

Abstract
Acquired Gitelman's syndrome (GS) associated with Sjögren syndrome (SS) is rare. A 50‐year‐old woman was admitted to our department because of nausea, acratia and sicca complex. Laboratory tests after admission showed renal failure, hypokalaemia, metabolic alkalosis, hypomagnesaemia and hypocalciuria, all of which met the diagnostic criteria for GS. Diagnostic evaluation identified primary SS as the cause of the acquired GS. Light microscopy of the renal tissue from the patient showed severe membranoproliferative glomerunephritis and tubulointerstitial nephritis. Immunohistochemical staining of the renal tissue showed the absence of sodium‐chloride co‐transporter (NCCT) in distal convoluted tubules. Genetic analysis of chromosomal DNA extracted from the patient's peripheral blood showed SLC12A3 gene heterozygous mutation. The reported case was comprehensively analyzed on the basis of the clinical features, and laboratory, pathological and genetic test findings. The patient has achieved a complete remission after meticulous care and appropriate treatment.
Keywords: Gitelman's syndrome, Sjögren syndrome, SLC12A3 mutation, sodium‐chloride cotransporter
Case Report
A 50‐year‐old woman was admitted to our department because of nausea, acratia, dry eyes and dry mouth for 2 months. She previously had been healthy without hypotension and hypertension, except that she began wearing false teeth at the age of 42 years because of rampant dental caries. No diuretics were used before diagnosis.
Laboratory results after admission are indicated in Table 1. Hypokalaemic metabolic alkalosis, hypomagnesaemia and hypocalciuria met the criteria for Gitelman's syndrome (GS). Serum creatinine was 227umol/L. Plasma renin activity and aldosterone level during rest periods were 1.2 μg/L per h and 331.92 pmmol/L respectively. No hypoproteinaemia, dyslipidaemia and thyroid dysfunction were detected.
Table 1.
Laboratory results of the patient on admission and after treatment
| 1 Day before Admission | 3 Month After Treatment | 6 Month After Treatment | 1 Year After Treatment | Normal Range | |
|---|---|---|---|---|---|
| Treatment | Prednisone + CTX | Prednisone + CTX + ARB + Aldactone | Prednisone + CTX + ARB + Aldactone | ARB + Aldactone | |
| Blood | |||||
| Urea nitrogen (mmol/L) | 19.9 | 21.0 | 11.1 | 8.7 | 2.5–6.5 |
| Creatinine (mmol/L) | 227 | 121 | 87 | 90 | 36–125 |
| Sodium (mmol/L) | 145 | 141 | 142 | 144 | 136–145 |
| Potassium (mmol/L) | 2.9 | 2.8 | 4.0 | 3.84 | 3.5–5.3 |
| Chloride (mmol/L) | 107 | 103 | 105 | 106 | 96–108 |
| Calcium (mmol/L) | 2.82 | 2.66 | 2.57 | 2.32 | 2.1–2.6 |
| Phosphate mmol/L) | 1.4 | 1.2 | 0.9 | 1.0 | 1.0–1.6 |
| Magnesium (mmol/L) | 0.7 | 0.8 | 1.1 | 0.9 | 0.8–1.2 |
| pH | 7.45 | 7.48 | 7.38 | 7.40 | 7.35–7.45 |
| Bicarbonate (mmol/L) | 27.4 | 32.9 | 23.2 | 24.5 | 21.4–27.3 |
| Carbon dioxide (mmol/L) | 24.5 | 34.6 | 26.3 | 27.1 | 24–32 |
| C3 (g/L) | 0.55 | 0.95 | 0.91 | 0.93 | 0.79–1.52 |
| C4 (g/L) | 0.22 | 0.18 | 0.23 | 0.21 | 0.16–0.38 |
| Aldosterone level (mmol/L) | ND | 331.92 | ND | ND | 22.7–138.5 |
| Urine | |||||
| Calcium (mmol/24 h) | 0.4 | 0.5 | 0.8 | ND | 2.7–7.5 |
| Potassium (mmol/24 h) | 45 | 43 | 58 | ND | 25–125 |
| 24 h urinary protein(g/24 h) | 0.61 | 0.2 | 0.18 | 0.14 | 0.028–0.141 |
ND, not done.
Immunologic tests showed that antinuclear antibody and double‐stranded DNA antibody were negative, and anti‐Ro /SS‐A antibody was positive. Serum complement C3 and C4 were 0.55 g/L (reference 0.79–1.52 g/L) and 0.24 g/L (reference 0.16–0.38 g/L), respectively. Schirmer's test was positive, showing less than 5 mm of tear flow at 5 min; and tear break‐up time test was positive. A lip biopsy showed lymphocytic infiltration (grade 4) of the minor salivary glands. Based on the clinical and laboratory test results, primary SS was diagnosed according to the classification of the 2015 American College of Rheumatology (ACR) annual meeting.
Light microscopy and immunofluorescence microscopy of kidney biopsies suggested membranoproliferative glomerulonephritis (MPGN) with crescent formation and severe tubulointerstitial nephritis (TIN). Immunohistochemical staining of the renal tissue showed the absence of the sodium‐chloride cotransporter (NCCT) in the distal convoluted tubules, which is consistent with the diagnosis of GS (Fig. 1).
Figure 1.
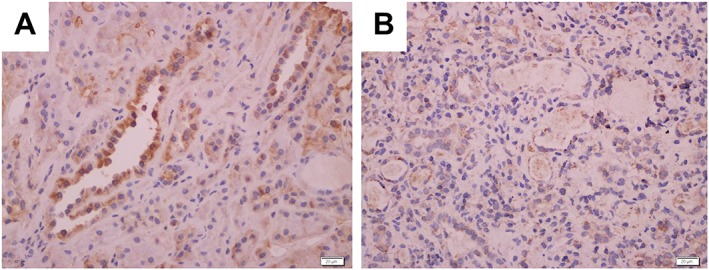
Immunohistochemical staining of the sodium‐chloride cotransporter (NCCT) in (B) renal tubules of the patient compared with (A) control tissue from the kidney of a normal person shows that NCCT in the distal convoluted tubules has less reactivity than control, which is consistent with the diagnosis of Gitelman syndrome.
After obtaining informed consent from the patient, genomic DNA was extracted from peripheral leukocytes for SLC12A3 whole genome sequencing (Jinyu, Shanghai). Surprisingly, the patient has a heterozygous C to A base pair substitution at position 545 in exon4. This mutation causes a Thr to Lys substitution at position 180 (Fig. 2).
Figure 2.
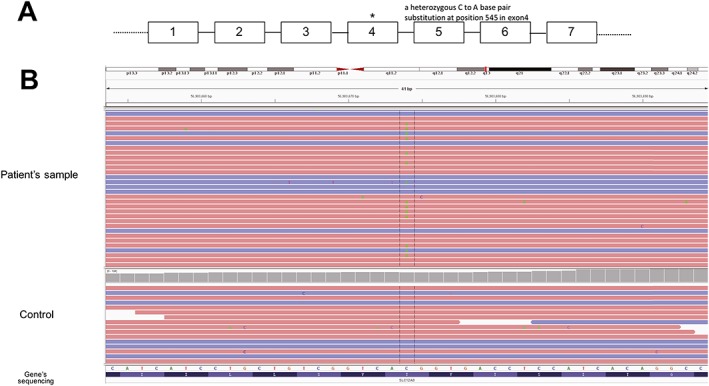
Genome sequencing of all exon regions of SLC12A3. (A) Schematic diagram of the SLC12A3 gene, with some exons (numbered boxes) and introns (lines). (B). Genome sequencing revealed a heterozygous mutation in C545A, which causes a Thr to Lys substitution at position 180. No additional mutation was found.
The patient was treated with prednisone at an initial dose of 1 mg/kg per day orally (which was tapered gradually until discontinuation), and intravenous dripping of cyclophosphamide (CTX) at a dose of 0.8 g per month. The treatment lasted for 12 months. In the first 3 months, serum creatinine, 24‐h urinary protein and immunologic parameters recovered rapidly, but hypokalaemic metabolic alkalosis, hypomagnesaemia and hypocalciuria still existed. After adding potassium chloride, spironolactone and angiotensin II receptor blocker to the therapeutic regimen, and 6‐month continuous treatment of immune‐suppression, the patient's blood gas and serum electrolytes recovered. Interestingly, after the patient stopped taking any medication, the symptoms of dry mouth and dry eye, with hypokalemia, hypercalcaemia and hypocalciuria emerged again, but without albuminuria and renal hypo‐function. This time, the patient was treated with low dose prednisone again. During the follow‐up, the serum electrolytes recovered again.
Discussion
Primary Sjögren syndrome (pSS) is an autoimmune disorder characterized by sicca symptoms (eye and mouth dry) caused by lymphoplasmacytic infiltration of the exocrine (salivary and lachrymal) glands. TIN is the most common renal complication of pSS1. MPGN secondary to cryoglobulinaemia, as a consequence of polyclonal B cell activation, is the second most frequent renal presentation in pSS2. The patient reported herein was confirmed to have pSS by the clinical manifestations, laboratory tests, and lip biopsy. The renal biopsy also showed severe renal involvement, with a combination of TIN and MPGN. Besides, she had unexplained electrolyte disorders. Taking into account the presence of hypokalaemia, metabolic alkalosis, hypomagnesaemia, hypocalciuria and increased aldosterone indicating the activation of the renal‐angiotensin‐aldosterone system, we suspected the patient as having GS.
Gitelman's syndrome is a kidney disorder characterized by hypokalaemic metabolic alkalosis with hypocalciuria and hypomagnesaemia due to loss of function mutation of SLC12A3, which encodes the apical furosemide‐sensitive NCCT located in the distal convoluted tubule3. GS mainly develops with a homozygous mutation or compound heterozygosity in SLC12A3.
A wide variety of conditions such as surreptitious diuretic use, laxative abuse, and cyclic vomiting can induce pseudo‐GS4. Unlike primary GS, pseudo‐GS is reversible when the contributing factors are removed5. As our patient did not have any of the conditions mentioned above, the diagnosis of acquired GS was suspected. pSS is an autoimmune disease that can lead to several acquired renal tubular disorders, including renal tubular acidosis, Bartter syndrome, and Gitelman syndrome6. Acquired GS with autoimmune disease is rare, and only eight cases have been reported in the literature7, 8, 9, 10, 11, 12, 13, 14, of which four cases were associated with pSS. One case was associated with anti‐SSA positive associated immunity. Among them, three cases underwent a genetic analysis, and no mutations in SLC12A3 were found in two of the cases. However, the heterozygous mutation in SLC12A3 was detected in the case associated with anti‐SSA positive related immunity11. It changed 1018 arginine to a stop codon in SLC12A3 and resulted in the elimination of the C‐terminal in the NCCT. The compound heterozygosity of SLC12A3 could not be excluded thoroughly, due to the possible mutations in the intronic regions. The researcher presumed that the heterozygous mutation could be responsible for the latent hypo‐function of NCCT. Interestingly, our genetic analysis of whole genome sequence also showed the patient did have a heterozygous mutation in Thr180Lys which changes the transmembrane region of NCCT. A Japanese group proved that the Thr180Lys mutation was a common mutation in Japanese patients with GS15. Also, it was estimated that the prevalence of heterozygotes based on phenotypic expression is approximately 1% in the Swedish and Italian populations16, 17. Recently, a case report18 described a Japanese patient diagnosed as GS was found compound heterozygous mutation involving Thr180Lys (maternal allele) and Ser615Leu (probably paternal allele) on the NCCT protein, while his parents with heterozygous mutation were healthy. To some extent, this report also confirmed that only heterozygous mutation of 539C > A in SLC12A3 itself might not result in GS. Another study has also indicated that the heterozygous mutation in SLC12A3 had a significant effect on salt homeostasis and blood pressure19. Our report is the second report for an acquired GS with a heterozygous mutation in SLC12A3, indicating heterozygous mutation with SS can cause GS.
To figure out the underlying autoimmune mechanism of SS‐induced acquired Gitelman Syndrome, Kim et al.13 used the immunohistochemical method to show the absence of NCCT expression in the distal convoluted tubules. Interestingly, they also incubated the patient's serum with the normal mouse renal tissue and found that the pattern was similar to that of rabbit polyclonal anti‐NCCT antibody incubated to the normal mouse renal tissue, suggesting that anti‐NCCT antibody may exist in the serum of patients diagnosed with acquired GS and pSS. To their disappointment, they failed to detect the presence of autoantibodies reactive to endogenous kidney protein by immunoblotting method13. Back to our case, we also performed the immunohistochemical staining on the renal tissue of this patient. In contrast to the normal renal tissue, we detected the absence of NCCT expression in the renal tissue of this patient, which is consistent with the findings of previous studies. Our case report may provide additional strong support to acquired GS associated with SS.
Furthermore, steroids in combination with other treatments have proved effective in our patient. The prognosis of this disease seems to be as good as that of the previous case reports. There is also strong evidence that this patient has acquired GS rather than primary GS. This case should also serve to remind clinicians that pSS‐associated GS is underreported as a matter of fact6. Patients who are diagnosed with pSS and present any electrolyte discrepancies should be suspected as having renal involvement. Especially, appropriate testing is required to detect asymptomatic tubulointerstitial involvement to avoid the occurrence of chronic kidney disease2. Also, genetic analysis should be done to exclude primary Gitelman syndrome. The homozygous mutation and compound heterozygosity in SLC12A3 can be attributed to impairment of NCCT and development of GS, while the heterozygous mutation in SLC12A3 might be responsible for the potential hypo‐function of NCCTs.
Acknowledgments
This work was supported by funds from Shanghai Municipal Natural Science Foundation (14ZR1442200 to XG, 14401972702 to YX), and the three year plan of action for the development of traditional Chinese medicine in Shanghai (ZY3‐CCCX‐3‐3009 to YW).
Gu, X. , Su, Z. , Chen, M. , Xu, Y. , and Wang, Y. (2017) Acquired Gitelman syndrome in a primary Sjögren syndrome patient with a SLC12A3 heterozygous mutation: A case report and literature review. Nephrology, 22: 652–655. doi: 10.1111/nep.13045.
The copyright line for this article was changed on 6 August 2018 after original online publication.
Contributor Information
Yanqiu Xu, Email: xuyanqiu6@medmail.com.cn.
Yi Wang, Email: dryiwang@126.com.
References
- 1. Bossini N, Savoldi S, Franceschini F et al. Clinical and morphological features of kidney involvement in primary Sjogren's syndrome. Nephrol. Dial. Transplant. 2001; 16 (12): 2328–36. [DOI] [PubMed] [Google Scholar]
- 2. Francois H, Mariette X. Renal involvement in primary Sjogren syndrome. Nat. Rev. Nephrol. 2016; 12 (2): 82–93. [DOI] [PubMed] [Google Scholar]
- 3. Gitelman HJ, Graham JB, Welt LG. A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans. Assoc. Am. Phys. 1966; 79: 221–35. [PubMed] [Google Scholar]
- 4. Yoshihara M, Sayo A, Mayama M, Oguchi H. Pseudo Gitelman Syndrome Associated With Pregnancy. Obstet. Gynecol. 2015; 126 (4): 877–80. [DOI] [PubMed] [Google Scholar]
- 5. Mount D, Sayegh MH, Singh AK. Core concepts in the disorders of fluid, electrolytes, and acid‐base balance. New York (NY): Springer, 2013. [Google Scholar]
- 6. Kim YK, Song HC, Kim YS, Choi EJ. Acquired Gitelman syndrome. Electrolyte Blood Press. 2009; 7 (1): 5–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen YC, Yang WC, Yang AH, Lin SH, Li HY, Lin CC. Primary Sjogren's syndrome associated with Gitelman's syndrome presenting with muscular paralysis. Am. J. Kidney Dis. 2003; 42 (3): 586–90. [DOI] [PubMed] [Google Scholar]
- 8. Hu DC, Burtner C, Hong A, Lobo PI, Okusa MD. Correction of renal hypertension after kidney transplantation from a donor with Gitelman syndrome. Am. J. Med. Sci. 2006; 331 (2): 105–9. [DOI] [PubMed] [Google Scholar]
- 9. Casatta L, Ferraccioli GF, Bartoli E. Hypokalaemic alkalosis, acquired Gitelman's and Bartter's syndrome in chronic sialoadenitis. Br. J. Rheumatol. 1997; 36 (10): 1125–8. [DOI] [PubMed] [Google Scholar]
- 10. Schwarz C, Barisani T, Bauer E, Druml W. A woman with red eyes and hypokalemia: a case of acquired Gitelman syndrome. Wien. Klin. Wochenschr. 2006; 118 (7‐8): 239–42. [DOI] [PubMed] [Google Scholar]
- 11. Kusuda T, Hosoya T, Mori T et al. Acquired Gitelman Syndrome in an Anti‐SSA Antibody‐positive Patient with a SLC12A3 Heterozygous Mutation. Intern. Med. 2016; 55 (21): 3201–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hinschberger O, Martzolff L, Ioannou G, Baumann D, Jaeger F, Kieffer P. Acquired Gitelman syndrome associated with Sjögren's syndrome and scleroderma. Rev. Med. Interne 2011; 32 (8): e96–8. [DOI] [PubMed] [Google Scholar]
- 13. Kim YK, Song HC, Kim WY et al. Acquired Gitelman syndrome in a patient with primary Sjögren syndrome. Am. J. Kidney Dis. 2008; 52 (6): 1163–7. [DOI] [PubMed] [Google Scholar]
- 14. Bansal R, Ranga VK. Acquired Gitelman's syndrome: an oxymoron? Int. Urol. Nephrol. 2011; 43 (1): 233–6. [DOI] [PubMed] [Google Scholar]
- 15. Monkawa T, Kurihara I, Kobayashi K, Hayashi M, Saruta T. Novel mutations in thiazide‐sensitive Na‐Cl cotransporter gene of patients with Gitelman's syndrome. J. Am. Soc. Nephrol. 2000; 11 (1): 65–70. [DOI] [PubMed] [Google Scholar]
- 16. Simon DB, Nelson‐Williams C, Bia MJ et al. Gitelman's variant of Bartter's syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide‐sensitive Na‐Cl cotransporter. Nat. Genet. 1996; 12 (1): 24–30. [DOI] [PubMed] [Google Scholar]
- 17. Rudin A. Bartter's syndrome. A review of 28 patients followed for 10 years. Acta Med. Scand. 1988; 224 (2): 165–71. [PubMed] [Google Scholar]
- 18. Ueda K, Makita N, Kawarazaki H et al. A novel compound heterozygous mutation of Gitelman's syndrome in Japan, as diagnosed by an extraordinary response of the fractional excretion rate of chloride in the trichlormethiazide loading test. Intern. Med. 2012; 51 (12): 1549–53. [DOI] [PubMed] [Google Scholar]
- 19. Cruz DN, Simon DB, Nelson‐Williams C et al. Mutations in the Na‐Cl cotransporter reduce blood pressure in humans. Hypertension 2001; 37 (6): 1458–64. [DOI] [PubMed] [Google Scholar]