, Tan SM, Cheema GS, Oamar B, Xu D, et al. B lymphocyte stimulator overexpression in patients with systemic lupus erythematosus: longitudinal observations. Arthritis Rheum 2003;48:3475–86. [DOI] [PubMed] [Google Scholar]
- 5. Zhang J, Roschke V, Baker KP, Wang Z, Alarcón GS, Fessler BJ, et al. Cutting edge: a role for B lymphocyte stimulator in systemic lupus erythematosus. J Immunol 2001;166:6–10. [DOI] [PubMed] [Google Scholar]
- 6. Furie R, Petri M, Zamani O, Cervera R, Wallace DJ, Tegzová D, et al. A phase III, randomized, placebo‐controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum 2011;63:3918–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Navarra SV, Guzman RM, Gallacher AE, Hall S, Levy RA, Jimenez RE, et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo‐controlled, phase 3 trial. Lancet 2011;377:721–31. [DOI] [PubMed] [Google Scholar]
- 8. European Medicines Agency . Summary of product characteristics. URL: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002015/WC500110150.pdf.
- 9. US Food and Drug Administration . URL: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/125370s016lbl.pdf.
- 10. Iaccarino L, Bettio S, Reggia R, Zen M, Frassi M, Andreoli L, et al. Effects of belimumab on flare rate and expected damage progression in patients with active systemic lupus erythematosus. Arthritis Care Res (Hoboken) 2017;69:115–23. [DOI] [PubMed] [Google Scholar]
- 11. Iaccarino L, Andreoli L, Bocci EB, Bortoluzzi A, Ceccarelli F, Conti F, et al. Clinical predictors of response and discontinuation of belimumab in patients with systemic lupus erythematosus in real life setting: results of a large, multicentric, nationwide study. J Autoimmun 2018;86:1–8. [DOI] [PubMed] [Google Scholar]
- 12. Dashiell‐Aje E, Harding G, Pascoe K, DeVries J, Berry P, Ramachandran S. Patient evaluation of satisfaction and outcomes with an autoinjector for self‐administration of subcutaneous belimumab in patients with systemic lupus erythematosus. Patient 2018;11:119–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sheikh SZ, Hammer AE, Fox NL, Groark J, Struemper H, Roth D, et al. Evaluation of a novel autoinjector for subcutaneous self‐administration of belimumab in systemic lupus erythematosus. Int J Clin Pharmacol Ther 2016;54:914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Struemper H, Murtaugh T, Gilbert J, Barton ME, Fire J, Groark J, et al. Relative bioavailability of a single dose of belimumab administered subcutaneously by prefilled syringe or autoinjector in healthy subjects. Clin Pharmacol Drug Dev 2016;5:208–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cai WW, Fiscella M, Chen C, Zhong ZJ, Freimuth WW, Subich DC. Bioavailability, pharmacokinetics, and safety of belimumab administered subcutaneously in healthy subjects. Clin Pharmacol Drug Dev 2013;2:349–57. [DOI] [PubMed] [Google Scholar]
- 16. Yapa SW, Roth D, Gordon D, Struemper H. Comparison of intravenous and subcutaneous exposure supporting dose selection of subcutaneous belimumab SLE phase 3 program. Lupus 2016;25:1448–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Van Vollenhoven RF, Petri MA, Cervera R, Roth DA, Ji BN, Kleoudis CS, et al. Belimumab in the treatment of systemic lupus erythematosus: high disease activity predictors of response. Ann Rheum Dis 2012;71:1343–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stohl W, Schwarting A, Okada M, Scheinberg M, Doria A, Hammer AE, et al. Efficacy and safety of subcutaneous belimumab in systemic lupus erythematosus: a fifty‐two–week randomized, double‐blind, placebo‐controlled study. Arthritis Rheumatol 2017;69:1016–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. World Medical Association . WMA Declaration of Helsinki: ethical principles for medical research involving human subjects. 2008. URL: https://www.wma.net/wp-content/uploads/2016/11/DoH-Oct2008.pdf. [PubMed]
- 20. Furie RA, Petri MA, Wallace DJ, Ginzler EM, Merrill JT, Stohl W, et al. Novel evidence‐based systemic lupus erythematosus responder index. Arthritis Rheum 2009;61:1143–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Buyon JP, Petri MA, Kim MY, Kalunian KC, Grossman J, Hahn BH, et al. The effect of combined estrogen and progesterone hormone replacement therapy on disease activity in systemic lupus erythematosus: a randomized trial. Ann Intern Med 2005;142:953–62. [DOI] [PubMed] [Google Scholar]
- 22. Petri M, Buyon J, Kim M. Classification and definition of major flares in SLE clinical trials. Lupus 1999;8:685–91. [DOI] [PubMed] [Google Scholar]
- 23. Petri M, Kim MY, Kalunian KC, Grossman J, Hahn BH, Sammaritano LR, et al. Combined oral contraceptives in women with systemic lupus erythematosus. N Engl J Med 2005;353:2550–8. [DOI] [PubMed] [Google Scholar]
- 24. Enck P, Klosterhalfen S, Weimer K, Horing B, Zipfel S. The placebo response in clinical trials: more questions than answers. Philos Trans R Soc Lond B Biol Sci 2011;366:1889–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weimer K, Colloca L, Enck P. Age and sex as moderators of the placebo response: an evaluation of systematic reviews and meta‐analyses across medicine. Gerontology 2015;61:97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Weimer K, Colloca L, Enck P. Placebo effects in psychiatry: mediators and moderators. Lancet Psychiatry 2015;2:246–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ginzler EM, Wallace DJ, Merrill JT, Furie RA, Stohl W, Chatham WW, et al. Disease control and safety of belimumab plus standard therapy over 7 years in patients with systemic lupus erythematosus. J Rheumatol 2014;41:300–9. [DOI] [PubMed] [Google Scholar]
- 28. Pascoe K, Lobosco S, Bell D, Hoskin B, Chang DJ, Pobiner B, et al. Patient‐ and physician‐reported satisfaction with systemic lupus erythematosus treatment in US clinical practice. Clin Ther 2016;39:1811–26. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials